Analisi di dati RNA-Seq
Alberto Ferrarini
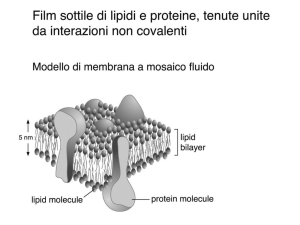
Il dogma centrale della biologia
molecolare
DNA
Replicazione
RNA
Trascrizione
Traduzione
PROTEIN
Geni sono trascritti da DNA ad mRNA che lascia il nucleo e viene tradotto in
proteine.
2
Il trascrittoma
• Il set completo di tutti gli mRNA di un organismo in un dato
momento.
• Il trascrittoma è dinamico e cambia a seconda delle condizioni
considerate. Differenti condizioni danno luogo a differenti profili di
espressione genica.
Trascrittomica: lo studio del trascrittoma; l’analisi del trascrittoma in
diverse condizioni permette di inferire quali geni siano potenzialmente
coinvolti in un dato processo di sviluppo, risposta a stress, ecc…
Analisi di espressione genica
Prima delle tecnologie
“omiche”
• Uno o pochi geni analizzati
per volta tramite analisi
Northern o PCR
quantitativa/semiquantitativa
Oggi
• Da poche migliaia di geni a
trascrittomi completi
analizzati in un singolo
esperimento.
Microarray
Next Generation
Sequecing (NGS)
4
Evoluzione delle tecnologie di analisi
del trascrittoma
1995- Sviluppati i primi
microarray basati su
spotting di molecole di
cDNA
Quantitative Monitoring of Gene
Expression Patterns with a Complementary
DNA Microarray- Schena et. al.
2002- High density
oligo microarrays
2008- RNA-Seq:
sequenziamento dei
messaggeri basato su
tecnologie NGS
Sequenziamento del trascrittoma
Campioni di interesse
Tessuto normale
Tessuto tumorale
Isolamento
dell’RNA/mRNA
Frammentazione
chimica
Immagine modificata da:
http://www.nature.com/nrc/journal/v6/n4/full/nrc1838.html
Sequenziamento
AGTCGTGGATCCAT
AGTCGTGGATCCAT
AGTCGTGGATCCAT
AGTCGTGGATCCAT
AGTCGTGGATCCAT
AGTCGTGGATCCAT
AGTCGTGGATCCAT
AGTCGTGGATCCAT
AGTCGTGGATCCAT
AGTCGTGGATCCAT
Milioni di read paired-end
Conversione a cDNA e
ligazione degli adattatori
Perché sequenziare l’RNA?
• Studi funzionali:comparazione
dell’espressione genica tra diverse condizioni
(sano-malato, diversi tessuti, risposta ad uno
stimolo, ecc…)
• Studio delle isoforme di espressione
• Identificazione di trascritti non annotati
• Studio RNA editing
• Identificazione di trascritti di fusione
Protocollo di analisi dati RNA-Seq
reads
Allineamento su un
genoma di riferimento
genome
Assegnamento delle read
ai geni annotati
Known gene
Rilevazione di eventuali
geni “nuovi” non annotati
Unknown gene
Quantificazione dell’espressione
e analisi statistica
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., & Wold, B. (2008).
Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature
methods, 5(7), 621-8. doi: 10.1038/nmeth.1226.
Assegnamento delle read ai geni e
quantificazione dell’espressione genica
• Il numero di read che mappano su un gene è
proporzionale al livello di espressione
• I valori di espressione ottenuti dall’RNA-Seq deriva
dalla conta diretta delle read che mappano su un
gene: misura digitale
• Non richiede la conoscenza a priori delle posizioni dei
geni
• Intervallo dinamico più ampio comparato a
microarray
Disegno sperimentale: numero di replicati
•
•
•
•
Tre o più repliche biologiche
Non sono generalmente richieste repliche tecniche
della stessa libreria ad RNA
La correlazione R2 (Pearson) tra i livelli di espressione
degli RNA rilevati in comune tra 2 replicati biologici
dovrebbe essere tra 0.92 e 0.98.
Esperimenti con correlazioni inferiori a 0.9 devono
venire ripetuti o spiegati.
Disegno sperimentale: profondità di
copertura richiesta
Numero di ORF rilevate al variare
della profondità
Numero di siti di inizio della
trascrizione al variare della profondità
•Analisi di epressione differenziale: sono raccomandate 30 o più milioni di read
paired-end (uomo).
•Esperimenti destinati alla scoperta e caratterizzazione di nuovi geni/isoforme o
finalizzati ad una quantificazione molto solida delle isoforme richiede coperture
maggiori (fino a 100-200 M di frammenti)
http://encodeproject.org/ENCODE/protocols/dataStandards/RNA_standards_v1_2011_May.pdf
Wang, Z., Gerstein, M., & Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nature Rev. Retrieved from
http://www.nature.com/nrg/journal/v10/n1/authors/nrg2484.html
Disegno sperimentale: profondità di
copertura richiesta
Il numero di read richieste dipende anche dal tipo di RNA che vogliamo
caratterizzare.
Tarazona, S., Garcia-Alcalde, F., Dopazo, J., Ferrer, a., & Conesa, a. (2011). Differential expression in RNA-seq: A matter of depth. Genome Research.
doi:10.1101/gr.124321.111
Problematiche connesse con l’analisi di
dati RNA-Seq
• Allineamento delle read ottenute da librerie a
cDNA su sequenze genomiche (per metodi
basati su genoma di riferimento).
• Assemblaggio de novo delle read ottenute da
librerie a cDNA in putativi trascritti (per
metodi che non utilizzano il genoma di
riferiemento).
• Quantificazione dei livelli di epressione
• Analizzare l’espressione differenziale
Garber, M., Grabherr, M. G., Guttman, M., & Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using
RNA-seq. Nature methods, 8(6), 469–77. doi:10.1038/nmeth.1613
Metodi di ricostruzione del
trascrittoma
Metodi
guidati dal
genoma
Metodi
indipendenti
dal genoma
Garber, M., Grabherr, M. G., Guttman, M., & Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using
RNA-seq. Nature methods, 8(6), 469–77. doi:10.1038/nmeth.1613
Nel caso di metodi basati sul
genoma il primo passaggio è
l’allineamento delle read
ottenute dai frammenti al
genoma di riferimento
Allineamento di read RNA-Seq ad un
genoma di riferimento
esoni
genoma
introni
mRNA
In un esperimento RNA-Seq le read vengono generate dal sequenziamento delle estremità di
frammenti da 200-300 bp dell’RNA messaggero da cui le sequenze introniche sono state
rimosse dal macchinario di splicing durante la maturazione dell’mRNA.
Alcuni frammenti saranno a cavallo delle giunzioni esone-esone
Allineamento di read RNA-Seq ad un
genoma di riferimento
esoni
genoma
introni
mRNA
Read derivanti da frammenti contenuti completamente in singoli esoni mapperanno
correttamente con una distanza tra le read compatibile con le dimensioni della libreria
Allineamento di read RNA-Seq ad un
genoma di riferimento
esoni
genoma
introni
mRNA
Coppie di read
mappanti su 2 esoni
diversi avranno una
dimensione dell’inserto
non compatibile con le
dimensioni della
libreria
Dimensioni libreria
Allineamento di read RNA-Seq ad un
genoma di riferimento
esoni
genoma
introni
mRNA
Read a cavallo di una
giunzione esone-esone non
potranno essere mappate
correttamente dagli
algoritmi standard.
Allineamento di read RNA-Seq ad un
genoma di riferimento
esoni
genoma
introni
mRNA
Read a cavallo di una
giunzione esone-esone non
potranno essere mappate
correttamente dagli
algoritmi standard.
Allineamento di read RNA-Seq ad un
genoma di riferimento
esoni
genoma
introni
mRNA
Idealmente la read dovrebbe
essere spezzata in uno spliced
alignment che tenga conto
dell’introne
• Non mappare le read sovrapposte a giunzioni
esone-esone porterebbe alla sottostima
dell’espressione dei geni con tanti esoni
Utilizzo di un database di giunzioni di
splicing
Un database di giunzioni
custom viene costruito
unendo le estremità degli
esoni.
Read spliced vengono rilevate
allineando le read non
mappanti sul database di
giunzioni.
Database
custom di
giunzioni note.
[…]
Una limitazione di questo aproccio è che può rilevare
solo giunzioni note.
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., … Burge, C. B. (2008). Alternative isoform regulation in human tissue transcriptomes.
Nature, 456(7221), 470–6. doi:10.1038/nature07509
Metodi computazionali per allineamento
splittato di read su un genoma di riferimento
Gli approcci per l’allineamento delle read su un
genoma di riferimento si dividono in:
• approccio exon-first
• approccio seed-extent
Approccio exon-first
• Nell’approccio exon-first
vengono prima allineate
tutte le read sul genoma.
• Le read che non mappano
utilizzate per trovare siti
di splicing candidati.
• Software:
– Tophat
– MapSplice
– SpliceMap
Garber, M., Grabherr, M. G., Guttman, M., & Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using
RNA-seq. Nature methods, 8(6), 469–77. doi:10.1038/nmeth.1613
TopHat
Pipeline scritta in Python e C++ basata su Bowtie e la libreria SeqAn
Versione pubblicata quando le read erano tendenzialmente < 50 bp
Identificazione ab initio dei siti di
splicing (fino a versione 0.8.3)
• Bowtie mappa le read sul genoma
con un massimo di 2 mismatch
nel seed e 10 allineamenti
multipli (serve a riportare geni
con copie multiple).
• Le read allineate vengono quindi
assemblate in un consenso a cui
vengono aggiunte 45 basi dalle
regioni fiancheggianti.
• Vengono quindi identificati i
possibili siti donatori e accettori
di splicing canonici (GT-AG) verso
le estremità di queste regioni.
• Le read non mappanti vengono
mappate sui putativi siti di
splicing.
TopHat 1.0
• Dalla versione 1.0 sfrutta le maggiore
lunghezza delle read
Maggiore sensibilità
Unmappable read
Reference genome
Trapnell, C., Pachter, L., & Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq.
Bioinformatics (Oxford, England), 25(9), 1105-11. doi: 10.1093/bioinformatics/btp120.
30
TopHat 1.0
• Dalla versione 1.0 sfrutta le maggiore
lunghezza delle read
Maggiore sensibilità
• Read non mappate da 75 basi (o più
lunghe) vengono splittate in 3 o più subread da 25 basi che vengono mappate
indipendentemente.
Unmappable read
25nt
Reference genome
Trapnell, C., Pachter, L., & Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq.
Bioinformatics (Oxford, England), 25(9), 1105-11. doi: 10.1093/bioinformatics/btp120.
31
TopHat 1.0
• Dalla versione 1.0 sfrutta le maggiore
lunghezza delle read
Maggiore sensibilità
• Read non mappate da 75 basi (o più
lunghe) vengono splittate in 3 o più subread da 25 basi che vengono mappate
indipendentemente.
• Read con segmenti che possono essere
mappati solo in maniera non contigua
Marcati come possibili read intronspanning
Unmappable read
25nt
Reference genome
Trapnell, C., Pachter, L., & Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq.
Bioinformatics (Oxford, England), 25(9), 1105-11. doi: 10.1093/bioinformatics/btp120.
32
TopHat 1.0
• Dalla versione 1.0 sfrutta le maggiore
lunghezza delle read
Maggiore sensibilità
• Read non mappate da 75 basi (o più
lunghe) vengono splittate in 3 o più subread da 25 basi mappate
indipendentemente.
• Read con segmenti che possono essere
mappati solo in maniera non contigua
Marcati come possibili read intronspanning
• Il set di tutte le possibili combinazioni
dondatore-accettore viene descritto da:
L1+L2=k; 1 < L1 < k-1; L2 = k-L1
Unmappable read
25nt
L1
L2
Reference genome
Trapnell, C., Pachter, L., & Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq.
Bioinformatics (Oxford, England), 25(9), 1105-11. doi: 10.1093/bioinformatics/btp120.
33
TopHat 1.0
• Dalla versione 1.0 sfrutta le maggiore
lunghezza delle read
Maggiore sensibilità
• Read non mappate da 75 basi (o più
lunghe) vengono splittate in 3 o più subread da 25 basi mappate
indipendentemente.
• Read con segmenti che possono essere
mappati solo in maniera non contigua
Marcati come possibili read intronspanning
• Il set di tutte le possibili combinazioni
dondatore-accettore viene descritto da:
L1+L2=k; 1 < L1 < k-1; L2 = k-L1
• k basi a monte del sito donatore
concatenate con k basi a valle
dell’accettore
Unmappable read
25nt
donor
site
acceptor
site
Reference genome
Trapnell, C., Pachter, L., & Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq.
Bioinformatics (Oxford, England), 25(9), 1105-11. doi: 10.1093/bioinformatics/btp120.
34
TopHat 1.0
• Dalla versione 1.0 sfrutta le maggiore
lunghezza delle read
Maggiore sensibilità
• Read non mappate da 75 basi (o più
lunghe) vengono splittate in 3 o più subread da 25 basi mappate
indipendentemente.
• Read con segmenti che possono essere
mappati solo in maniera non contigua
Marcati come possibili read intronspanning
• Il set di tutte le possibili combinazioni
dondatore-accettore viene descritto da:
L1+L2=k; 1 < L1 < k-1; L2 = k-L1
• k basi a monte del sito donatore
concatenate con k basi a valle
dell’accettore
Unmappable reads
Allineamento delle
read non allineabili al
database di giunzioni
Trapnell, C., Pachter, L., & Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq.
Bioinformatics (Oxford, England), 25(9), 1105-11. doi: 10.1093/bioinformatics/btp120.
35
Limiti dei sistemi exon-first
Il genoma umano contiene circa 14,000 pseudogeni e molti pseudogeni hanno una
sequenza simile ad un gene annotato read possono mappare sia sul gene che sul
corrispondente pseudogene
• L’allineamento su pseudogeni processati favorito rispetto all’allineamento sul gene nel
caso di read a cavallo di giunzioni esone-esone.
La maggior parte delle read a cavallo di giunzioni “assorbite” da pseudogeni
Garber, M., Grabherr, M. G., Guttman, M., & Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using RNA-seq.
Nature methods, 8(6), 469–77. doi:10.1038/nmeth.1613 Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., & Salzberg, S. L. (2013).
TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14(4), R36. doi:10.1186/gb-201314-4-r36
Basato su Bowtie2 (migliore sensibilità per Indel)
Attua una serie di strategie per migliorare la sensibilità e la
specificità di allineamento.
Riduce il problema di allineamenti scorretti dovuti a
pseudogeni
Workflow di TopHat2
1) transcriptome mapping
c
c
• Se viene fornita un’annotazione (consigliato)
TopHat2 allinea le read contro le sequenze del
trascrittoma.
aumenta la sensibilità e specificità verso
trascritti noti.
TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14(4), R36. doi:10.1186/gb-2013-14-4-r36
Workflow di TopHat2
1) transcriptome mapping
c
• Le read che non mappano sui trascritti
annotati vanno al passaggio successivo
TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14(4), R36. doi:10.1186/gb-2013-14-4-r36
Workflow di TopHat2
2) genome mapping
c
c
• Nel secondo passaggio le read che vengono mappate in modalità end-toend sul genoma di riferimento
solo le read che mappano completamente su un esone vengono
allineate
TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14(4), R36. doi:10.1186/gb-2013-14-4-r36
Workflow di TopHat2
2) genome mapping
c
c
• Read che non mappano completamente sul
genoma vanno al passaggio successivo.
TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14(4), R36. doi:10.1186/gb-2013-14-4-r36
Workflow di TopHat2
3) spliced mapping
• Read non mappate nel
secondo passaggio
vengono utilizzate per
cercare i segnali di
splicing (GT-AG, GC-AG,
AT-AC).
• E’ stato inoltre integrato
algoritmo per
identificare breakpoint
di fusione (da TopHatFusion)
Workflow di TopHat2
3) spliced mapping
• Nell’ultima fase di questo passaggio vengono
riallineate le read che si sovrappongono
minimamente con sequenze introniche
Soglia basata sulla edit-distance
• TopHat2 consente di indicare una soglia (t)
basata sulla edit distance:
– Se una read allinea in un passaggio ma con una
edit distance ≥ t essa verrà riallineata nei passaggi
successivi per cercare un eventuale allineamento
migliore.
– Se viene settata una soglia t = 0 tutte le read che
mappano nel passaggio 1 verranno riallineate nei
passaggi successivi aumenta la sensibilità e la
specificità.
Effetto in presenza di pseudogeni
• Allineamento contro
trascrittoma noto assegna
tutte le read possibili ai
trascritti noti evitando che
allineino contro gli
pseudogeni corrispondenti
• Riallineamento basato su
edit distance consente di
rimappare read sovrapposte
a siti di splicing ignoti
mappate scorrettamente a
pseudogeni nel passaggio 2.
Approccio seed-extent
• Nell’approccio seed-extend
viene memorizzato un indice
di k-mer del genoma.
• Le read vengono divise in kmer e confrontate con l’indice
del genoma.
• I k-mer mappati vengono
quindi estesi e l’allineamento
può includere siti di splicing.
• Software:
– GSNAP
– QPALMA
• Sistemi seed-extent sono
accurati ma generalmente
molto più lenti di sistemi exonfirst
Garber, M., Grabherr, M. G., Guttman, M., & Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using
RNA-seq. Nature methods, 8(6), 469–77. doi:10.1038/nmeth.1613
GSNAP
Genomic Short-read Nucleotide Alignment Program
Allineatore creato per identiticare varianti complesse e
siti di splicing da read NGS.
Allineamento sul reference “space”
• GSNAP utilizza una tabella di
hash dei possibili 12-mer sul
genoma (spaziati di 3 nt).
• SNP in un 12-mer genomico
vengono rappresentate
duplicando le posizioni nella lista
per tutte le combinazioni di alleli
maggiori e minori nel 12-mer.
• Alleli maggiori vengono
rappresentati in un genoma
compresso mentre gli alleli
minori vengono rappresentati in
un altro genoma compresso.
Wu, T. D., & Nacu, S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics (Oxford, England),
26(7), 873–81. doi:10.1093/bioinformatics/btq057
Rilevazione di varianti ed eventi di
splicing
• GSNAP può utilizzare 2
tipi di evidenze per
identificare i siti di
splicing:
Wu, T. D., & Nacu, S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics (Oxford, England),
26(7), 873–81. doi:10.1093/bioinformatics/btq057
Rilevazione di varianti ed eventi di
splicing
• GSNAP può utilizzare 2
tipi di evidenze per
identificare i siti di
splicing:
1. Modello probabilistico
di siti donatoriaccettori
Wu, T. D., & Nacu, S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics (Oxford, England),
26(7), 873–81. doi:10.1093/bioinformatics/btq057
Rilevazione di varianti ed eventi di
splicing
• GSNAP può utilizzare 2
tipi di evidenze per
identificare i siti di
splicing:
1. Modello probabilistico
di siti donatoriaccettori
2. Database di estremità
esone-introne note
Wu, T. D., & Nacu, S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics (Oxford, England),
26(7), 873–81. doi:10.1093/bioinformatics/btq057
Rilevazione di varianti ed eventi di
splicing
• GSNAP può utilizzare 2
tipi di evidenze per
identificare i siti di
splicing:
1. Modello probabilistico di
siti donatori-accettori
2. Database di estremità
esone-introne note
•
Eventi di splicing
possono anche essere
intercromosomali
(fusioni geniche)
Wu, T. D., & Nacu, S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics (Oxford, England),
26(7), 873–81. doi:10.1093/bioinformatics/btq057
Controllo qualità di dati RNA-Seq
●
RseQC è un pacchetto software che fornisce
dei moduli per controllare la qualità delle
sequenze RNASeq allineate
Livello di duplicazione
read_duplication.py calcola i livelli di duplicazione a livello di allineamento e a livello
di sequenza.
Read duplicate nei dati RNA-Seq
• Read duplicate non vengono normalmente
rimosse dai dati RNA-Seq:
– Duplicati di PCR non sono distinguibili da
frammenti uguali dovuti a elevati livelli di
espressione
Distribuzione delle read
Distribuzione delle read tra le diverse feature (CDS,UTR, Introni, …)
Distribuzione tra le feature
900000
3000
800000
700000
2500
600000
2000
Read intergeniche
500000
1500
400000
1000
300000
200000
500
100000
0
0
cds
5'UTR
3'UTR
intron
intergenic
TSS_up_5kb
TES_down_1kb
TES_down_10kb
TSS_up_10kb
TSS_up_1kb
TES_down_5kb
upstream
downstream
Bias possono intervenire se i rapporti CDS/UTR/Introni non vengono mantenuti
Analisi del gene body coverage
Permette di rilevare bias nel coverage rispetto alla posizione nel gene body
3’ end bias
Un bias al 3’ potrebbe indicare un campione degradato.
Distanza tra paia di read
Distanza tra due paia di read tenendo in considerazione la posizione degli introni.
Saturazione degli RPKM
●
●
●
RPKM_saturation.py
Stima dell'errore come
percentuale comparata
all'RPKM ottenuto da
tutte le read.
Q1, Q2, Q3, Q4 sono i 4
quartili di espressione.
Reads Per Kilobase of gene per Million
mapped reads (RPKM)
• Valore di espressione normalizzato dividendo
le conte grezze per la lunghezza in kilobasi dei
geni e per i milioni di read totali mappate per
campione:
– Geni più lunghi hanno una maggiore probabilità di
essere sequenziati
– Il numero di read ottenute può variare a seconda
della run di sequenziamento
Comparazione delle giunzioni rilevate
con l’annotazione
Total splicing Events: 160912
Known Splicing Events:
155526
Partial Novel Splicing Events:
3396
Novel Splicing Events:1941
Total splicing Junctions:
4326
Known Splicing Junctions: 3871
Partial Novel Splicing Junctions: 259
Novel Splicing Junctions: 196
splice event: Una read RNA-Seq,
specialmente se lunga, può venire splittata 2
o più volte; ogni volta viene contata come
splicing event
splice junction: eventi di splicing multipli
riguardanti lo stesso introne.
Saturazione delle giunzioni
Fornisce una misura di quanto la profondità utilizzata è stata in grado di
saturare le giunzioni di splicing note e novel importante se si è interessati
all’analisi dello splicing alternativo.
Giunzioni note a saturazione
Giunzioni note non sono
saturate