Per emoglobinopatie si intendono quelle condizioni
morbose, caratterizzate da un’alterazione strutturale
ereditaria di una delle catene globiniche: le anomalie
cliniche associate derivano dalle proprietà fisiche
dell’emoglobina abnorme.
Per talassemie si intendono quelle condizioni
morbose, caratterizzate da difetti ereditari della sintesi
(ovverossia una ridotta o assente sintesi) di una o più
delle catene globiniche
Considerate nel loro insieme, esse costituiscono le
malattie monogeniche più comuni nella razza umana.
Esse sono diffuse soprattutto tra le popolazioni
mediterranee, del medio-oriente, del continente
indiano, Indonesia, isole del Pacifico, Tailandia, Cina
meridionale e Africa centrale, ma sono anche comuni
in quei paesi, dove più forte è stata l’immigrazione da
queste aree geografiche.
Epidemiologia:
Numerosissimi lavori epidemiologici, in
mancanza di chiare evidenze sperimentali,
hanno mostrato lo stretto legame tra la
diffusione di questi disordini ereditari e la
malaria, nel senso che i portatori di questi geni
alterati sono più resistenti all’infezione
malarica.
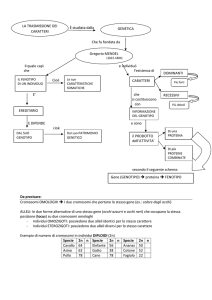
Controllo genetico dell’emoglobina umana
• il fenotipo ematologico delle a-talassemie si
manifesta già alla nascita
gene a2 viene trascritto più del gene a1
(rapporto 3:1)
• il
• il fenotipo ematologico delle b-talassemie e
delle emoglobinopatie b (ad es. HbS, HbC etc.),
si rende evidente solo dopo i primi mesi di vita.
Genetica delle Talassemie
Fisiopatologia delle talassemie
L’emoglobina adulta normale (o HbA) è un tetramero,
costituito da 2 catene a e 2 catene b.
In condizioni normali il rapporto
a/b = 1
Nelle b-talassemie questo rapporto, per la diminuzione del
denominatore, è superiore a 1, e vi sarà pertanto un eccesso di
catene a.
Nelle a-talassemie il rapporto sarà inferiore a 1, per
diminuzione del denominatore e vi sarà pertanto un eccesso
di catene b nella vita adulta o g nella vita fetale.
b-Talassemie
Vengono distinte in due gruppi principali:
• b0 = assenza totale di produzione di catene beta
• b+ = deficit parziale di produzione di catena beta
Rari sono i difetti genetici delezionali, tranne uno (619-bp)
estremamente diffuso nel subcontinente indiano.
Più comuni sono le mutazioni non delezionali o puntiformi
(>200 descritte in letteratura): esse possono interessare la
trascrizione, il processamento o la translazione dell’RNA
messaggero.
E’ importante sottolineare, che poche mutazioni rappresentano
la quasi totalità di quelle identificate in una particolare area
geografica.
a-Talassemie
La genetica delle a-talassemie è più complicata, essendovi 4
geni a, 2 (a2 e a1) per ogni cromosoma 16.
Si parla di a0 talassemia quando nessuna catena a viene
prodotta dal cromosoma interessato, ovverosia entrambi i geni a
sono inattivati.
Si parla di a+ talassemia, quando l’attività di un singolo gene a
è deficitaria: esistono forme da mutazioni delezionali, più
comuni, e non delezionali o puntiformi, più rare.
È importante sottolineare che il fenotipo ematologico è più
severo, se il difetto genetico interessa il gene a2, e se si tratta di
forme non delezionali.
d-Talassemie
Non sono forme importanti da un punto di vista
clinico.
Si distinguono in d0 o d+, a seconda che il difetto
genetico determina assenza o ridotta sintesi di catene
delta.
Sono importanti da conoscere, perché, determinando
una riduzione dei livelli di HbA2, mascherano lo stato
di portatore di b-talassemia.
db-Talassemie
Il difetto genetico consiste nella delezione del gene d e b sullo
stesso cromosoma, lasciando, per lo più, intatto il gene g
adiacente.
Una forma particolare è caratterizzata dalla presenza di una
variante emoglobinica, detta Lepore: essa è costituita da 2
catene a normali e 2 catene non a, costituite dai primi 50-80
residui aminoacidici della catena d e dagli ultimi 60-90 residui
aminoacidici della catena b.
L’emoglobina Lepore è pertanto il prodotto di un gene db da
fusione, che è a sua volta dovuto ad un processo di “crossingover” non omologo.
Questo gene ha una ridotta attività trascrizionale: si spiega così
perché l’emoglobina Lepore rientri tra i difetti talassemici.
Persistenza ereditaria dell’emoglobina fetale
(o HPFH)
Si tratta di difetti genetici, delezionali o non
delezionali, caratterizzati dalla persistenza della
produzione di emoglobina fetale in età adulta, in
assenza di modificazioni importanti del fenotipo
ematologico. La loro importanza risiede nella capacità
modulante del fenotipo clinico, quando sono associati
con altre talassemie o emoglobinopatie.
Clinica delle b -talassemie
b -Talassemia maior (o morbo di Cooley)
È il quadro clinico risultante dalla condizione di
omozigosi o eterozigosi composta per mutazioni btalassemiche severe.
Questi pazienti stanno bene alla nascita; si presentano,
in genere nei primi mesi di vita, con un quadro clinico
ingravescente di pallore, ittero, epato-splenomegalia e
crescita stentata.
Il fenotipo ematologico sarà caratterizzato da:
• anemia microcitica grave
• lieve reticolocitosi
L’osservazione dello striscio periferico mostrerà:
• anisopoichilocitosi severa
• ipocromia
• cellule a bersaglio
• punteggiatura basofila
• presenza costante di eritroblasti
L’elettroforesi dell’emoglobina mostra:
• aumentati livelli di emoglobina fetale
• assenza o livelli molto ridotti di emoglobina adulta
normale A
• livelli di emoglobina A2 bassi, normali o elevati.
Diagnosi
Questo fenotipo ematologico, unitamente allo
studio dei genitori (entrambi portatori di btalassemia) permette, abbastanza agevolmente, di
formulare la diagnosi di talassemia maior.
Terapia
La sopravvivenza e la qualità di vita dei soggetti affetti da
Morbo di Cooley è notevolmente migliorata negli ultimi
decenni.
La terapia di questi pazienti si basa essenzialmente su:
• regolare regime trasfusionale
• somministrazione quotidiana
rimuovere l’eccesso di ferro
di
ferro
chelanti,
per
• prevenzione e/o trattamento delle complicanze a carico dei
vari organi o apparati, soprattutto cuore, fegato, ossa e
ghiandole endocrine.
Trapianto di midollo
L’esperienza accumulata negli ultimi anni ha
dimostrato che il trapianto di midollo da fratello
compatibile rappresenta l’unica opzione terapeutica,
attualmente a disposizione, per una cura definitiva di
questa affezione.
È possibile anche l’utilizzazione del trapianto di midollo
da donatore compatibile non familiare.
Prospettive future
• Terapia genica
Portatori di b-talassemia (o b-talassemia minor)
Sono soggetti, in cui uno solo dei geni b è mutato.
Sono clinicamente sani, tutt’al più, lievemente pallidi e subitterici.
Il fenotipo ematologico è, per lo più, caratterizzato da:
• lieve eritrocitosi
• lieve anemia
• microcitosi e ipocromia
L’esame dello striscio periferico mostra:
- modesta anisopoichilocitosi
- cellule a bersaglio e punteggiatura basofila.
PORTATORI DI b-TALASSEMIA
• portatore “classico”:
Hb A2 =
(3,3%
Hb F = N/
(1%
7%)
5%)
• portatori di db talassemia:
Hb A2 = N/
Hb F =
(5%
15%)
• portatori con Hb A2 normale:
- microcitosi e ipocromia
- coeredità di mutazioni che diminuiscono
la funzione sia del gene b che del gene d; la
mutazione del gene d può essere presente
sullo stesso cromosoma o sul cromosoma
opposto
• portatori silenti:
- assenza o quasi di microcitosi e ipocromia
- livelli di Hb A2 normali o “borderline”
b-Talassemia intermedia
Questi pazienti presentano un quadro clinico, che è più severo di
quello dei portatori di b-talassemia, ma meno grave della btalassemia maior, con una necessità occasionale di emotrasfusioni.
Complicanze:
• emocromatosi, da aumentato assorbimento di ferro
• crescita stentata
• alterazioni scheletriche
• litiasi biliare
• crisi aplastiche ed emolitiche
• ipersplenismo
• ulcere agli arti inferiori, etc
Meccanismi molecolari delle b-talassemie intermedie
Sono essenzialmente tre:
• presenza di mutazioni b-talassemiche lievi: esse interessano,
per lo più, il “promoter” del gene b, riducendo la trascrizione
del gene adiacente
• coeredità di a-talassemia, che, riducendo lo sbilanciamento
globinico, attenua il quadro clinico
• coeredità di determinanti genetici (o HPFH), che
aumentando la produzione di catene g, riducono lo
sbilanciamento globinico, attenuando così il quadro clinico.
Il fenotipo ematologico ricalca quello descritto
per il morbo di Cooley, anche se più attenuato.
L’estrema variabilità dei dati ematologici e
laboratoristici rendono spesso indispensabili
l’esecuzione in questi soggetti e nei loro
familiari, di indagini complesse di biologia
molecolare, per chiarire la diagnosi.