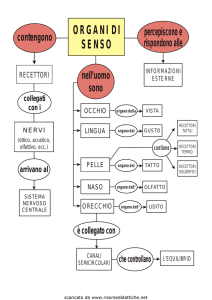
SISTEMA NERVOSO CENTRALE
Encefalo e Midollo Spinale formano il sistema
nervoso centrale (SNC) o asse cerebrospinale (o
nevrasse o tubo neurale) che è in grado di
raccogliere, trasmettere e integrare le informazioni.
Custodito
all'interno
della
scatola
cranica
(encefalo) e del canale vertebrale (midollo spinale),
il SNC, viene protetto ulteriormente e nutrito dal
sistema delle meningi, dal liquor cerebrospinale e
dal proprio sistema di vascolarizzazione (barriera
emato-encefalica,
retinica)
emato-liquorale
e
emato-
La conduzione degli impulsi lungo l’assone è di tipo elettrico ed è provocata dagli scambi
di ioni Na+ e K+ attraverso la membrana neuronale
(La conduzione da un neurone ad un altro o da un neurone ed un effettore cellulare non
neuronale è di tipo chimico – mediata dall’azione di neurotrasmettitori e Ca2+)
SNP: le connessioni tra neuroni avvengono tra assone presinaptico e processo dendritico
postsinaptico
SNC: organizzazione sinaptica più complessa:
• contatto funzionale tra due neuroni può avvenire tra assone e corpo cellulare, tra
assone e processo dendritico, tra corpo cellulare e corpo cellulare o fra dendrite e
dendrite
• neurotrasmettitori continuamente liberati e interazioni rapidamente interrotte in
modo che gli stessi recettori possano essere attivati in modo ripetuto
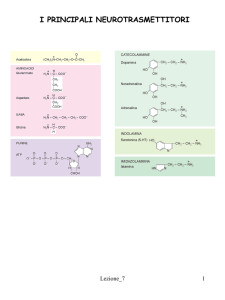
Neurotrasmettitori e Neuromodulatori del SNC
NEUROMODULATORI
Agiscono più lentamente dei neurotrasmettitori ed hanno effetti più ampi. Possono essere
liberati dallo stesso terminale dei neurotrasemettitori (si parla di Co-trasmissione).
Il loro ruolo è proprio quello di modulare gli effetti dei neuotrasmettitori
es.. neuropeptidi come la sostanza P e la somatostatina, prostaglandine e NO
❖ Acetilcolina
❖ Amine biogene (catecolamine e serotonina 5-HT, istamina derivato imidazolo ma indicata come amina
biogena)
❖ Aminoacidi (glutammato e acido gamma-aminobutirrico GABA)
❖ Neuropeptidi (endorfine, encefaline e dinorfine)
❖ Purine (ATP e adenosina)
❖ Acido arachidonico e suoi metaboliti (eicosanoidi e cannabinoidi endogeni)
3
Ach: a livello del SNC ha effetti principalmente eccitatori
Amine biogene
Serotonina: esistono 7 famiglie di recettori serotoninergici e la maggior parte si trova nel
cervello. Tutti, tranne il 5-HT3 – recettore canale, sono accoppiati a proteine G.
Modula l’umore, gli stati emotivi, il sonno, l’appetito, la pressione arteriosa, il riflesso del
vomito e svolge un ruolo anche nella percezione del dolore
Dopamina: rappresenta il neurotrasmettitore di molti neuroni centrali. 5 tipi recettoriali
accoppiati a proteine G e classificati come D1-simili (stimolano l’adenilaciclasi) e D2-simili
(inibiscono l’adenilatociclasi)
Noradrenalina
Istamina: 3 sottotipi recettoriali (H1, H2 e H3). Sono state identificate poche vie
istaminergiche centrali. Tuttavia gli antagonisti H1 sono ampiamente usati come sedativi e
antiemetici
Aminoacidi
GABA: è il principale neurotrasmettitore inibitorio del SNC. GABAA GABAB GABAC (retina)
Glutammato: è il principale neurotrasmettitore eccitatorio del SNC. 6 recettori
(NmetilDaspartato – NMDA)
Glicina: azione simile al GABA. Interneuroni del midollo spinale
Neuropeptidi (agiscono sui recettori degli oppioidi e spesso come co-trasmettitori)
Β-Endorfina e altre Endorfine: attivano un gran numero di neuroni centrali
(ipotalamo, amigdala, talamo e locus ceruleus)
Met-Encefalina e Leu-Encefalina
Dinorfine: sono sette peptidi con sequenze aminoacidiche simili. Coesistono con le
encefaline. La sostanza P è presente nei neuroni centrali e in alta concentrazione
nei gangli delle radici dorsali. Viene rilasciata mediante stimoli afferenti di dolore
intenso
Purine (sono mediatori e neurotrasmettitori in una grande varietà di tessuti)
Adenosina: nel SNC è un neurotrasmettitore inibitorio. Recettori A1, A2a, A2b, A3
ATP: recettori P2x e P2y e…………
Acido arachidonico e metaboliti (eicosanoidi e cannabinoidi endogeni): agiscono
come messaggeri intracellulari o possono interagire con recettori sulla membrana
cellulare
FARMACI DEL SISTEMA NERVOSO CENTRALE
❖ ANESTETICI GENERALI
❖ ANALGESICI OPPIOIDI
❖ NEUROLETTICI o TRANQUILLANTI MAGGIORI
❖ SEDATIVO-IPNOTICI
❖ ANTICONVULSIVANTI
❖ NEUROANALETTICI o STIMOLANTI SNC
❖ REGOLATORI DEL COMPORTAMENTO
ANESTETICI GENERALI
Definizioni
• Anestesia = perdita o assenza, reversibile, di sensibilità
al dolore e della coscienza
Stato di incoscienza indotto da farmaci durante il quale possono essere tollerate manovre chirurgiche
senza percezione del dolore e senza che siano provocate nel paziente reazioni difensive vegetative o
muscolari
• Sedazione = grado blando di depressione del SNC in cui il
paziente rimane sveglio e non eccitato
• Analgesia = solo perdita della sensibilità dolorosa, rimane
lo stato di coscienza
• Neuroleptoanalgesia = uso combinato di tranquillanti ed
analgesici
Gli anestetici generali inducono, in maniera reversibile, analgesia, amnesia e, in genere, ipnosi
e rilassamento muscolare che permettono il mantenimento della stabilità fisiologica del
paziente e l’attenuazione dello stress chirurgico
Scelta dell’anestetico
1. Tipo di intervento e durata
2. Specie
3. Sensibilità di specie o di razza (levriero*) o in
seguito a particolari stati fisiologici
4. Interazioni farmacologiche
4. Temperatura dell’animale e dell’ambiente
5. Premedicazione, induzione, anestesia, risveglio
*Per esempio il barbiturico tiopentale, un tempo utilizzato come agente induttore
dell’anestesia, a causa di una particolare modalità di redistribuzione nel tessuto adiposo (i
levrieri hanno una minore % di tessuto adiposo) e di una minor capacità di
metabolizzazione epatica per la riduzione dell’attività di un enzima (del citocromo p450)
riscontrata nei levrieri, può provocare risvegli molto più rallentati e imprevedibili rispetto
alle altre razze.
Ad oggi è possibile evitare di utilizzarlo sostituendolo con il propofol che, pur avendo
anch’esso un metabolismo più lento nei levrieri, ha margini di sicurezza molto più ampi
Pianificazione dell’intervento
• Effettuare analisi per valutare lo stato di salute
dell’animale
• Lasciare l’animale a digiuno
• Fluidoterapia se paziente debilitato
• Ossigenoterapia se paziente è in carenza di O2
• Vena pervia
• Medicazione preanestetica
Vie di somministrazione degli anestetici generali
•
•
•
•
Inalatoria (alotano, isofluorano)
Endovena (barbiturici)
Intramuscolare (a. dissociativi)
Orale (solo per sedazione)
Gli anestetici deprimono in ordine:
•
•
•
•
Corteccia
coscienza
Mesencefalo
coordinazione
Midollo spinale
riflessi spinali
Midollo allungato
centri bulbari
(respiro e frequenza cardiaca)
Stadi dell’anestesia
• 1° stadio = di eccitamento volontario
(c’è analgesia e coscienza)
• 2° stadio = eccitamento involontario
(solo nella fase finale c’è perdita di coscienza)
• 3° stadio = dell’anestesia chirurgica
(depressione della corteccia, mesencefalo e MS)
• 4° stadio = paralisi del midollo allungato
(depressione dei centri bulbari
morte)
Oggi, con le miscele di anestetici disponibili, non
si notano più i segni clinici che differenziano i
diversi stadi
Stadio I: fase di analgesia, di induzione o dell’eccitamento volontario.
Va dall’induzione alla perdita di coscienza, si manifesta con irrequietezza motoria ed
atassia, permangono ancora capacità mentali che dissolvono progressivamente fino
all’incoscienza.
Stadio II: fase del delirio, dell’eccitazione involontaria o della reattività incontrollata.
Va dalla perdita di coscienza alla comparsa dell’automatismo del respiro, si manifesta con
eccitamento generalizzato.
Stadio III: fase chirurgica.
Viene distinto in quattro piani: interventi chirurgici lievi, di grado medio, invasivi, molto
devastanti.
Stadio IV: fase della paralisi bulbare o della paralisi respiratoria per eccesso di
dosaggio dell’anestetico.
Perdura per tutto il tempo che va dall’arresto del respiro a quello dell’arresto cardiaco.
Anestetici generali
Inalatori
Iniettabili
Anestetici inalatori
Inalazione
Alveoli polmonari
Legge di Dalton
Legge di Henry
Capillari-sangue
Tessuti lipidici
SNC
Gli anestetici inalatori sono
ampiamente utilizzati in medicina
veterinaria in virtù delle loro
caratteristiche farmacocinetiche
che
permettono
tempi
di
induzione e di risveglio veloci e
rapidi nonché aggiustamenti del
livello di anestesia dell’animale.
Essi sono liquidi a basso punto di
ebollizione (etere, cloroformio,
alotano,
metossiflurano,
enflurano, isoflurano) oppure gas
(protossido d’azoto, xenon).
Raggiunto il polmone, essi
diffondono dagli alveoli al
sangue e poi in tutto il corpo,
incluso il SNC. Gli anestetici
inalatori
presentano
diverse
caratteristiche
di
solubilità
nell’acqua, sangue e grassi ed è
quindi necessario somministrarli
a concentrazioni diverse nell’aria
inspirata
per
ottenere
concentrazioni anestetiche nel
SNC.
Legge di Dalton
La pressione totale esercitata da una miscela di gas
ideali è uguale alla somma delle pressioni parziali
che sarebbero esercitate dai gas se fossero presenti
da soli in un eguale volume.
La legge di Henry
Un gas che esercita una pressione sulla superficie
di un liquido, vi entra in soluzione finché avrà
raggiunto in quel liquido la stessa pressione che
esercita sopra di esso. A temperatura costante,
la solubilità di un gas è direttamente
proporzionale alla pressione che il gas esercita
sulla soluzione
La potenza di un anestetico inalatorio è
espressa dalla MAC (concentrazione
alveolare minima) che ad 1 atm di P fa
scomparire la reazione difensiva nel 50%
dei pazienti sottoposti ad uno stimolo
dolorifico
Principali anestetici inalatori
•
•
•
•
•
•
•
Alotano
Protossido d’azoto
Metossifluorano
Isofluorano
Enfluorano
Cloroformio
Etere
Non più utilizzati
N2O
Il protossido d’azoto è un gas, incolore,
non infiammabile e non irritante. Ha un
basso potere anestetico. È stato il primo
anestetico generale utilizzato
Vantaggi
• Non è un anestetico
depressante del SNC
• Ha azione eccitatoria
(gas esilarante)
• Rapida induzione e
rapido risveglio
• Scarsi effetti collaterali
• Non determina nessuna
alterazione a livello
cardiopolmonare, renale
od epatico
Svantaggi
• Causa ipossia
• Ha un bassissimo potere
anestetico
• Scarso miorilassamento
• In genere si associa
all’alotano per diminuirne
la dose o ad altri anestetici
iniettabili
L’alotano si presenta liquido,
incolore, non infiammabile.
E’ fotosensibile e la sua
metabolizzazione avviene a
livello epatico
Vantaggi
• Utilizzato in tutte le specie
animali anche uccelli e
specie esotiche
• Induzione e risveglio rapidi
• Scarsi effetti collaterali
• Non genera vomito, non è
epatotossico ma può dare
nefrotossicità
• Buon miorilassamento
Svantaggi
• Non presenta alcun effetto
analgesico post-operatorio
• Depressione del SNC e
cardiopolmonare
proporzionale al grado
dell’anestesia
• Riduce il legame e la
disponibilità di Ca++
• Depressione del centro
ipotalamico della
termoregolazione
Il metossifluorano si presenta come
un liquido incolore, non
infiammabile, ed è fotosensibile.
Viene metabolizzato a livello epatico
Vantaggi
• Maggiore potere anestetico
in assoluto
• Rilassamento muscolare
ottimo e duraturo
• Analgesia pre-post
operatoria eccellente
• Non è epatotossico, può
dare nefrotossicità
• Uso preferito nei piccoli
animali
Svantaggi
• Fase di induzione e di
risveglio molto lente
• Azione depressante sul
SNC e sui centri del respiro
proporzionale al grado
dell’anestesia
• Riduce la disponibilità di
Ca++
• Depressione del centro
termoregolatore
L’enfluorano si presenta come un
liquido incolore, non infiammabile, e
non è fotosensibile. Non viene
metabolizzato ma eliminato con la
respirazione
Vantaggi
• Non è un anestetico
depressante del SNC
• Ha azione dissociativa
• Approvato l’utilizzo negli
equini
Svantaggi
• Deprime la funzione
cardiovascolare e quella
respiratoria
• Produce tremori
muscolari
• Risveglio più lento di
quello con alotano
• Leggermente irritante per
le vie respiratorie
• Costoso
L’isofluorano è un isomero
dell’enfluorano (stessa formula bruta). Si
presenta come un liquido incolore,
non infiammabile. Viene scarsamente
metabolizzato e principalmente
eliminato con la respirazione
Vantaggi
• Solubilità inferiore a quella di •
alotano e enfluorano e
•
metossifluorano
• MAC inferiore a quella di
•
enfluorano, ma superiore a
quella di alotano
• Rapida induzione e risveglio
• Buon miorilassamento
•
• Approvato l’uso nei mammiferi e
scelta d’elezione per pazienti
•
cardiopatici o anziani
Svantaggi
Depressore del SNC
Non presenta proprietà
analgesiche
Deprime scarsamente la
funzione cardiovascolare e
quella respiratoria (<
rispetto all’enfluorano)
Leggermente irritante per
le vie respiratorie
Costoso
Principali classi di anestetici iniettabili
• Barbiturici (tiopentale)
• Anestetici dissociativi (ketamina)
Svantaggi : profondità dell’anestesia meno controllabile
eliminazione preceduta dalla metabolizzazione
Formula dei barbiturici
durata d’azione
• Ad azione lunga
(fenobarbitale)
• Ad azione intermedia
(amobarbitale)
• Ad azione breve
(pentobarbitale)
• Ad azione ultrabreve
(tiopentale sodico)
Usati come sedativi
ed anticolvulsivanti
Usati per anestesia
Sono facilmente decomponibili ed alcuni tiobarbiturici
molto instabili, si inattivano dopo 36 ore a T ambiente
Assorbimento distribuzione ed
escrezione
O o IV
Permanenza in circolo breve
Passaggio BEE veloce
Passaggio nei tess.lipidici
Metabolizzazione epatica
ed escrezione renale
SNC
Tutti i barbiturici deprimono il SNC. Deprimono la
corteccia sensoriale riducono l’attività motoria e
producono sedazione a bassi dosaggi ed anestesia
ad alti dosaggi. Effetto anticonvulsivante a dosi
non anestetiche solo per fenobarbitale
Basso potere analgesico
Azione GABAergica
Apparato respiratorio
Depressione respiratoria dose-dipendente, nella
sedazione la depressione del respiro è simile a
quella fisiologica del sonno, aumentando i
dosaggi si assiste ad una riduzione della
frequenza, della profondità e del volume
respiratorio.
Gatti = broncospasmi
Apparato cardio-vascolare e renale
A dosi elevate inducono tachicardia, riduzione
della contrattilità miocardica, della gittata
cardiaca, della P arteriosa e delle resistenze
vascolari periferiche. Nessun effetto diretto sul
rene, i danni eventuali sono legati all’ipotensione
Apparato scheletrico e fegato
Riducono la sensibilità della placca motrice
all’Ach ma senza significato clinico. Non inducono
un buon miorilassamento addominale. Non sono
epatotossici nei soggetti sani. Il metabolismo è a
livello epatico ed alcuni (fenobarbitale) sono
degli induttori. Danno ipotermia
Indicazioni terapeutiche
Fenobarbital a scopo di sedazione, negli
stati di ipereccitabilità, in associazione con
analgesici (es prurito). Anche per attacchi
epilettici.
PentoB. e tiopentale in anestesiologia o in
caso di intervento d’urgenza
Effetti collaterali
Farmaci con scarsa manegevolezza. Effetto
depressante a carico del respiro: la frequenza può
essere aumentata o ridotta ma quello che si riduce è il
volume respiratorio/min. mucose cianotiche ed
ipotermia. Morte a breve con i B. a breve durata
d’azione. Utilizzo analettici respiratori e ossigeno.
Attenzione in pazienti con lesioni epatiche e renali
Anestetici dissociativi
in medicina veterinaria
• Feniciclidina cloroidrato
(polvere d’angelo) e derivati
• Chetamina cloroidrato
• Tiletamina cloroidrato
Non più usati
Gatto e primati
Gatto e cane
Ketamina: assorbimento distribuzione ed
escrezione
IV o IM
Permanenza in circolo breve
Distribuzione tes.adiposo,
fegato, polmone e cervello
Metabolizzazione epatica
ed escrezione renale dei metaboliti
Ha un ampio indice terapeutico, di circa 5
volte superiore a quello dei barbiturici
SNC
Antagonismo non competitivo con recettori NMDA. La
K. induce anestesia con rottura funzionale del SNC
depressione del sistema talamo-corteccia ed
attivazione del sistema limbico, stato di incoscienza ed
analgesia ma mantenimento dei riflessi. Potenzia i
sistemi inibitori del SNC, impedendo il reuptake del
GABA. Blocca anche i sistemi di trasporto NA e
dopamina
App. cardio-vascolare
e respiratorio
Depressione del sistema cardiovascolare e
contemporaneamente del SN simpatico per cui:
aumento della gittata cardiaca, della P. aortica e
polmonare, e della frequenza cardiaca, ma il
mecc. d’azione non risulta chiarito. Nessun
effetto inibitorio sui centri del respiro ma
permangono anche i riflessi faringeo e laringeo
App. muscolo-scheletrico
In quasi tutte le specie produce un aumento del
tono muscolare e rigidità degli arti. Di solito i
pazienti presentano perdita della sensibilità
dolorifica e midriasi, ma sono conservati i riflessi
pupillari, corneali e di deglutizione. Può essere
associata a miorilassanti.
FARMACI NELLA PREANESTESIA
ANTICOLINERGICI: -ATROPINA
-GLICOPIRROLATO
TRANQUILLANTI: - DERIVATI FENOTIAZINICI ( acepromazina, clorpromazina, propionilpromazina)
- BENZODIAZEPINE ( diazepam, zolazepam, midazolam)
- BUTIRROFENONI ( droperidolo, azaperone)
SEDATIVI: ALFA 2 AGONISTI ( medetomidina, detomidina, xilazina, romifidina)
ANALGESICI NARCOTICI - OPPIACEI
“ NON ESISTONO AGENTI ANESTETICI SICURI,
NE’ PROCEDURE ANESTETICHE SICURE.
ESISTONO SOLO ANESTESISTI SICURI “
( R. Smith)
IL DOLORE
BASI MOLECOLARI DELLA NOCICEZIONE
Nocicezione: ricezione da parte del sistema nervoso centrale di segnali evocati dalla
attivazione di recettori sensitivi periferici (nocicettori)
Non tutti gli stimoli nocivi che attivano i nocicettori vengono necessariamente avvertiti come
dolorosi
Dolore: esperienza sensoriale ed emozionale spiacevole associata a danno tissutale, in atto
o potenziale, o descritta in termini di danno
(definizione della IASP – International Association for the Study of Pain adottata dall’O.M.S.)
Il dolore non può essere descritto come un fenomeno meramente sensoriale, bensì deve essere
valutato come la composizione di una parte percettiva (la nocicezione), che costituisce la modalità
sensoriale che permette la ricezione ed il trasporto al sistema nervoso centrale di stimoli
potenzialmente lesivi per l’organismo, e di una parte esperienziale (quindi del tutto soggettiva), che
comporta un processo di elaborazione e di astrazione dei segnali afferenti sensitivi
Le vie nervose e biochimiche attivate dalla sensazione dolorifica
creano una serie di risposte molto complesse e articolate che
coinvolgono il sistema endocrino e il sistema immunitario
si spiegano così fenomeni come il trauma postoperatorio, quell'insieme di fenomeni,
cioè, che incorrono dopo stimolazione nocicettiva prolungata e che comprendono
essenzialmente maggiore suscettibilità alle infezioni, orientamento catabolico del
metabolismo e squilibri omeostatici
La nocicezione inizia nelle terminazioni periferiche delle fibre afferenti primarie C e Aδ
(nocicettori) che hanno il corpo cellulare nei gangli delle radici dorsali e i rami centrali in
contatto sinaptico coi neuroni del corno dorsale del midollo spinale
I nocicettori termici e meccanici (meccanocettori e termocettori) hanno fibre Aδ di piccolo
diametro, dotate di una sottile guaina mielinica, che conducono alla velocità di 5-30 m/s
I nocicettori polimodali vengono attivati da stimoli meccanici di intensità elevata, da stimoli
chimici e da stimoli termici come il calore intenso o il freddo ed hanno fibre C, di piccolo
diametro, amieliniche, che conducono alla velocità di 0,5-2 m/s
▪
FIBRE MIELINICHE
cute e mucose
Alta velocità
Bassa soglia
Aδ 2-5 micron
5-30 m/sec
Sono responsabili della trasmissione del PRIMO DOLORE
(dolore rapido) come stimoli MECCANICI e TERMICI INNOCUI
▪
FIBRE AMIELINICHE
distribuzione superficiale e profonda
Bassa velocità
Alta soglia
C 0.5-1 micron
0.5-2 m/sec
Sono responsabili della trasmissione del SECONDO DOLORE
(dolore lento e sensibilità termica)
Nelle terminazioni periferiche gli stimoli
nocicettivi di diverse modalità
(meccanica, termica, chimica) vengono
trasformati in segnali elettrici o
potenziali generatori (trasduzione) che
attivano la scarica di potenziali d’azione
condotti lungo gli assoni (conduzione) e
sono propagati tramite la liberazione di
neurotrasmettitori al sistema nervoso
centrale (SNC) (trasmissione)
La regolazione spinale di un segnale algogeno
è complessa: un segnale riflesso torna
immediatamente agli organi prossimi alla
fonte del segnale algogeno (ghiandole,
muscoli vasali, viscerali e locomotori).
Nello stesso tempo il segnale attraverso il
midollo (neurone afferente) giunge nel
cervello dove viene integrato, controllato ed
elaborato. La risposta torna lungo un
neurone efferente al relativo interneurone e
(in base alla 'decisione' del cervello) amplifica
o smorza la risposta nel neurone attivato.
Il primo riflesso è dunque una risposta pronta
e incondizionata.
Poi la risposta viene corretta secondo il
……..non solo SNA
"parere" del cervello e può essere amplificata
fino al panico o smorzata fino alla letargia
dolore fisiologico: dolore evocato da stimoli di elevata intensità, potenzialmente in
grado di provocare danni tissutali
• ha lo scopo di proteggere i tessuti mediante l’attivazione di meccanismi di protezione
• non si accompagna a fenomeni di ipersensibilità (finché il dolore rimane fisiologico esiste una relazione
diretta più o meno proporzionale tra l’intensità dello stimolo applicato e la risposta evocata)
dolore patologico: sensazione di dolore evocata da stimoli successivi al danno del
tessuto periferico (più intensa di quella provocata dagli stessi stimoli su tessuti integri).
Non solo le lesioni tissutali, ma anche quelle che interessano il sistema nervoso
centrale e periferico, possono dar luogo a dolore patologico
Mentre il dolore fisiologico viene trasmesso esclusivamente da fibre di piccolo calibro, alla genesi del
dolore patologico, contribuiscono anche le fibre nervose di grande calibro: per esempio, in corso di
infiammazione, queste ultime possono iniziare a produrre sostanza P, cosa che in condizioni normali
avviene solo nelle fibre di piccolo calibro. I processi di trasduzione, conduzione e trasmissione vanno
incontro a modificazioni dinamiche, attività-dipendenti, che risultano in aumenti sia nella sensibilità e
nell’attività dei nocicettori e dei neuroni centrali
La sensibilizzazione periferica si verifica
quando le terminazioni dei nocicettori
sono esposte all’azione di sostanze liberate
o prodotte in seguito ai danni tissutali e
processi flogistici (protoni, purine,
bradichinina, NGF Nerve Growth Factor o fattore
di crescita neuronale, serotonina, istamina,
ecc.). Si manifesta come abbassamento
della soglia di attivazione delle
terminazioni periferiche con conseguente
attivazione di un numero maggiore di
nocicettori (reclutamento) e un aumento
della risposta a stimoli soprasoglia.
Le alterazioni si traducono in
un’aumentata frequenza di scarica nelle
fibre afferenti primarie che dà luogo ad un
aumento nella liberazione dei
neurotrasmettitori. Questi attivano i
meccanismi sinaptici che inducono e
mantengono l’iperattività e
l’ipereccitabilità dei neuroni postsinaptici
(sensibilizzazione centrale)
I meccanismi della sensibilizzazione centrale sono comuni al dolore infiammatorio e al
dolore neuropatico
L’attività afferente però mostra:
• differente substrato anatomico: fibre afferenti primarie intatte nel primo caso,
danneggiate e in parte degenerate nel secondo caso
• differente substrato fisiologico: sensibilizzazione dei nocicettori e reclutamento nel primo
caso, scariche anormali (injury discharges) nella sede del danno al nervo periferico nel
secondo caso
La sensibilizzazione si traduce in alterati stati di percezione del dolore come:
iperalgesia primaria (aumentata percezione di stimoli dolorifici nella zona di lesione)
iperalgesia secondaria (percezione di dolore in zone circostanti la lesione)
allodinia (percezione di dolore per stimoli normalmente innocui)
I meccanismi cellulari e molecolari che sottendono la sensibilizzazione coinvolgono cambiamenti nell’attività delle funzioni di
proteine come canali ionici, recettori, enzimi etc che funzionano come secondi messaggeri, e fattori di trascrizione come cfos e c-jun che sono considerati terzi messaggeri
PRINCIPI CLASSIFICATIVI
Dal punto di vista semeiologico, il dolore nocicettivo può essere classificato in:
a. dolore somatico: gli stimoli nocicettivi originano al livello della cute e delle mucose;
qualitativamente si tratta di un dolore netto o sordo, che il paziente riesce a localizzare
bene
b. dolore somatico profondo: gli stimoli nocicettivi provengono dai legamenti, dai
muscoli, dai tendini, dal periostio o dalle capsule articolari
c. dolore viscerale: deriva dall’attivazione dei nocicettori e dei nervi afferenti viscerali e
viene descritto dal paziente come una sensazione spiacevole, indefinita, profonda,
crampiforme, che spesso precipita nei dermatomeri superficiali dipendenti dai neuroni
che innervano i visceri coinvolti; tra gli stimoli in causa ricordiamo lo spasmo viscerale e
la distensione delle anse intestinali
d. dolore neuropatico: derivante dal danno diretto dei recettori periferici, nervi o
strutture del sistema SNC: viene descritto come urente o disestesico e spesso compare
in un’area di deficit sensitivo
Russell Portenoy ha descritto anche un dolore psicogeno, non spiegabile sulla base delle
lesioni organiche ma che riceve un concreto e significativo contributo da fattori
soggettivi psicologici
(la percezione cosciente del dolore ha luogo unicamente nella corteccia cerebrale: al 'dolore puro' della corteccia somestetica
primaria vengono aggiunte le sfumature emotive da parte della corteccia cerebrale prefrontale)
Dal punto di vista temporale, il dolore può
essere classificato in:
a. acuto: si presenta in maniera repentina, ha
una probabile durata limitata (inferiore ai tre
mesi), è spesso associato a segni clinici che
dimostrano un coinvolgimento del sistema
nervoso autonomo (tachicardia, ipertensione,
diaforesi); in genere ha una correlazione
causale e temporale identificabile con un
danno tissutale o con una malattia
b. cronico: si presenta con sintomatologia
persistente per tre mesi ed oltre; porta ad
alterazioni significative delle capacità funzionali
del paziente, compromettendone la qualità di
vita
VIE DISCENDENTI DEL CONTROLLO DEL DOLORE
Oltre alla via ascendente, il sistema preposto alla nocicezione comprende anche vie
discendenti di controllo del dolore
Due gruppi di fibre della modulazione inibitoria del dolore:
• sistema oppioide
• sistema noradrenergico
Recettori per gli oppioidi endogeni (encefaline e endorfine) e per le amine (noradrenalina,
serotonina) sono variamente distribuiti nel SNC sia a livello spinale che sopraspinale
(soprattutto nella sostanza grigia periacqueduttale, nella formazione reticolare e nel nucleo del raphe magno)
I meccanismi inibitori centrali sono attivati per modulare il dolore a livello spinale
attraverso le vie discendenti. I neurotrasmettitori sono la noradrenalina, la serotonina, le
endorfine
Sulla membrana presinaptica dei neuroni di primo ordine i neurotrasmettori oppioidi e
noradrenergici condividono lo stesso meccanismo d'azione: l'interazione agonistarecettore iperpolarizza le fibre afferenti impedendo la propagazione dello stimolo
doloroso e il rilascio dei mediatori eccitatori glutammato, sostanza P e neurokinine.
In aggiunta sembra che gli oppioidi endogeni intervengano nella modulazione del dolore anche con meccanismi di inibizione postsinaptica
ANALGESICI OPPIOIDI
Oppiacei: farmaci derivati da alcalodi dell’oppio
(morfina, codeina)
Oppioidi: tutte le sostanze (naturali, sintetiche, semisintetiche) che
producono effetti morfinosimili, che vengono bloccati da
antagonisti specifici (es. naloxone). Comprendono vari neuropeptidi e
anologhi sintetici la cui struttura può essere molto differente da quella
della morfina.
I peptidi oppioidi endogeni sono localizzati
all’interno dei neuroni del corno dorsale del
midollo spinale e qui determinano analgesia
mediante l’interazione con specifici recettori
1. m
2.
3. d
4. s
MECCANISMO D’AZIONE
•Tutti gli oppioidi agiscono mediante interazione con uno
specifico recettore identificato nelle famiglie µ, k, δ, s
•Di questi, la famiglia di recettori µ appare la più
importante per ciò che riguarda gli effetti analgesici e di
depressione respiratoria.
•Questa famiglia consiste in due sottotipi: µ1 e µ 2.
•Molto probabilmente il recettore µ1 media gli effetti
dell’analgesia, mentre il recettore µ2 è coinvolto nell’azione
di depressione respiratoria, bradicardia e dipendenza fisica.
MECCANISMO D’AZIONE
•Dai recettori K dipende l’effetto di sedazione e la
dinorfina è il ligando endogeno. Sono attribuibili anche
effetti di analgesia (sovraspinale e spinale), disforia,
diuresi, miosi
•Ai recettori sigma sono attribuiti gli effetti maniacali e
gli effetti di tipo psicotico. I ligandi endogeni sono le
encefaline (questi non sono antagonizzati dal naloxone)
Tutti i recettori oppioidi sono legati a proteine Gi con conseguente:
• Inibizione dell’adenilato ciclasi
• Facilitazione dell’apertura dei canali per il K+
• Inibizione dell’apertura dei canali voltaggio dipendenti per il Ca2+
Endogenous opiates can cause post or pre synaptic inhibition
Classificazione analgesici oppioidi
Agonisti
Dolore moderato
Codeina
Ossicodone
Deidrocodeina
Destropropossifene Tramadolo
Agonisti-antagonisti
Pentazocina
Butorfanolo
Meptazinolo
Dolore molto intenso
Breve emivita
Morfina
Meperidina
Lunga emivita
Metadone
Levorfanolo
Fentanile
Agonisti parziali
Buprenorfina
Oppioidi endogeni
Oppioidi: effetti sul SN
Effetti mediati da interazione con i recettori
µ: analgesia, sedazione, euforia e
depressione respiratoria
Analgesia
Attribuibile alle complesse interazioni che coinvolgono recettori
presenti a livello centrale, nel midollo spinale e nei tessuti periferici
L’azione si manifesta selettivamente sulle vie nervose coinvolte
nel dolore (le altre vie sensoriali/motorie rimangono intatte)
Midollo spinale:-siti di azione
A. Livello presinaptico sulle afferenze dei nocicettori con
diminuzione rilascio sostanza P
B. Iperpolarizzazione degli interneuroni della substantia gelatinosa
con diminuzione della trasmissione dell’impulso nocicettivo
Recettori oppiodi interessati: tipo µ1
Sedazione
• Conseguenza più comune alla somministrazione di
oppioidi ma specie dipendente
• Se somministrati in monoterapia, gli oppioidi possono
produrre sonno (facile risveglio)
• Se associati a sedativi-ipnotici sonno prolungato e
profondo
• Sedazione maggiore con derivati fenantrenici (morfina,
ossi e idrocodone, ossi e idromorfone)
• Sedazione minore con gli agenti sintetici (fentanile,
meperidina)
Depressione respiratoria
Tutti gli oppiodi inducono significativa depressione dei centri
respiratori del midollo allungato: tutte le fasi e i parametri
respiratori risultano alterati, anche se nel cane questa fase segue
una fase di stimolazione dei centri del respiro.
La ridotta risposta alla CO2 è:
• dose-dipendente
• tollerata in pazienti che non presentano difficoltà
respiratorie
• non tollerata in pazienti con asma, malattia polmonare
cronica ostruttiva (COPD), cuore polmonare e aumentata
pressione intracranica
Depressione della tosse
• Azione posseduta da tutti gli oppioidi
• Efficace soprattutto la codeina
•L’azione di sedazione si può associare ad accumulo delle secrezioni
nel lume bronchiale (no x tosse produttiva)
Pupilla
Variabilità di specie:
Gatto, pecora e cavallo: midriasi
Cane, ratto e uomo: miosi
Rigidità muscoli del tronco
• Dovuta ad azione sopraspinale
• Diminuisce compliance toracica; fa peggiorare la
ventilazione
• Più frequente con i preparati più lipofilici (fentanil,
sulfentanil, alfentanil) se somministrati e.v.
• Reversibile con la somministrazione di antagonisti
• Il mantenimento dell’analgesia associato alla rigidità
muscolare spesso richiede l’impiego dei bloccanti
neuromuscolari.
Effetti gastrointestinali
•Stomaco:
•Tono aumentato
• Secrezione gastrica (HCl) diminuita
•Motilità diminuita
• Piccolo intestino e grande intestino:
• Tono aumentato con spasmi
• Onde propulsive peristaltiche diminuite
Ciò determina:
1. Ritardo nel passaggio delle feci (costipazione)
2. Promuove il riassorbimento di acqua (costipazione)
•Sfinteri :
•Tono aumentato e percezione sensoriale diminuita
Azione Emetica e
Termoregolazione
Emesi
- Stimolazione chemiocettori midollo allungato (CTZ)
- Presente anche una componente periferica, quella
vestibolare
Variabilità di specie: più sensibile è il cane, mentre il gatto è il
meno sensibile. Suini, polli bovini e cavalli non rispondono
Termoregolazione
Variabilità di specie:
Gatto, capra, bovino e cavallo: ipertermia
Cane e coniglio: ipotermia
Apparato urinario
Funzione renale: depressa
• Liberazione di ADH
• Diminuzione flusso renale
• Aumento del tono mm vescicale, ureterale e degli sfinteri
Effetti neuroendocrini
Gli oppioidi promuovono il rilascio di
• ADH
• Prolattina
•ACTH
• Gli oppioidi inibiscono il rilascio di
• LH
Assorbimento
• Gli analgesici oppioidi sono generalmente ben assorbiti
per via s.c. o i.m.
•Assorbimento gastrointestinale: veloce
- Alcuni– notevole effetto di primo passaggio:
• ad es. morfina
Distribuzione
• Legame proteico variabile (morfina 35%; metadone 89%)
• Concentrazione tissutale più elevata nei tessuti ad
alta perfusione
• I muscoli scheletrici rappresentano il più importante
compartimento di riserva
• Gli oppiodi più lipofili (fentanil), si concentrano nel
tessuto adiposo
Distribuzione
• Gli agenti anfoteri (morfina) presentano grande
difficoltà nell’attraversare la barriera E-E
• migliore penetrabilità: eroina, codeina
•Animali neonati sono più sensibili agli oppioidi per
presenza di barriera E-E non sviluppata, la sensibilità
diminuisce con lo sviluppo dell’animale (gli oppiodi usati
in analgesia ostetrica, attraversano la placenta, possono
indurre depressione respiratoria del nuovo nato)
Metabolismo
• Fegato: metaboliti più polari
• Gli oppioidi con un gruppo idrossilico sono con
maggiore frequenza coniugati con acido glucuronico
(morfina, levorfanolo)
•Gli oppioidi con un legame esterico sono idrolizzati da
esterasi tissutali (eroina, remifentanil)
La N-demetilazione: è una via metabolica minore
- accumulo del metabolita demetilato
Ossidazione (epatica) è la via principale del metabolismo
degli oppioidi fenilpiperidinici: fentanil; alfentanil;
sufentanil
FARMACI PSICOTROPI
CLASSIFICAZIONE NEUROLETTICI o ANTIPSICOTICI
· Reserpina
· Fenotiazine
· Tioxanteni
· Butirrofenoni
· Difenil-butil-piperidine
· Neurolettici atipici
Neurolettici
tipici o
convenzionali
NEUROLETTICI
Caratteristiche comuni
✓Alleviano i sintomi della psicosi
✓Aiutano a prevenire le crisi acute
✓Non guariscono la malattia
✓Controllano prevalentemente i sintomi positivi
(allucinazioni, deliri)
✓Notevole carico di effetti tossici
✓Il 30% dei pazienti risponde scarsamente alla terapia
Neurolettici tipici
Meccanismo d’azione (inibizione competitiva - vari gradi a seconda delle classi)
Blocco dei recettori
muscarinici
Blocco dei
Blocco dei
recettori alfa- recettori
adrenergici dopaminergici
Blocco dei
recettori H1
dell’Istamina
Farmaci neurolettici tipici
Indicazioni terapeutiche nell’uomo
✓Sindromi schizofreniche (episodi acuti e forme croniche)
✓Sindromi schizo-affettive
✓Fase maniacale delle sindromi bipolari
✓Psicosi organiche
✓Psicosi associate all’invecchiamento (M. di Alzheimer)
✓Emesi
Farmaci neurolettici tipici
Effetti collaterali
Sul SNC
Sul SNA
Sul sistema Endocrino
Farmaci neurolettici tipici
Effetti collaterali
Sul SNC
Blocco dei recettori
Dopaminergici
-Parkinsonismo
-Discinesia tardiva
Blocco dei recettori
Muscarinici
-Stato tossico
confusionale
Farmaci neurolettici tipici
Effetti collaterali
Sul SNA
Blocco dei recettori
Muscarinici
Blocco dei recettori
Alfa-Adrenergici
-Perdita dell’accomodazione
-Ipotensione ortostatica
-Distensione urinaria
-Impotenza
-Stipsi
-Mancanza di
eiaculazione
-Secchezza delle fauci
Farmaci neurolettici tipici
Effetti collaterali
Sul Sistema Endocrino
Blocco dei recettori
Dopaminergici
Aumento della prolattina
-Amenorrea
-Galattorrea
-Ginecomastia
-Infertilità ed impotenza
Fenotiazine
·
·
·
CLORPROMAZINA
primo farmaco antipsicotico
blocca i recettori della dopamina D2
blocco modesto dei recettori Ach e 5-HT
Effetti comportamentali:
- catalessia
- inibizione attività motoria spontanea
- inibizione della reattività e dell’interesse
EFFETTI SU ALTRI RECETTORI
•Bloccano i recettori H1 dell’istamina
(proprietà sedative ed antiemetiche)
•Bloccano i recettori dell’Ach (offuscamento
della vista, aumento della pressione
intraoculare, secchezza delle fauci e degli
occhi, costipazione e ritenzione urinaria)
•L’antagonismo muscarinico è positivo per
gli effetti collaterali di tipo extrapiramidale
•Bloccano i recettori alfa-adrenergici
(ipotensione ortostatica)
IMPIEGHI CLINICI - UOMO
• Comportamento maniacale in pazienti
schizofrenici o affetti da psicosi maniaco
depressiva, nelle psicosi tossiche e
postraumatiche e senili
• Turbe del pensiero, allucinazioni e deliri
• Ritiro autistico
• Apatia
• Appiattimento affettivo e riduzione del
linguaggio
• Nausea e vomito
Efficacia clinica
·60-70% dei pazienti sono responsivi
·miglioramento clinico dopo settimane o
mesi (raggiungimento dello steady-state)
•Necessarie terapie di mantenimento per
evitare ricadute
•10% dei pazienti non migliora
•20% manifesta tolleranza
•Migliori risultati con dosi più basse
FARMACOCINETICA DELLE
FENOTIAZINE
• Assorbimento irregolare dopo
somministrazione orale
• Somministrazioni intramuscolo short o long
acting (formulazioni depot) (per pz agitati)
• La relazione tra concentrazione plasmatica ed
effetto clinico varia da soggetto a soggetto
• Sono metabolizzati a livello epatico
• Circa 90% legame prot plasmatiche
• Emivita: 15-30 ore
• Escrezione urinaria
EFFETTI COLLATERALI E TOSSICI
Sindromi extrapiramidali acute
• Agitazione psicomotoria, insonnia ed
ipereccitabilità (=ACATISIA)
• Distonie acute (contrazioni intermittenti o
prolungate dei muscolo del collo, tronco,
mandibola, lingua)
• Parkinsonismo: rigidità, bradicinesia, tremori,…
Discinesie tardive
EFFETTI COLLATERALI E TOSSICI
•Sonnolenza fino a letargia
•Crisi convulsive
•Catatonia, stupore, febbre, alterazione della pressione,
aritmie (=sindrome maligna da neurolettici)
•Ittero, diabete
•Agranulocitosi,
•Reazioni dermatologiche (lesioni cutanee,
fotosensibilità)
•Aumento dei livelli di prolattina: turgore mammario,
galattorea, amenorrea, sterilità, impotenza
•Effetti anti-colinergici
L’effetto sedativo è diverso da quello esercitato dai barbiturici poiché non modifica in
maniera apprezzabile le risposte motorie integrate degli animali e, inoltre, il risveglio
degli animali appare facilmente conseguibile
Occasionalmente possono produrre eccitazione (soprattutto se ev), particolarmente
evidente nel cavallo
Tioxantenici
• Sono strutturalmente correlati alle
fenotiazine
• Flupentixolo e Clorprotixene sono i
principali tioxantenici
• Hanno le stesse proprietà farmacologiche
delle fenotiazine
• Maggiore intensità degli effetti
anticolinergici
• Minore incidenza di effetti collaterali
extrapiramidali
• Lieve azione sedativa
• Lieve attività antidepressiva
• Farmacocinetica = Fenotiazine
Butirrofenoni
aloperidolo
• Primo butirrofenone studiato: Aloperidolo
• Non hanno azione antistaminica, anticolinergica,
antiadrenergica (debole)
• Lievi turbe a carico del SNA
• Mancanza di sedazione (utile per integrazione sociale)
• Insonnia (per blanda tolleranza e dipendenza fisica)
• Ben assorbiti per os, max picco plasmatico entro 1-6 h
dall’ingestione, eliminati lentamente da urine e feci
L’aloperidolo (es. SERENASE) è un neurolettico appartenente al gruppo
dei butirrofenoni. L’aloperidolo è un potente antagonista della dopamina;
ha affinità simile per tutti i sottotipi recettoriali della dopamina: è quindi un
antagonista dopaminergico non selettivo. Il farmaco ha anche attività
antagonista verso i recettori alfa-adrenergici, mentre non presenta attività
antistaminergica o anticolinergica.
Si ritiene che l’effetto del farmaco sul delirio e le allucinazioni sia legato
all’antagonismo dopaminergico a livello delle regioni meso-corticali e
limbica.
L’antagonismo a livello dei gangli basali è probabilmente la causa degli
effetti collaterali motori di tipo extrapiramidale (distonia, acatisia e
parkinsonismo).
L’aloperidolo presenta un efficace effetto sedativo psicomotorio che
contribuisce all’azione favorevole sulla mania ed altre sindromi da
agitazione.
L’aloperidolo si è mostrato utile anche nel trattamento del dolore cronico,
un effetto forse dovuto all’azione a livello limbico.
Gli effetti antidopaminergici più periferici spiegano l’attività contro nausea
e vomito (antagonismo a livello della chemoreceptor trigger zone, CTZ),
l’aumentato rilascio di prolattina (tramite l’antagonismo nei confronti
dell’attività di inibizione, mediata dalla dopamina, del rilascio della
prolattina da parte dell’adenoipofisi) ed il rilasciamento degli sfinteri
gastrointestinali
L’aloperidolo è metabolizzato nel fegato mediante: glucuronidazione ed il sistema
del citocromo P450 (in particolare CYP 3A4 o CYP 2D6). L’inibizione di queste vie
metaboliche da parte di un altro farmaco o una diminuzione dell’attività enzimatica
del CYP 2D6 può provocare l’aumento della concentrazione di aloperidolo e un
aumento del rischio di eventi avversi tra cui il prolungamento del QT
(cardiotossicità).
E’ escreto nelle urine e, attraverso la via biliare, nelle feci. L’emivita varia tra 13 e
40 ore. Il legame con le proteine plasmatiche è molto elevato (90%). Si distribuisce
ampiamente nell’organismo (alto volume di distribuzione) e attraversa la barriera
ematoencefalica.
Difenil – butil - piperidine
Penfluridolo, Fluspirilene, Pimozide
Disordini extrapiramidali molto
frequenti
Sedazione, sonnolenza, debolezza e
malessere generale
Dolori muscolari, spasmi, galattorrea
Pimozide (es. ORAP) è particolarmente indicato come farmaco di base
nel corso di una terapia antipsicotica di mantenimento di lunga durata in
pazienti psicotici cronici ed acuti, sensibili agli effetti antipsicotici
specifici dei neurolettici.
ORAP è anche indicato come terapia d'attacco in pazienti ambulatoriali
od ospedalizzati da poco tempo o riammessi in clinica purché
l'agitazione psicomotoria, l'aggressività, o stati ansiosi particolarmente
gravi non costituiscano i sintomi dominanti del quadro clinico.
ORAP è indicato, infine, nei casi limite tra forme schizofreniche e forme
nevrotiche (per es. stati paranoidi e schizoidi) che implicano difficoltà di
rapporti sociali.
L'impiego del prodotto ad alte dosi deve essere limitato agli Ospedali e
Case di Cura con le indicazioni ridotte al trattamento dei casi resistenti.
Come molti neurolettici gli effetti avversi sono: sintomi
extrapiramidali, discinesia tardiva, crisi epilettiche, disturbi
endocrini, disturbi della regolazione della temperatura del corpo,
disturbi cardiovascolari.
Neurolettici atipici
Denominati atipici in quanto caratterizzati da
minori effetti collaterali (disturbi extrapiramidali,
discinesia tardiva, etc.)
Neurolettici atipici (benzamidi)
Clozapina, Sulpiride, Risperidone,…
Selettivi per i recettori D2 della dopamina
Molto efficaci nelle psicosi confusionali acute
Effetti più o meno intensi di tipo parkinsoniano
No catalessia
Aumento lieve del turnover di dopamina
No interferenze sui recettori Ach, GABA e H1
Interferenza con i recettori 5-HT
Utili per allucinazioni
Buon assorbimento per os, max picco plasmatico
entro 4h dall’ingestione, metabolismo epatico,
emivita media: 20h
Antipsicotici non convenzionali
• Farmaci di scelta nel trattamento della
schizofrenia, depressione con tratti psicotici,
disturbi psicoaffettivi, maniacalità
• Migliore efficacia, maneggevolezza e
tollerabilità (…maggiore costo!)
• Migliorano i disturbi cognitivi (linguaggio,
memoria) e le performance motorie (migliore
qualità di vita)
Farmaci antipsicotici atipici
Meccanismo d’azione
Blocco selettivo dei recettori D2-Like presinaptici,
in particolare sui D4 delle zone limbiche
Inibizione del tono dopaminergico nelle
strutture limbiche ma non del sistema
nigro-striatale
Riduzione dei sintomi psicotici senza o con
ridotta interferenza col sistema
extrapiramidale
Neurolettici tipici (blocco non selettivo dei recettori D2 )
Neurolettici atipici (Blocco
selettivo dei recettori D4 presinaptici)
Neurolettici atipici
Caratteristiche comuni
➢Affinità per i recettori D2-like della dopamina
➢Affinità per i recettori 5-HT2 della serotonina
➢Maggiormente efficaci sui sintomi negativi (apatia,
depressione, autoesclusione) e cognitivi
➢Riduzione o assenza dei disturbi motori ( parkinsonismo,
discinesia tardiva, etc.)
Neurolettici atipici
Indicazioni terapeutiche
➢Forme di psicosi schizofreniche refrattarie
ai trattamenti farmacologici convenzionali
➢Intolleranza ai disturbi motori provocati dai
neurolettici tipici
➢Psicosi in corso di Malattia di Parkinson
Attualmente, i neurolettici di maggior interesse in Medicina Veterinaria sono i derivati
fenotiazinici (clorpromazina, acepromazina, promazina), i quali, come gli agonisti 2adrenergici presinaptici (xilazina, detomidina, medetomidina), vengono impiegati nella
medicazione preanestetica, riducendo notevolmente le quantità di anestetico
necessario, nella esecuzione di esami clinici invasivi e per ridurre l’ansietà e
l’eccitamento provocati da situazioni particolari (es. sovraffollamento, trasporto,
fenomeni autolesivi che si accompagnano ad otiti,, pruriti ed eczemi)
Pur essendo molto simili per struttura molecolare, meccanismo d’azione ed effetti,
sono molto meno impiegati i derivati butirrofenonici e tioxantenici.
Gli unici di
interesse sono il droperidolo e l’azaperone (utilizzato come tranquillante nel suino e
nella neuroleptoanalgesia in associazione a metomidato)
La reserpina (Rawolfia serpentina) è stata abbandonata nella pratica clinica veterinaria