LA GENETICA DEI TUMORI
Single disease?
No, group of multifactorial diseases
(100 types of cancer have been classified: leukemias/lymphomas,
carcinomas/adenocarcinomas, sarcomas)
Mutazioni
oncogeni)
dominanti
(attivazione
di
(Inattivazione
di
Malattia genetica
Mutazioni recessive
oncosoppressori)
Aploinsufficienza (alterata funzione di geni
regolatori della risposta al danno)
Stile di vita (es. fumo, dieta, comportamento
sessuale, etc)
Ambiente (es. esposizione a carcinogeni,
mutageni, UV, etc)
Malattia ambientale
Vita riproduttiva (contraccezione, età primo
parto, trattamenti per la fertilità, etc)
Altro (difetti metabolici produzione ROS,
prodotti mutageni, etc)
La base genetica del cancro
1.
eredità clonale
2.
induzione di tumore da parte di virus
3.
il tumore può essere indotto da agenti mutageni
4.
familiarità di alcuni tipi di tumori
5.
associazione tra tumori e alterazioni cromosomiche
Il tumore è la seconda causa di morte in Italia
(rilevazione ISTAT 2010)
Tuttavia…..
- Nel corpo umano ci sono circa 1011 cellule che replicano (su circa 1014 totali)
-Il tasso di mutazione spontanea nell’uomo è di 10-4 - 10-5/gene/generazione;
Nonostante l’alto numero di mutazioni che presumibilmente si verificano, non
si sviluppa un tumore
-molte di queste mutazioni sono riparate e corrette (tasso ridotto tra 10-9 e 1011);
- altri meccanismi protettivi (checkpoint, apoptosi, senescenza, sistema
immunitario, etc)
- necessità di più mutazioni
- elevati tempi di latenza
…un tumore maligno e' un evento rarissimo!
No single mutation can convert a normal cell into a malignant one !!!
Il tumore è un processo multistep
(modello di Fearon e Vogelstein per lo sviluppo del carcinoma colorettale)
Epitelio normale
Displasia
Carcinoma in situ
Carcinoma metastatico
Il cancro e' una malattia (epi)genetica
Mutazioni
modificazioni epigenetiche
Attivazione di oncogeni
Inattivazione di oncosoppressori
(>100)
(>30)
Trasformazione neoplastica
“Stop” and “go” signals
STOP
In tumors
Oncogene as the accelerator
Tumor suppressor as the brake
Oncogeni:
Fattori di crescita (c-sis/PDGF)
Recettori per fattori di crescita (ErbB2, HER/neu)
Trasduttori del segnale (Ras, Src, Abl)
Fattori trascrizionali (β
β-catenina, myc)
Effettori coinvolti nella proliferazione (ciclina D1)
Oncosoppressori:
Regolatori del ciclo cellulare (INK4a, pRb, p53)
Proteine dei “checkpoint” (p53, ATM, CHK1 e 2)
Enzimi di riparazione del DNA (BRCA1 e 2)
Cancer and apoptosis
oncogene
oncosuppressor
citocromo c/
Via intrinseca
Via estrinseca
Apoptosi
GENI DELLA PROGRESSIONE TUMORALE
“Metastasis inducers”:
Regolatori negativi delle giunzioni cellulari (Twist, Snail, metalloproteasi)
Fattori angiogenici (VEGF)
“Metastasis suppressors”:
Componenti o regolatori delle adesioni cellulari (E-caderina, inibitori delle
metalloproteasi)
The hallmarks of cancer
From Hanahan and Weinberg, 2000
The hallmarks of cancer
e.g. activate
Ha-Ras oncogene
e.g. produce
survivor factors like IGF
e.g. produce VEGF
From Hanahan and Weinberg, 2000
e.g. turn on
telomerase
e.g. lose
Rb suppressor
e.g. inactivate
E-cadherin
Cancers are microcosms of evolution
(Merlo et al., Nat Rev Cancer, 2006)
“…mutant cells compete for space and resources, evade predation by
the immune system and can ever cooperate to disperse and colonize
new organs…………”
“…mutant clones can expand or contract by natural selection and
genetic drift, regardless of any negative effect on the
organism…………”
“…dispersion is an advantage for escape from deteriorating local
conditions caused by population growth and the over-consumption
of resources…………”
Differenziamento e malignità…
…sono inversamente proporzionali
malignità
differenziamento
L’esempio del carcinoma tiroideo
Tiroide normale
Carcinoma follicolare
Carcinoma anaplastico
Carcinoma papillifero
Cancer stem cells
La cellula trasformata origina da cellule differenziate (a) o staminali (b)?
Probabilmente da entrambe!!!
Mutazioni dominanti degli oncogeni
Oncogeni (>100):
Fattori di crescita
Recettori per fattori di crescita
Trasduttori del segnale
Fattori trascrizionali
Effettori coinvolti nella proliferazione
Fattori anti-apoptotici
Regolatori negativi delle giunzioni cellulari
Fattori angiogenici
Inibitori di oncosoppressori
Retrovirus e oncogeni virali (v-onc)
(1911)
1911 Peyton ROUS : scopre il primo virus tumorale come agente
eziologico del sarcoma dei polli che prende il suo nome (sarcoma di Rous).
Il virus contiene l’omologo del gene cellulare src
Identificazione di altri 20 oncogeni virali tra cui:
v-src
v-erbB
v-abl
v-sis
v-fms
v-fos
v-Ha-Ras
Src
EGF-R
Abl
PDGF
CSF-1
Fos
Ras
v-onc sono omologhi a geni cellulari (c-onc) detti proto-oncogeni
Il 15% circa dei tumori umani ha una probabile eziologia virale
Virus oncògeni umani
VIRUS A RNA:
RETROVIRUS
Virus umano T-linfotropico (HTLV-1) Adult T-cell leukemia/lymphoma (ATL)
LENTIVIRUS
Virus dell’immunodeficienza umana (HIV-1) Linfomi associati all’AIDS
VIRUS A DNA:
PAPOVAVIRUS
Virus umano del papilloma (HPV) Carcinoma del collo dell'utero (sierotipi ad
alto rischio: 16, 18, 31)
HERPESVIRUS
Virus di Epstein-Barr (EBV) Linfoma di Burkitt, Carcinoma nasofaringeo
Virus erpetico umano di tipo 8 (HHV-8) Sarcoma di Kaposi
HEPADNAVIRUS
Virus dell'epatite B e C (HBV e HCV) Carcinoma epatico
Meccanismo diretto Oncoproteine virali possono inattivare
oncosoppressori cellulari
E1B
Adenovirus
E7
E1A
Virus del papilloma
E6
Rb
p53
Large T
SV40
Meccanismo indiretto L’infezione cronica favorisce lo sviluppo
del tumore (es. HBV)
Attraverso quali meccanismi un proto-oncogene diventa oncogene?
Mutazioni puntiformi
Delezioni/Inserzioni
Amplificazione genica
Traslocazione cromosomica
Una mutazione puntiforme –un oncogene: l’es. di Ras
Guanine nucleotide
Exchange Factors
GTPase Activating
Proteins
Gly12
Val
da The Biology of Cancer, Garland Science, 2006
Inhibition
of
apoptosis
stimulation
of cell
proliferation
stimulation
of cell
proliferation
Stimulation
of cell
motility
Modificata da The Biology of Cancer, Garland Science, 2007
Amplificazione genica: es. di N-Myc nel neuroblastoma
4, 9 o 13
2p
= homogeneous staining chromosomal regions
How do oncogenes amplify?
A
B
Double minutes
C
Traslocazioni cromosomiche fusione genica
CML
FISH image of bcr/abl positive
rearranged metaphase
From Snustad & Simmons “Principles of Genetics” 2006
BCR= Breakpoint Cluster Region
Fusion gene
N-term
bcr
abl
C-term
Constitutively active c-Abl tyrosine kinase
activation of cell cycle-controlling proteins
inhibition of DNA repair
induction of genomic instability
Traslocazioni cromosomiche over-espressione
Burkitt’s lymphoma
Figure 24.7 A reciprocal translocation involved in Burkitt’s lymphoma.
Only the translocation chromosome (14q+) that carries both the c-myc oncogene
and the immunoglobulin heavy chain genes (IgH) is shown.
From Snustad & Simmons “Principles of Genetics” 2006
Il linfoma di Burkitt è una neoplasia di linfociti B maturi,
maturi endemica nell’Africa equatoriale,
strettamente associata all’infezione del virus di Epstein Barr (agente eziologico?).
Costituisce il 70% dei tumori infantili.
Esiste anche una forma sporadica, presente in Europa e nelle Americhe, e una associata
ad immunodeficienza (es.HIV)
Esordisce come tumefazioni delle ossa mascellari e degenerazioni delle mucose
circostanti.
La traslocazione più comune è la t(8;14) che coinvolge c-myc e il gene per la catena
pesante delle immunoglobuline.
Le varianti t(2;8) e t(8;22) coinvolge c-myc e il gene per le catene leggere κ e λ delle
immunoglobuline, rispettivamente.
Chr 8
Promoter
Exon 1
Exon 2
Chr 14
Enhancer
CH
JH
t(8;14)
Enhancer
CH
Exon 2
Exon 3
DH
VH
Exon 3
myc
IgH
IgH-myc
cellula normale
cellula tumorale
Mutazioni recessive degli oncosoppressori
e perdita dell’eterozigosità
I principali tumor suppressor: Rb …..
X
Cyc/CDK
UV, IR, stalled replication
…e p53
Sensors
ATM/ATR
CHK1/2
Transducers
Effectors
others
BRCA1/2
DNA repair
Cell cycle arrest
p53
Apoptosis
Esempi di mutazioni a carico di oncosoppressori
Mutazioni puntiformi - missenso
- nonsenso
- con frameshift
- con effetto sullo splicing
Delezioni/Inserzioni
Rb/FHIT
Traslocazioni cromosomiche
FHIT
Perdita di eterozigosi
P53, Rb, BRCA1, etc
Silenziamento per metilazione del DNA
INK4a, BRCA1
INK4a, BRCA1
Mutazioni recessive loss-of-function (es. Rb, APC, BRCA1)
Mutazioni dominanti loss-of function (es. p53)
Mutazioni dominanti gain-of-function (es. p53)
Aploinsufficienza dosaggio genico (es. p21, enzimi di riparazione del DNA)
La genetica della perdita di funzione degli oncosoppressori
Oncosoppressori attivi come monomeri
(es. pRb)
Oncosoppressori attivi come multimeri
(es. p53)
wild-type
mutante
funzione mantenuta nell’eterozigote
(ma trasmette la predisposizione allo sviluppo di
tumore)
Etero-tetrameri
inattivi
Tetrameri attivi
funzione compromessa nell’eterozigote
In alcuni casi guadagno di funzione
Mutazioni recessive
Mutazioni dominanti
Il gene p53
Genetica:
Mutato in più del 50% dei tumori umani
Le mutazioni somatiche sono circa due ordini di grandezza più frequenti delle mutazioni
germline (riconosciute ~ 1700 diverse mutazioni di p53 ; le più comuni cadono negli esoni
5-8, codificante per il DNA-binding domain).
La mutazione germline è in realtà molto rara e avviene in una sindrome (Li-Fraumeni
syndrome), in cui gli individui eterozigoti hanno un’alta probabilità, se esposti ad agenti
danneggianti o agenti mutageni, di sviluppare tumori (“cancer-prone”).
Sistemi modello:
Linee cellulari p53-/-: ipersensibili al danno, difettive per l’induzione di apoptosi,
sviluppano instabilità genetica
Topo p53-/- : vitale, ma altamente “cancer-prone”
La proteina p53: domini strutturali e mutazioni
SV40 TAg
MDM2
HPV E6
Ad E1B
Box I-V: altamente conservate tra i membri della famiglia p53
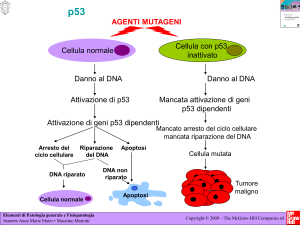
p53 guardiano del genoma
DNA
Damage
p53 p53
p53p53
p53
p21
Transient cell cycle arrest
p53
Re-entry in the
cell cycle
p53 p53
p53p53
DNA Repair
GADD45
p53 p53
p53p53
Apoptosis
Bax, PUMA, etc
p53 mutants
wild-type p53
mutant p53
Loss-of-function
Gain-of-function
Il gene Rb-1
Genetica:
Frequenza del retinoblastoma: 1/14.000 nascite le
cellule tumorali sono prive di entrambi gli alleli Rb-1
funzionali
Variante familiare (40% dei casi)
Variante sporadica (60% dei casi)
Assetto più frequente:
(i) piccola delezione trasmessa dalla linea germinale
(ii) mutazione di senso sull’allele omologo: si origina un clone somatico
Predisposizione allo sviluppo di tumori dominante:
dominante un cromosoma "difettivo"
predispone i portatori a sviluppare tumori
Sviluppo di tumori recessivo:
recessivo il tumore si sviluppa in completa assenza del
prodotto, quando entrambi gli alleli sono deleti e/o mutati
BRCA1/2
Genetica:
Mutazioni di BRCA1 (chr 17q21) 85% probabilità di sviluppare carcinoma mammario
ereditari, 40-50% probabilità di sviluppare carcinoma ovarico
Mutazioni generalmente frameshift o nonsense produzione di una proteina tronca
Carattere autosomico dominante LOH in tutti i tumori con mutazioni di BRCA1
Genetica:
Mutazioni di BRCA2 (chr 13q12) 80% probabilità di sviluppare carcinoma mammario
ereditario, 10% probabilità di sviluppare carcinoma ovarico; alta incidenza di altri tumori
(es.pancreas)
Carattere autosomico dominante ad alta penetranza LOH in tutti i tumori con
mutazioni di BRCA2
CHK2
p53
P
BRCA1
ATM
Rad51
P
TAD
BRCA2
BRCA2
TAD
Rad51
UV, γ-ray
ATM/ATR
CHK2
P P
BRCA1
Cell-cycle control
Regulation of gene expression
DNA-damage repair
APC
Genetica:
Mutazioni nella “mutation cluster region” di APC sono correlate con l’insorgenza della
Poliposi Adenomatosa Familiare (FAP) proteina tronca non più in grado di fosforilare la
β-catenina
La FAP è una malattia autosomica dominante con penetranza completa.
Myc, cyclin D1
FHIT
Genetica:
Mutazioni nel gene FHIT (fragile histidine triad gene) sul chr3 presenti nel 90% di
pazienti fumatori
Aumentato rischio di sviluppare vari tipi di tumori (polmone, mammella, esofago,
stomaco, reni) e una forma ereditaria di carcinoma prostatico
Sono stati descritte delezioni, traslocationi (3;8), integrazioni da parte del virus HPV e
metilazione del promotore (in fumatori cronici).
Enzima con attività idrosilasica, regola l’apoptosi e la riparazione del DNA
Ereditarietà dei tumori
RETINOBLASTOMA:
Eredità autosomica dominante anche se il carattere è recessivo a livello cellulare
LOH
LOH
tumore
tumore
Teoria del “doppio colpo”
(1971 - Alfred Knudson)
Potential mechanisms of loss of heterozygosity (LOH)
Modified from Wijnhoven et al., 2001
Meccanismi di LOH
= oncosoppressore mutato
Syndrome
Primary tumor
Chromosome
location
Cloned gene
Proposed function of gene
product
Familial retinoblastoma
Retinoblastoma
13q14.3
RB1
Cell cycle and transcriptional
regulation
Li-Fraumeni Syndrome (LFS)
Sarcomas, breast
cancer
17p13.1
p53 (TP53)
Transcription factor; response to
DNA damage and stress;
apoptosis
Familial adenomatous
polyposis (FAP)
Colorectal cancer
5q21
APC
Regulation of β-catenin
Hereditary nonpolyposis
colorectal cancer (HNPCC)
Colorectal cancer
2p16, 3p21 2q32, 7p22
MSH2, MLH1
PMS1, PMS2
DNA mismatch repair
Neurofibromatosis type
1 (NF1)
Neurofibromas
17q11.2
NF1
GAP for p21 ras proteins
Neurofibromatosis type 2
(NF2)
Acoustic neuromas,
meningiomas
22q12.2
NF2
Linkage to cytoskeleton
Wilms’ tumor
Wilms’ tumor
11p13
WT1
Transcriptional repressor
Familial breast cancer 1
Breast cancer
17q21
BRCA1
Repair of double-strand breaks
Familial breast cancer 2
Breast cancer
13q12
BRCA2
DNA repair
von Hippel-Lindau (VHL)
syndrome
Renal cancer
3p25
VHL
Regulates transcriptional
elongation by RNA polymerase II
Familial melanoma
Melanoma
9p21
p16 (CDKN2)
Inhibitor of CDKs
Ataxia telangiectasia
Lymphoma
11q22
ATM
DNA repair
Bloom's syndrome
Solid tumors
15q26.1
BLM
DNA helicase
Pedigree di una forma di retinoblastoma familiare
In most families, the tumour predisposition segregates as an
autosomal dominant trait with high (90%) penetrance
Rare instances of familial retinoblastoma with low penetrance and variable
expressivity have been described.
A typical pedigree of Li-Fraumeni syndrome
bilateral breast cancer
diagnosed at age 40
breast cancer
at age 32
brain tumor
at age 35
soft tissue sarcoma at age 19
and breast cancer at age 33
osteosarcoma leukemia
at age 8
at age 2
soft tissue sarcoma
at age 3
Caratteristiche generali dei tumori familiari ereditari
Stesse (o correlate) forme di tumori in due o più persone della stessa famiglia
Comparsa precoce del tumore in una o più persone della stessa famiglia
Tumori bilaterali in organi doppi
Tumori primari multipli nello stesso individuo
Trasmissione autosomica dominante della suscettibilità allo sviluppo di tumori
http://www.sanger.ac.uk/genetics/CGP/Census/
Molecular signatures for specific cancers
~350 cancer genes/ ~ 25000 total genes
microRNA e tumori
microRNA come oncogeni e geni oncosoppressori
miRNA profiling come STRUMENTO CLINICO
DIAGNOSIS
Metastatic cancer of
unknown primary site
PROGNOSIS
Disease identification
mRNA profiling
miRNA profiling
Disease evolution
Good prognosis
miRNA profiling
Bad prognosis
potere predittivo
La terapia anti-tumorale
Chemio/radio-terapia (induzione di morte cellulare)
Inibitori di tirosine-chinasi
Agenti differenzianti (es. agenti demetilanti il DNA, retinoidi etc.)
Terapie farmacologiche (es. inibitori dell’angiogenesi, inibitori del
proteasoma, etc)
Vettori per “target-are” nel tumore farmaci o geni
Terapia genica (es. trasduzione di p53)