Introduzione
La sindrome di Lynch (LS) è una malattia ereditaria a
trasmissione autosomica dominante, che determina la
predisposizione a sviluppare un cancro del colon retto
(CRC) (LS I) e/o in altre sedi (LS II) con un’incidenza
stimata del 2.8% -3.6% dei CRC ed una prevalenza
di 1:500, 1:1000 (1-4).
Nel 1966 Lynch ha pubblicato il primo lavoro in cui descrive due famiglie con le caratteristiche della sindrome, definita anche Hereditary Non Polyposis Colorectal
Cancer (HNPCC). Nel 1991 un gruppo internazionale
di ricercatori ha stilato i Criteri di Amsterdam I per definire in quali pazienti sospettare la sindrome, successivamente modificati nel 1999, con l’aggiunta dei tumori
extracolici (tabella 1). Nel corso degli anni 90 è stata
chiarita la patogenesi della sindrome, causata da una
mutazione a carico dei geni del mismatch repair (principalmente MLH1 ed MSH2, meno frequentemente
MSH6, più raramente PMS2) che codificano per proteine coinvolte nell’identificazione e riparazione degli errori
di mismatch del DNA. Una mutazione a carico di questi
geni determina un fenotipo di replicazione degli errori
nella cellula, inizialmente definito RER, oggi, più propriamente, instabilità dei microsatelliti (MSI) (4,5). Circa
RL
Vittoria Stigliano
Lupe Sanchez-Mete
U.O. Gastroenterologia
ed Endoscopia Digestiva
Istituto Nazionale Tumori
Regina Elena di Roma
il 40% dei casi di famiglie con criteri di Amsterdam positivi non presenta fenotipo MSI sul tessuto tumorale,
ed è definito stabile (MSS) all’analisi dei microsatelliti.
Secondo recenti linee guida, queste forme familiari sono oggi classificate Familial Colorectal Cancer Type X.
Si riserva la definizione di LS solo ai soggetti portatori di
mutazione germinale a carico dei geni del MMR (4-6).
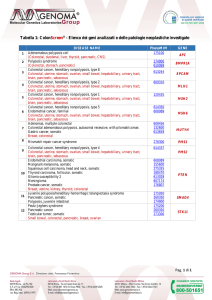
tab. 1: criteri di Amsterdam II (1999) (6)
Almeno 3 familiari con CRC o cancri
nello spettro della LS (endometrio,
piccolo intestino, uretere, pelvi renale)
Ciascun familiare deve essere di I grado
rispetto agli altri due
Almeno due generazioni successive
devono essere affette
Almeno un tumore deve essere diagnosticato
prima dei 50 anni
Giorn Ital End Dig 2010;33:263-267
La sindrome di Lynch (LS)
è una malattia ereditaria associata
a mutazione a carico dei geni
del mismatch repair. I pazienti affetti
da tale sindrome hanno un aumentato
rischio di sviluppare un cancro
del colon retto e tumori extracolici
multipli in età giovanile (dall’età
di 20-25 anni), con carcinogenesi
accelerata. L’identificazione dei pazienti
affetti da tale patologia è necessaria
per la definizione di adeguati
programmi di sorveglianza clinica.
Revisione della Letteratura
> rassegna biennale
Cancro colorettale
ereditario: la sindrome
di Lynch
Se presenti CRC la FAP deve essere esclusa
263
RL
Revisione della Letteratura
> rassegna biennale
Identificazione dei soggetti
a rischio e diagnosi
Dalla definizione dei criteri di Amsterdam sono state
messe a punto varie metodologie di screening clinico
e molecolare per identificare e selezionare i soggetti
da sottoporre a test genetico in termini di un ottimale
costo-efficacia.
Il sospetto diagnostico di LS sorge nel caso di pazienti
affetti da CRC in età giovanile (<50 anni) e/o con storia
familiare positiva oppure in pazienti con cancri primitivi multipli del colon e/o altri organi nello spettro della
sindrome (1,4).
Nel 1997 un gruppo di ricercatori ha messo a punto i
Criteri di Bethesda, successivamente revisionati e modificati nel 2004 (tabella 2), utilizzati per identificare i
tab. 2: criteri di Bethesda modificati (2004) (6)
1. Soggetto affetto da CRC di età < 50 anni
2. Presenza di CRC o altri tumori associati alla LS
(endometrio, stomaco, ovaio, pancreas, uretere,
pelvi renale, vie biliari, cervello, piccolo intestino,
adenomi ghiandole sebacee, cheratoacantomi),
sincroni e/o metacroni, indipendentemente dall’età
3. CRC con fenotipo MSI-H diagnosticato in soggetto
di età < 60 anni
4. Paziente con CRC ed un parente di I grado
con tumori associati alla LS, con uno dei cancri
diagnosticati in età < 50 anni
5. Paziente con CRC con due o più parenti di I grado
con tumori associati alla LS, indipendentemente
dall’età
Vittoria Stigliano et al > La sindrome di Lynch
264
pazienti da sottoporre ad analisi dell’instabilità dei
microsatelliti su tessuto tumorale. Tali criteri, sebbene abbiano un’alta sensibilità (fino al 94%), non sono
sufficientemente specifici (fino al 51%) per garantire
un’adeguata selezione. I criteri di Amsterdam II viceversa hanno un’alta specificità (fino al 98%) ma bassa
sensibilità (fino al 65%) (4,6-8).
In considerazione dei limiti diagnostici di tali criteri,
sono stati recentemente messi a punto dei modelli
predittivi che permettono di calcolare la probabilità
di un determinato paziente di essere portatore di una
mutazione a carico dei geni del MMR. Ad oggi sono stati messi a punto 5 modelli principali: il Leiden,
l’MMR predict, il PREMM 1,2 l’MMRpro ed il modello AIFEG. Un recente studio canadese (7) ha per
la prima volta messo a confronto i primi 4 dei suddetti
5 modelli su 725 pazienti consecutivi con diagnosi di
CRC in età <75 anni, indipendentemente dal rischio
familiare. I risultati sono stati corretti per la dimensione di ciascuna delle famiglie esaminate, in considerazione del fatto che le famiglie più ampie e informative
possono avere una stima del rischio più elevata. Il
test con la migliore performance è risultato l’MMR
predict (sensibilità 94%, specificità 91%). Infatti, in
questo studio la stima dei pazienti da sottoporre a
indagine genetica valutata con MMR predict, rispetto
alla valutazione con i criteri di Bethesda, si è ridotta
dal 50% all’11%, con un notevole abbattimento dei
costi e tempi di diagnosi. Il limite di questi modelli è
che forniscono bassi valori di rischio per i portatori di
mutazioni a carico dei geni MSH6 e PMS2.
Lo screening basato sull’anamnesi (Criteri di Amsterdam e Bethesda, modelli predittivi) permette di selezionare i pazienti da sottoporre a screening tissutale
per identificare quelli a cui effettuare il test genetico
(più indaginoso e costoso). I test di screening molecolare su tessuto ricercano due caratteristiche tipiche
della sindrome, determinate dalla mutazione dei geni
del MMR, con metodiche semplici e a basso costo:
l’analisi dell’instabilità dei microsatelliti (MSI) e la ricerca della perdita di espressione della proteina corrispondente al gene mutato mediante l’analisi immunoistochimica (IHC) (5). In un recente studio Hampel
e De la Chapelle (9), hanno esaminato 550 soggetti
consecutivi affetti da CRC indipendentemente dal rischio familiare e li hanno sottoposti a screening molecolare su tessuto (IHC e MSI). I risultati dello studio
dimostrano l’utilità di questi test nell’identificare la LS
anche in soggetti a rischio intermedio per CRC, infatti il 3.8% dei pazienti con CRC esaminati, è risultato
portatore di mutazione a carico di un gene del MMR,
il 100% dei tumori di pazienti portatori di mutazione è
risultato MSI-H (oltre il 30% di instabilità nei marcatori
esaminati) e nel 94% dei casi il gene mutato era stato
correttamente identificato dall’analisi immunoistochimica. Da rilevare che, se fossero stati utilizzati i Criteri
di Bethesda per selezionare i pazienti, il 28% dei casi
di LS non sarebbe stato diagnosticato.
A tutt’oggi non vi è consenso sull’utilità di proporre i test
di screening tissutali a tutti i pazienti operati per CRC.
Nella pratica clinica i modelli predittivi dovrebbero essere utilizzati come test di pre-screening sui pazienti
affetti da CRC per selezionare quelli da sottoporre a
test molecolare tissutale, indipendentemente dall’età
e dal rischio familiare. Solo i pazienti con analisi molecolare suggestiva per LS (MSI –H ed alterata espressione delle proteine del MMR su tessuto) dovrebbero
essere sottoposti al test genetico, con un notevole abbattimento di costi e riduzione dei tempi di diagnosi.
RL
Revisione della Letteratura
> rassegna biennale
Sorveglianza per il cancro del colon
Nei soggetti portatori di mutazione a carico dei geni del
MMR, il rischio lifetime di sviluppare un cancro del colon retto (CRC) è stato stimato essere fino all’80% vs il
5% della popolazione generale (tabella 3). Tali soggetti
devono pertanto essere sottoposti ad una sorveglianza endoscopica intensiva. Una recente consensus (5)
sul management clinico della LS, sottolinea l’efficacia
della sorveglianza endoscopica che, come dimostrato
in numerosi trials, determina una significativa riduzione
del rischio e della mortalità per CRC. Studi osservazionali (10,11) hanno dimostrato l’insorgenza di cancri
intervallari a 2 anni dopo una precedente pancolonscopia negativa, pertanto le linee guida correnti hanno ridotto l’intervallo di sorveglianza ad 1-2 anni. In particolare, quelle del NCCN (National Comprehensive Cancer
Network) del 2010 indicano di effettuare, nei soggetti
portatori di mutazione a carico dei geni del MMR, una
colonscopia ogni 1-2 anni dall’età di 20-25 anni o 10
anni prima del caso più giovane in famiglia (12).
Ad oggi non è ancora chiaro in quali soggetti effettuare
una colonscopia ogni anno invece che ogni due. Negli
ultimi due anni tre importanti studi hanno riconsiderato
la problematica. A tal proposito, Vasen et al (13) in un
recentissimo studio hanno selezionato dal registro tumori ereditari dei Paesi Bassi, tutti i pazienti con criteri di
Amsterdam II positivi e li hanno divisi in due gruppi, pazienti affetti da LS e pazienti con CRC familiare di tipo X e
sottoposti a colonscopia ogni 2 anni. Il 4,4% dei soggetti
con LS ha presentato un CRC (90% Dukes A o B, 62%
nel colon destro) vs l’1,7% dei pazienti con CRC familiare di tipo X. È stato inoltre evidenziato un trend di rischio
aumentato, non statisticamente significativo, nei soggetti
con età >40 anni e mutazione a carico dei geni MLH1 o
MSH2. Engel C et al (14) sostengono invece la necessità
di effettuare una colonscopia di sorveglianza annuale in
soggetti con LS ed evidenziano una frequenza di CRC
a 1 anno dell’1.8%. Gli autori non hanno riscontrato
differenze significative in relazione al gene mutato ma,
come atteso, hanno evidenziato un aumento del rischio legato all’età, sia in soggetti con LS e pregresso
CRC sia in soggetti mutati senza pregresso CRC. Da
rilevare che lo studio è limitato dal breve periodo di
follow-up considerato (48 mesi).
Al momento attuale non c’è consensus se effettuare
una sorveglianza annuale o biennale. In tutti i pazienti
devono essere considerati i seguenti fattori: la compliance, l’età, la mutazione associata alla sindrome e,
ovviamente, la presenza di adenomi intervallari.
Sorveglianza per tumori extracolici
Il rischio lifetime di sviluppare tumori extracolici nei
soggetti portatori di mutazione è stimato essere circa il
37.5% (15). Organi bersaglio individuati sono l’endometrio, l’ovaio, l’urotelio, lo stomaco il piccolo intestino, il
cervello, il tratto bilio-pancreatico (tabella 3). Nella popolazione generale il rischio di sviluppare un tumore in tali
organi è stimato essere molto basso (<1%-2,7%). Studi
precedenti hanno riportato un rischio cumulativo per tumori extracolici maggiore nei soggetti portatori di mutazione MSH2 vs MLH1 e delle donne rispetto agli uomini
(15). L’endometrio è risultato l’organo più colpito con un
rischio di cancro stimato del 20-60% che aumenta fino al 71% dei casi nelle portatrici di mutazione a carico
del gene MSH6. Il rischio per cancro dell’ovaio sembra
essere più basso (9-12%). La sorveglianza ginecologica
è stata ormai standardizzata dalle linee guida internazionali (12) e prevede un videat ginecologico ed un’ecografia trans vaginale con prelievo endometriale (endocyte)
ogni anno, sebbene
ad oggi non ne sia
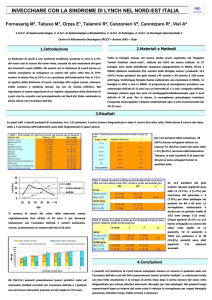
tab. 3: rischio di cancro fino all’età di 70 anni nei soggetti con HNPCC
stata dimostrata
confrontati con la popolazione generale*
l’efficacia in termini
di migliore sopravHNPCC
(LS)
vivenza (15,16). Il
Rischio popolazione
Cancro
cancro dell’urotelio
generale
Rischio
Età media diagnosi
è stimato essere fiColon
5.9%
80%
44 anni
no al 28% dei casi
e la sorveglianza
Endometrio
2.7%
20-60%
46 anni
annuale mediante
Stomaco
< 1%
11-19%
56 anni
esame chimico, ciOvaio
1.6%
9-12%
42.5 anni
tologico delle urine
associato o meno
Fegato e vie biliari
< 1%
2-7%
Non riportato
ad ecografia addoVie urinarie
< 1%
4-5%
55 anni
minale è fortemente
consigliata (15,16).
Piccolo intestino
< 1%
1-4%
49 anni
Il rischio lifetime di
Cervello
< 1%
1-3%
50 anni
cancro gastrico è
* da Kohlmann W, Gruber SB. HNPCC. In GeneReviews. www.genetests.org
Giorn Ital End Dig 2010;33:263-267
Programmi di sorveglianza
265
RL
Revisione della Letteratura
> rassegna biennale
riportato dal 1.6% al 19% dei casi, con alta incidenza
nei paesi asiatici (15,17). Recenti studi (17,18) hanno
dimostrato che il cancro gastrico associato alla LS è
di tipo intestinale, MSI-H e non esprime all’IHC il gene
del MMR corrispondente alla mutazione germinale. Le
linee guida internazionali (12) sono sempre più concordi nell’effettuare una gastroscopia ogni 1-3 anni nei
soggetti portatori di mutazione.
Non c’è ancora consenso invece sulla sorveglianza
del piccolo intestino, sebbene recenti studi riportino
un rischio di cancro lifetime del 2,5%-7,2% (15,19,20).
Negli ultimi anni, studi prospettici di confronto tra metodiche radiologiche hanno dimostrato la validità delle
videocapsula endoscopica nella sorveglianza del piccolo intestino in pazienti affetti da poliposi familiare
(adenomatosa e amartomatosa) seguita dall’enteroscopia in casi selezionati (21,22). Per stabilire la validità in termini di costo-efficacia della sorveglianza del
tenue nella LS sono però necessari ulteriori studi.
Il rischio lifetime di cancro del pancreas è ancora dubbio. Due recenti studi (23,24), riportano un rischio lifetime fino al 4% (vs 0,5% nella popolazione generale),
con rischio relativo elevato tra i 20 ed i 40 anni. La
diagnosi precoce del cancro del pancreas rappresenta ancora oggi un obbiettivo difficile da raggiungere. Numerose tecniche di imaging radiologiche sono
state valutate nel corso degli anni, con sensibilità variabili: ecografia addome 67%; TC 77%; PET TC 9095%; ecoendoscopia (EUS) 99%; RMN 87.5% con
incremento dell’accuratezza diagnostica fino al 90%
nella colangio-RMN. Ad oggi l’EUS sembra essere la
metodica con maggiore sensibilità e specificità rispetto ad altre ed ha il vantaggio di avere un alto valore
predittivo negativo, che raggiunge quasi il 100% (24).
Trattamento chirurgico
Vittoria Stigliano et al > La sindrome di Lynch
266
La decisione riguardante il trattamento chirurgico da
offrire ai pazienti con CRC e LS è difficile. In tali pazienti il rischio di sviluppare un CRC metacrono è alto
per cui nella scelta della strategia chirurgica i trattamenti proposti sono la resezione segmentaria del colon o la colectomia totale con ileo-retto-anastomosi.
Quest’ultima è stata proposta come trattamento di
scelta, in quanto un’estesa colectomia potrebbe ridurre il rischio di cancri metacroni. Nel confrontare tali
metodiche è necessario considerare, oltre la sopravvivenza anche la qualità di vita del paziente. Infatti, i
pazienti sottoposti a colectomia totale possono presentare 5 o più scariche diarroiche durante il giorno
e nel 30% dei casi si associa incontinenza diurna o
notturna. Dopo una colectomia segmentaria vi sono
invece minime variazioni dell’alvo (25,26).
Maeda et al in un recente lavoro (25), confrontano i
due tipi di intervento chirurgico tra pazienti con CRC
e LS e riportano le differenze nella sopravvivenza
e nella qualità di vita. Tale studio dimostra che, nei
pazienti giovani, sopravvivenza e qualità di vita sono
approssimativamente equivalenti nei due tipi di intervento, mentre, nei pazienti di età >50 anni la colectomia segmentaria diventa l’intervento di prima scelta.
In entrambi i casi il paziente dovrà essere sottoposto
ad un follow-up intensivo. Studi effettuati nello stesso anno (1,26,27) confermano tali raccomandazioni
e suggeriscono, inoltre, di considerare nelle donne
affette da LS, la possibilità di effettuare un’isterectomia totale profilattica dopo la decisione di non avere
ulteriori gravidanze.
Chemioprevenzione
Vari studi sono stati pubblicati sulla chemioprevenzione nel CRC. Tra i vari farmaci utilizzati, l’aspirina ed i
suoi analoghi (inibitori della COX-2) sembrano avere la
migliore efficacia, determinando una possibile riduzione del rischio di sviluppare adenomi o cancro colorettale. Burn, nel 2008, ha pubblicato lo studio CAPP2
(28) sulla chemioprevenzione con aspirina (600 mg/
die) e amidi resistenti (30 g/die) in 937 pazienti affetti
da LS. L’analisi post-trial (3), dopo 10 anni di followup, ha evidenziato una riduzione significativa dell’incidenza di nuovi CRC e tumori dell’endometrio. I dati
di tale studio, sicuramente a tutt’oggi l’unico valido
sull’argomento, indicano la possibilità di un nuovo e
preventivo approccio nella LS.
Sopravvivenza
I pazienti con CRC affetti da LS sembrerebbero avere una migliore prognosi rispetto ai CRC sporadici. In
passato sono stati pubblicati numerosi studi contraddittori sulla sopravvivenza nei soggetti affetti da tale
sindrome, ma due recenti studi italiani (29,30) sul confronto tra pazienti operati per CRC con o senza LS,
sembrano confermare tale dato.
Questa differenza è dovuta a caratteristiche peculiari
della neoplasia: l’instabilità dei microsatelliti, che determina una riduzione dell’espressione dell’endothelial growth factor e quindi della densità microvascolare con una bassa tendenza a sviluppare metastasi
a distanza e un difetto del MMR che, verosimilmente
provoca l’accumulo di mutazioni nei geni necessari alla
sopravvivenza della cellula neoplastica riducendone la
vitalità. Tale ipotesi riportata nel lavoro di Russo et al (30)
dovrà essere confermata in futuro da ulteriori studi.
RL
Corrispondenza
Vittoria Stigliano
U.O. Gastroenterologia ed Endoscopia Digestiva
Istituto Nazionale Tumori Regina Elena
Via Elio Chianesi, 53 - 00144 Roma
Tel. + 39 06 52665015
Fax + 39 06 52666259
e-mail: [email protected]
Bibliografia
1.Jasperson KW, Tohy TM, Neklason DW et al. Hereditary and
familial colon cancer. Gastroenterology 2010;138:2044-2058.
2.Rahner N, Steinke V, Schlegelberger B et al. Clinical utility gene
card for: Lynch syndrome (MLH1, MSH2, MSH6, PMS2). European
Journal of Human Genetics (2010) advance online publication, 27
January 2010.
3.Boland CR, Shike M. Report from the Jerusalem Workshop on
Lynch syndrome-hereditary non polyposis colorectal cancer.
Gastroenterology 2010;138:2197.e1-2197.e7.
4.Lynch HT, Lynch PM, Lanspa SJ et al. Review of the Lynch
syndrome: history, molecular genetics, screening, differential
diagnosis and medico-legal ramifications. Clin Genet
2009;76:1-18.
5.Boland CR, Ajay G. Microsatellite instability in colorectal cancer.
Gastroenterology 2010;138:2073-87.
6.Vasen HFA, Möslein G, Alonso A et al. Guidelines for the clinical
management of Lynch syndrome (HNPCC) J. Med. Genet.
published online 30 Mar 2007.
7.Green RC, Parfrey PS, Woods MO et al. Prediction of Lynch
syndrome in consecutive patients with colorectal cancer. J Natl
Cancer Inst 2009;101:331-340.
8.Barrow E, Alduaji W, Robinson L et al. Colorectal cancer in HNPCC:
cumulative lifetime incidence, survival and tumour distribution.
A report of 121 families with proven mutation. Clin Genet
2008;74:233-42.
9.Hampel H, Frankel WL, Martin E et al. Feasibility of screening
for Lynch syndrome among patients with colorectal cancer. J Clin
Oncol 2008 26:5783-88.
10.Vasen HF, Nagengast FM, Khan PM et al. Interval cancers in
Hereditary non-polyposis colorectal cancer (Lynch syndrome).
Lancet 1995;345:1183-84.
11.Mecklin JP, Aarnio M, Laara E et al. Development of colorectal
tumours in colonoscopic surveillance in Lynch syndrome.
Gastroenterology 2007;133:1093-8.
12.National Comprehensive Cancer Network (NCCN) Practice
Guidelines in Oncology_ Lynch Syndrome, version 1.2010.
Online at www.nccn.org.
13.Vasen HF, Abdirahman M, Brohet R et al. One to 2-year
surveillance intervals reduce risk of colorectal cancer
in families with Lynch syndrome. Gastroenterology
2010;138:2300-6.
14.Engel C, Rahner N, Schulmann K et al. efficacy of annual
colonoscopic surveillance in individuals with hereditary
non polyposis colorectal Cancer. Clin Gastroenterol Hepatol
2010;8:174-182.
15.Barrow E, Robinson L, Alduaji W et al. Cumulative lifetime
incidence of exracolonic cancers in Lynch syndrome: a
report of 121 families with proven mutation. Clin Genet
2009;75:141-149.
16.Koornstra JJ, Mourits MJE, Sijmons RH et al. Agement of
extracolonic tumours in patients with Lynch syndrome. Lancet
Oncol 2009;10:400-8.
17.Gylling A, Abdel-Rahman WM, Juhola M et al. Is gastric cancer
part of the tumour spectrum of Hereditary Non-polyposis colorectal
cancer? A molecular genetic study. Gut 2007;56:926-33.
18.Capelle LG, Van Grieken NCT, Lingsma HF et al. Risk and
epidemiological time trends of gastric cancer in Lynch syndrome
carriers in the Netherlands. Gastroenterology 2010;138:487-92.
19.Ten Kate GL, Kleibeuker JH, Nagengast FM et al. Is surveillance
of the small bowel indicated for Lynch syndrome families? Gut
2007;56;1198-201.
20.Koornstra JJ, Kleibeuker JH, Vasen HF. Small-bowel cancer in
Lynch syndrome: is it time for surveillance? Lancet Oncol 2008
Sep;9(9):901-5.
21.Günther U, Bojarski C, Buhr HJ et al. Capsule endoscopy in smallbowel surveillance of patients with hereditary polyposis syndromes.
Int J Colorectal Dis. 2010 Jun 11.
22.Schulmann K, Hollerbach S, Kraus K et al. Feasibility and diagnostic
utility of video capsule endoscopy for the detection of small bowel
polyps in patients with hereditary polyposis syndromes. Am J
Gastroenterol. 2005 Jan;100(1):27-37.
23.Kastrinos F, Mukherjee B, Tayob N et al. risk of pancreatic cancer in
families with Lynch syndrome. JAMA 2009;302(16):1790-95.
24.Saftoiu A, Vilmann P. Role of endoscopic ultrasound in the
diagnosis and staging of pancreatic cancer. J Clin Ultrasound
2009Jan;37(1):1-17.
25.Maeda T, Cannom RR, Beart Jr RW et al. Decision model
of segmental compared with total abdominal colectomy
for colon cancer in Hereditary Nonpolyposis Colorectal
Cancer. J Clin Oncol 2010 28;1175-80.
26.Natarajan N, Watson P, Silva-Lopez E et al. Comparison of extended
colectomy and limited resection in patients with Lynch syndrome.
Dis Colon Rectum 2010;53 (1):77-82.
27.Baglietto L, Lindor NM, Dowty JG et al. Risks of Lynch syndrome
cancers for MSH6 mutation carriers. J Natl Cancer Inst
2010;102:193-201.
28.Burn J, Bishop T, Mecklin JP et al. Effect of aspirin or resistant
starch on colorectal neoplasia in the Lynch syndrome. N Engl Med
2008;359:2567-78.
29.Stigliano V, Assisi D, Cosimelli M et al. Survival of Hereditary
non polyposis colorectal cancer patients compared with
sporadic colorectal cancer patients. J Exp Clin Cancer Res
2008;27:39-43.
30.Russo A, Sala P, Alberici P et al. Prognostic relevance of MLH1
and MSH2 mutations in hereditary non-polyposis colorectal cancer
patients. Tumori 2009;95:731-738.
Giorn Ital End Dig 2010;33:263-267
Revisione della Letteratura
> rassegna biennale
267