Copyright “Tutti i diritti sono riservati”
Disordini da alterata secrezione o azione dell’AMH
Soara Menabò 1, Antonio Balsamo 2
1
Laboratorio di Genetica Molecolare, Dipartimento Salute della Donna, del Bambino e
dell’Adolescente, Programma Endocrinologia Pediatrica, AOU S.Orsola-Malpighi, Bologna;
2
Dipartimento di Scienze Mediche e Chirurgiche, Unità Operativa di Pediatria, Programma di
Endocrinologia, Azienda Ospedaliera-Universitaria di Bologna, Bologna.
Fisiopatologia e genetica
L’ormone Anti-Mülleriano (AMH) è una proteina secreta dalle cellule del Sertoli, che nel
feto maschio è responsabile della regressione dei dotti di Müller (che vanno a costituire utero,
tube e parte alta della vagina). L’azione più specifica, mediata dal recettore di tipo II, è
l’induzione dell’apoptosi nei dotti di Müller fetali omolaterali: ogni testicolo sopprime lo
sviluppo delle strutture Mülleriane solo dalla sua parte (1).
Il gene AMH, localizzato in 19p13.3, è formato da 5 esoni e codifica per una proteina di 535
aminoacidi membro della famiglia TGF-β. Le mutazioni sono per lo più missenso, disperse in
tutto il gene (non ci sono hotspot mutazionali). I pazienti sono in prevalenza omozigoti e la
mutazione è privata.
Il gene AMHR2, localizzato in 12q13, è formato da 11 esoni e codifica per una proteina
recettoriale trans-membrana. Circa la metà dei pazienti presenta la stessa mutazione, una
delezione di 27-bp nell’esone 10 (2).
La regolazione dell’AMH è finemente controllata da diversi fattori, poiché l’espressione
deve avvenire in uno specifico lasso temporale, tra l’8° e la 10° settimana di gestazione. L’SRY,
per esempio, è importante per l’attivazione di SOX9, il quale assieme a SF1 e DAX1 stimolano in
modo diretto l’espressione di AMH nei testicoli fetali. Ma intervengono anche altri fattori quali
GATA e FSH. Poiché il recettore è espresso anche nei neuroni degli embrioni di topo,
probabilmente gioca un ruolo anche nello sviluppo sessuale dimorfico del cervello.
L’espressione di AMH avviene anche nelle femmine, ma con un timing completamente
diverso: viene infatti secreto dalle cellule della granulosa dei follicoli ovarici e può essere usato
come marcatore del numero di cellule follicolari di riserva per predire l’età della menopausa (3).
L’AMH è probabilmente coinvolto anche nell’inizio della pubertà in entrambi i sessi (2).
1
Copyright It.DSD Gruppo di Studio – “Tutti i diritti sono riservati”
www.gruppodistudio-it-dsd.org
Copyright “Tutti i diritti sono riservati”
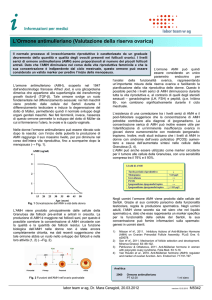
I livelli di AMH nel sangue variano in base a età e sesso:
-
nei maschi resta alto fino a prima della pubertà, poi scende ed è basso nel maschio
pubere e adulto;
-
nelle femmine avviene il contrario, infatti resta indosabile fino alla pubertà e poi
aumenta.
Clinica
La sindrome da Persistenza dei Dotti di Müller (PMDS, OMIM 261550) include due
forme diverse:
-
La PMDS di tipo I è causata da difetti di secrezione dell’AMH, riconducibili a
mutazioni nel gene AMH (OMIM 600957);
-
la PMDS di tipo II è causata da difetti di azione per mancata responsività del recettore,
attribuibili a mutazioni nel gene AMHR2 (OMIM 600956), che codifica per il recettore
di tipo II dell’AMH (4).
Si tratta di una sindrome rara a trasmissione autosomica recessiva con prevalenza ignota.
Circa un 13-15% di pazienti con PMDS rimane senza diagnosi genetica, non avendo mutazioni
nè nel gene AMH nè nel gene AMHR2. Le femmine con entrambi gli alleli mutati per uno di
questi geni non presentano alterazioni dei genitali.
La manifestazione fenotipica è identica per entrambi i tipi: maschi XY con normale
virilizzazione, sviluppo dei genitali esterni e caratteri sessuali secondari, con presenza di un utero
rudimentale. Alcuni hanno criptorchidismo bilaterale, altri pazienti hanno un testicolo disceso
che ha trascinato la tuba omolaterale nel canale inguinale, generando un’ernia e con il testicolo
controlaterale localizzato a livello addominale. È stata riportata infertilità (azoospermia) in
diversi pazienti.
Diagnosi
Generalmente, se non si presenta criptorchidismo, la diagnosi di PMDS avviene solo in
modo accidentale (ecografia addominale, interventi chirurgici, ecc.). Il criptorchidismo invece
può far sì che la diagnosi avvenga fin dalla nascita.
Il dosaggio di AMH rispecchia il tipo di difetto: se è basso o indosabile è indicativo di difetti
sul gene AMH, se è normale o alto è indicativo di difetti sul gene del recettore AMHR2. Sono
riportati tuttavia casi eccezionali di livelli normali in pazienti con mutazione nel gene AMH; si
tratta di mutazioni che affliggono la bioattività ma non la secrezione (5).
2
Copyright It.DSD Gruppo di Studio – “Tutti i diritti sono riservati”
www.gruppodistudio-it-dsd.org
Copyright “Tutti i diritti sono riservati”
Terapia
La rimozione chirurgica dei residui mülleriani, un tempo evitata per non danneggiare i vasi
deferenti che sono spesso intimamente aderenti a queste strutture, è invece ora raccomandata, dal
momento che si è osservata degenerazione neoplastica (carcinoma) in alcuni pazienti (6).
Bibliografia
1. Josso N, Lamarre I, Picard JY, et al. Antimüllerian hormone in early human
development. Early Hum Dev 1993, 33: 91–9.
2. Josso N, Belville C, di Clemente N, Picard JY. AMH and AMH receptor defects in
Persistent Müllerian Duct Syndrome. Hum Reprod Update 2005, 11: 351–6.
3. Belville C, Josso N, Picard JY. Pesistence of müllerian derivatives in males. Am J Med
Genet 1999, 89: 218–23.
4. Imbeaud S, Belville C, Messica-Zeitoun L, et al. A 27 base-pair deletion of the antimüllerian type II receptor gene is the most common cause of the Persistent Müllerian
Duct Syndrome. Hum Mol Genet 1996, 5: 1269–77.
5. Menabò S, Balsamo A, Nicoletti A, et al. Three novel AMH gene mutations in a patient
with persistent mullerian duct syndrome and normal AMH serum dosage. Horm Res
2008, 70: 124-8.
6. Lindhardt Johansen M, Hagen CP, Johannsen TH, et al. Anti-Müllerian hormone and its
clinical use in pediatrics with special emphasis on disorders of sex development. Int J
Endocrinol 2013, 2013: 198698.
3
Copyright It.DSD Gruppo di Studio – “Tutti i diritti sono riservati”
www.gruppodistudio-it-dsd.org