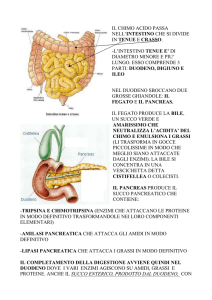
Cenni di fisiopatologia dell’ Apparato gastro‐intestinale La digestione e il successivo assorbimento dei nutrienti introdotti con la dieta dipendono da due meccanismi, l'azione di ormoni ed enzimi digestivi e da movimenti peculiari del tubo gastroenterico. Quest'ultimo è costituito, con piccole variazioni distrettuali, dalle seguenti strutture: 1) Sierosa 2) Muscolatura longitudinale 3) Plesso mioenterico di Auerbach (motorio) 4) Muscolatura circolare 5) Plesso sottomucoso di Meissner (sensoriale) 6) Muscolaris mucosae 7) Sottomucosa 8) Mucosa Esistono quindi 3 strati di tessuto muscolare liscio di differente orientamento, responsabile dei movimenti peristaltici e due plessi nervosi che combinati con l'azione del sistema nervoso autonomo determinano questo movimento e la conseguente progressione del cibo. Dalla bocca allo stomaco Dopo l'azione triturante dei denti il primo enzima che si incontra è l'amilasi salivare o ptialina che inizia una prima digestione dei carboidrati. La saliva secerne inoltre immunoglobuline della classe IgA (dette appunto salivari), fondamentali per l'immunità acquisita del neonato. Le IgA sono prodotte anche dalle cellule mucosali dell'apparato respiratorio e dagli enterociti; deficit primario di questi anticorpi determinano patologie a carico dell'apparato respiratorio e gastro‐intestinale, così come la xerostomia ( letteralmente: bocca secca), sintomo tipico della sindrome di Sjiogren, provoca infezioni a carico del cavo orale. La produzione di saliva dalle tre ghiandole deputate (parotide, sottomandibolare e sottolinguale) è determinata dal nervo vago ed ogni sostanza che blocca l'azione colinergica ( come l'atropina, potente inibitore del sistema parasimpatico) ha come effetto la soppressione dello stimolo salivare. Con l'azione volontaria della deglutizione il contenuto viene spinto verso l'esofago da onde peristaltiche sino ad arrivare nella regione cardiale dove il plesso mioenterico ha il compito di rilasciare l'omonimo sfintere e permettere l'ingresso del cibo (ormai bolo) all'interno dello stomaco. L'assenza congenita del plesso di Auerbach in questo o in altri distretti del tubo gastro‐intestinale determina l'arresto della progressione del contenuto e viene conosciuta con il termine di acalasia; a carico dell'intestino crasso questa paralisi determina il megacolon congenito. Nello stomaco il bolo subisce l'azione del succo gastrico, mescolanza di ormoni, enzimi digestivi ed acidi, ognuno dei quali prodotto da una specifica popolazione di cellule: Gastrina.......................cellule dell'antro Pepsina............... …....cellule principali Muco...........................cellule della regione pilorica e del collo delle ghiandole gastriche Acido cloridrico..........cellule del fondo e della porzione parietale delle ghiandole gastriche. Fattore intrinseco Lipasi gastrica La gastrina è un ormone che ha l'azione di favorire la produzione di acido cloridrico, di pepsina, del fattore intrinseco ed enzimi del succo pancreatico; aumenta sotto l'influenza del sistema vagale ed è inibito dal simpatico, inoltre influenza la motilità gastrica e quella intestinale. La pepsina, enzima proteolitico e l'acido cloridrico svolgono la principale azione digestiva; come già detto sono sotto il controllo della gastrina. Il muco è costituito da una glicoproteina e svolge la ben nota azione protettiva sulle cellule della mucosa gastrica; la sua produzione è influenzata dai glucorticoidi. Il fattore intrinseco è una mucoproteina fondamentale per l'assorbimento della Vit. B12, un suo deficit provoca anemia megaloblastica. Questi fattori sono comunque in rapporto continuo con influenze di varia provenienza: emozionali (l'odore, la presenza del cibo, oppure la paura o l'ansia), gastriche (la presenza stessa del bolo all'interno dello stomaco stimola la produzione di gastrina), intestinali (il duodeno produce un ormone, l'enterogastrone che inibisce la produzione di gastrina se sono presenti grassi o carboidrati), alimentari (alcool e caffeina sono noti stimolatori della secrezione gastrica). Malattie legate all'alterato equilibrio tra gli elementi costituenti il succo gastrico sono l'ulcera peptica e la sindrome di Zollinger Ellison, quest'ultima provocata da un adenoma pancreatico gastrina secernente. Discussa è l'azione della gastrina nella patogenesi del cancro gastrico o gli effetti dell'ipergastrinemia dopo inibizione della secrezione peptica indotta dagli inibitori di pompa protonica (si ricordi che la loro azione si esplica sul blocco del trasporto attivo degli ioni H+ e Cl‐
dalle cellule parietali al lume gastrico). Intestino tenue, pancreas esocrino, fegato Il bolo, da qui chiamato chimo, dallo stomaco viene letteralmente spruzzato nella porzione di duodeno che si chiama bulbo ed è questa la sede preferita dell'ulcera duodenale. Qui si mescola con la bile e il succo pancreatico e prosegue con l'intestino tenue. Per convenzione si denomina digiuno i primi 2/5 a partire dal legamento del Treitz, ileo i successivi 3/5 sino alla valvola ileo‐
cecale. La lunghezza media del tenue è di circa 3 metri. La mucosa presenta aspetti peculiari: sono presenti numerosi noduli linfatici e a livello dell'ileo formano grappoli denominati placche di Peyer; inoltre lungo tutta la lunghezza alla radice dei villi che tappezzano le cellule mucosali e ne aumentano l'area di assorbimento fino a 300 mq (grazie anche alla presenza di ulteriori microvilli lungo il perimetro) esistono delle ghiandole tubulari semplici (cripte di Lieberkuhn) secernenti enzimi proteolitici, disaccaridasi (lattasi, maltasi) ed altri deputati alla scissione degli acidi nucleici Le cellule enterocromaffini stimolate dall'aumento della pressione intraluminale secernono serotonina la cui azione sembra quella di abbassare la soglia di attivazione dei recettori del plesso sottomucoso, favorendo così l'onda peristaltica; a livello duodenale le ghiandole tubulari del Brunner secernono muco che ha l'azione di proteggere le cellule del bulbo dal contenuto gastrico. Come per lo stomaco la motilità intrinseca del tenue è sotto il controllo dei plessi mioenterico e sottomucoso, entrambi scarsamente modificabili dall'azione stimolante vagale e inibente del simpatico. La resezione o l'impedimento della sua funzione di ampi tratti di tenue conduce ad una sindrome da malassorbimento tanto più grave quanto è il tratto interessato; l'abnorme presenza di grassi e proteine indigerite in questi pazienti è la causa della steatorrea che si riscontra nelle feci. Da ricordare la sprue tropicale, la malattia celiaca o enteropatia da glutine (nota 1), il morbo di Crohn e l'azione dei farmaci citotossici per blocco del turn over cellulare che nel tenue si rinnova ogni 2‐3 giorni. Nei traumi o nelle infezioni importanti la motilità intestinale si blocca e si assiste all'ileo paralitico; l'ileo meccanico è provocato invece da ostacoli alla progressione del chimo: entrambe sono condizioni gravi, nel secondo caso il dolore intenso e l'attivazione dei sintomi vagali (sudorazione profusa, ipotensione, vomito fecaloide) possono orientare la diagnosi. Pancreas E’ una ghiandola alveolare composta che secerne nella sua porzione esocrina numerosi enzimi digestivi, i più importanti di natura proteolitica (tripsina, chimo tripsina) ma non mancano amilasi, lipasi ed enzimi che aggrediscono gli acidi nucleici ed anioni bicarbonato, solfato e fosfato che alzano il pH del chimo a livelli alcalini. Questi enzimi si attivano nel duodeno ad opera delle enterochinasi; un’attivazione intrapancreatica indotta per esempio dalla bile per ostruzione delle vie comuni di escrezione provoca pancreatite con immissione in circolo di grandi quantità di enzimi. La funzione escretrice del pancreas è sotto il controllo di due ormoni secreti dalla mucosa duodenale sotto l’effetto del contenuto acido che proviene dallo stomaco. Questi ormoni sono due polipeptidi, la secretina e la pancreozimina. La prima induce produzione di succo pancreatico alcalino povero di enzimi, la seconda stimola la secrezione di grandi quantità di enzimi; entrambe le sostanze agiscono anche sull’immissione in duodeno della bile incrementano la produzione di insulina dalle beta cellule della porzione endocrina. Anche il pancreas è condizionato dal sistema vagale che ha un’azione stimolatrice, e già come avviene nello stomaco, bloccato dall’atropina. La particolare collocazione anatomica del pancreas, accerchiato da strutture come il duodeno, il fegato, le vie biliari extraepatiche determina, quando è affetto da processi infiammatori o neoplastici, ittero da ostruzione o processi flogistici essudativi a carico dei tessuti circostanti. Fegato Elencare le innumerevoli funzioni di questa ghiandola richiederebbe una trattazione a parte, nello specifico ci occuperemo di quelle attività legate principalmente alla digestione degli alimenti e in quel meccanismo che va sotto il nome di circolo entero‐epatico. La bile, secreta dagli epatociti scorre lungo le vie intraepatiche sino al dotto epatico comune; dopo l’incontro con il dotto cistico si deposita e si concentra nella cistifellea. Di qui, sotto l’effetto della colecistochinina, polipeptide con struttura identica alla pancreozimina, prosegue lungo il coledoco sino all’ampolla di Vater. Qui una struttura valvolare, lo sfintere di Oddi, regola il flusso all’interno del lume intestinale. La bile è una soluzione complessa che contiene in gran parte acqua, acidi biliari (acido colico, desossicolico, chinodessosicolico e litocolico) coniugati con sali di Na e K e con aminoacidi come la glicina e la taurina, pigmenti derivati dalla scissione dell’emoglobina (bilirubina e biliverdina), colesterolo, e in misura minore sali inorganici e un fosfolipide, la lecitina, quest’ultima con la funzione di abbassare la tensione superficiale; lecitina entra anche nella composizione del surfattante polmonare. I Sali biliari così coniugati (glicocolico e taurocolico) hanno la funzione di complessare in micelle ed emulsionare i lipidi intestinali preparandoli alla digestione da parte dei villi intestinali e attraverso il vaso chilifero del villo stesso entrano come chilomicroni nel sistema linfatico e di lì nel torrente circolatorio. La mancanza della bile nel lume intestinale provoca steatorrea e acolia fecale. Queste caratteristiche sono tipiche delle malattie infiammatorie (epatite) e particolarmente di quelle ostruttive (colestasi). In questo ultimo caso la causa può essere l’alterata composizione della bile con formazione di calcoli di colesterolo (squilibrio tra Sali biliari, lecitina e colesterolo) o di bilirubinato di Calcio (per deconiugazione dalla beta glicuronidasi da parte dei batteri e formazione di bilirubina libera). La bilirubina è responsabile anche della formazione dell’ittero cutaneo e sclerale quando la sua concentrazione supera i 2 mg/100ml. Si parla di itteri a bilirubina diretta o coniugata quando è coinvolto il fegato, di ittero a bilirubina indiretta o non coniugata nelle malattie ad aumentata distruzione dei globuli rossi o nei difetti di coniugazione (ittero del neonato) Circolo entero‐epatico: alcune delle sostanze contenute nella bile vengono riassorbite dal lume intestinale e attraverso il sistema portale ritornano al fegato; qualcosa di simile avviene a carico del rene da parte del sistema tubulare. In particolare viene riassorbita la quota non coniugata della bilirubina e gli urobilinogeni, derivati della digestione batterica della bilirubina; questi ultimi sono in parte assorbiti e riescreti dal fegato, in parte entrano nel circolo sanguigno e compaiono nell’urina caratterizzandone il colore. Anche i sali biliari, dopo che hanno svolto la loro azione emulsionante vengono riassorbiti a livello dell’ileo e riescreti dal fegato con la bile. Solamente il 5% non viene riassorbito; nel morbo di Crohn, o ileite regionale, una delle cause della diarrea è l’azione irritante dei sali biliari non riassorbiti per le lesioni della mucosa dell’ileo terminale Colon La funzione principale del grosso intestino è il riassorbimento dell’acqua e dei sali minerali. La valvola ileo‐cecale sotto l’influsso delle onde peristaltiche si rilascia e permette l’ingresso di piccole quantità di chimo; maggiore quantità sono liberate quando il cibo lascia lo stomaco per un’azione riflessa (riflesso gastro‐colico). Nel colon la muscolatura longitudinale è raccolta in tre nastri che ne accorciano la lunghezza e le conferiscono l’aspetto austrato; la mucosa non contiene villi e le ghiandole sono piccole introflessioni della mucosa produttrici di muco; follicoli linfatici sono presenti nella regione cecale e in particolare nell’appendice. Il colon non produce alcun enzima digestivo La motilità è sostenuta dai plessi mioenterici e sottomucosi, la loro assenza provoca il morbo di Hirschsprung o megacolon agangliare; nel 40% dei soggetti si associa distensione vescicale e dilatazione degli ureteri. Si pensa che un deficit dell’attività parasimpatica, corresponsabile del movimento di queste strutture sia alla base del coinvolgimento delle vie urinarie. Il colon riassorbe anche vitamine ed aminoacidi ma non le proteine, i carboidrati e i grassi. La presenza di residui abbondanti di queste sostanze accanto alla grande quantità di batteri è causa di processi digestivi con presenza di amine potenzialmente tossiche (tiramina, istamina), di altre (indolo, scatolo) responsabili dell’odore fecale, di idrogeno solforato e metano, gas del flatus intestinale. E’ riassorbita anche l’ammoniaca, metabolita terminale della digestione proteica e una sua mancata riconversione in urea da parte del fegato è causa di gravi sintomi nervosi (iperammoniemia) Sempre i batteri sono responsabili, quando in eccesso, del deficit di assorbimento della Vit. B12 per iperconsumo; essi però sono fondamentali per l’assorbimento della vit K e di varie vitamine del complesso B. La malattia emorragica del neonato è prodotta dal deficit di Vit K per la sterilità del colon alla nascita e viene prevenuta dalla profilassi obbligatoria per via parenterale. Il deficit di motilità si chiama costipazione ed è causa spesso per aumento della pressione endoaddominale, di numerose estroflessioni chiamate diverticoli facilmente preda di processi flogistici (diverticolosi del colon). La diarrea al contrario può provocare quando protratta disidratazione e shock ipovolemico con perdita di sali minerali; la forma acuta specialmente nei primi 2 anni di vita può essere grave quando conduce ad un calo ponderale intorno al 10%; le forme croniche sono principalmente a carico delle malattie infiammatorie della mucosa con perdita, oltre che di acqua, di sangue e muco (colite ulcerosa, morbo di Crohn) Cenni del metabolismo dei nutrienti Una pur sommaria trattazione della fisiologia dell’apparato gastrointestinale non può prescindere dalla conoscenza dei processi metabolici dei carboidrati, delle proteine e dei grassi. Metabolismo dei Carboidrati: Nella dieta i Carboidrati sono costituiti da amidi (polimeri del glucosio) e zuccheri semplici, questi costituiti da disaccaridi (saccarosio, lattosio) o monosaccaridi (fruttosio) I processi digestivi dell’amilasi salivare, dell’acidità gastrica, dell’amilasi pancreatica e delle disaccaridasi delle cripte ghiandolari del tenue scindono queste molecole sino all’unico monosaccaride assorbibile, il glucosio. Questo, grazie alla presenza dell’insulina prodotta dalle beta cellule del pancreas endocrino entra nella cellula e le fornisce l’energia necessaria a tutti i processi metabolici attraverso un processo denominato glicolisi, con la produzione di molecole ad alto legame energetico, l’ATP. Questo processo, utilizzato per il 70%, in presenza di ossigeno (glicolisi aerobica) degrada il glucosio sino ad acido piruvico e ad Acetil CoA, quest’ultimo crocevia fondamentale del metabolismo intermedio comune. Di qui la glicolisi prosegue con l’ingresso dell’Acetil CoA nel ciclo dell’acido citrico (o ciclo di Krebs) con produzione di altre molecole di ATP e liberazione di acqua e anidride carbonica. Questo processo si autoalimenta in presenza dei substrati necessari, ma in assenza di ossigeno la glicolisi si ferma allo stadio di acido lattico. Una via alternativa alla glicolisi è quella del fosfogluconato o dei pentoso‐fosfati, ed è utilizzata per il 30% della degradazione del glucosio, specie dal fegato e dal sistema adiposo. Se manca il glucosio o questo non può entrare per difetto di insulina (diabete), si attua la trasformazione dei grassi in prodotti intermedi del catabolismo del glucosio e conseguente produzione di altre molecole di ATP (gluconeogenesi); in questo processo però si vengono a formare dei cataboliti acidi (acido acetico, acido acetacetico e acido beta idrossibutirrico), denominati corpi chetonici e in grado di provocare una grave acidosi nel diabete scompensato; Al contrario, se il glucosio della dieta è in eccesso questo viene polimerizzato in glicogeno nel fegato e riconvertito al bisogno, oppure trasformato in grasso di deposito attraverso l’Acetil CoA. Metabolismo dei lipidi Si distinguono grassi neutri (trigliceridi, composti da glicerolo e tre molecole di acidi grassi), fosfolipidi (esteri del glicerolo con due molecole di acido grasso, un radicale fosforico e una base azotata) e colesterolo. La digestione dei grassi neutri inizia con la lipasi gastrica e pancreatica degradando i trigliceridi nei loro costituenti, ma l’azione principale è svolta dai sali biliari che permettono con la loro attività emulsionante l’assorbimento da parte dei villi delle cellule parietali del tenue. Qui vengono nuovamente sintetizzati in trigliceridi, complessate con molecole lipoproteiche, esteri del colesterolo e fosfolipidi e trasportati dal sistema linfatico come chilomicroni. Nel fegato i trigliceridi vengono nuovamente degradati in glicerolo ed acidi grassi: il primo si trasforma in gliceraldeide e quindi in glucosio, i secondi sono scissi sino ad Acetil CoA ed entrano nel ciclo dell’acido citrico. Questo processo va sotto il nome di beta ossidazione. I Fosfolipidi sono assorbiti in modo non dissimile dai trigliceridi e vengono sintetizzati praticamente in ogni cellula costituendo la base strutturale delle membrane, della mielina delle cellule nervose e della tromboplastina, una cefalina che induce i processi della coagulazione. Il colesterolo è assorbito dall’epitelio intestinale e viaggia nel torrente linfatico come chilomicroni; nelle cellule del fegato è il substrato per i sali biliari, in altri organi per la produzione degli ormoni surrenalici e delle gonadi. E’ inoltre costituente delle membrane cellulari e si trova nello strato corneo della pelle come agente protettivo. Può essere sintetizzato dagli acidi grassi, sempre a partire dall’Acetil CoA. Metabolismo delle proteine Le sostanze proteiche, molto differenti per struttura molecolare, hanno in comune i loro costituenti principali, gli aminoacidi. Essi sono in numero di ventuno, dieci dei quali non possono essere sintetizzati da altre sostanze (aminoacidi essenziali). Gli altri undici possono essere prodotti dal metabolismo glicidico intermedio attraverso la transaminazione, il trasferimento di un gruppo aminico su un chetoacido attraverso sostanze donatrici di radicali aminici. Così ad esempio dall’acido piruvico si forma l’alanina attraverso la donazione del gruppo aminico da parte della glutamina. Le transaminasi più conosciute sono la GOT (glutamico‐ossalacetica) e la GPT (glutamico‐piruvica) e sono espressione dello stato di salute delle cellule epatiche e non solo. La digestione proteica inizia nello stomaco per l’azione dell’acido cloridrico e la pepsina che le scinde in polipeptidi; si prosegue nel duodeno con l’azione degli enzimi pancreatici e termina nel tenue con l’azione delle di peptidasi. Gli aminoacidi vengono assorbiti con un meccanismo attivo che ha nel Na il suo “carrier” principale, così come avviene per l’assorbimento del glucosio. Nell'infanzia il tenue assorbe proteine intere, utili come le gammaglobuline del colostro e del latte materno nei primi giorni di vita ma talvolta dannose come le proteine del latte o dell'uovo perché possono innescare il processo dell'allergia alimentare. (nota 1) Nel fegato e nelle altre cellule vengono utilizzati per la sintesi delle innumerevoli proteine plasmatiche e di struttura. Quando in eccesso, gli aminoacidi possono essere utilizzati per la produzione di energia attraverso il processo inverso della transaminazione (desaminazione) prima descritta e la trasformazione in metaboliti che entrano nel ciclo dell’acido citrico e possono quindi essere trasformati, sempre con il coinvolgimento dell’Acetil CoA in carboidrati o in grassi neutri (gluconeogenesi). Questo processo di transaminazione degli aminoacidi produce ammoniaca che il fegato converte in urea attraverso un sistema metabolico (ciclo dell'urea) e che a sua volta viene eliminata dal rene. Nota 1 Le Allergie Alimentari (2) Le allergie alimentari sono reazioni avverse agli agli alimenti scatenate con un meccanismo immunologico (3) e si differenziano dalle altre reazioni, di intolleranza o indotte da tossine. Interessano il 2‐4% dei bambini fino a 3 anni, ma il problema sembra coinvolgere una percentuale superiore di popolazione per sovrastima del fenomeno. Si distinguono reazioni IgE mediate, non IgE mediate (citotossiche in primo luogo) e forme miste; le prime sono responsabili degli effetti dopo poche ore dall'ingestione dell'alimento, le altre possono avere una latenza anche di giorni. Gli alimenti coinvolti sono latte e uovo in testa alla classifica, seguono: grano soia riso pesce pomodoro arachidi Nell'età pediatrica i sintomi sono spesso di tipo gastro‐intestinale, con vomito e diarrea in primo luogo nelle forme IgE mediate; la stipsi, il reflusso gastroesofageo, la distensione addominale, la diarrea muco‐ematica sono appannaggio delle forme non IgE mediate le quali possono presentare anche difficoltà di diagnosi differenziale con malattie di altra natura. Nel latte le proteine interessate sono le lattalbumine e le lattoglobuline, meno frequente l'allergia alla caseina; si pensa che il meccanismo immunologico venga scatenato dall'assorbimento di intere molecole proteiche da parte dell'intestino tenue del lattante e la sua successiva sensibilizzazione all'alimento; nell'uovo l'allergia è data dall'albume poiché il tuorlo contiene principalmente colesterolo e fosfolipidi. Nell'adulto le allergie alimentari, quando persistono, interessano altri distretti quali la cute, con manifestazioni di tipo ponfoide (orticaria) o eczematoso, o le vie respiratorie, con asma o oculorinite. I test di diagnosi (4) sono lo Skin prick test e il Rast e valutano la presenza di IgE specifiche per gli alimenti sospetti; nelle forme non IgE mediate può essere d'aiuto l'Atopy Patch test che valuta la reazione cellulo‐mediata della risposta allergica. Non vi sono riscontri scientifici sull'utilità diagnostica di test complementari (ALCAT‐test, biorisonanza, kinesiologia, analisi del capello, test elettrodermici). Più raramente vengono utilizzati altri esami, come il dosaggio dei livelli fecali dei marcatori di flogosi intestinale (calprotectina, proteina cationica eosinofila), o indagini istologiche o endoscopiche; questi esami sono utili nella diagnosi differenziale con altre patologie, quali la celiachia o le patologie infiammatorie croniche (Crohn, colite ulcerosa). La terapia si fonda sull'eliminazione dell'alimento sospettato; dopo qualche settimana, specie se associata alla scomparsa della sintomatologia si esegue il test di provocazione orale che viene universalmente considerato il gold standard della diagnosi. Nota 2 La celiachia (5) Dal greco “koilìa” che significa cavità, ventre, deve la sua scoperta ad un evento bellico. Il pediatra olandese Willelm K. Dicke notò alla fine della seconda guerra mondiale che alcuni suoi pazienti affetti da patologie intestinali croniche, a seguito della dieta a base di patate indotta dalla carestia avevano visto regredire i loro sintomi, ripresentatisi puntualmente all'ingestione del frumento. Negli anni la malattia ritenuta erroneamente di interesse pediatrico, con l'affinamento delle tecniche di diagnosi è risultata presente anche nella fascia adulta però con sintomatologia spesso extraintestinale. La frequenza nella popolazione è di circa 1/100 ma solo il 15% delle diagnosi viene effettuata. Si distingue una celiachia sintomatica ed una pauci o asintomatica, responsabile delle mancate diagnosi. La causa è un'intolleranza alla frazione proteica del frumento, segale, avena, orzo: la gliadina. Questa proteina si lega agli antigeni di istocompatibilità DQ2 e DQ8 presenti sulla superficie delle cellule parietali che insieme ai linfociti T helper (CD4+) provoca a livello della cellula mucosale la liberazione di numerose citochine ad azione pro‐infiammatoria con la conseguenza di una proliferazione di cellule linfocitarie, l'appiattimento dei villi e l'apoptosi cellulare. La conseguenza è una condizione di malassorbimento, steatorrea e marcata perdita di peso, considerando che il tenue è la struttura che assorbe la gran parte dei carboidrati, proteine e grassi della dieta. Il coinvolgimento del sistema di istocompatibilità rende ragione della familiarità della malattia: i portatori degli antigeni DQ2 e DQ8 hanno maggiore probabilità di contrarre la malattia tant'è che i fratelli dei celiaci hanno il 30% di avere la celiachia e i genitori il 10%. Sintomi tipici, oltre quelli citati sono un anemia ferro‐carenziale, ipoplasia dello smalto dentale, afte del cavo orale ed osteoporosi. Nelle forme dell'adulto si associano malattie quali le tiroiditi, il diabete tipo 1, la già citata malattia di Sjiogren, la sindrome di Down. La diagnosi un tempo si basava sul miglioramento dei sintomi con la dieta da esclusione; sono disponibili da qualche anno dei test che svelano la presenza degli anticorpi verso la gliadina (classe IgA, IgG), anticorpi antiendomisio, e quelli verso le transglutaminasi tissutali, responsabili del difetto enzimatico che sta alla base della mancata scissione della gliadina. La diagnosi istologica previa indagine endoscopica del duodeno prima e dopo il test di provocazione orale è però ancora l'esame più attendibile per una malattia che richiede l'esclusione della maggior parte dei cereali per tutta la vita. La mancata aderenza alla dieta o una diagnosi tardiva può condurre a complicanze quali il linfoma intestinale, una forma ulcerativa del tenue e all'istaurarsi di una celiachia refrattaria alla dieta da esclusione; è importante sapere che un alimento si intende privo di glutine quando questo è presente sotto la quota di 20 parti per milione; particolare attenzione va posta nella preparazione di alimenti idonei e nella presenza di derivati del frumento (amido) nella preparazione di innumerevoli prodotti commerciali e di molti farmaci in compresse. Cereali alternativi che non contengono gliadina sono il mais, il grano saraceno, il riso, il miglio, per citare i più comuni. La terapia medica si basa sull'assunzione di ferro, acido folico e vit. B12 nelle forme con anemia, di terapie specifiche nelle complicanze a carico della tiroide, nel diabete e nella S. di Sjiogren. Nelle forme refrattarie si sono usati farmaci immunosoppressori; l'aderenza alla dietoterapia è data dal controllo periodico dell'assenza degli anticorpi specifici. Bibliografia essenziale 1) Guyton‐ Trattato di Fisiologia Umana, Piccin editore 1990 2) Mansueto P. et al. ‐ Food allergy in gastroenterologic disease: review of literature – Word J. Gastroenterolol. 2006; 12, 7744‐52 3) Berni Canani R. et al – The diagnosis of food allergy in children – Curr. Opin. Pediatr. 2008; 20, 584‐9 4) Berni Canani R, et al. ‐ Il Laboratorio in gastroenterologia pediatrica – Area Pediatrica 2009; 7, 3‐10 5) M. Mazzetti di Pietralata et al. ‐ Patologie da Alimentazione e Nutrizione – Atti del Convegno 23‐25 Marzo 1994 ‐ Roma