- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
SPETTROSCOPIA DI RISONANZA MAGNETICA AL CARBONIO (13C)
La spettroscopia NMR riguarda l’assorbimento di radiazioni elettromagnetiche da parte di
nuclei atomici magneticamente attivi immersi in un campo magnetico.
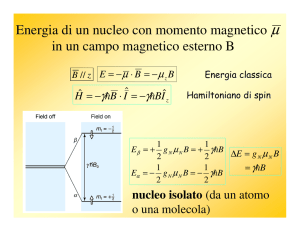
Ogni nucleo atomico che possiede o massa atomica A dispari o numero atomico Z dispari
o entrambi dispari, è magneticamente attivo ossia possiede un momento angolare di spin
P (e un momento magnetico nucleare μ).
Il momento angolare di spin (e il momento magnetico nucleare μ) di un nucleo posto in un
campo magnetico può assumere due o più orientamenti ad ognuno dei quali è associato
un diverso livello energetico.
Le transizioni tra i livelli energetici generate dall'interazione con una radiofrequenza
opportunamente rilevate danno luogo ad uno spettro di risonanza magnetica.
Il numero di possibili orientamenti è quantizzato ed è pari a 2I+1 dove I è il numero
quantico di spin del nucleo.
L’isotopo dell’idrogeno 1H ha spin = 1/2 e in un campo magnetico può assumere due
orientamenti.
1
H ha una abbondanza naturale quasi del 100%, per cui in pratica tutti i nuclei di idrogeno
presenti in una molecola "contribuiscono" alla creazione del segnale NMR.
La frequenza di risonanza è data da:
L’isotopo più abbondante del carbonio è il
12
C non è magneticamente attivo (I = 0).
L'isotopo 13C invece ha numero quantico di spin 1/2 come il protone e quindi si comporterà
come l'idrogeno dal punto di vista della risonanza magnetica nucleare con alcune
importanti differenze:
abbondanza naturale dell’isotopo attivo all’NMR
rapporto giromagnetico
intervallo di osservazione
accoppiamenti scalari (accoppiamenti spin-spin) omo- e eteronucleare
intensità relativa dei picchi di risonanza e tempi di rilassamento
sequenze particolari di registrazione degli spettri NMR (disaccoppiato, off-resonance
e effetto NOE)
spostamenti chimici (chemical shift)
-1-
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
isotopo Abbondanza Numero quantico Stati di spin Frequenza di risonanza
naturale (%) di spin nucleare I
a 42.276 Gauss (MHz)
1
H
99.88
1/2
±1/2
180
13
C
19
31
1.1
1/2
±1/2
45.3
F
100
1/2
±1/2
169.2
P
100
1/2
±1/2
73.2
l'abbondanza naturale del
13
C è solo dell'1.1% per cui solo questa piccola
percentuale di nuclei “contribuisce” alla creazione del segnale NMR con una sensibilità
molto inferiore rispetto all’idrogeno (in pratica occorrono più acquisizioni).
Inoltre anche il valore del rapporto giromagnetico γ del nucleo in esame influisce sulla
sensibilità della tecnica:
ampiezza segnale NMR ∝ γ3
γ (13C)/ γ (1H) ≈ 1/4
γ (1H) = 26.75 x 107 rad s-1T-1
γ (13C) = 6.73 x 107 rad s-1T-1
Sperimentalmente l'ampiezza di un segnale NMR è proporzionale circa al cubo della
costante giromagnetica. Poiché il rapporto giromagnetico γ del
13
C è circa 1/4 di quello
dell’1H (6.73 x 107 rad s-1 T-1 vs 26.75 x 107 rad s-1 T-1) il segnale di un
13
C sarà (1/4)3 =
1/64 del segnale di un 1H, inoltre tenendo conto dell’abbondanza naturale pari a circa
l’1.1%, si avrebbe un segnale per il
13
C circa 6000 volte inferiore al segnale 1H a parità di
quantità del campione. A causa della bassa sensibilità dell'esperimento NMR applicato al
13
C solo strumenti a trasformata di Fourier possono registrare spettri significativi in tempi
ragionevoli. Per questo motivo la tecnica
13
C NMR è stata sviluppata sviluppata dopo a
quella 1H NMR.
Nella tecnica a impulsi tutte le frequenze di interesse vengono messe in risonanza nello
stesso istante, utilizzando un impulso della giusta radiofrequenza molto potente e molto
breve. Dopo l’irraggiamento occorre attendere che l’eccesso di energia venga disperso dal
sistema di spin attraverso vari processi di rilassamento prima di ripetere l’impulso di
radiofrequenza. Questa tecnica permette di ottenere molti accumuli in poco tempo.
Tra due impulsi successivi, “si dovrebbe” ristabilire l’equilibrio di partenza tra gli spin di tutti
gli atomi in esame attraverso processi di rilassamento di varia natura.
Un meccanismo di rilassamento importante per i nuclei di
13
C è dovuto alle interazioni
dipolo-dipolo tra due nuclei 1H e 13C e la sua velocità dipende da diversi fattori:
-2-
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
Numero di protoni legati al 13C: T1 (CH3) < T1 (CH2) < T1 (CH) < T1 (C) ossia il tempo
di rilassamento T1 diminuisce all’aumentare del numero dei protoni direttamente legati al
nucleo 13C.
Dimensioni e simmetria della molecola: molecole piccole o molto simmetriche hanno
tempi di rilassamento T1 più lunghi di molecole grandi o complesse.
Intorno chimico: atomi di carbonio diversi hanno tempi di rilassamento diversi.
La variabilità del tempo di rilassamento T1 a seconda del tipo di atomi di carbonio è
responsabile del fatto che l’integrazione dei picchi negli spettri
13
C di routine NON è
correlata al rapporto degli atomi di carbonio al contrario del protone che avendo tempi di
rilassamento T1 brevi genera segnali con intensità proporzionali al numero di nuclei
coinvolti.
NB negli spettri 13C:
se l’intervallo tra i vari impulsi è troppo breve rispetto ai tempi di rilassamento alcuni
segnali possono scomparire del tutto.
se l’intervallo tra i vari impulsi è maggiore o uguale al trempo di rilassamento di tutti i
nuclei
13
C presenti nella molecola, l’intensità dei segnali è proporzionale al numero dei
nuclei coinvolti. In tal caso però i tempi di acquisizione aumentano notevolmente: non
adatto per analisi di routine.
Il rapporto giromagnetico γ del
13
C è circa 1/4 di quello dell’1H (vedi prima) per cui a
parità di campo magnetico esterno applicato la frequenza di risonanza del carbonio sarà
circa 1/4 di quella dell’1H. (infatti come ricordato in precedenza ν = γ B0/2π)
L’'intervallo di osservazione è molto più ampio di quello dell’idrogeno e va
comunemente da 0 a 240 ppm. Come composto di riferimento a δ=0 ppm si usa il TMS
come per l’idrogeno.
Costanti di accoppiamento
H
H
H C
H
13
H
C OH
H
H
13
H
H
H
C
C OH
H
H
H
13
C
H
13
C OH
H
0.012% del totale
(probabilità: 0.0112)
1.1% del totale
(probabilità: 0.011)
-3-
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
Gli accoppiamenti
13
C-13C non sono in genere osservabili in quanto è estremamente
improbabile che due nuclei
13
C si trovino adiacenti nella stessa molecola data la bassa
abbondanza isotopica (ovviamente ciò può non essere vero in sostanze arricchite
isotopicamente).
Costanti di accoppiamento visibili in spettri di sostanze NON arricchite isotopicamente:
spettro 1H-NMR
spettro 13C-NMR
J(H/H)
sì
J(C/C)
no
J(H/F)
sì
J(C/F)
sì
J(H/P)
sì
J(C/P)
sì
J(H/C)
no
J(C/H)
sì
…..
1
J(13C-1H) ≈ 110–320 Hz;
13
…..
2
J(13C-1H) ≈ 0–60 Hz;
13
C-H
C-C-H
3
J(13C-1H) ≈ 0–10 Hz
13
C-C-C-H
Però rimangono gli accoppiamenti con i protoni ed eventuali altri nuclei
magneticamente attivi. Poiché le costanti di accoppiamento 1J C-H hanno valori molto
elevati (1JCH = 110–320 Hz, ma anche le 2JCH = 0-60 Hz e 3JCH = 0-10 Hz possono essere
significative) spesso le linee relative ai vari segnali sono sovrapposte e lo spettro risulta
molto complicato. Per semplificare gli spettri del carbonio si può utilizzare la tecnica della
doppia risonanza in cui un canale supplementare irradia alla frequenza di risonanza
dell'intero spettro protone annullando gli accoppiamenti con il carbonio (vedi dopo). In
queste condizioni lo spettro finale risulta formato da soli singoletti uno per ogni atomo di
carbonio non chimicamente equivalente nella molecola (a meno di casuali coincidenze di
spostamento chimico).
Spostamenti chimici (chemical shift)
scala 1H NMR ≈ 12–0 (TMS) ppm;
scala 13C NMR ≈ 220–0 (TMS) ppm
-4-
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
Gli spostamenti chimici nella spettroscopia del carbonio dipendono dagli stessi parametri
che influenzano il protone, quali ibridazione del carbonio ed elettronegatività del
sostituente, anche se si estendono per un maggior numero di ppm (da 0 a 240 ppm circa).
Fattori che influenzano il chemical shift:
Ibridazione degli atomi di C. I carboni ibridati sp3 risuonano a campi più alti
(frequenze più basse) rispetto a quelli ibridati sp2. A metà strada di trovano i carboni
ibridati sp, che risentono del fenomeno dell’anisotropia diamagnetica
Effetto induttivo. I sostituenti elettronegativi provocano una deschermatura sul
carbonio in posizione α, in maniera proporzionale alla loro elettronegatività
Effetto mesomero. La delocalizzazione delle cariche nei sistemi insaturi (anelli
aromatici, carbonili α,β insaturi) provoca delle variazioni dei chemical shift. Sostituenti
elettron attrattori (COOR, COR, CN, NO2) riducono la densità di carica nelle posizioni orto
-5-
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
e para dell’anello benzenico, deschermando i corrispondenti atomi di carbonio. Sostituenti
elettron donatori e con doppietti elettronici delocalizzabili sul sistema aromatico aumentano
la densità di carica sulle posizioni orto e para, schermando i corrispondenti atomi di
carbonio.
Effetti sterici. quando due atomi di C si trovano tra loro in posizione gauche o
eclissata, la compressione sterica provoca la schermatura di entrambi i nuclei di 5–10 ppm
Il concetto di additività degli effetti del sostituente è valido anche per questo tipo di spettri.
Quando sono presenti più fattori si può ricorrere a delle tabelle di correlazione per
prevedere l’entità del chemichal shift.
Disaccoppiamento protonico
A causa degli elevati valori degli accoppiamenti 1JC-H (110-320 Hz) e dei valori a volte
significativi di 2JC-H (0-60 Hz) e 3JC-H (0-10 Hz) gli spettri
13
C accoppiati con il protone
spesso presentano multipletti complessi e sovrapposti, difficili da interpretare.
Per superare questo inconveniente si ricorre al disaccoppiamento protonico a banda larga,
che permette di rimuovere l’accoppiamento con tutti i protoni presenti nella molecola. In tali
condizioni ogni atomo di carbonio chimicamente non equivalente presente nel composto
genera un unico segnale sottile, fatta eccezione per eventuali sovrapposizioni dovute a
fortuite coincidenze di chemical shift. Si ottiene così una notevole semplificazione dello
spettro accompagnata dalla perdita delle informazioni relative alla molteplicità dei segnali
(struttura fine).
Il disaccoppiamento di spin
Se non è influenzato dell'esterno, un protone rimane nel suo stato di spin α o β. Se però
viene irradiato con continuità alla sua frequenza di risonanza, la radiazione
elettromagnetica causa un continua variazione del suo stato di spin da α a β e viceversa.
Si ha in pratica una serie continua di assorbimenti ed emissioni stimolate.
Consideriamo ora un sistema AX, e supponiamo di registrare uno spettro mentre si irradia
costantemente il nucleo del protone A. In queste condizioni il nucleo X non si trova più
nelle vicinanze di un protone nello stato α o nello stato β, ma nelle vicinanze di un protone
-6-
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
che cambia continuamente il suo stato di spin. Il nucleo X risente quindi dello momento
magnetico medio del nucleo A, che è zero.
Senza disaccoppiamento, il nucleo X è schermato o deschermato e l’insieme di nuclei X
genera un doppietto.
Con il disaccoppiamento ogni nucleo X è prima schermato, poi deschermato, e così via per
cui risente di un valore di campo medio.
In definitiva, se si irradia con continuità sul nucleo A, il nucleo X non risente più
dell’accoppiamento con il nucleo A, e risuona come singoletto, cioè come se il nucleo
A non ci fosse (anche se eventuali accoppiamenti con altri nuclei continuano ad
esistere!).
Nello stesso tempo, il segnale del nucleo A non è più presente nello spettro, poiché la
continua irradiazione fa sì che le popolazioni degli stati α e β diventino identiche.
-7-
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
L’altezza relativa del singoletto sarà maggiore dell’altezza delle singole componenti del
doppietto.
Con il disaccoppiamento è possibile stabilire quali protoni sono accoppiati ad un certo
protone. Questa informazione si ricava oggi più facilmente dall'esperimento NMR
bidimensionale COSY.
Broadband proton decoupled (disaccoppiamento a banda larga)
Negli spettri
13
C NMR di routine viene applicato un segnale continuo con una banda larga
di frequenze radio che eccita tutti i nuclei 1H e cancella così tutti gli accoppiamenti dovuti
all’interazione 1H/13C. Per cui ogni carbonio genererà un linea singola a meno di eventuali
eteronuclei o isotopi dell’idrogeno magneticamente attivi, quali 2H,
19
F etc etc., che
continueranno ad accoppiare con il carbonio.
Off-resonance decoupling (disaccoppiamento parziale)
Sequenza che permette di mantenere solo gli accoppiamenti
13
C-1H che avvengono
attraverso un solo legame anche se con valori della costante di accoppiamento 1JC-H minori
del reale. In tali condizioni il tipo di segnale di ogni C dipende dal numero di protoni ad
-8-
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
esso legati in accordo con la regola n+1. Per cui un CH3 darà un quartetto (q), un CH2 un
tripletto (t), un CH un doppietto (d) e un C quaternario un singoletto (s).
Effetto nucleare Overhauser (Nuclear Overhauser Effect, NOE)
L’effetto NOE è una variazione dell’intensità del segnale di un nucleo (es protone o
carbonio) in seguito all’irradiazione di un protone che si trova spazialmente vicino al nucleo
osservato.
Nel disaccoppiamento a banda larga si applica una forte sorgente di radiofrequenza che
satura i livelli dei protoni e che, oltre a eliminare la struttura fine del
13
C provoca un
aumento dell’intensità dei segnali dei 13C spazialmente vicini a protoni
Nel caso di atomi di carbonio non legati a protoni l’incremento è praticamente zero
Per molecole organiche di dimensioni medio-piccole il rilassamento dipolare 13C-1H relativo
agli atomi di carbonio legati a protoni è molto efficiente e può produrre incrementi dei
segnali prossimi al 200%.
L’effetto NOE è molto utile perché aumenta la sensibilità dell’esperimento
13
C, anche se,
allo stesso tempo, contribuisce a rendere senza significato l’integrazione dei picchi.
Deuterio (D, 2H)
Composti marcati con deuterio vengono utilizzati per vari scopi nella spettroscopia NMR:
es nello spettro 1H NMR la marcatura può essere usata per identificare un particolare
gruppo, semplificare lo spettro 1H di una sostanza, avere solventi “trasparenti” che non
interferiscano con la misura e su cui fare il lock. …
Solventi per l'NMR
Il solvente più usato è il CDCl3, comuni sono anche D2O, CD3OD, C6D6, etc. Anche i
solventi deuterati contengono una piccola percentuale di atomi di H, e danno luogo ad un
segnale residuo (cfr tabelle nell’articolo allegato: J. Org. Chem. 1997, 62, 7153)
Il segnale residuo è sempre nella stessa posizione (per CDCl3 a δ 7.26 ppm), e può essere
usato per calibrare lo spettro.
-9-
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
Altri solventi utilizzati sono:
δ del picco residuo
del solvente
1
H NMR
13
C NMR
C6D60 CD3OD0
D2O0
7.160
3.310
4.790
39.5000 128.100
49.000
CDCl30
(CD3)2SO
(d6-DMSO)
7.260v
2.500v
77.000v
Omogeneità del campo magnetico esterno
Negli spettri NMR i segnali possono e “devono” essere molto stretti (< 1Hz).
Poiché la frequenza di risonanza dipende dal campo magnetico, il campo magnetico deve
essere estremamente uniforme in tutto il volume del campione e costante nel tempo
specie nel caso di esperimenti lunghi:
Se il campo magnetico non è perfettamente uniforme e costante i segnali si slargano. in
pratica occorre che la frequenza di risonanza non cambi nemmeno di 1 Hz (su centinaia di
MHz!!).
Il magnete superconduttore non riesce da solo a produrre un campo magnetico così
omogeneo.
Si usano allora delle bobine (anche più di 20) posizionate intorno al campione che
producono dei piccoli campi magnetici non omogenei, e che possono essere regolate in
modo da compensare ogni disomogeneità del campo magnetico. Questa regolazione si
chiama shimming, e deve essere fatta per ogni singolo campione.
- 10 -
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
Per mantenere il campo magnetico costante nel tempo si usa .un sistema di lock a
deuterio che sfrutta il deuterio dei solventi deuterati.
Durante l’acquisizione di uno spettro, oltre ad irraggiare alla frequenza di risonanza del
nucleo di interesse, si irraggia il campione alla frequenza di risonanza del deuterio: per
piccole fluttuazioni del campo magnetico, il deuterio va fuori risonanza, in tal caso lo
strumento aggiusta l’intensità del campo in modo da riportare il deuterio in risonanza
Accoppiamenti con il deuterio (D, 2H)
Il deuterio è un nucleo magneticamente attivo.
Ricordiamo che:
Numero quantico magnetico:
m = 2I+1
m corrisponde al numero totale di spin che un nucleo con spin I può assumere
Numero quantico di spin nucleare di 1H:
IH = 1/2
molteplicità del segnale del nucleo accoppiato al 1H:
n+1
Numero quantico di spin nucleare di 2H:
ID = 1
molteplicità del segnale del nucleo accoppiato al D:
2n+1
n 0 molteplicità 0 intensità relative
I = 1/2
0
s
1
molteplicità0
s
intensità relative
I=1
1
1
d
1001
t
1001001
2
t
1002001
q
1002003002001
3
q
1003003001
settetto
1003006007006003001
4
quint
1004006004001
nonetto
1004010016019016010004001
JH-D ≈ 1-2 Hz
Per cui negli spettri
13
JC-D ≈ 20-25 Hz
C NMR registrati in CDCl3 il tripletto di intensità 1:1:1 che risuona a
circa 77 ppm è il segnale dovuto al C del CDCl3 che accoppia con l’atomo di deuterio. Tale
segnale NON va riportato nella descrizione dello spettro.
- 11 -
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
Conseguenze del disaccoppiamento protonico a banda larga:
collasso dei multipletti in singoletti
guadagno in sensibilità
interpretazione più semplice (un segnale per ogni tipo di C presente
nella molecola a parte coincidenze fortuite di chemical shift)
perdita di informazioni associata all’accoppiamento 1H-13C che possono
però essere recuperate con opportune sequenze di acquisizione dello spettro
13
C (es spettro disaccoppiato off-resonance, DEPT, APT, spettro
bidimensionali come HMQC)
Effetto Overhauser nucleare (NOE)
aumento di intensità dei C legati o spazialmente vicini a idrogeni
INTENSITA’ RELATIVE DEI SEGNALI NON PROPORZIONALI AL
NUMERO DI NUCLEI CHE LI GENERANO
test del protone legato (APT, Attached Proton Test):
Impostando opportunamente i valori di alcuni intervalli nella sequenza d'impulso, è
possibile registrare gli atomi di carbonio quaternari e metilenici con fase positiva e i metili e
i metini con fase negativa.
Poiché il segno della fase è arbitrario, l'ordine può essere invertito.
- 12 -
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
C↓
13
(CHx5) ↑
CD ↓
↓ CH2
↑ CH3
C NMR (50 MHz, CDCl3) = δ 144.2 (s, Cipso), 128.4 (d, 2 C; Cmeta), 127.9 (d, 2 C; Corto),
125.7 (d, Cpara), 29.0 (t, CH2), 15.6 (q, CH3) ppm.
- 13 -
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
Descrizione dello spettro 13C NMR
13
C NMR: Specificare a che frequenza è stato registrato lo spettro ed il solvente utilizzato.
Riportare nell’ordine:
i chemical shift approssimati alla prima cifra decimale,
se noto indicare che molteplicità avrebbe il segnale nello spettro disaccoppiato parziale
(off-resonance decoupling)(s, d, t, q)
Se il segnale è dovuto alla risonanza di n carboni equivalenti (con n>1) scrivere nC
parentesi.
e quando possibile
l’assegnazione indicando in modo univoco quali atomi di carbonio della molecola
risuonano a quella data frequenza.
Far precedere il primo chemical shift dal simbolo δ e concludere la descrizione con ppm.
13
C NMR (50 MHz, CDCl3) = δ 66.0 (t, 2 C; CH2), 15.4 (q, 2 C; CH3) ppm.
- 14 -
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
13
C NMR (50 MHz, CDCl3) = δ 32.1 (t, 2 C; C-3 e C-5), 29.3 (t, C-4), 22.9 (t, 2 C; C-2 e C-
6), 14.1 (q, 2 C; C-1 e C-7) ppm.
13
C NMR (50 MHz, CDCl3) = δ 149.9 (s, 3 C; Cipso), 119.4 (d, 3 C; CH), 35.0 (s, 3 C; CMe3),
31.6 (q, 9 C; CH3) ppm.
- 15 -
- Lab. Chim. Org. II- 09-10 – DM207/04 semestreII
13
C NMR (50 MHz, CDCl3) = δ 143.4 (d, 4 C; =C), 75.3 (d, 2 C; CH2CH), 50.3 (t, CH2) ppm.
Esempi di valori di chemical shift e loro calcolo approssimato
Alcani: δ ≤ 60 ppm
- 16 -