SINDROMI GENETICHE
E CROMOSOMICHE
E PATOLOGIE DEL CAVO ORALE
IN ETÀ EVOLUTIVA
a cura di
Giovanni D’Alessandro e Gabriela Piana
Bononia University Press
www.buponline.com
www.buponline.com
SINDROMI GENETICHE
E CROMOSOMICHE
E PATOLOGIE DEL CAVO ORALE
IN ETÀ EVOLUTIVA
a cura di
Giovanni D’Alessandro e Gabriela Piana
Bononia University Press
www.buponline.com
Si ringrazia la Fondazione del Monte di Bologna e Ravenna per il generoso sostegno dato all’attività di
ricerca del Servizio di Assistenza Odontoiatrica per Disabili in età evolutiva della Clinica Odontoiatrica
del Dipartimento di Scienze Biomediche e Neuromotorie dell’Alma Mater Studiorum – Università di
Bologna.
Bononia University Press
Via Farini 37 – 40124 Bologna
www.buponline.com
e-mail: [email protected]
© 2014 Bononia University Press
Tutti i diritti riservati
ISBN 978-88-7395-937-3
Impaginazione: Sara Celia
Prima edizione: luglio 2014
www.buponline.com
Indice
Prefazione
Guido Cocchi
5
Introduzione
Giovanni D’Alessandro, Gabriela Piana
7
Capitolo 1
Controllo del dolore e dell’ansia
S. Bagattoni, N. Alkhamis, M.G. Currà, T. Tagariello, G. D’Alessandro, G. Piana
15
Capitolo 2
Cardiopatie congenite
M. Taddei, S. Bagattoni, T. Tagariello, G. D’Alessandro, G. Piana
27
Capitolo 3
Sindrome di Down T. Tagariello, N. Alkhamis, G. D’Alessandro, G. Piana
39
Capitolo 4
Sindrome di Klinefelter
I. Cremonesi, M.G. Currà, T. Tagariello, G. D’Alessandro, G. Piana
53
Capitolo 5
Sindrome da Delezione 22q11.2
S. Bagattoni, N. Alkhamis, T. Tagariello, G. D’Alessandro, G. Piana
61
Capitolo 6
Sindrome di Turner
I. Cremonesi, S. Bagattoni, T. Tagariello, G. D’Alessandro, G. Piana
71
www.buponline.com
Capitolo 7
Sindrome dell’X Fragile
S. Bagattoni, N. Alkhamis, T. Tagariello, G. D’Alessandro, G. Piana
81
Capitolo 8
Sindrome di Williams
I. Cremonesi, S. Bagattoni, G. D’Alessandro, G. Piana
89
Capitolo 9
Sindrome di Angelman
N. AlKhamis, T. Tagariello, L. Armuzzi, G. D’Alessandro, G. Piana
99
Capitolo 10
Sindrome di Prader Willi
I. Cremonesi, S. Bagattoni, T. Tagariello, G. D’Alessandro, G. Piana
109
Capitolo 11
Sindrome di Silver Russel
N. Alkhamis, F. Skendo, G. D’Alessandro, G. Piana
119
Capitolo 12
Sindrome di Noonan
L. Armuzzi, I. Cremonesi, T. Tagariello, G. D’Alessandro, G. Piana
129
Capitolo 13
Sindrome di Marfan
M. Taddei, T. Tagariello, G. D’Alessandro, G. Piana
139
Capitolo 14
Neurofibromatosi
L. Kondo, F. Skendo, M. Taddei, G. D’Alessandro, G. Piana
151
Capitolo 15
Sindrome di Alagille
S. Bagattoni, F. Skendo, I. Cremonesi, G. D’Alessandro, G. Piana
161
Capitolo 16
Displasie Ectodermiche
F. Battelli, M. Montanari, I. Cremonesi, N. Alkhamis, G. D’Alessandro, G. Piana
171
Capitolo 17
Distrofie Muscolari
I. Cremonesi, N. Alkhamis, T. Tagariello, G. D’Alessandro, G. Piana
183
www.buponline.com
Prefazione
La salute orale rappresenta un elemento di fondamentale importanza nello stato di salute generale e contribuisce a garantire una buona qualità di vita della persona. In particolare nei bambini affetti da sindromi
cromosomiche e genetiche, l’acquisizione di stili di salute orale adeguati fin dalle primissime età della
loro vita rappresenta una chiave di successo nel prevenire l’insorgenza delle patologie orali più diffuse. È
bene ricordare che spesso nei pazienti con patologie responsabili di disabilità in età evolutiva, a causa della
ridotta collaborazione, le terapie odontoiatriche sono di più difficile esecuzione e la necessità di eseguire le
terapie in narcosi con ospedalizzazione può comportare stress psicologici per le famiglie e costi economici
elevati per la società. Inoltre, nel bambino con patologia congenita rara, l’insorgenza di patologie odontoiatriche può comportare rischi per la salute generale.
Particolari manifestazioni cliniche odontoiatriche sono caratteristicamente associate a singole sindromi,
è quindi importante conoscerle per saperle riconoscere e attuare programmi di prevenzione e terapia
adeguati.
È quindi fondamentale che questi bambini siano presi in carico sin dalla primissima infanzia presso
strutture odontoiatriche a loro dedicate e che l’odontoiatra infantile e l’ortodontista possiedano tutte le
conoscenze di base relative al vasto e complesso capitolo delle sindromi genetiche o cromosomiche.
Il testo Sindromi genetiche e cromosomiche e patologie del cavo orale costituisce un valido strumento di aggiornamento e un concreto aiuto per tutti gli operatori sanitari che nella loro pratica clinica quotidiana
hanno l’obiettivo di promuovere la salute di questi bambini.
Guido Cocchi
www.buponline.com
www.buponline.com
Introduzione
Giovanni D’Alessandro, Gabriela Piana
Il mondo scientifico nell’aprile del 2003, dopo la
prima bozza pubblicata nel 2001, ha annunciato di
aver completato la mappatura del genoma umano;
questo risultato ha aperto enormi prospettive in
campo biologico e medico, dal punto di vista sia
delle diagnosi che delle terapie.
Alterazioni del patrimonio genetico, quali alterazioni nel numero o nella struttura dei cromosomi
e mutazioni di uno o più geni, in alcuni casi sono
incompatibili con la vita, determinando l’interruzione più o meno precoce della gravidanza e, in
altri, causano quadri patologici.
Le malattie geneticamente determinate sono
classificate in due gruppi: cromosomiche e genetiche.
Le malattie cromosomiche sono causate da alterazioni nel numero (aneuploidie) o nella struttura
dei cromosomi (delezione, inversione, duplicazione, traslocazione). Considerando che ogni cromosoma è composto da migliaia di geni, alterazioni di
numero e di struttura determinano quadri patologici in genere molto gravi, caratterizzati da malformazioni di apparati e di organi, da ritardo di crescita e da ritardo mentale. Un esempio di malattia
cromosomica è la sindrome di Down, causata da
trisomia del cromosoma 21.
Le malattie genetiche sono causate da mutazioni
di un singolo gene; possono essere ereditate secondo le leggi di Mendel o manifestarsi ex-novo.
L’uso di avanzati metodi di biologia molecolare ha
consentito di arricchire la mappa dei geni-malattia
responsabili di quadri sindromici.
La diagnosi di queste patologie, primariamente
clinica, è confermata dallo studio degli alberi genealogici familiari, dall’analisi del cariotipo, da
indagini biochimiche.
In determinati casi è possibile la diagnosi prenatale attraverso analisi citogenetiche e biochimiche di
cellule fetali prelevate mediante villocentesi o amniocentesi. Anomalie cromosomiche o genetiche
nei genitori o in un precedente figlio sono un’indicazione alla diagnosi prenatale. Considerando che
in almeno il 50% degli aborti spontanei precoci il
feto presenta anomalie cromosomiche, anche aborti spontanei ricorrenti sono un’indicazione per la
diagnosi prenatale e per lo studio dei cromosomi
parentali.
La quasi totalità delle sindromi genetiche e cromosomiche sono definibili come malattie rare.
Secondo l’Unione Europea sono rare le malattie
che colpiscono non più di 5 persone su 10.000.
Secondo le stime, nell’Unione Europea, tra 7.000 e
www.buponline.com
8
Introduzione
8.000 malattie rare diverse colpiscono e colpiranno
29 milioni di persone.
Attualmente la mancanza di politiche sanitarie
specifiche e l’insufficienza di conoscenze scientifiche comportano ritardi nelle diagnosi e difficoltà di accesso alle cure, che hanno come conseguenze nel paziente danni fisici, psicologici e
intellettuali e trattamenti inadeguati. Inoltre i
servizi di diagnosi, terapia e riabilitazione erogati
dai diversi Sistemi Sanitari Nazionali ai pazienti
affetti da malattie rare variano molto in termini di
disponibilità e di qualità. Di qui la necessità di una
cooperazione a livello europeo che permetta di
intervenire efficacemente, mettendo in comune le
conoscenze ed utilizzando le risorse nel modo più
efficiente.
La Commissione delle Comunità Europee (Bruxelles, 11/11/2008) ha proposto un approccio
integrato alle malattie rare e ha dato chiare indicazioni sulle attività comunitarie presenti e future per
migliorare la prevenzione, la diagnosi e la cura,
individuando tre linee di azione:
1. migliorare il riconoscimento e la visibilità delle malattie rare;
2. appoggiare l’azione degli Stati membri per
quanto riguarda le malattie rare;
3. sviluppare sul piano europeo la cooperazione,
il coordinamento e la regolamentazione nel campo delle malattie rare.
In Europa le malattie rare costituiscono una della
priorità nel Programma di Azione Comunitaria in
materia di Sanità Pubblica 2007-2013 ma solo pochi Paesi europei hanno sviluppato piani d’azione
nazionali o intrapreso iniziative in tale senso.
In Italia i Piani Sanitari Nazionali da anni hanno
indicato fra le priorità la tutela dei soggetti colpiti
da malattie rare. Su indicazione del PSN 19982000, con il D.M. 279/2001 è nata la Rete Nazionale Malattie Rare con la realizzazione di una
rete nazionale costituita da Presidi (individuati dalle Regioni) per la prevenzione, la sorveglianza, la
diagnosi e la terapia di queste patologie. Tuttavia, a
causa delle diverse modalità di raccolta, i dati epidemiologici disponibili sulla frequenza delle malattie croniche ad elevata complessità assistenziale,
comprese le malattie rare, sono molto variabili e
la loro frequenza (espressa come prevalenza nella
fascia di età 0-17 anni compiuti) è soltanto stimata
(Tabelle 1 e 2).
www.buponline.com
9
Introduzione
Tabella 1: stima della prevalenza in Italia (ISTAT)
Stima della prevalenza nella fascia di età 0 - 17 anni compiuti (circa 10 milioni in Italia) di alcune patologie
esemplificative e stima prevalenza globale delle malattie genetiche e/o disabilità complesse.
Condizione
Prevalenza
Condizione
Prevalenza su 10
milioni
2.860
Acondroplasia
400
Neurofibromatosi tipo 1
Sindrome di Angelman
625
Sindrome di Noonan
5.000
Sindrome di Beckwith-Wiedemann
730
1.785
Sindrome di Down
10.000
Sindrome Oculo-auricolovertebrale
Sindrome di Prader-Willi
Sindrome Del 22
5.000
Spina bifida
2.000
Sindrome FRA – X
2.000
Sindrome di Stickler
1.000
Malattie metaboliche
3.600
Sindrome di Turner
2.000
Malattie neuro-muscolari
3.300
Sindrome di Williams
Sindrome di Marfan
1.666
Totale delle condizioni in lista
43.431
Totale stimato considerando anche condizioni non in lista,
comprese quelle senza diagnosi precisa
50.000
800
666
• In Italia secondo i dati ISTAT al 31-12-2005 su 58.750.000 abitanti, 9.979.005 hanno un’età compresa tra 0-17
anni compiuti
• Cassidy SB & Allanson JE (Eds). Management of Genetic Syndromes. Wiley Press 2001
• Stima eseguita tenendo conto che: (a) la prevalenza totale delle condizioni inserite nella lista della tabella 1 (del tutto
parziale e limitata alle condizioni più comuni) è del 4,3 per mille; (b) la stima di alcune di essa può essere imprecisa
(talora per difetto, ma anche per eccesso – es. Sindrome di Noonan); (c) la prevalenza della sola paralisi cerebrale,
abbastanza costante nel tempo e nelle diverse popolazioni, è intorno al 2 per mille; (d) secondo il Sistema Informativo
del Ministero della Pubblica Istruzione, la prevalenza di bambini con disabilità certificata che hanno frequentato la
scuola nel biennio 2002-2003 (6-15 anni) è stata del 20 per mille; (e) circa 1 bambino su 4 che frequenta la scuola e
ha una disabilità certificata ha una condizione complessa.
www.buponline.com
10
Introduzione
Tabella 2: numero di disabilità complesse per Regione
Numero bambini e adolescenti con disabilità complesse per Regione
Regione
Nati vivi
Pop 0-17 a
Pop Totale
% 0-17 a
Piemonte
Valle d’Aosta
Lombardia
Trentino-Alto Adige
Veneto
Friuli-Venezia Giulia
Liguria
Emilia-Romagna
37.251
1.161
92.480
10.719
46.264
10.083
11.957
38.518
646.444
19.515
1540349
189.442
785.458
173.740
214.526
619.299
4.341.733
123.978
9.475.202
985.128
4.768.313
1.208.278
1.610.134
4.187.557
14,9%
15,7%
16,3%
19,2%
16,6%
14,4%
13,3%
14,8%
N con Disabilità
complesse
3.232
98
7.702
947
3.927
869
1.073
3.096
Toscana
Umbria
Marche
Lazio
Abruzzo
Molise
Campania
Puglia
Basilicata
Calabria
Sicilia
Sardegna
ITALIA
31.390
7.732
13.440
50.833
11.200
2.527
62.599
38.715
4.908
18.228
50.791
13.226
554.022
528.368
131.188
241.823
890.983
214.905
53.195
1.245.190
789.010
107.284
382.983
1.002.426
265.613
10.041.741
3.619.872
867.878
1.528.809
5.304.778
1.305.307
320.907
5.790.929
4.071.518
594.086
2.004.415
5.017.212
1.655.677
58.751.711
14,6%
15,1%
15,8%
16,8%
16,5%
16,6%
21,5%
19,4%
18,1%
19,1%
20,0%
16,0%
17,1%
2.642
656
1.209
4.455
1.075
266
6.226
3.945
536
1.915
5.012
1.328
50.209
Pop 0-17 a = Popolazione residente al 31 Dicembre 2005 (0-17 anni)
Pop Totale = Popolazione residente al 31 Dicembre 2005
N disabilità complesse = Numero di bambini e adolescenti per Regione assumendo una prevalenza di 1 su 200 nella
fascia di età 0-17 anni
Lo studio e le ricerche epidemiologiche sull’incidenza, sulle cause e sulla variabilità territoriale
quantitativa e qualitativa delle patologie congenite hanno avuto uno sviluppo scientifico serio e
produttivo da quando sono stati istituiti i Registri
delle Malformazioni Congenite.
Nel mondo oggi sono attivi vari Registri, tra cui
l’European Registration of Congenital Anomalies and Twins (EUROCAT), un network di regi-
stri europei sotto il controllo della Comunità Europea, e l’International Clearinghouse for Birth
Defect Monitoring Systems (ICBDMS), un network di registri mondiali affiliato all’Organizzazione Mondiale della Sanità (OMS) che coinvolge
attualmente 28 Registri di 35 nazioni appartenenti
a tutti i continenti.
In Italia, a partire dal 2001, le Regioni hanno iniziato a individuare i Presidi per l’assistenza ai pa-
www.buponline.com
11
Introduzione
zienti affetti da malattie rare e attualmente sono
attivi e operativi 6 Registri per le Malformazioni
Congenite (MC) che, con metodologie simili anche se non del tutto sovrapponibili, consentono
una copertura di buona parte dell’Italia. Dal 1984
è attivo un Coordinamento Nazionale dei Registri
delle MC con sede a Roma presso l’Istituto Superiore di Sanità (ISS).
Il Registro Nord Est Italia delle Malformazioni
Congenite (N.E.I.), attivo nelle Regioni Veneto,
Friuli-Venezia Giulia e Trentino-Alto Adige, riguarda 31 MC selezionate il cui rilevamento interessa il periodo prenatale e neonatale. Monitorizza
circa 56.000 nati/anno con una copertura di circa
il 97% del territorio. È membro di EUROCAT e
di ICBDMS.
L’Indagine Malformazioni Congenite Emilia
Romagna (IMER) monitorizza circa 25.000 nati/
anno con una copertura di circa il 95% del territorio regionale, coinvolgendo 45 centri. Da anni
conduce numerosi studi, tra cui quelli sull’impatto
della diagnosi prenatale sulla prevalenza alla nascita delle patologie cromosomiche e quelli sulla ricorrenza familiare. È membro di EUROCAT e di
ICBDMS.
Il Registro Toscano Difetti Congeniti (R.T.D.C.)
monitorizza circa 25.000 nati/anno. È membro di
EUROCAT e di ICBDMS.
L’Indagine Umbra Malformazioni Congenite (I.U.M.C.) si basa sul rilevamento di tutte le
anomalie congenite, principalmente nel periodo
neonatale ma anche in fase pre- e post-natale. La
copertura territoriale è intorno al 95%. Partecipa al
Coordinamento Nazionale e ad EUROCAT.
Il Registro Campano Difetti Congeniti (B.D.Re.
Cam.) coinvolge numerosi Ospedali della regione;
monitorizza circa 50.000 nati/anno con una copertura di circa il 75% del territorio. È membro di
EUROCAT e di ICBDMS.
L’Indagine Siciliana Malformazioni Congenite
www.buponline.com
12
Introduzione
(I.S.MA.C.), organizzato su base volontaria, copre l’intero territorio regionale e monitorizza circa 60.000 nati/anno. Partecipa al Coordinamento
Italiano, a ICBDMS e ad EUROCAT.
Dal Luglio 2002 è stato istituito nell’ambito della
conferenza Stato-Regioni un gruppo tecnico interregionale permanente, al quale partecipano il
Ministero della Salute e l’ISS, il cui obiettivo è rappresentato dall’ottimizzazione del funzionamento
delle reti regionali e dalla salvaguardia del principio di equità dell’assistenza per tutti i cittadini. Il
10 maggio 2007 è stato siglato il secondo accordo
tra il Governo, le Regioni e le Province autonome
di Trento e Bolzano sul riconoscimento di Centri
di coordinamento regionali e/o interregionali, di
Presidi assistenziali sovraregionali per le patologie a
bassa prevalenza e sull’attivazione dei registri regionali ed interregionali delle malattie rare.
L’interesse a livello internazionale e nazionale rivolto allo studio delle malformazioni congenite
evidenzia come le malattie rare rappresentino un
rilevante problema di salute pubblica ad alto
impatto sociale.
Perché elaborare un testo odontoiatrico sulle
sindromi genetiche e cromosomiche?
I pazienti affetti da sindromi genetiche e cromosomiche presentano quadri clinici generalmente
ben definiti, con patologie e segni specifici che
coinvolgono più organi ed apparati; in particolare,
in questi pazienti la prevalenza di anomalie e di
malformazioni dell’apparato stomatognatico e
del distretto cranio-facciale, di patologia cariosa
e di patologia parodontale è statisticamente più
elevata rispetto alla restante popolazione.
Tuttavia, le conoscenze scientifiche sulle dismorfosi dento-scheletriche e sulle alterazioni dell’apparato stomatognatico di numerosi quadri sindromici
sono carenti e/o prevalentemente rappresentate da
case report. Le malocclusioni scheletriche e dentali,
in particolare, sono causa sia di un aggravamento
delle condizioni di salute generale (in quanto determinano alterazioni della masticazione, della respirazione e della fonetica ed aumentano il rischio
di ammalare di patologie parodontale e cariosa) sia
di un peggioramento delle possibilità relazionali e
della qualità della vita a causa di problemi estetici.
Le patologie cariosa e parodontale possono essere
ricondotte ad una insufficiente igiene orale domiciliare (perché sono scarse la considerazione delle
problematiche del cavo orale rispetto alla patologia
sistemica, la manualità e la collaborazione) e/o direttamente correlate alla patologia di base (ipoplasie dello smalto, condizioni di defic immunitario,
…) e/o a condizioni associate (respirazione orale,
deglutizione atipica, …) e/o a farmaci assunti per
le problematiche sistemiche (antiepilettici, sciroppi
ad alto contenuto glucidico, ….) e/o ad una alimentazione cariogenica.
La salute orale è una componente importante del
benessere individuale e, nel caso di gravi alterazioni funzionali ed estetiche, come quelle presenti
nei soggetti affetti da sindromi genetiche e cromosomiche, assume un ruolo fondamentale non solo
per il paziente ma anche per la famiglia. Nonostante il crescente interesse per la salute orale da
parte degli operatori sanitari, delle famiglie, delle
associazioni e delle istituzioni, persistono grandi
difficoltà nel garantire questo benessere primario,
soprattutto nelle fasce di popolazione in condizione di vulnerabilità sanitaria.
Queste evidenze sottolineano la necessità, al momento della diagnosi, di intercettare i momenti
patogenetici delle patologie orali, di individuare
e attuare interventi specifici di prevenzione primaria e secondaria odontoiatrica e ortopedicoortodontica utilizzando percorsi di salute orale
specifici che permettano di prevenire l’instaurarsi
delle patologie nell’obiettivo di apportare un miglioramento delle funzioni, della vita di relazione e della qualità della vita della persona.
Il testo nasce con l’obiettivo di inquadrare, in un
www.buponline.com
13
Introduzione
gruppo di sindromi, le alterazioni e le patologie
del distretto oro-cefalico e di formulare il piano
di trattamento più appropriato, dando sistematicità alle conoscenze acquisite dall’esperienza clinica e supportate da un costante confronto con la
letteratura scientifica internazionale. Le attività
di ricerca e clinica sono state condotte presso il Servizio di Assistenza Odontoiatrica per Disabili del
Dipartimento di Scienze Odontostomatologiche
dell’Università degli Studi di Bologna, nell’ambito
del Progetto di Dottorato in Odontoiatria per Disabili del Corso di Dottorato in Scienze Mediche
Generali e dei Servizi dell’Università degli Studi di
Bologna.
Un sintetico inquadramento genetico e medico
è il punto di partenza per focalizzare l’attenzione
sulle caratteristiche di pertinenza odontoiatrica tipiche di ciascun quadro sindromico. La genetica
e l’anatomo-patologia, elementi fondamentali per
capire le dinamiche eziopatogenetiche delle alte-
razioni del distretto cranio-facciale e dell’apparato
stomatognatico, l’anamnesi medica e odontoiatrica, gli esami obiettivi intra- ed extraorali e gli esami
strumentali guidano la diagnosi delle alterazioni
odontoiatriche di pazienti in età evolutiva affetti
da sindromi genetiche e cromosomiche. La definizione di piani di trattamento mirati garantisce la
fattibilità delle terapie e l’ottenibilità dei risultati, garantiti da interventi di prevenzione specifici
per ciascuna patologia.
Il testo, rivolto a Pediatri, Odontoiatri, Igienisti
dentali e Studenti si pone l’obiettivo di fornire un
supporto sintetico e pratico sulle problematiche
odontoiatriche e sulla loro gestione relativamente a pazienti affetti da sindromi cromosomiche
e genetiche, nella convinzione dell’importanza di
un approccio interdisciplinare che veda coinvolti il
paziente, la sua famiglia e tutti gli operatori sanitari
che se ne prendono cura.
www.buponline.com
www.buponline.com
1. Controllo del dolore e dell’ansia
S. Bagattoni, N. Alkhamis, M.G. Currà, T. Tagariello, G. D’Alessandro, G. Piana
L’esperienza clinica evidenzia che il successo di un
intervento terapeutico dipende non solo dalle capacità tecniche dell’operatore ma anche dal livello
di collaborazione del paziente. Numerose ricerche
si sono occupate dell’argomento nell’obiettivo di
individuare tecniche di tipo farmacologico e/o psicologico in grado di migliorare la collaborazione
del paziente. In campo odontoiatrico un’analgesia
efficace, in grado di garantire una terapia senza dolore, abbassa i livelli di ansia e di paura del paziente
e risulta di fondamentale importanza per creare un
legame positivo tra l’odontoiatra e il paziente.
Nel paziente in età evolutiva e/o scarsamente collaborante e/o affetto da patologie sistemiche la scelta
della tecnica di controllo del dolore necessita di
una valutazione globale delle condizioni mediche e
psichiche, nell’obiettivo di individuare la necessità
di tecniche analgesiche alternative all’anestesia locale e la loro effettiva possibilità di applicazione. In
considerazione dei minori rischi medici, dei minori costi sociali e individuali e del valore educativo
insito in una seduta odontoiatrica senza dolore ma
con il paziente vigile, è sempre preferibile il trattamento in anestesia locale, limitando la sedazione
cosciente e l’anestesia generale ai casi in cui la collaborazione sia assente o scarsissima.
L’esperienza clinica dimostra come nella maggior
parte dei bambini con disabilità di tipo psichico
e/o fisico e/o medico sia possibile ottenere la collaborazione necessaria per lo svolgimento di una seduta odontoiatrica utilizzando tecniche di approccio psicologico (tell-show-do, desensibilizzazione,
rinforzi positivi, …) unite ad un’efficace analgesia
ottenuta con anestetici locali. Solo nel caso in cui
il livello collaborativo del soggetto renda non attuabile il trattamento in anestesia locale, si ricorre
a tecniche alternative (sedazione cosciente o profonda, anestesia generale) che necessitano dell’intervento dell’anestesista.
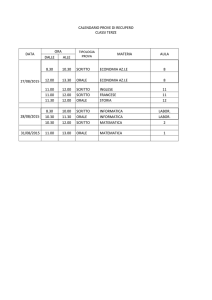
ANESTESIA LOCALE
L’anestesia locale è la perdita temporanea di
sensibilità in una parte del corpo prodotta dall’applicazione, topica o per mezzo di iniezione, di un
agente farmacologico (AAPD, 2009a).
Gli agenti farmacologici disponibili per l’analgesia si
differenziano in due categorie principali, a seconda
della formulazione chimica: gli esteri (tetracaina,
procaina, benzocaina), metabolizzati a livello plasmatico, e gli amidici (lidocaina, mepivacaina, pri-
www.buponline.com
16
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Tabella 1: Anestetici locali più utilizzati in odontoiatria
Nome
Nome
comm.le
Conc.
Vasocostrittore
1:100.000
Adrenalina
Lidocaina
Xylocaina
2%
Mepivacaina
Carbocaina
verde
3%
Mepivacaina
Carbocaina
rossa
2%
dose max
7 mg/kg
6,6 mg/kg
1:100.000
Adrenalina
6,6 mg/kg
7mg/kg
Articaina
Ultracaina
D-S
4%
1:200.000
Adrenalina
locaina, articaina), metabolizzati a livello epatico. I
casi di allergia documentati sono rarissimi verso gli
anestetici amidici, più frequenti verso gli esteri.
Gli anestetici locali (AL) attualmente utilizzati in
odontoiatria appartengono alla classe degli amidici.
Nella tabella 1 sono descritti gli anestetici locali più
frequentemente utilizzati in odontoiatria e i relativi
dosaggi in funzione del peso (Haas DA, 2003).
In pazienti affetti da grave insufficienza epatica è
consigliabile ridurre la dose complessiva dell’anestetico amdico somministrato.
La tossicità degli anestetici locali è in funzione della
modalità di somministrazione, del sito di inoculazione (iniezione intravasale accidentale), delle condizioni cliniche del paziente (insufficienza renale o
epatica) e sono dose dipendenti. I segni di tossicità
possono essere rilevanti ed includono agitazione,
tremori-convulsioni, bradicardia fino alla depressione miocardia e respiratoria. Il rischio delle reazioni
tossiche va ridotto mantenendosi entro i parametri
di sicurezza per posologia e tecnica di iniezione.
5 mg/kg
in sogg
pediatrici
es: bambino di 15 kg:
(1 tubofiala= 1,8 ml)
7 mg/Kg x 15 Kg= 105mg
2% lidocaina = 20 mg/ml
105mg= 5.25 ml
Dose max 2.9 tubofiala
6.6 mg/Kg x 15Kg= 99mg
3% mepivacaina = 30mg/
ml
99mg=3.3 ml
Dose max 1.8 tubofiala
5 mg/Kg x 15 Kg= 75mg
4% articaina = 40mg/ml
75mg=1,88 ml
Dose max 1tubofiala
In seguito alla somministrazione di un anestetico locale si possono avere manifestazioni cliniche
come iperventilazione, nausea, vomito, sudorazione, disorientamento, lieve bradicardia, che rientrano nel gruppo delle reazioni vaso-vagali per attivazione del sistema nervoso autonomo.
Le reazioni di ipersensibilità di tipo allergico
rappresentano una quota numericamente trascurabile nell’ambito delle reazioni avverse agli anestetici locali, non superando l’1%. Tuttavia, queste reazioni sono clinicamente rilevanti per la loro
imprevedibilità e potenziale gravità. Dal punto di
vista clinico il quadro può essere caratterizzato da
notevole variabilità ed interessare diversi organi
ed apparati. Si possono avere manifestazioni cutanee di tipo orticarioide con comparsa di rush
eritemato-pomfoide, pruriginoso, diffuso che
può associarsi ad angioedema a carico di uno o
più sedi (palpebre, labbra, lingua, …). L’apparato
respiratorio può essere interessato con sintomi di
rinorrea, broncospasmo e difficoltà respiratoria; a
carico dell’apparato cardiovascolare si può verifica-
www.buponline.com
17
Controllo del dolore e dell’ansia
re ipotensione severa. L’anafilassi sistemica rappresenta l’evento clinico più drammatico e potenzialmente letale (Finucane BT, 2006).
Gli AL sono agenti vasodilatatori e vengono facilmente assorbiti in circolo, con riduzione dell’effetto analgesico locale. Al fine di limitare la porzione
che va in circolo e ridurre il rischio di tossicità,
si usano AL con l’aggiunta di vasocostrittori (es.
adrenalina). L’uso di vasocostrittori, oltre a ridurre
il sanguinamento nell’area, consente di aumentare
la dose massima totale iniettata di quasi il 40%
(AAPD, 2009a; Sisk AL, 1992).
Non sono documentate in letteratura reazioni
avverse di tipo allergico all’adrenalina (Haas
DA, 2002); esistendo un rischio reale di allergia
nei confronti di agenti antiossidanti (solfiti) presenti nelle soluzioni anestetiche contenenti vasocostrittore, nei pazienti con documentata allergia
ai solfiti è controindicata la somministrazione di
anestetici con vasocostrittore.
Il vasocostrittore è controindicato nelle seguenti
situazioni cliniche: tachicardia parossistica od aritmia associata ad alta frequenza, grave insufficienza coronarica, grave ipertensione, tireotossicosi,
glaucoma ad angolo irido-corneale stretto, diabete
scompensato, feocromocitoma.
Il vasocostrittore può interagire con numerosi
farmaci. L’azione simpaticomimetica dell’adrenalina può essere potenziata dalla contemporanea
assunzione di MAO-inibitori o di antidepressivi
triciclici. L’adrenalina può inibire la liberazione
di insulina nel pancreas e diminuire l’azione degli ipoglicemizzanti orali. Alcuni narcotici da inalazione, come l’alotano, possono sensibilizzare il
cuore alle catecolamine e provocare aritmie.
Nei pazienti con gravi patologie cardiovascolari
l’utilizzo del vasocostrittore è riservato agli interventi lunghi e ai casi in cui l’utilizzo dell’anestetico
senza vasocostrittore non sia stato in grado di determinare un adeguato controllo del dolore: provocare dolore durante il trattamento odontoiatrico
per un’anestesia non efficace o di breve durata crea
una situazione di stress che causa la secrezione endogena di catecolamine in concentrazioni superiori a quelle introdotte con la soluzione anestetica,
esponendo un soggetto scompensato a maggiori
rischi cardiovascolari.
La presenza di un processo infiammatorio acuto
(ascesso), caratterizzato da una acidificazione tissutale, diminuisce l’efficacia dell’anestetico locale.
In questi casi può essere indicato l’utilizzo di una
soluzione anestetica priva di vasocostrittore, poiché a pH più elevato.
In età evolutiva, essendo elevati la frequenza cardiaca, la perfusione tissutale e il metabolismo basale, il passaggio in circolo della soluzione anestetica è molto rapido e, di conseguenza, è indicato
l’uso di vasocostrittori.
L’applicazione di un anestetico topico, garantendo l’analgesia della mucosa orale in una profondità
di circa 2-3 mm, riduce il fastidio causato dalla
penetrazione dell’ago nei tessuti (Meechan JG,
2008). Per aumentarne l’efficacia deve essere applicato su mucosa asciutta e lasciato in sede per
almeno 1 minuto (Ram D et al., 2002).
Gli anestetici topici più utilizzati in odontoiatria
sono:
- lidocaina, che presenta un tempo di inizio di
azione di 3-5 minuti (Ram D et al., 2002) e ha
una bassa incidenza di reazioni tossiche; essendo assorbita a livello sistemico si combina con la
quantità di anestetico iniettato (Manani G, 2003);
- benzocaina, che ha un effetto molto rapido; sono
descritte reazioni localizzate di tipo allergico in
caso di uso prolungato;
- tetracaina, che presenta un tempo di inizio di
azione di circa 60 sec (Ram D et al, 2002).
L’atto anestetico può essere vissuto dal paziente
come un atto invasivo e deve essere sempre preceduto da un approccio psicologico individualizzato; in particolare in odontoiatria infantile si
informa il bambino che è necessario “addormen-
www.buponline.com
18
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
tare il dente”, non utilizzando mai le parole “ago”,
“siringa”, “puntura” e per distrarlo si focalizza
l’attenzione sull’anestetico di contatto, la “pomata
magica”.
La scelta dell’anestetico locale (AL) da utilizzare si
effettua nel singolo paziente in base all’anamnesi medica, all’area anatomica, alla tipologia dell’intervento e alla sua presunta durata (Haas DA, 2003).
Nel bambino è consigliato l’utilizzo di lidocaina 2%
con epinefrina 1:100.000 (Haas DA, 2003).
Prima di usare un AL, l’odontoiatra esegue una
corretta anamnesi medica per ridurre il rischio di
complicanze correlate ad una eventuale patologia
di base e stabilisce il corretto dosaggio in relazione al peso corporeo, in grado di ottenere un’anestesia efficace per la durata di tempo prevista per
l’intervento e di ridurre i rischi di tossicità (AAPD,
2009a; Blanton PL et al., 2003).
Le tecniche di infiltrazione più comunemente utilizzate in odontoiatria infantile comprendono:
- anestesia plessica o terminale: l’anestetico iniettato al di sotto della mucosa raggiunge i rami terminali liberi del plesso alveolare;
- anestesia tronculare: l’anestetico determina il
blocco completo della sensibilità di un tronco nervoso; è utilizzata prevalentemente per gli elementi
mandibolari;
- anestesia intraligamentosa e anestesia intrapulpare, utilizzate come complementari.
È sempre necessario effettuare l’aspirazione, al
fine di evitare l’iniezione intravasale della soluzione anestetica, esponendo il paziente a effetti tossici
correlati al sovradosaggio, nella consapevolezza di
falsi negativi relativamente frequenti utilizzando
aghi molto sottili.
Le tecniche di anestesia per gli elementi dentari
decidui e permanenti sono descritte nelle tabelle
2 e 3.
In dentizione decidua è possibile ottenere l’anestesia degli elementi mandibolari posteriori decidui
con l’anestesia terminale in quanto la teca mandibolare ossea è più sottile ed è composta da una
maggiore quantità di tessuto spugnoso; questa tecnica riduce il problema delle lesioni da morsicatura
post-intervento.
In caso di anestesia tronculare al nervo alveolare
inferiore bisogna considerare che il forame mandibolare in dentizione decidua è localizzato al di
sotto del piano occlusale, tra i 6 e i 12 anni circa,
allo stesso livello del piano occlusale e dopo i 12
anni coincide con quello dell’adulto (Ram D et al.,
2002).
Per evitare la complicanza della morsicatura nel
post-intervento, per il primo molare permanente
nella prima fase della dentizione mista, è consigliata l’infiltrazione plessica. L’utilizzo dell’articaina, a
rapido inizio di azione e ad elevata diffusione nei
tessuti, riduce ulteriormente il ricorso al blocco del
nervo alveolare inferiore per l’anestesia dei primi
molari permanenti.
L’anestesia locale, ad eccezione dell’anestesia intraligamentosa, non rappresenta una manovra per
cui è indicata la profilassi antibiotica nei pazienti
a rischio di endocardite batterica.
Tabella 2: Tecniche di anestesia per gli elementi dentari decidui
Elementi dentari decidui
Tecnica di anesthesia
Incisivi, canini superiori
Anestesia plessica
Molari superiori
Incisivi, canini inferiori
Anestesia plessica
Anestesia plessica
Anestesia plessica
Anestesia tronculare
Molari inferiori
www.buponline.com
19
Controllo del dolore e dell’ansia
Tabella 3: Tecniche di anestesia per gli elementi dentari permanenti
Elementi dentari permanenti
Tecnica di anesthesia
Incisivi, canini superiori
Anestesia plessica
Premolari superiori
Anestesia plessica
I, II molare superiore
Anestesia plessica
Incisivi, canini inferiori
Anestesia plessica
Anestesia tronculare
Anestesia al foro mentoniero
Premolari inferiori
Anestesia tronculare
Anestesia al foro mentoniero
I, II molare inferiore
Anestesia plessica
Anestesia tronculare
Gli aghi utilizzati in odontoiatria si differenziano
per calibro e lunghezza (Manani G, 2003). Gli aghi
attualmente in commercio hanno calibri variabili da
25 a 30 Gauge e lunghezze fra 12 mm e 35 mm. Nei
bambini i più utilizzati sono aghi 30 gauge ultracorti
e corti da 12 mm, 16 mm e 21 mm per le anestesie
plessiche e l’ago 27 gauge, di diametro maggiore per
favorire l’aspirazione e ridurre il rischio di frattura,
da 25 mm per le anestesie tronculari.
La rottura dell’ago è un evento raro che può verificarsi in caso di flessione dell’ago o di movimenti
impropri (AAPD, 2009a; Manani G, 2003; Ram
D et al., 2002).
Le fasi di una corretta procedura per l’esecuzione
di un’anestesia locale in un bambino sono descritte
nella tabella 4 (AAPD 2009a; Manani G, 2003;
Ram D et al., 2002).
Tabella 4: L’anestesia locale: procedure operative
Preparare tutto il materiale fuori dalla portata visiva del bambino.
Sistemare il soggetto in posizione supina, al fine di ridurre le conseguenze di una sincope vasovagale; se la posizione
sdraiata crea ansia, lo schienale deve essere almeno retroinclinato in modo da facilitare all’odontoiatra l’accesso al
punto d’iniezione e da rendere al bambino più difficile la visione della siringa (può essere utile anche coprire gli
occhi).
Asciugare la mucosa e applicare con cotton fioc un anestetico topico sotto forma di pomata di sapore gradevole
(un sapore sgradevole rischia di sensibilizzare negativamente il bambino, aumentandone l’ansia e abbassandone la
soglia del dolore).
Nei casi in cui si prevedano reazioni motorie o movimenti incontrollati da parte del paziente, attuare una
“contenzione dolce” da parte di un genitore/accompagnatore (che si siede sulla poltrona e tiene in braccio il
bambino) e utilizzare l’apribocca di gomma.
www.buponline.com
20
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Tendere i tessuti.
Dare continui rinforzi positivi (per es. “grazie”, “sei proprio bravo”, “tieni aperta la bocca molto bene”, “sei
veramente coraggioso”, etc.) e considerando prevedibile e ammissibile una reazione di pianto, inserire l’ago e
iniettare solo qualche goccia di anestetico locale;
durante l’approfondimento, prima dell’infiltrazione, effettuare sempre l’aspirazione per evitare l’iniezione
endovenosa accidentale (nella consapevolezza di falsi negativi relativamente frequenti utilizzando aghi molto
sottili);
iniettare lentamente (il tempo di iniezione non deve mai essere inferiore a 60 secondi per ogni ml di soluzione
iniettata).
Estrarre l’ago e riporre la siringa fuori dal campo visivo del paziente, massaggiare la zona e somministrare al
paziente rinforzi positivi verbali.
Attendere sempre 3-5 minuti prima di iniziare la procedura e verificare l’efficacia dell’anestesia (es. test termico al
freddo in caso di terapia conservativa, utilizzo di specillo sulla mucosa in caso di estrazione)
Al termine della seduta raccomandare ai genitori/tutori di vigilare sul paziente per evitare lesioni da morsicatura,
particolarmente in caso di anestesia tronculare al nervo alveolare inferiore.
Inserire nella cartella i dati relativi all’anestesia (tipo di farmaco, dosaggio, tecnica, eventuali complicanze).
SEDAZIONE COSCIENTE E SEDAZIONE PROFONDA
La sedazione cosciente è la riduzione dello stato
di coscienza con depressione minima delle funzioni cognitive; non determina la perdita dei riflessi e
della funzione respiratoria e il soggetto mantiene la
capacità di rispondere a stimoli verbali e a stimolazioni somatiche.
La sedazione profonda è uno stato di depressione della coscienza in cui la perdita dei riflessi è
parziale, mentre la funzione respiratoria è mantenuta.
L’obiettivo primario della sedazione è la riduzione dell’ansia e delle risposte comportamentali
ed emozionali a questa correlate; il controllo del
dolore è garantito dall’utilizzo dell’anestesia locale.
La sedazione in odontoiatria viene ottenuta principalmente con la somministrazione di farmaci per
via orale, endovenosa e inalatoria.
I vantaggi rispetto all’anestesia generale sono rappresentati dai minori rischi sistemici, dalla possibi-
lità di somministrazioni del farmaco ripetute, anche ravvicinate, da tempi più rapidi di dimissione
e dal minor costo. Una minima collaborazione
durante le fasi iniziali, quali il posizionamento
volontario della mascherina (nell’induzione inalatoria) o l’ingestione di sciroppi dal gusto poco
gradevole (nel caso di sedazione per via orale) è
comunque richiesta al paziente e la sua assenza
può rappresentare un limite. L’odontoiatra che
ricorre alla sedazione, specie in soggetti affetti da
patologie sistemiche, deve possedere approfondite conoscenze sui principi attivi disponibili e sulla
terapia delle complicanze a cui possono dare luogo;
deve inoltre essere fornito delle strumentazioni
per il monitoraggio continuo dei parametri vitali
(AAPD, 2009b; AAPD, 2006).
Per la sedazione vengono utilizzati numerosi principi attivi, elencati e descritti nella Tabella n.4.
Non esiste un protocollo universalmente accettato
(Matharu L et al., 2006); in letteratura sono presenti numerosi studi difficilmente comparabili poi-
www.buponline.com
21
Controllo del dolore e dell’ansia
ché i farmaci presi in esame sono molteplici, spesso
somministrati in combinazione, con tempi e modalità differenti. In tutti gli studi viene sottolineata la necessità di un monitoraggio continuo della
frequenza cardiaca e della saturazione dell’ossigeno
tramite pulsossimetro e viene evidenziata la possibilità di effetti collaterali che, nella maggior parte
dei casi, non provocano situazioni di emergenza,
se non in caso di sovradosaggio (Matharu L et al.,
2006; Manani G, 2003).
Tabella 5: Principali farmaci utilizzati in odontoiatria per la sedazione
Via di
somministra-zione
Orale
Classe
farmacologica
Benzodiazepine
Farmaco
(dose mg/kg)
Midazolam
(0,1-0,5 mg/kg)
Diazepam (0,20,4 mg/kg)
Ipnotici non
barbiturici
Rettale
Vantaggi
Basso rischio di
complicanze
Rapida induzione
Rapido recupero delle
funzioni cognitive
Amnesia
Basso rischio di
complicanze
Rapida induzione
Rapido recupero delle
funzioni cognitive
Amnesia
Svantaggi
Gusto molto amaro
Rischio di vomito
e quindi di
sottodosaggio
Sconsigliato nei
pazienti di età
inferiore a 6 anni
Niaprazina (1
mg/kg)
Basso rischio di
complicanze
Da somministrasi 3060 minuti prima della
seduta
Possibile uso
domiciliare
Assenza di effetto
analgesico
Controindicato in
caso di allungamento
dell’intervallo Q-T
all’ECG
Possibile interazione
con altri farmaci
sedativi e neurolettici
Raro ma possibile
effetto paradosso
Midazolam (0,30,5 mg/kg)
Facile
somministrazione
Assorbimento
incompleto
Diazepam (0,040,2 mg/kg)
Facile
somministrazione
Efficacia inferiore al
Midazolam
Assorbimento
incompleto
www.buponline.com
22
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Endonasale
Midazolam (0,10,2 mg/kg)
Rapida induzione
Basso rischio di
complicanze
Facile
somministrazione
Intramuscolare
Midazolam
(0,1-0,2 mg/kg)
Rapida induzione
Inalatoria
Endovenosa
Protossido
d’azoto+ossigeno
(40-70% NO2 +
minimo 30% O2)
Benzodiazepine
Midazolam (0,10,2 mg/kg)
Necessità di vie nasali
pervie
L’iniezione provoca
dolore
Facile
somministrazione
Rapida induzione e
rapido risveglio
Buon effetto
analgesico
Possibilità di
modulare il livello di
sedazione
Maschera raramente
accettata dai pazienti
con età inferiore a 3
anni
Necessità di ambiente
idoneo con cappa di
aspirazione
Controindicata in
pazienti con alterata
diffusione polmonare
ed in soggetti con
alterazioni anatomiche
facciali nell’area di
applicazione della
maschera
Basso rischio di
complicanze
Rapida induzione e
rapido recupero
Possibilità di
associazione con altri
farmaci analgesici/
anestetici
Amnesia
Amnesia
Possibile effetto
paradosso nel
bambino
Necessita di un
anestesista-rianimatore
Diazepam
0,1-0,4 mg/kg
www.buponline.com
Frequente effetto
paradosso nel
bambino
Necessita di un
anestesista-rianimatore
23
Controllo del dolore e dell’ansia
Propofol
Oppioidi
1-3 mg/kg
Remifentanyl
0,05-0,2 mcg/
kg/min
ANESTESIA GENERALE
L’anestesia generale (AG) è uno stato di assenza della
coscienza farmacologicamente indotto, accompagnato da una parziale o completa perdita dei riflessi protettivi e dall’inabilità ad emettere risposte finalizzate a
stimolazioni somatiche e a comandi verbali.
Il ricorso all’AG rappresenta l’unica alternativa
nei pazienti con livello di collaborazione molto
scarso o nullo, nei quali l’approccio cognitivo/
comportamentale si sia rivelato non efficace. L’AG
è anche indicata nei pazienti che necessitano di una
bonifica del cavo orale in tempi rapidi, perchè le
patologie odontoiatriche sono responsabili di dolore persistente e di intensità elevata (es: bambini
con Early Childhood Caries) oppure sono potenziale causa di complicanze sistemiche severe in pazienti affetti da gravi patologie (es: oncoemopatie,
pazienti in attesa di trapianto) (Glassman P, 2009).
L’AG è controindicata in pazienti ASA IV, ASA
V, in pazienti affetti da gravi patologie sistemiche
transitorie e in pazienti affetti da patologie respiratorie acute severe.
Gli svantaggi dell’AG sono i rischi medici che
inevitabilmente comporta, l’elevato costo e la difficoltà nel reperire in breve tempo strutture specializzate (Glassman P, 2009).
Rapida induzione e
rapido risveglio
Possibilità di
somministrazioni in
continuo
Sconsigliato in
pazienti con età
inferiore a 2 anni
Necessita di un
anestesista-rianimatore
Rapida induzione
Buon effetto
analgesico
Necessita di un
anestesista-rianimatore
Contoindicato in boli
L’intubazione d’elezione è quella naso-tracheale,
in quanto non ostacola le manovre odontoiatriche.
È consigliato l’uso di tubi zaffati e non cuffiati per
evitare la caduta di corpi estranei nelle prime vie
aeree o l’inalazione dell’aerosol.
Nei casi in cui sia indicata l’intubazione laringotracheale, è fondamentale l’utilizzo di tubi modificati (tubi nord-sud), per consentire un comodo
accesso alle arcate dentali.
Quando un paziente viene trattato in AG, in una
sola seduta vengono effettuate tutte le terapie di
cui necessita.
Per gli interventi dolorosi (per es.terapie endodontiche, estrazioni) è indicata l’infiltrazione
di anestetico locale con vasocostrittore. Questa
procedura permette un efficace controllo della
sedazione in termini di dosaggio farmacologico
e riduce il sanguinamento locale. Inoltre procura
un’antalgia postoperatoria, diminuendo la dose
di analgesico necessario nel post-operatorio. Dal
momento che gli anestetici locali, in dosi eccessive, possono agire da depressori cardiaci e del
SNC, è possibile la loro interazione con i sedativi.
È raccomandato calcolare in mg/Kg la dose massima somministrabile e registrare le dosi somministrate.
Durante tutte le terapie la cavità orale viene mante-
www.buponline.com
24
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
nuta aperta posizionando un apribocca di gomma
di misura appropriata.
Si lavora per quadranti, portando a termine in un
singolo quadrante tutte le terapie. Per prime vengono effettuate le terapie conservative (terapie endodontiche, otturazioni, sigillature di solchi, fessure
e fori ciechi) utilizzando la diga di gomma, che
garantisce un campo operatorio privo di contaminazione, sanguinamento e umidità. Il ricorso a tale
dispositivo non comporta un allungamento dei
tempi anestesiologici, facilita le procedure, le rende
più sicure e garantisce migliori risultati nel tempo.
Terminata la parte conservativa viene rimossa la
diga e vengono eseguite l’igiene orale e le terapie
chirurgiche (terapie parodontali ed estrazioni).
L’igiene orale professionale precede gli interventi
chirurgici per limitare il rischio di contaminazione
dei siti e favorire una corretta guarigione. I siti chirurgici vengono suturati utilizzando dei fili riassorbibili in modo da evitare al paziente un controllo
odontoiatrico per la rimozione dei punti.
Nel caso in cui sia necessaria la terapia conservativa di elementi del settore frontale, viene isolato il
settore frontale e tutti gli elementi vengono trattati
contemporaneamente per garantire migliori risultati estetici.
Portate a termine tutte le terapie viene effettuata
l’applicazione di gel al fluoro sugli elementi dentali
e di clorexidina sui siti chirurgici.
Nei casi in cui sia necessario effettuare estrazioni
“precoci” degli elementi decidui posteriori è da valutare la possibilità, in funzione alla collaborazione
del paziente, di predisporre mantenitori di spazio.
Quando possibile, in una seduta preliminare preanestesiologica a livello ambulatoriale vengono rilevate impronte su cui vengono realizzati i dispositivi che vengono cementati al termine della seduta
operatoria. La seduta in anestesia generale può essere l’occasione per la rilevazione di impronte delle
arcate dentali e di cera occlusale finalizzati ad una
diagnosi ortodopedico-ortodontica.
L’intervento odontoiatrico in anestesia generale è
vantaggioso da un punto di vista clinico perché
permette di effettuare tutte le terapie necessarie in
un’unica seduta ma non lo è da un punto di vista
educativo e formativo, mancando l’interazione e la
comunicazione tra l’odontoiatra e il paziente e il
ricordo da parte del paziente dell’esperienza vissuta, utile se positiva. L’intervento in AG deve essere
quindi considerato come il punto di partenza per
un programma di controlli periodici ravvicinati,
nel corso dei quali attuare igiene orale professionale
e intercettare le lesioni allo stadio iniziale, quando
le terapie sono più semplici e di rapida esecuzione,
quindi più accettate dal paziente.
BIBLIOGRAFIA
AAPD, American Association of Paediatric Dentistry. Guideline on Use of Local Anesthesia for
Pediatric Dental Patients. 2009a
AAPD, American Association of Paediatric Dentistry. Guideline on Appropriate Use of Nitrous
Oxide for Pediatric Dental Patients. 2009b
AAPD, American Association of Paediatric Dentistry. Guideline on Use of Anesthesia Personnel in the Administration of Office-based Deep
Sedation/General Anesthesia to the Pediatric
Dental Patient. 2007
AAPD, American Association of Paediatric Dentistry. Guideline for Monitoring and Management of Pediatric Patients During and After
Sedation for Diagnostic and Therapeutic Procedures. 2006
Blanton PL and Jeske AH. Avoiding complications
in local anesthesia induction: anatomical considerations. J Am Dent Assoc. 2003;134:88893
Finucane BT. Allergies to local anesthetics- the real
truth. Can J Anesth. 2006;50:869-74,
Glassman P. A review of guidelines for sedation,
www.buponline.com
25
Controllo del dolore e dell’ansia
anesthesia, and alternative interventions for
people with special needs. Spec Care Dentist.
2009;29:9-16
Haas DA. An update on local anesthetics in dentistry. J Can Dent Assoc. 2002;268:546-51
Manani G. Anestesia in odontostomatologia.
Ed.Idelson-Gnocchi. 2003
Matharu L and Ashley PF. Sedation of anxious children undergoing dental treatment. Cochrane
Database Syst Rev. 2006;(1):CD003877
Meechan JG. Intraoral topical anesthesia. Periodontol 2000. 2008;46:56-79
Ram D and Peretz B. Administering local anaesthesia to paediatric dental patients - current
status and prospects for the future. Int J Paediatr Dent. 2002;12:80-9
Sisk AL. Vasoconstrictors in local anesthesia for
dentistry. Anesth Prog 1992; 9:187-93
www.buponline.com
www.buponline.com
2. Cardiopatie congenite
M. Taddei, S. Bagattoni, T. Tagariello, G. D’Alessandro, G. Piana
DEFINIZIONE, EPIDEMIOLOGIA ED ETIOLOGIA
Le cardiopatie congenite (CC) rappresentano una
variegata classe di patologie del cuore e dei grossi vasi.
L’incidenza di CC è relativamente elevata, approssimativamente 6-8 su 1000 nati vivi, in assenza di predilezione razziale, etnica, geografica (Hoffman JI et al.,
2002); sono particolarmente colpiti il genere femminile e i nati prematuri o sottopeso o da madri con problemi di gestazione nel primo trimestre di gravidanza.
L’etiologia delle CC è multifattoriale: ambientale
(virus, in particolare della rosolia, farmaci, stupefacenti, alcool, radiazioni) e/o genetica; in molti casi
non è possibile individuare una causa. Le CC possono far parte di quadri patologici più ampi, come
sindromi cromosomiche (sindromi di Down, da
delezione del cromosoma 22q11.2 q, dell’X fragile,
di Turner, di Williams, etc.) e genetiche (sindromi
di Alagille, di Noonan, di Marfan, etc.).
MANIFESTAZIONI CLINICHE SISTEMICHE
Dal punto di vista fisio-patologico le CC si possono classificare in: cianogene (CCc), non cianogene (CCnc) e ostruttive (CCo).
Le forme cianogene sono caratterizzate da uno
shunt destro-sinistro prevalente (Kornosky JL et al.,
2008).
La più frequente (~75%) è la tetralogia di Fallot,
caratterizzata da difetto del setto interventricolare,
stenosi polmonare, ipertrofia ventricolare destra e
destrorotazione dell’aorta, a cui possono associarsi
deformazioni dell’apparato muscolo-scheletrico, alterazioni respiratorie (ridotta capacità respiratoria,
dispnea), trombosi e ascessi cerebrali, difetti coagulativi (aumento del tempo di sanguinamento), ritardo mentale e difetti immunologici (aumento della
produzione di immunoglobuline, in particolare di
classe M, aumentata formazione di immunocomplessi circolanti, deficit della fagocitosi dei neutrofili,
diminuzione dei T-linfociti).
Altre CCc sono la trasposizione dei grossi vasi, la
stenosi polmonare in associazione a pervietà del forame ovale e atresia della tricuspide ed il cuore univentricolare (Starr JP, 2010; Apitz C et al., 2009).
I bambini affetti da CCc presentano spesso una
notevole riduzione della capacità respiratoria che
comporta ridotta resistenza allo sforzo fisico, dispnea, tendenza ad assumere posizioni di ortopnea
compensatoria e ritardo di crescita e di sviluppo.
La ridotta capacità respiratoria si rivela con segni
www.buponline.com
28
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
clinici: murmuri sistolici, ippocratismo digitale, unghie a vetrino di orologio. Sono manifestazioni del
basso livello di ossigenazione del sangue il pallore
cutaneo e la cianosi delle labbra e di tutte le mucose
(“bambini blu”) (Cordina RL et al., 2010).
Le forme non cianogene sono caratterizzate da uno
shunt sinistro-destro prevalente. La forma più comune è il difetto del setto ventricolare. La sintomatologia e il quadro clinico e strumentale dipendono dalle
dimensioni del difetto e dall’entità dello shunt e delle
resistenze polmonari. I segni clinici variano dalla presenza di soli soffi sistolici per piccoli difetti, a ridotta
capacità respiratoria, polipnea, scarsa resistenza
allo sforzo, scarso accrescimento staturo-ponderale
ed episodi di flogosi broncopolmonare per difetti
maggiori (Beghetti M et al., 2010).
Le forme ostruttive sono caratterizzate da diminuzione del flusso del sangue nei vasi. Ne è un
esempio la coartazione aortica. I pazienti affetti
da CCo hanno un aspetto fisico normale ma sono
caratterizzati da un maggiore sviluppo del torace
e degli arti superiori (anisosfigmia tra arti superiori e inferiori) e presentano ipertensione arteriosa
nel distretto vascolare prossimale alla coartazione
(Kenny D et al., 2011).
L’analisi della letteratura evidenzia che il cavo orale di
bambini affetti da patologie cardiache è frequentemente sede di patologie di pertinenza odontoiatrica: ipoplasie dello smalto, lesioni cariose,
gengivite marginale severa, malocclusioni sono
descritte con una prevalenza superiore a gruppi di
controllo di bambini sani (Micheletti A et al., 2010;
Tasioula V et al., 2008; Franco E et al, 1996; Piana
G et al., 1996; Hallet KB et al., 1992). Le motivazioni che supportano questi dati epidemiologici non
sono del tutto chiare. In molti casi le manifestazioni
odontoiatriche sono associate ad una sindrome di
base, in altri è ipotizzabile una scarsa considerazione
della salute orale da parte di coloro che si prendono
cura dei bambini con CC, spesso concentrati sulla
gravità della malattia sistemica.
Le cardiopatie congenite possono predisporre al rischio di insorgenza di endocardite batterica (EB),
infezione acuta dell’endocardio e delle valvole cardiache causata da agenti infettivi (Knirsch W et al.;
2011; Allen U, 2010; Micheletti A et al., 2010;
Dajani AS et al., 1997; Etienne J, 1994).
Le alterazioni anatomiche associate a una CC possono causare anomale turbolenze del flusso ematico che traumatizzano le strutture valvolari e le
superfici endocardiche contigue. L’endotelio danneggiato espone al sangue circolante le fibre collagene e lo stroma connettivale e diviene sede di
deposito, adesione ed aggregazione di piastrine; il
deposito di fibrina favorisce il consolidarsi di tali
aggregati e la formazione di vegetazioni trombotiche sterili, realizzando la condizione di endocardite
trombotica non batterica. In caso di batteriemia
i microrganismi sono in grado di impiantarsi nei
depositi di piastrine e fibrina (molto più recettivi
alla colonizzazione batterica dell’endotelio integro)
e di moltiplicarsi dando luogo alla formazione di
vegetazioni di dimensioni variabili costituite da
masse amorfe di piastrine, fibrina, microrganismi
e cellule infiammatorie, realizzando la condizione
di endocardite batterica. Con la risoluzione della
flogosi acuta si instaurano processi cicatriziali con
fibrosi, jalinizzazione e successiva endotelizzazione
dei tessuti coinvolti nell’infezione (Allen U, 2010;
Khader RN et al., 2007).
Gli agenti eziologici più frequentemente implicati nell’eziopatogenesi dell’EB sono: Streptococcus
bovis, sanguis, mitis e mutans e Stafilococchus epidermidis e aureus (Tornos P et al., 2011). Sia streptococchi che stafilococchi sono ceppi batterici di
frequente riscontro a livello del cavo orale e delle
alte vie respiratorie; in particolare, il pattern microbico oro-faringeo a crescita lenta H.A.C.E.K.
(Haemophilus, Actinobacillus, Cardiobacter, Eiknella e Kingella) in pazienti cardiopatici congeniti può
causare EB (Mocchegiani R et al., 2009; Steelman
R et al., 2000).
www.buponline.com
29
Cardiopatie congenite
I sintomi dell’EB sono quelli di un’infezione acuta:
febbre alta, con brividi all’inizio e con sudorazione al termine dell’accesso, astenia, vomito, cefalea,
diarrea e inappetenza. L’auscultazione cardiaca rileva
rumori anomali e soffi, dovuti alle lesioni prodotte
dai microrganismi sulle valvole cardiache. Una complicanza tipica è l’embolia: nelle zone valvolari lese si
formano trombi che, distaccandosi, formano emboli
infetti che entrano nel circolo ematico. Sono segni
obiettivi aspecifici di endocardite: petecchie cutanee
(frequenti negli stati settici), noduli di Osler (formazioni eritematose e dolenti dei polpastrelli delle
mani), lesioni di Janeway (macule rossastre, indolenti,
di 3-5 mm sulla superficie palmare di mani e piedi,
che tendono a schiarirsi alla compressione), macchie
retiniche di Roth (di origine microembolica), emorragie subungueali a scheggia, ippocratismo digitale (secondario a vasculite e microembolie), splenomegalia
moderata (dolente solo in caso di infarto splenico),
epatomegalia (espressione di scompenso cardiaco).
Questi segni diventano diagnostici se correttamente
integrati nel quadro clinico e supportati dai risultati
delle indagini elettrocardiografiche e di laboratorio
(Wilson W et al., 2008; Dajani AS et al., 1997). In alcuni casi sono le complicanze cardiache (ulcerazioni
o perforazioni con conseguente insufficienza valvolare e pericardite suppurativa), renali (glomerulonefrite
diffusa o focale) e di tipo embolico (a livello di milza,
reni, cervello) a richiamare l’attenzione sull’infezione
cardiaca primaria.
L’analisi della letteratura evidenzia che:
sono potenzialmente responsabili dell’insorgenza
di batteriemie le patologie infettive a livello del
cavo orale (gengiviti, ascessi di origine endodontica e parodontale) e gli interventi odontoiatrici, in
particolare estrazioni (10-100%), chirurgia parodontale (36-88%), scaling e root planing (8-80%),
igiene orale professionale (40%), posizionamento
di diga di gomma o di matrice responsabile di sanguinamento (9-32%), procedure endodontiche (>
20%) (Wilson W et al., 2008);
l’incidenza di batteriemia ad origine dal cavo orale
è direttamente proporzionale al grado di infiammazione e infezione dei tessuti orali;
la batteriemia prodotta in caso di sanguinamento
causato da infiammazione gengivale durante lo spazzolamento nelle pratiche di igiene orale domiciliare
(20-68%) e con la masticazione (7-51%), definita
batteriemia random o spontanea, caratterizzata da
una bassa intensità e da una breve durata, è sovrapponibile a quella indotta dalle terapie odontoiatriche ma
si ripete quotidianamente e più volte nell’arco di una
stessa giornata (Wilson W et al., 2008).
Nei pazienti affetti da CC le patologie orali non
solo influiscono in modo significativamente negativo sulle condizioni di salute generale e sulla qualità di vita ma comportano un aumento del rischio
di sviluppare l’EB.
CARATTERISTICHE ORO-FACCIALI
La frequenza di anomalie dentali, in particolare
anomalie di struttura dello smalto e della dentina
e di eruzione (ritardo dell’eruzione dei denti della
serie sia decidua che permanente) nei pazienti con
CC è superiore a quella in soggetti sani. Le ipoplasie dello smalto (Figura 1), in forma diffusa o
Figura 1: Ragazzo di 12 anni affetto da CCc. Ipoplasia
dello smalto diffusa
www.buponline.com
30
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Figura 2: Bambina di 7 anni affetta da CCc. Carie destruenti di elementi decidui e permanenti
localizzata, possono essere causate da fattori eziologici sistemici pre/perinatali (nascita prematura e
sottopeso) e postnatali (basso livello di ossigenazione del sangue). Secondo alcuni autori sono cause
di ipoplasie anche l’insufficiente assorbimento gastrointestinale di minerali e il ridotto metabolismo
della vitamina D (Stecksen-Blicks C et al., 2004;
Jowett NI et al., 2000).
La prevalenza di lesioni cariose a livello degli elementi decidui e permanenti è elevata (Figura 2). I
fattori predisponenti sono le ipoplasie dello smalto, lo scarso livello di igiene orale domiciliare, le
abitudini alimentari scorrette (in particolare assunzione di bevande contenenti zuccheri durante
le ore notturne) e l’utilizzo per lunghi periodi di
sciroppi contenenti zuccheri cariogenici (StecksenBlicks C et al., 2004).
Quadri di gengivite generalizzata (figura 3), con
tendenza al sanguinamento in seguito a pressione
di lieve entità, sono caratteristiche delle CCc tipo
Tetralogia di Fallot. È ipotizzabile che il basso grado di saturazione dell’ossigeno e il maggior tasso di
emoglobina siano in grado di influenzare la condizione di infiammazione gengivale. È stata evidenziata in questi pazienti una particolare microflora orale, il pattern H.A.C.E.K. (Haemophilus,
Actinobacillus, Cardiobacter, Eiknella e Kingella),
responsabile di patologia parodontale indipenden-
Figura 3: Ragazza di 11 anni affetto da CCc. Gengivite
cronica e accumuli di placca e tartaro
Figura 4: Bambino di 6 anni affetto da CCc. Marcate fissurazioni della lingua e papille gustative filiformi e fungiformi appiattite, assottigliate e arrossate
temente dalla cianosi, dal livello di saturazione
dell’ossigeno e dal tipo di difetto cardiaco (Mocchegiani R et al., 2009; Steelman R et al., 2000)
(figura 4).
In letteratura nei bambini affetti da CCc, in particolare da Tetralogia di Fallot, sono descritte numerose manifestazioni orali quali cianosi delle labbra
e della mucosa orale, marcate fissurazioni della
www.buponline.com
31
Cardiopatie congenite
lingua e papille gustative filiformi e fungiformi appiattite, assottigliate e arrossate (Stecksen-Blicks C
et al., 2004).
Le patologie ortopedico-ortodontiche di più frequente riscontro nel bambino con CC sono riconducibili ad alterazioni della dinamica respiratoria:
la riduzione della capacità respiratoria favorisce
una meccanica respiratoria prevalentemente orale,
potenzialmente responsabile di alterazioni a livello
sia locale che sistemico.
A livello del distretto oro-facciale la ridotta pressione esercitata dalla lingua sul palato, unitamente
alla pressione centripeta delle guance, ne determina la contrazione del diametro traverso: la volta del
palato assume il tipico aspetto ogivale e l’ampiezza delle fosse nasali risulta ridotta, con aumento della resistenza al flusso aereo. Per consentire il
passaggio dell’aria, la mandibola ruota in basso e
posteriormente modificando il proprio vettore di
crescita e il terzo inferiore del volto risulta allungato. L’aspetto tipico del viso del respiratore orale
è descritto come facies adenoidea: viso allungato,
occhiaie, narici ipotoniche, incompetenza labiale,
pallore ed espressione stanca. Il palato ogivale si accompagna frequentemente ad una malocclusione
caratterizzata da morso crociato posteriore mono
o bi-laterale.
A livello sistemico la respirazione orale si rende
responsabile di alterazioni posturali, disturbi del
sonno, difficoltà di concentrazione e scarso rendimento scolastico. La correzione della respirazione
orale richiede la collaborazione di più specialisti.
In particolare all’otorinolaringoiatra è affidato il
compito di risolvere le cause di ostruzione nasale
farmacologicamente o chirurgicamente e all’ortodontista quello di ripristinare precocemente una
corretta anatomia del palato (Cordina RL et al.,
2010; Roberts GJ et al., 2000).
LINEE GUIDA DI TERAPIA
Essendo i bambini affetti da CC stressati da frequenti controlli medici e da manovre fastidiose e/o
dolorose, è ipotizzabile che il loro livello di collaborazione anche in ambiente odontoiatrico sia scarso.
Per questo motivo è fondamentale un approccio
psicologico individualizzato che enfatizzi il più
possibile “l’aspetto ludico” della visita odontoiatrica e consenta l’instaurarsi di un buon rapporto con
lo staff odontoiatrico.
Se la cooperazione del paziente non è ottimale,
nelle prime sedute è indicato ridurre sia l’intensità
che la durata degli interventi, limitandosi a terapie
indolori o alla risoluzione di emergenze/urgenze e
attendere che il livello di collaborazione del paziente
migliori per poter affrontare tutte le terapie necessarie. A questo scopo di grande utilità sono le “sedute
di avvicinamento”, che permettono di conquistare
la fiducia del bambino e di abituarlo all’ambiente
odontoiatrico, agli operatori e agli strumenti.
Tutte le terapie odontoiatriche potenzialmente
fonte di fastidio/dolore debbono essere eseguite in
anestesia locale, al fine di evitare al paziente stress,
responsabile dell’increzione di catecolamine. È
opportuno concordare con il cardiologo l’utilizzo
di anestetici con vasocostrittore, tenendo sempre
presente che la quantità di adrenalina endogena
secreta dalle ghiandole surrenali alla percezione
del dolore dovuto ad una anestesia non efficace o
di breve durata è superiore a quella contenuta in
una tubofiala di anestetico. In pazienti scarsamente collaboranti a rischio di trauma post-operatorio
(morsicatio buccarum), in particolare in caso di
anestesia tronculare all’arcata inferiore, può essere
indicato un anestetico senza adrenalina, quindi a
più breve durata (30-90 min). La quantità di anestetico deve essere dosata in funzione del peso del
paziente, della tipologia e della durata dell’intervento previsto. Nel caso in cui si utilizzi un anestetico senza adrenalina possono essere indicate ulte-
www.buponline.com
32
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
riori somministrazioni nel corso del trattamento.
Considerando la paura che il bambino può provare
nei confronti dell’ago è necessario mettere in atto
tutte le manovre che possano agevolare il rapporto
dentista-bambino (utilizzare un anestetico topico
di odore gradevole, manovrare gli strumenti fuori
dal campo visivo, ecc.). Solo l’anestesia intraligamentosa rende necessaria la profilassi antibiotica
dell’EB (Wilson W et al., 2007).
Oltre alle considerazioni sempre valide in tema di
salute orale e prevenzione delle malattie del cavo
orale, nel caso dei bambini con CC il mantenimento di un buon livello di salute orale è di importanza
fondamentale per la prevenzione dell’EB (Jowett NI
et al., 2000; Tong DC et al., 2000). È infatti più
probabile che una EB si sviluppi in seguito all’esposizione frequente a batteriemie random correlate alle
attività quotidiane di spazzolamento e masticazione,
specie se effettuate su tessuti infiammati, piuttosto
che a batteriemie isolate causate da un intervento
odontoiatrico. Essendo quindi la profilassi antibiotica prima di interventi odontoiatrici in grado di prevenire solo un numero estremamente basso di casi
di EB, in un’ottica di prevenzione risulta molto più
rilevante il mantenimento della salute orale.
Per promuovere la salute orale del bambino cardiopatico è necessario che i genitori adottino, fin
dai primissimi mesi di vita, stili di vita corretti per
quanto riguarda l’alimentazione, l’igiene orale e l’utilizzo di fluoro e motivino il bambino, appena il suo
livello di maturazione lo consenta, all’autoassistenza
(Cheuk DK et al., 2004). Per questo è necessario il
coinvolgimento tempestivo di tutti gli operatori sanitari coinvolti. Le attuali tecniche di diagnosi ecografica precoce permettono di individuare le patologie
cardiache congenite alla 19ª-20ª settimana di gestazione; è quindi auspicabile informare la gestante sulla
malattia del/della figlio/a, sull’importanza della salute
orale e sulla tecniche di prevenzione odontoiatrica e
indirizzarla ad un odontoiatra per la valutazione delle
condizioni di salute orale e per le terapie del caso. È
infatti documentato come la carica batterica orale del
bambino sia sovrapponibile a quella materna a causa
della trasmissione dei batteri tramite gesti spontanei
comunemente ritenuti innocui come baci, effusioni,
ciuccio (mettendolo in bocca per “pulirlo”) e pappe
(utilizzando le posate per assaggiarle, soffiando per
raffreddarle). Nelle donne in gravidanza in cui sia
stata fatta diagnosi di cardiopatia congenita del feto
è opportuno eseguire uno screening microbiologico,
indicatore di rischio di malattia cariosa, per individuare le concentrazioni salivari di Streptococcus mutans e di Lactobacillus mediante test microbiologici
(tipo deep-slide test) rapidi, di semplice esecuzione,
di facile lettura e poco costosi. Quando positivi (concentrazioni > 100.000 CFU/ml), è necessario che la
gravida bonifichi il cavo orale, migliori l’igiene orale
domiciliare e utilizzi collutori a base di clorexidina
(Sauders CP et al., 1997).
Nei bambini affetti da CC è di primaria importanza attuare tutte le misure di prevenzione domiciliare (alimentazione corretta, igiene orale, utilizzo
di fluoro) e ambulatoriale (visite odontoiatriche
periodiche, applicazione topica di fluoro, sedute di
igiene professionale bimestrali/trimestrali, sigillature dei solchi e delle fessure).
La posologia del fluoro assunto per via sistemica
non presenta alcuna peculiarità in questa categoria
di pazienti: tempi e dosaggi sono quindi gli stessi
utilizzati nei bambini con anamnesi medica negativa. Non sono descritte in letteratura interazioni tra
i farmaci che i pazienti cardiopatici più frequentemente assumono e il fluoro.
Per quanto concerne la patologia cariosa, è necessario non solo prevenirla ma anche intercettare e
trattare le lesioni il più precocemente possibile sottoponendo il bambino con CC a visite di controllo
ogni sei mesi fin dai primi anni di vita. Le terapie di
lesioni iniziali sono di più facile esecuzione, quindi
meglio accettate dal bambino, garantiscono risultati
duraturi nel tempo e riducono il rischio di complicanze endodontiche responsabili di batteriemia.
www.buponline.com
33
Cardiopatie congenite
Nei pazienti ad alto rischio di patologia cariosa per
prevenire lesioni a livello di solchi profondi e fessure, la sigillatura è sempre indicata a livello di
tutti gli elementi dentali posteriori permanenti e
da valutare per i decidui.
È necessario programmare controlli periodici per
valutare il livello di salute orale del bambino, lo status dei restauri eseguiti, intercettare eventuali lesioni cariose e/o infiammazione gengivale e motivare
prima i genitori, poi il bambino, sull’importanza
della prevenzione.
Nei pazienti a rischio di EB, gli interventi odontoiatrici responsabili di batteriemia debbono essere
eseguiti dopo aver effettuato la profilassi antibiotica (vedi tabelle 1, 2 e 3) (Oliver R et al., 2008;
Balmer R et al., 2003; Al-Karaawi ZM et al., 2001).
Se un paziente a rischio, al momento dell’intervento
odontoiatrico, si trova già in regime di terapia antibiotica per os con un antibiotico indicato anche per
la profilassi dell’endocardite batterica, è prudente
a scopo profilattico scegliere un farmaco sostitutivo
appartenente ad una classe differente piuttosto che
incrementare il dosaggio del farmaco già assunto a
causa della possibile comparsa nei giorni precedenti
di ceppi batterici resistenti. Se possibile, è comunque
preferibile rimandare l’intervento di almeno dieci
giorni dal termine della terapia per consentire il ripristino della usuale flora batterica. Se invece il paziente
è già in regime di terapia antibiotica parenterale, la
stessa terapia può continuare regolando la posologia
in maniera tale che il farmaco venga somministrato
30-60 minuti prima dell’intervento odontoiatrico; gli
elevati dosaggi del farmaco somministrato per via parenterale coprono un eventuale rischio correlato allo
sviluppo di ceppi resistenti (Wilson W et al., 2008).
Nei pazienti che necessitano di profilassi antibiotica,
per ridurre l’assunzione di antibiotico, è opportuno
impostare il piano di trattamento per quadranti,
se lo consente il livello di collaborazione del paziente
(parametro fondamentale per stabilire la durata delle
sedute).
La somministrazione di antibiotici nei pazienti che
necessitano di profilassi antibiotica è indispensabile anche in caso di lesioni di origine traumatica che
comportino un danno a livello delle fibre del legamento alveolo-dentale (dalla sublussazione all’avulsione). In caso di lesioni della mucosa orale o di
sanguinamento del labbro la profilassi antibiotica
non è necessaria. (Wilson et al., 2008).
Non è da sottovalutare la possibilità che la somministrazione di antibiotici ad un paziente a rischio di
endocardite infettiva possa interagire con i farmaci
comunemente assunti per la patologia cardiaca. Per
esempio, l’assunzione di diuretici come lo spironolattone o il clorotiazide (utilizzati nell’insufficienza
cardiaca congestizia) può alterare la concentrazione plasmatica e l’emivita dell’antibotico rendendolo inefficace. Da qui la necessità di una raccolta
anamnestica accurata e di un consulto con il medico curante per concordare la terapia più indicata
per il paziente.
Per risolvere le conseguenze a livello del cavo orale delle problematiche respiratorie è indicato, per
rimodellare la volta del palato, aumentare lo spazio aereo nasale e diminuire la resistenza al flusso
aereo, ripristinando una corretta respirazione, un
trattamento ortopedico-ortodontico (mediante utilizzo di apparecchiature quali il disgiuntore
rapido) da attuare il più precocemente possibile,
appena il livello di collaborazione del bambino lo
consente. Qualora permangano respirazione orale
e/o deglutizione atipica, il logopedista, attraverso
esercizi miofunzionali, può favorire il corretto
posizionamento della lingua ed aumentare la tonicità della muscolatura perilabiale.
In una fase successiva, al termine della dentizione
mista, eventuali concomitanti malocclusioni vengono valutate, attraverso lo studio cefalometrico
delle radiografie del cranio in proiezione laterolaterale e postero-anteriore, e trattate con apparecchiature ortodontiche fisse.
www.buponline.com
34
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Tabella 1: Condizioni cardiache associate al più alto rischio di esiti avversi da endocardite per i quali è raccomandata la profilassi antibiotica in occasione di procedure odontoiatriche
o Valvole cardiache artificiali
o Pregresso episodio di endocardite batterica
o Cardiopatie congenite *
1.
Cardiopatie congenite cianogene non risolte compresi shunt e condotti palliativi
2.
Durante i primi sei mesi dopo la completa risoluzione di difetti cardiaci congeniti con
dispositivi protesici posizionati chirurgicamente o tramite interventi di cateterismo. §
3.
Cardiopatie congenite risolte con difetti residui a livello del o adiacenti al sito del dispositivo
protesico (che inibisce l’endotelizzazione)
o Trapiantati cardiaci che sviluppano valvulopatie
* Eccetto per le condizioni elencate sotto, la profilassi antibiotica non è più consigliata per altre forme di
cardiopatie congenite.
§ La profilassi è raccomandata perché l’endotelizzazione dei materiali protesici si verifica entro sei mesi dopo
le procedure chirurgiche di risoluzione dei difetti cardiaci congeniti.
Tabella 2: Indicazioni alla profilassi antibiotica in occasione di interventi odontoiatrici
Interventi a profilassi raccomandata
Tutte le procedure che richiedono la manipolazione del tessuto gengivale o della regione periapicale del dente o la
perforazione della mucosa orale
* Detartrasi
* Estrazione dentaria
* Iniezione anestetica intraligamentosa
* Posizionamento di bande ortodontiche (non di brackets)
* Posizionamento di impianti dentali
* Reimpianto di denti avulsi
* Strumentazione endodontica oltre apice
* Trattamenti parodontali (chirurgia dei tessuti duri e molli, scaling e root planing)
Interventi a profilassi non raccomandata:
* Applicazione topica di fluoro
* Esfoliazione di elemento deciduo
* Iniezioni anestetiche locali (tranne l’intraligamentosa) attraverso tessuti non infetti
* Posizionamento di diga
* Posizionamento di dispositivi protesici rimovibili
* Posizionamento di dispositivi ortodontici
* Posizionamento di brackets ortodontici
* Rimozione di suture
* Radiografie (endorali, ortopantomografie, teleradiografie…)
* Rilevamento di impronte
* Terapia conservativa con o senza fili di retrazione gengivale
* Terapia endodontica
www.buponline.com
35
Cardiopatie congenite
Tabella 3: Regimi di profilassi antibiotica
Situazione
Antibiotico
Profilassi standard
(Penicillina)
Amoxicillina
Regime °
Adulti: 2.0 g
Bambini٭: 50 mg/kg
Per OS, 30-60 min prima dell’intervento
Ampicillina
Adulti: 2.0 g
Bambini: 50 mg/kg
I.M./I.V., 30-60 min prima dell’intervento
Impossibilitati all’assunzione orale
Cefazolina o Ceftriaxone
Allergici alla Penicillina
Adulti: 600 mg
Bambini: 20 mg/kg
Per OS, 30-60 min prima dell’intervento
Clindamicina
Cefalexina*§
Adulti: 2.0 g
Bambini: 50 mg/kg
Per OS, 30-60 min prima dell’intervento
Azitromicina o
Claritromicina
Allergici alla penicillina e
impossibilitati all’assunzione orale
Adulti: 1.0 g
Bambini: 50 mg/kg
I.M./I.V., 30-60 min prima dell’intervento
Adulti: 500 mg
Bambini: 15 mg/kg
Per OS, 30-60 min prima dell’intervento
Clindamicina
Adulti: 600 mg
Bambini: 15 mg/kg
I.M./I.V., 30-60 min prima dell’intervento
Cefazolina o ceftriaxone§
Adulti: 1 g
Bambini: 25 mg
I.M./I.V., 30 min prima dell’intervento
° la dose complessiva nei bambini non deve mai
superare quella degli adulti;
٭Fino a 30 kg;
* Le cefalosporine non dovrebbero essere somministrate ad individui con reazione di ipersensibilità
di tipo immediata alle penicilline (orticaria, angioedema, anafilassi);
§ o altre cefalosporine di prima e seconda generazione ad assunzione orale in dosaggi equivalenti
www.buponline.com
36
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
BIBLIOGRAFIA
Al-Karaawi ZM, Lucas VS, Gelbier M, Roberts
GJ. Dental procedures in children with severe
congenital heart disease: a theoretical analysis
of prophylaxis and non-prophylaxis procedures. Heart. 2001;85:66-8
Allen U. Infective endocarditis: Updated guidelines. Paediatr Child Health. 2010;15:205-12
Apitz C, Webb GD, Redington AN. Tetralogy of
Fallot. Lancet. 2009;374:1462-71
Balmer R, Bu’Lock FA. The experiences with
oral health and dental prevention of children
with congenital heart disease. Cardiol Young.
2003;13:439-43
Beghetti M, Tissot C. Pulmonary hypertension in congenital shunts. Rev Esp Cardiol.
2010;63:1179-93
Cheuk DK, Wong SM, Choi YP, Chau AK, Cheung YF. Parents’ understanding of their child’s
congenital heart disease. Heart. 2004;90:435-9
Cordina RL, Celermajer DS. Chronic cyanosis
and vascular function: implications for patients
with cyanotic congenital heart disease. Cardiol
Young. 2010;20:242-53
Dajani AS, Taubert KA, Wilson W, Bolger A. Prevention of bacterial endocarditis: recommendation by A.H.A. JADA. 1997;128:1142-51
Etienne J. Prevention of infectious endocarditis.
Ann Fr Anesth Reanim. 1994;13:67-72
Franco E, Saunders CP, Roberts GJ, Suwanprasit
A. Dental disease, caries related microflora and
salivary IgA of children with severe congenital
cardiac disease: an epidemiological and oral
microbial survey. Ped Dent. 1996;18:228-35
Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol.
2002;39:1890-900
Hallet KB, Radford DJ, Seow WK. Oral health of
children with congenital heart disease: a controlled study. Ped Dent. 1992;14:224-30
Jowett NI, Cabot LB. Patients with cardiac disease:
considerations for dental practitioners. BDJ.
2000; 189:297-302
Kenny D, Hijazi ZM. Coarctation of the aorta: from fetal life to adulthood. Cardiol J.
2011;18:487-95
Khader RN, Rosenberg M. American Heart Association: The 2007 American Heart Association guideline for the prescription of antibiotic
prophylaxis: a brief overview. J Mass Dent Soc.
2007;56:34-6
Knirsch W, Nadal D. Infective endocarditis
in congenital heart disease. Eur J Pediatr.
2011;170:1111-27
Kornosky JL, Salihu HM. Getting to the heart of
the matter: epidemiology of cyanotic heart defects. Pediatr Cardiol. 2008;29:484-97
Micheletti A, Negura D, Piazza L, Saracino A,
Butera G, Arcidiacono C, Carminati M, Calaciura R, Chessa M. Infective endocarditis in
patients with congenital heart disease. Pediatr
Med Chir. 2010;32:270-3
Mocchegiani R, Nataloni M. Complications of infective endocarditis. Cardiovasc Hematol Disord Drug Targets. 2009;9:240-8
Oliver R, Roberts GJ, Hooper L, Worthington
HV. Antibiotics for the prophylaxis of bacterial
endocarditis in dentistry. Cochrane Database
Syst Rev. 2008
Piana G, Culot C, Montanari MC, Baietti AM.
L’endocardite nelle cardiopatie congenite: il ruolo dell’odontoiatra. Dent Cad 1996;1:48-52
Roberts GJ, Lucas VS, Omar J. Bacterial endocarditis and orthodontics; J R Coll Surg Edinb.
2000; 45:141-5
Sauders CP, Roberts GJ. Dental attitudes, knowledge, and health practices of parents of children with congenital heart disease. Arch Dis
Child. 1997;76:539-40
Starr JP. Tetralogy of fallot: yesterday and today.
World J Surg. 2010;34:658-68
www.buponline.com
37
Cardiopatie congenite
Stecksen-Blicks C, Rydberg A, Nyman L, Asplund
S, Svanberg C. Dental caries experience in children with congenital heart disease: a case-control study. Int J Paediatr Dent. 2004;14:94-100
Steelman R, Einzing S, Balian A, Thomas J, Rosen
D, Gustafson R, Gochenour L. Increased susceptibility to gingival colonization by specific
HACEK microbes in children with congenital
heart disease. J of Clin Ped Dent. 2000;25:91-4
Tasioula V, Balmer R, Parsons J. Dental health and
treatment in a group of children with congenital heart disease. Pediatr Dent. 2008;30:323-8
Tong DC, Rothwell BR. Antibiotic prophylaxis in
dentistry a review and practice recommenda-
tions. J Am Dent Assoc. 2000;131:366-74
Tornos P, Gonzalez-Alujas T, Thuny F, Habib G.
Infective endocarditis: the European viewpoint. Curr Probl Cardiol. 2011;36:175-222
Wilson W, Taubert KA, Gewitz M, Lockhart PB,
Baddour LM, Levison M, Bolger A, Cabell
CH, Takahashi M, Baltimore RS, Newburger JW, Strom BL, Tani LY, Gerber M, Bonow
RO, Pallasch T, Shulman ST, Rowley AH,
Burns JC, Ferrieri P, Gardner T, Goff D, Durack DT; American Heart Association. Prevention of infective endocarditis: Guidelines from
the American Heart Association. J Am Dent
Assoc. 2008;139:3-24
www.buponline.com
www.buponline.com
3. Sindrome di Down
Sinonimi: T21, Trisonomia G, Mongolismo
Codici: ICD9-CM:758.0; 758.9
T. Tagariello, N. Al Kamhis, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Sindrome di Down (SD), descritta per la prima volta nel 1866 dal medico britannico John
Langdon Down, è la più frequente e la più nota
delle malattie cromosomiche; è caratterizzata da
patologie sistemiche e da ritardo mentale (Down
JLH, 1866).
L’incidenza è stimata fra 1 su 800 e 1 su 1.000
nati vivi per anno, in assenza di predilezione razziale, etnica, geografica e di genere (Shin M et al.,
2009; Centers for Disease Control and Prevention,
2006). L’incidenza aumenta proporzionalmente
all’età materna, variando da 1 su 1.400 in madri
con età inferiore a 25 anni a 1 su 60 in madri con
età superiore a 40 anni (Frydman A et al., 2012).
Gli indici di morbilità e mortalità sono maggiori rispetto alla popolazione normale. Le principali cause di morte sono cardiopatie congenite,
infezioni (in particolare respiratorie), ostruzioni
intestinali, ipotiroidismo e leucemie (Roizen NJ
et al., 2003). Attualmente, grazie ai progressi nelle diagnosi e nelle terapie delle patologie associate,
l’aspettativa di vita risulta compresa tra 55 e 65
anni. Tuttavia nella SD si instaura un processo di
invecchiamento precoce ed accelerato, responsa-
bile di un precoce deterioramento delle condizioni
neurologiche, psichiatriche e fisiche (Day SM et
al., 2005; Yang Q et al., 2002).
GENETICA E DIAGNOSI
La SD è causata da un’alterazione numerica del
cromosoma 21. Sono descritti tre diversi assetti
del cariotipo. Il più frequente (92-95%) è rappresentato dalla trisomia 21 libera: il cromosoma 21
in sovrannumero origina da una non-disgiunzione
casuale durante la meiosi ed è presente in tutte le
cellule. Il secondo, molto più raro (3-4%), è la traslocazione: il cromosoma soprannumerario o una
sua parte è legato ad un altro cromosoma, più frequentemente il cromosoma 14. Il terzo, ancora più
raro (2-4%), è il mosaicismo, caratterizzato dalla
contemporanea presenza di due linee cellulari differenti: una normale a 46 cromosomi ed una anomala
a 47 cromosomi per la presenza di un cromosoma
21 soprannumerario originato da una non-disgiunzione post-zigotica (Stoll C et al., 1998).
La diagnosi di SD può essere prenatale, tramite lo
studio del cariotipo su villocentesi o amniocentesi,
o dopo la nascita, in base alla constatazione del-
www.buponline.com
40
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
le caratteristiche cliniche del neonato, poi confermata dall’indagine genetica (Roizen NJ et al.,
2003).
MANIFESTAZIONI CLINICHE SISTEMICHE
Molte sono le caratteristiche somatiche comuni ai pazienti affetti da SD (Tabella n.1), nonostante l’ampia gamma di variazioni fenotipiche
(Abanto J et al., 2011; Sureshbabu R et al., 2011;
Lott IT et al., 2010; Hawli Y et al., 2009; Roizen
NJ et al., 2003; American Academy of Pediatrics,
2001).
L’aspetto somatico (Figura 1) del soggetto con
SD è caratterizzato da bassa statura, brachicefalia
con occipite piatto, viso arrotondato con aspetto
“orientaleggiante”, occhi piccoli e distanziati con
taglio obliquo delle rime palpebrali (dal basso
in alto e dall’interno all’esterno), iride screziata
(macchie di Brush-Field), ponte nasale piatto,
orecchie piccole e dismorfiche con impianto basso, collo corto e tozzo, torace piatto, addome
Figura 1: Bambina di 4 anni affetta da SD, facies caratteristica: viso arrotondato con aspetto “orientaleggiante”,
ipotonia dei muscoli del volto, orecchie piccole con impianto
basso, protrusione linguale
Tabella 1: Manifestazioni cliniche in pazienti affetti da SD
APPARATO O FUNZIONE
ASPETTO SOMATICO
MANIFESTAZIONI CLINICHE
Bassa statura
Brachicefalia
Ponte nasale piatto
Volto di aspetto “orientaleggiante”
Orecchie piccole e dismorfiche
Collo corto e tozzo
Torace piatto
Addome espanso
Bacino basso e largo
Mani tozze
Brachidattilia
Lingua fissurata
www.buponline.com
41
Sindrome di Down
APPARATO
CARDIO-VASCOLARE
APPARATO OCULARE
APPARATO UDITIVO
APPPARATO ENDOCRINO
APPARATO
MUSCOLO-SCHELETRICO
SISTEMA IMMUNITARIO
SISTEMA EMATOLOGICO
SISTEMA NERVOSO
Difetti setto atrio-ventricolare
Difetti interventricolari
Ostium secundum
Persistenza del dotto arterioso di Botallo
Tetralogia di Fallot
Prolasso valvola mitrale
Insufficienza aortica
Cataratta congenita
Glaucoma
Disturbi rifrattivi
Strabismo
Nistagmo
Ipoacusia su base trasmissiva, neurosensoriale o mista
Otiti medie
Ipotiroidismo
Diabete
Ritardato sviluppo motorio
Ipotonia muscolare
Lassità ligamentosa
Dislocazioni articolari
Instabilità atlanto-assiale
Deficit immunitario
Malattia celiaca
Policitemia
Macrocitosi
Disordini mieloproliferativi transitori
Leucemia mieloide acuta
Leucemia linfoblastica acuta
Segni neurologici focali
Demenza senile
Crisi epilettiche
espanso, bacino basso e largo, mani tozze, brachidattilia, lingua fissurata (Down JLH, 1866).
Il ritardo mentale è sempre presente ma di entità molto variabile, da lieve a severo; sono presenti
anomalie di apprendimento, di memoria e di linguaggio, con compromissione del funzionamento
intellettivo (Lott IT et al., 2010). I pazienti con SD
manifestano frequentemente i segni clinici della
demenza senile in un’età più precoce rispetto alla
restante popolazione, a partire dalla quinta decade
di vita. In età adulta sono frequenti crisi epilettiche (58%), alterazioni della personalità (46%), segni neurologici focali (46%), apatia (36%), perdita
di competenze linguistiche (36%) (Margallo-Lana
ML et al., 2007; Roizen NJ et al., 2003).
Circa il 50% dei bambini con SD presenta una
cardiopatia congenita; le anomalie più frequenti
sono i difetti del setto atrioventricolare (45%) e
i difetti interventricolari (35%). Altre anomalie
sono i difetti interatriali tipo ostium secundum
www.buponline.com
42
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
(8%), la persistenza del dotto arterioso di Botallo
(7%), la tetralogia di Fallot (4%). Un’elevata percentuale di pazienti con SD in età adulta sviluppa
prolasso della valvola mitralica (46%) o insufficienza aortica (17%) (Geggel RL et al., 1993).
Sono frequenti patologie oftalmiche, in particolare cataratta congenita, glaucoma, disturbi rifrattivi
(35-76%), strabismo (25-57%), nistagmo (20%)
(Roizen NJ et al., 2003).
Il 38-78% dei pazienti con SD è affetto da patologie otorinolaringoiatriche quali ipoacusia
(trasmissiva, neurosensoriale o mista) e otiti medie
(Roizen NJ et al., 2003).
Il metabolismo basale spesso rallentato contribuisce ad una più alta prevalenza di obesità rispetto
alla popolazione generale (Rubin SS et al., 1998).
Sono di riscontro relativamente frequente endocrinopatie, in particolare ipotiroidismo (che può
essere presente già alla nascita con segni e sintomi
che si sviluppano lentamente) e diabete di tipo 2
(che si manifesta in circa l’1% dei bambini/adolescenti) (Anwar AJ et al., 1998; Karlsson B et al.,
1998).
Per quanto concerne l’apparato muscolo-scheletrico, i pazienti affetti da SD presentano bassa
statura, ritardo dello sviluppo motorio, ipotonia generalizzata, lassità legamentosa, iperflessibilità e dislocazioni articolari. In alcuni soggetti
è presente una sublussazione atlanto-assiale, che
nel 2% dei casi causa sintomi e segni di compressione midollare (iperreflessia, dolore al collo, torcicollo, cambiamenti nella deambulazione, perdita
di controllo degli sfinteri, tetraparesi) (Abanto J
et al., 2011; Hawli Y et al., 2009; Roizen NJ et
al., 2003). L’1-2% dei bambini/adolescenti con
SD sviluppa una patologia delle articolazioni simile all’artrite cronica giovanile (Roizen NJ et al.,
2003).
Nel soggetto con SD è presente un deficit del sistema immunitario responsabile di maggiore suscettibilità alle infezioni, che si manifestano più
frequentemente a livello degli apparati respiratorio
e gastrointestinale, delle mucose e della cute (Desai
SS, 1997).
I pazienti con SD possono essere affetti da malattia celiaca (4,6-7,1%) (Roizen NJ et al., 2003).
Per quanto concerne l’apparato ematopoietico, frequentemente i pazienti con SD sono affetti da macrocitosi (66%), policitemia nel neonato
(64%), disordini mieloproliferativi transitori, leucemia mieloide acuta, leucemia linfoblastica acuta
(Abanto J et al., 2011; Roizen NJ et al., 2003).
CARATTERISTICHE ORO-FACCIALI
Nella SD sono molto frequenti anomalie dentarie,
patologie parodontali e patologie ortopedico-ortodontiche.
Le anomalie dentarie riguardano eruzione, numero, dimensione, forma e struttura (Shore S et al.,
2010; De Moraes ME et al., 2007; Horbelt CV,
2007; Fiske J et al., 2001).
Per quanto concerne le anomalie di eruzione,
sono frequenti ritardi nella dentatura decidua
(oltre il 9° mese) e permanente (oltre gli 8 anni).
Sono frequenti anche anomalie nella sequenza
eruttiva della serie decidua (l’eruzione dei primi
molari prima dell’eruzione degli incisivi). La ritardata permuta fisiologica degli elementi decidui può
portare ad anomalie di posizione dei permanenti;
la più frequente è l’eruzione di incisivi inferiori
permanenti in posizione linguale, non preceduta
dall’esfoliazione del deciduo corrispondente (Desai
SS, 1997) (Figura 2).
Le anomalie di numero si presentano sia in difetto (agenesie nel 50%), sia, molto raramente, in
eccesso (soprannumerari nello 0,3%) (De Moraes
ME et al., 2007). L’agenesia più frequente riguarda gli incisivi laterali mandibolari (23.3%); sono
frequenti agenesie multiple (Kumasawa S et al.,
1997).
www.buponline.com
43
Sindrome di Down
senza di sigillo labiale, della presenza di malocclusioni
e di bruxismo (Figura 4). Sono di frequente riscontro
nel bambino quadri di gengivite marginale e nel
giovane-adulto quadri di gengivite acuta ulcero-necrotica (Morinushi T et al., 2006; López-Pérez R et
al., 2002). L’esordio della parodontopatia profonda
è precoce ed interessa per primi gli incisivi inferiori
(Khocht A et al., 2010; Amaral Loureiro AC et al.,
2007; Morgan J, 2007).
Emorragie spontanee e lesioni persistenti a livello
Figura 2: Anomalia di posizione dell’elemento permanente
(11) erotto in posizione palatale rispetto al corrispondente deciduo a causa del ritardo nell’esfoliazione del deciduo
(51)
Le dimensioni dei denti decidui in genere sono
maggiori rispetto alla norma, mentre quelle dei
permanenti sono normali o ridotte (microdonzia nel 35-55% dei casi); questa sproporzione può
portare alla comparsa di diastemi interdentali in
dentizione permanente (Bell E et al., 2001; Peretz
B et al., 1998; 1996).
Per quanto concerne le anomalie di forma (Figura 3), sono descritti denti conoidi a livello del
settore frontale (14,28%) e molari taurodonti, in
particolare il secondo molare inferiore (66%) (De
Moraes ME et al., 2007). I molari presentano solchi e fessure poco profondi (De Moraes ME et al.,
2007; Peretz B et al., 1998; Peretz B et al.,1996).
Le radici sono frequentemente piccole e di forma
conica (Desai SS, 1997).
Di riscontro relativamente frequente sono le ipoplasie dello smalto, in alcuni casi correlabili a cardiopatia congenita e/o a malattia celiaca.
I pazienti affetti da SD sviluppano frequentemente
malattia parodontale a causa del deficit immunitario (ridotta chemiotassi dei neutrofili, ridotta capacità
battericida intracellulare), dello scarso controllo dell’igiene orale domiciliare, della spinta linguale, dell’as-
Figura 3: Bambino di 4 anni affetto da SD. Anomalia di
forma: fusione di 82 e 83
Figura 4: Abrasioni degli elementi decidui da bruxismo in
bambina di 8 anni affetta da SD
www.buponline.com
44
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
gengivale possono essere segno di leucemia (Desai SS,
1997).
L’incidenza di lesioni aftose e di infezioni orali da
Candida albicans è elevata (Scully C et al., 2002;
Carlstedt K et al., 1996).
L’analisi della letteratura evidenzia come i pazienti
affetti da SD siano poco suscettibili alla patologia
cariosa per il pH salivare elevato, per la bassa concentrazione salivare di Streptococco mutans e per la
presenza di diastemi e di solchi e fessure poco profondi a livello dei molari (Anders PL et al., 2010;
Davidovich E et al., 2010). Tuttavia l’esperienza clinica evidenzia casi di carie della dentatura sia decidua
che permanente correlati all’assunzione di farmaci, in
particolare sciroppi, al consumo di cibi e bevande ad
alto contenuto di zuccheri, all’igiene orale domiciliare
insufficiente.
Di costante riscontro nel soggetto con SD sono le
patologie ortopedico-ortodontiche. Caratteristica
della sindrome è l’ipoplasia del terzo medio del viso
per il ridotto sviluppo dimensionale del mascellare
superiore nelle tre direzioni dello spazio (Suri S et
al., 2010; Alio JJ et al., 2008). L’ipoplasia ossea del
mascellare superiore, responsabile delle ridotte dimensioni sagittali e trasversali del palato, provoca un
prognatismo mandibolare relativo (Kieser J et al.,
2003).
Unitamente all’ipoplasia del terzo medio del volto, le
alterazioni del tono e della motilità della muscolatura oro-facciale, in particolare della lingua, contribuiscono a determinare la facies caratteristica (Shott
SR, 2006). L’ipotonia interessa tutta la muscolatura
del volto (muscoli orbicolare della bocca, zigomatico,
massetere, temporale) e del cavo orale, in particolare
dei muscoli della lingua e del velo palatino (Desai SS,
1997; Limbrock GJ et al., 1993).
La lingua ipotonica appare più grossa e più lunga; a
causa delle ridotte dimensioni del palato è appiattita
sul pavimento della bocca e spesso sporge sopra ad
un labbro inferiore ipotonico (pseudomacroglossia)
(Limbrock GJ et al., 1993) (Figura 5 a, b). La postura
Figura 5 a, b: Bambina di 4 anni affetta da SD. Protrusione linguale e pseudo macroglossia
bassa della lingua ipotonica non permette la fisiologica
espansione del palato. L’ipotonia dei muscoli orbicolari delle labbra determina mancanza di sigillo labiale,
provoca difficoltà nella suzione ed è responsabile, unitamente alla postura linguale bassa, dello sventagliamento del gruppo incisivo inferiore (Mizuno K et al.,
2001).
La lingua, nei due terzi anteriori, presenta spesso
fissurazioni di lunghezza e profondità variabili, in
cui possono persistere residui di cibo responsabili
di alitosi (Limbrock GJ et al., 1993).
La respirazione è prevalentemente di tipo orale
per la ridotta pervietà delle vie aeree nasali, legata
all’appiattimento della radice nasale e all’ipoplasia
del mascellare. La respirazione orale aggrava la riduzione dei diametri trasversali e causa l’aumento della
dimensione verticale del palato (Shott SR, 2006;
Clarke RW, 2005; Marcus CL et al., 1991).
La conformazione del naso, di piccole dimensioni
e con forma “a sella”, causa difficoltà alla fisiologica
espulsione del muco; il suo ristagno nelle cavità nasali predispone a processi infiammatori locali (favoriti dal deficit del sistema immunitario), responsabili
di ipertrofia adenoidea e di occlusione delle tube di
Eustachio. L’ostacolo alla normale areazione dell’orecchio medio è responsabile dell’insorgenza di otiti
ricorrenti e di diminuzione dell’udito.
Nei pazienti con SD sono di frequente riscontro
www.buponline.com
45
Sindrome di Down
(50-80%) le apnee ostruttive notturne (OSAS),
dovute a ostruzione delle vie aeree superiori e favorite dall’ipotonia della muscolatura del palato
molle (Waldman HB et al., 2009; Stebbens VA
et al., 1991; Marcus CL et al., 1991). I sintomi
sono russamento, arresto della respirazione, posizioni anomale e frequenti risvegli durante il
sonno, ipersonnia diurna. Nell’adulto con SD il
grado di severità delle OSAS è spesso aggravato
dall’obesità (Trois MS et al., 2009; Waldman HB
et al., 2009).
I quadri di malocclusione di più frequente riscontro nei soggetti Down sono: III classe con morso
inverso anteriore; morso aperto anteriore causato
dall’ipotono dei muscoli elevatori e periorali e dalla
pseudomacroglossia; morso crociato posteriore
mono o bilaterale (Oliveira AC et al., 2010; Suri S
et al., 2010; Winter K et al., 2008).
Nei soggetti con SD la malocclusione di III classe
tende ad un progressivo peggioramento col procedere dell’età, anche a causa dell’instaurarsi, durante l’adolescenza, di discinesie in avanzamento
della mandibola, favorite dalla lassità ligamentosa
dell’articolazione temporo-mandibolare (Faulks D
et al., 2008) (Figura 6a, b).
a
Figura 6a: Bambino di 7 anni affetto da SD. Morso inverso
anteriore. Ampi diastemi a livello dell’arcata inferiore
b
Figura 6b: Paziente di 14 anni affetto da SD: profilo
LINEE GUIDA DI TERAPIA
I pazienti con SD sono affetti da numerose alterazioni e patologie che rendono necessari interventi multispecialistici di prevenzione, diagnosi, follow-up e
terapia, che richiedono un team multi e interdisciplinare adeguatamente formato sulla sindrome.
Anche in assenza di segni o sintomi è fondamentale
indagare già durante le prime settimane di vita la
presenza di cardiopatia congenita, che spesso viene
risolta chiurgicamente entro il primo anno di vita. I
soggettti non affetti da cardiopatia congenita sono a
rischio di sviluppare patologie valvolari durante l’adolescenza o da giovani adulti; per tale motivo sono
necessarie visite cardiologiche periodiche di controllo anche in assenza di segni o sintomi di malattia
(Roizen NJ et al., 2003; Marder E et al., 2001).
Durante i primi mesi di vita sono necessarie una
valutazione oftalmologica per indagare la presenza di strabismo, cataratta, nistagmo e una otorinolaringoiatrica per indagare la presenza, di ipoacusia e di infezioni del tratto respiratorio (Roizen
NJ et al., 2003; American Academy of Pediatrics,
2001).
L’ipotiroidismo, congenito o acquisito, durante i
primi anni di vita necessita di terapia sostitutiva
www.buponline.com
46
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
con ormone tiroideo, che migliora ma non normalizza lo sviluppo e le funzioni cognitive (Hawli Y
et al., 2009).
Durante la crescita i pazienti con SD devono essere periodicamente sottoposti a visite ed esami
strumentali e di laboratorio per controllare crescita, sviluppo cognitivo, vista, patologie otorinolarigoiatriche, instabilità atlanto-assiale, funzionamento tiroideo (American Academy of Pediatrics,
2001). Deve essere inoltre indagata la presenza di
malattia celiaca, diabete mellito e patologie ematologiche.
In considerazione dell’ipotonia muscolare, del ritardato sviluppo motorio, delle alterazioni scheletriche, dei deficit di linguaggio e di apprendimento
è necessario un intervento fisioterapico e logopedico precoce da parte di operatori specificatamente
preparati sulla sindrome.
In caso di OSAS l’intervento deve essere tempestivo, in quanto le apnee notturne possono contribuire al ritardo di sviluppo e favorire l’ipertensione polmonare con rischio di scompenso cardiaco
congestizio. Escludendo le patologie risolvibili con
adeno-tonsillectomia, la terapia varia dall’applicazione di placche di riposizionamento occlusale alla
correzione chirurgica per il ripristino della pressione positiva nelle vie aeree (Sedaghat AR et al.,
2012; Sato et al., 2010; Shete MM et al., 2010;
Waldamn HB et al., 2009; Merrell JA et al., 2007).
Per adattare il paziente alla propria situazione e favorire il suo inserimento nella società può essere
utile un intervento psicologico.
Dal punto di vista odontostomatologico è necessario porsi tre obiettivi: la riabilitazione funzionale della lingua, la prevenzione delle parodontopatie e della patologia cariosa, la correzione
ortopedica della discrepanza scheletrica. In una
fase successiva, in base al grado di collaborazione
(in particolare all’igiene orale domiciliare) e alle necessità funzionali ed estetiche, può essere indicata
la terapia ortodontica.
La prima visita odontoiatrica deve avvenire precocemente, verso i 6 mesi di vita. In questa occasione i genitori vengono informati sulle patologie
orali più frequenti e sulla necessità di interventi
di prevenzione primaria mirati ed intensivi con
particolare riferimento a: - valutazione della funzionalità della muscolatura orofacciale, - alimentazione non cariogenica, che focalizzi i rischi derivanti
dall’assunzione frequente e prolungata di soluzioni
zuccherate con il biberon, - igiene orale domiciliare
corretta, - utilizzo di fluoro topico e quando indicato sistemico, - visite odontoiatriche periodiche
trimestrali/semestrali (Shore S et al., 2010).
Quando presente ipotonia della muscolatura orofacciale, è possibile migliorare la tonicità muscolare mediante un approccio di tipo miofunzionale,
unitamente alla fisioterapia dai 6 mesi ed alla logopedia dai 3 anni. Gli obiettivi sono stimolare la
postura alta della lingua, il movimento e la tonicità
della lingua, delle labbra e dei muscoli masticatori
per contenere la lingua all’interno del cavo orale e
realizzare un buon sigillo labiale, evitando la fuoriuscita di saliva.
Nei casi con ipotonia muscolare e protrusione linguale, risultati positivi sono descritti con la terapia
che combina la stimolazione manuale all’applicazione di placche palatine (placche Castillo-Morales modificate) dai primi mesi di vita (Korbmacher
HM et al., 2006; Korbmacher HM et al., 2005;
Korbmacher HM et al., 2004; Bäckman B et al.,
2003; Limbrock GJ et al., 1993; Limbrock GJ et
al 1991). Per contrastare l’assuefazione agli stimoli
si ricorre all’utilizzo sequenziale di placche palatine
in resina acrilica.
La placca tipo 1 è dotata di un bottone cavo modellato nella parte distale e di un bordo anteriore
(diviso in due parti simmetriche dall’incisura per il
frenulo labiale superiore) caratterizzato da due rigonfiamenti vestibolari con rastremature verticali.
Il bottone ha la funzione di stimolare la tonicità
della lingua e di modificarne la posizione bassa e
www.buponline.com
47
Sindrome di Down
protrusa. I rigonfiamenti vestibolari hanno un’azione specifica sul labbro superiore ma fungono
da stimolatori per tutta la muscolatura orbicolare
(Figura 7).
Sulla placca tipo 2, che sostituisce la n.1 dopo circa 6 mesi, viene aggiunta una pallina zigrinata mobile con la funzione di stimolo per la punta della
lingua (Figura 8).
Sulla placca tipo 3, che sostituisce la n.2 dopo altri
6 mesi, vengono inseriti dei bottoncini vestibolari
per la stimolazione della muscolatura circum-orale
(Figura 9).
Le placche vanno applicate tre volte al giorno per
5-30 minuti per volta per abituare la lingua allo
stimolo proposto (Faulks D et al., 2008). L’adesione al palato può essere facilitata dall’utilizzo di piccole quantità di crema adesiva per protesi totale.
Nella maggior parte dei casi le placche sono ben
accettate dal bambino, che modifica la postura della lingua ed aumenta la tonicità della muscolatura,
e di facile gestione per i genitori, che apprezzano i
risultati ottenuti.
All’eruzione dei primi denti decidui, è possibile
utilizzare placche che rispondono agli stessi criteri funzionali, caratterizzate da spazi per alloggiare
i denti e da elementi accessori di ritenzione (ad
esempio ganci ed arco vestibolare).
La presenza di ostruzioni anatomiche delle prime
vie aeree (grave deviazione del setto, stenosi delle
coane) rappresenta una controindicazione all’applicazione delle placche, per cui è necessaria una
valutazione otorinolaringoiatrica preliminare.
Per ottenere i migliori risultati si associa l’uso della
placca palatina a regolari sedute di fisioterapia,
basate sul massaggio e sulla stimolazione di risposte
riflesse, sia del distretto oro-facciale sia degli arti e
del tronco, al fine di correggere l’ipotonia generalizzata e le posture patologiche. Appena il livello di
collaborazione lo consente (generalmente intorno
ai 3 anni di età) è necessario l’intervento logopedico (Van Bysterveldt AK et al., 2010).
Figura 7: Placca CM tipo 1
Figura 8: Placca CM tipo 2
Figura 9: Placca CM tipo 3 (si evidenzia la presenza in arcata degli incisivi centrali superiori)
www.buponline.com
48
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Nella seconda infanzia le visite odontoiatriche
debbono essere effettuate ad intervalli frequenti
per sviluppare un buon livello di collaborazione,
individuando il metodo di comunicazione più
efficace. L’odontoiatra, consapevole che nel soggetto Down il livello di espressività può essere
inferiore al livello di comprensione, e informato
dai genitori/tutori del paziente sul tipo di comunicazione più appropriato, individua e attua le
strategie comportamentali idonee a instaurare
un buon livello di collaborazione.
È preferibile programmare gli appuntamenti nella
prima parte della giornata, quando il paziente è
più riposato e rilassato.
Nei pazienti affetti da cardiopatie congenite un
buon livello d’igiene orale è fondamentale per evitare batteriemie causate da sanguinamento gengivale, che comportano il rischio di endocardite
batterica (EB) (Khocht A et al.; 2010; Morgan J,
2007). In presenza di patologie cardiovascolari a
rischio di EB è necessario sottoporre il paziente
a profilassi antibiotica prima di effettuare procedure odontoiatriche che possano causare sanguinamento (Freeman SB et al., 2008) (vedi capitolo
2).
Deve essere sempre indagata la presenza di instabilità atlanto-assiale per i rischi derivanti dall’iperestensione del capo durante le manovre odontoiatriche (Sedaghat AR et al., 2012 ).
In caso di gengivite e parodontopatia profonda i
controlli sono trimestrali e prevedono sedute di
igiene orale professionale.
In presenza di ipoplasie dello smalto sono indicate applicazioni topiche di fluoro domiciliari e
ambulatoriali (gel o vernici ad alta concentrazione e a lento rilascio). Le sigillature dei solchi, delle fessure e dei fori ciechi dei molari permanenti
vengono eseguite previa valutazione del rischio di
carie e della morfologia della superficie occlusale
(Shore S et al., 2010; Pilcher ES, 2001).
In caso di alitosi, causata dal ristagno di cibo
all’interno delle fissurazioni della lingua, si consiglia di eseguire la pulizia della lingua con spazzolino o con puliscilingua.
Nella fase di dentizione mista, in genere intorno
agli 8 anni in considerazione del ritardo di eruzione, si evidenzia la necessità di una terapia di tipo
ortopedico-ortodontico per espandere il palato e
per intercettare la malocclusione scheletrica di III
classe, da attuare non appena il bambino fornisca
la collaborazione necessaria all’applicazione delle
apparecchiature (Musich DR, 2006). Si utilizzano due dispositivi ad azione ortopedica: l’espansore rapido del palato (REP) per correggere il
deficit trasversale derivante dall’ipoplasia palatina
e la maschera di Delaire, che, stimolando la protrazione del mascellare superiore, attenua il deficit
sagittale e verticale.
Il REP, inducendo la diastasi della sutura palatina
mediana non ancora ossificata, favorisce il processo di neoapposizione ossea e produce un ampliamento del diametro trasversale del mascellare
superiore, con abbassamento del pavimento del
naso e conseguente aumento delle cavità nasali
e diminuzione delle resistenze al flusso aereo. Il
REP favorisce quindi il ripristino di una corretta
respirazione nasale nei pazienti con respirazione
orale da ristrettezza palatina. L’aumento di diametro dell’arcata dento-alveolare, inoltre, migliora la
postura e la funzione della lingua, che trova una
cavità sufficientemente spaziosa per accoglierla.
La maschera di Delaire (Figura 10) è il dispositivo per la trazione ortopedica postero-anteriore
del mascellare superiore utilizzato nelle III classi
scheletriche da retrusione superiore e miste. L’apparecchiatura è costituita da una porzione extraorale mobile dotata di un appoggio frontale e di
uno mentale e da una porzione intraorale fissa,
collegate tra loro da elastici con forza ortopedica
(da 500 a 1000 g per parte). La trazione va applicata per non meno di 12 ore al giorno, non necessariamente consecutive, per il tempo necessario
www.buponline.com
49
Sindrome di Down
ad ottenere un overjet di almeno 4-5 mm che,
in caso di corretto utilizzo, è di circa 6 mesi. Il
meccanismo d’azione della maschera varia in base
all’età di applicazione. Nei bambini di età inferiore ai 7-8 anni stimola le suture posteriori (pterigopalatina e maxillo-palatina) non ancora ossificate,
quindi ottiene uno spostamento “in toto” verso
l’avanti del mascellare superiore. Dopo gli 8 anni
(nei soggetti Down anche più tardivamente in relazione al ritardo dell’ossificazione), procedendo
l’ossificazione delle suture, il dispositivo produce un movimento mesiale del complesso dentoalveolare e, in una fase successiva, un movimento
quasi esclusivamente dentale. Questo aspetto va
tenuto presente nella pianificazione della terapia:
la mesializzazione degli elementi dentali dell’arcata superiore non è sempre indicata e, qualora
lo fosse, esistono dispositivi più efficaci per ottenerla.
In fase di dentizione permanente può essere
effettuata la terapia ortodontica propriamente
detta con applicazione di dispositivi fissi per rispondere ad esigenze di tipo funzionale ed estetico; oltre a favorire una masticazione efficace ed
una corretta postura mandibolare, la riabilitazione “del sorriso” facilita l’inserimento sociale e
contribuisce all’autostima del paziente.
La possibilità di applicazione di apparecchiature
ortodontiche fisse è tuttavia subordinata al grado di collaborazione del paziente e della famiglia,
in particolare per quanto concerne l’igiene orale domiciliare. Un livello di igiene orale insufficiente rappresenta infatti una controindicazione
assoluta all’ortodonzia fissa a causa della maggior
suscettibilità dei soggetti Down alla malattia parodontale: lo spostamento ortodontico in presenza di placca batterica provoca un peggioramento
iatrogeno dello stato dei tessuti di sostegno del
dente.
BIBLIOGRAFIA
Abanto J, Ciamponi AL, Francischini E, Murakami C, Medeiros de Rezende NP, Gallottini
M. Medical problems and oral care of patients
with Down syndrome: a literature review. Spec
Care Dentist. 2011;31:197-201
Alio JJ, Lorenzo J, Iglesias C. Cranial base growth
in patients with Down syndrome: a longitudinal study. Am J Orthod Dentofacial Orthop.
2008;133:729-37
Amaral Loureiro AC, Oliveira Costa F, Eustàquio
da Costa J. The impact of periodontal disease on the quality of life of individuals with
Down syndrome. Downs Syndr Res Pract.
2007;12:50-4
American Academy of Pediatrics. Committee on
Genetics: American Academy of Pediatrics:
Health supervision for children with Down
syndrome. Pediatrics. 2001;107:442-9
Anders PL, Davis EL. Oral health of patients with
intellectual disabilities: a systematic review .
Spec Care Dentist. 2010;30:110-7
Anwar AJ, Walker JD, Frier BM. Type I diabetes
mellitus and Down‘s syndrome: prevalence,
management and diabetic complications. Diabet Med. 1998;15:160-3
Bäckman B, Grevér-Sjölander AC, Holm AK, Johansson I. Children with Down Syndrome:
oral development and morphology after use of
palatal plates between 6 and 18 months of age.
Int J Paediatr Dent. 2003;13:327-35
Bell E, Townsend G, Wilson D, Kieser J, Hughes
T. Effect of Down syndrome on the dimensions of dental crowns and tissues. Am J Hum
Biol. 2001;13:690-8
Carlstedt K, Krekmanova L, Dahllöf G, Ericsson
B, Braathen G, Modéer T. Oral carriage of
Candida species in children and adolescents
with Down‘s syndrome. Int J Paediatr Dent.
1996;6:95-100
www.buponline.com
50
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Centers for Disease Control and Prevention. Improved national prevalence estimates for 18
selected major birth defects – United States,
1999 – 2001. Morbidity and Mortality Weekly
Report. 2006;54:1301-5
Clarke RW. Ear, nose and throat problems in children with Down syndrome. Br J Hosp Med
(Lond). 2005;66:504-6
Davidovich E, Aframian DJ, Shapira J, Peretz B.
A comparison of the sialochemistry, oral pH,
and oral health status of down syndrome children to healthy children. Int J Paediatr Dent.
2010;20:235-41
Day SM, Strauss DJ, Shavelle RM, Reynolds RJ.
Mortality and causes of death in persons with
Down syndrome in California. Dev Med Child
Neurol. 2005;47:171-6
De Moraes ME, de Moraes LC, Dotto GN, Dotto
PP, dos Santos LR. Dental anomalies in patients with Down syndrome. Braz Dent J.
2007;18:346-50
Desai SS. Down syndrome: a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:279-85
Down JLH. Observation on ethnic classification of
idiots. London Hospital Reports. 1866; 3:25962
Faulks D, Collado V, Mazille MN, Veyrune JL, Hennequin M. Masticatory dysfunction in persons
with Down’s syndrome. Part 1: aetiology and incidence. J of Oral Rehabil. 2008;35:854-62a
Faulks D, Mazille MN, Collado V, Veyrune JL,
Hennequin M: Masticatory dysfunction in
persons with Down’s syndrome. Part 2: management. J of Oral Rehabil. 2008;35:863-9b
Fiske J, Shafik HH. Down’s syndrome and oral
care. Dent Update. 2001;28:148-56
Freeman SB et al. Ethnicity, sex, and the incidence
of congenital heart defects: a report from the
National Down Syndrome Project. Genet
Med. 2008;10:173-80
Frydman A, Nowzari H: Down syndrome-associated periodontitis: a critical review of the
literature. Compend Contin Educ Dent.
2012;33:356-61
Geggel RL, O’Brien JE, Feingold M. Development of valve dysfunction in adolescents and
young adults with Down syndrome and no
known congenital heart disease. J Pediatr.
1993;122:821-3
Hawli Y, Nasrallah M, El-Hajj Fuleihan G. Endocrine and musculoskeletal abnormalities in
patients with Down syndrome. Nat Rev Endocrinol. 2009;5:327-34
Horbelt CV. Down syndrome: a review of common physical and oral characteristics. Gen
Dent. 2007;55:399-402
Karlsson B, Gustafsson J, Hedov G, Ivarsson SA,
Anneren G. Thyroid dysfunction in Down’s
syndrome: relation to age and thyroid autoimmunity. Arch Dis Child. 1998;79:242-5
Khocht A, Janal M, Turner B. Periodontal health
in Down syndrome: Contributions of mental
disability, personal, and professional dental
care. Spec Care Dentist. 2010;30:118-23
Kieser J, Townsend G, Quick A. The Down syndrome patient in dental practice, part I: Pathogenesis and general and dental features. N Z
Dent J. 2003;99:5-9
Korbmacher HM, Limbrock JG, Kahl-Nieke B.
Long-term evaluation of orofacial function in
children with Down syndrome after treatment
with a stimulating plate according to Castillo
Morales. J Clin Pediatr Dent. 2006;30:325-8
Korbmacher HM, Moeller HC, Klocke A, Limbrock JG, Kahl-Nieke B. Cephalometric evaluation of children with Down syndrome after
early intervention with the stimulating plate.
Spec Care Dentist. 2005;25:253-9
Korbmacher HM, Limbrock JG, Kahl-Nieke
B. Orofacial development in children with
Down’s syndrome 12 years after early interven-
www.buponline.com
51
Sindrome di Down
tion with a stimulating plate. J Orofac Orthop.
2004;65:60-73
Kumasawa S, Miyagi A, Sakai N, Shindo J, Kashima I. Oligodontia: a radiographic comparison
of subjects with Down syndrome and normal
subjects. Spec Care Dentist. 1997;17:137-41
Limbrock GJ, Castillo-Morales R, Hoyer H, Stöver
B, Onufer CN. The Castillo-Morales approach
to orofacial pathology in Down syndrome. Int
J Orofacial Myology. 1993;19:30-7
Limbrock GJ, Fischer-Brandies H, Avalle C. Castillo-Morales orofacial therapy: treatment of
67 children with Down syndrome. Dev Med
Child Neurol. 1991;33:296-303
López-Pérez R, Borges-Yáñez SA, Jiménez-García
G, Maupomé G. Oral hygiene, gingivitis, and
periodontitis in persons with Down syndrome.
Spec Care Dentist. 2002;22:214-20
Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol.
2010;9:623-33
Marcus CL, Keens TG, Bautista DB, von Pechmann WS, Ward SL. Obstructive sleep apnea
in children with Down syndrome. Pediatrics.
1991;88:132-9
Marder E, Dennis J. Medical management of children with Down’s syndrome. Current Paediatrics. 2001;11:57-63
Margallo-Lana ML, Moore PB, Kay DW, Perry
RH, Reid BE, Berney TP, Tyrer SP. Fifteenyear follow-up of 92 hospitalized adults with
Down’s syndrome: incidence of cognitive decline, its relationship to age and neuropathology. J Intellect Disabil Res. 2007;51:463-77
Merrell JA, Shott SR. OSAS in Down syndrome:
T&A versus T&A plus lateral pharyngoplasty.
Int J Pediatr Otorhinolaryngol. 2007;71:1197203
Mizuno K, Ueda A. Development of sucking behavior in infants with Down’s syndrome. Acta
Paediatr. 2001;90:1384-8
Morgan J: Why is periodontal disease more prevalent and more severe in people with Down syndrome? Spec Care Dentist. 2007;27:196-201
Morinushi T, Lopatin DE, Nakao R, Kinjyo S. A
comparison of the gingival health of children
with Down syndrome to healthy children residing in an institution. Spec Care Dentist.
2006;26:13-9
Musich DR. Orthodontic intervention and patients with Down syndrome. Angle Orthod.
2006;76:734-5
Oliveira AC, Pordeus IA, Torres CS, Martins MT,
Paiva SM. Feeding and nonnutritive sucking
habits and prevalence of open bite and crossbite in children/adolescents with Down syndrome. Angle Orthod. 2010;80:748-53
Peretz B, Shapira J, Farbstein H, Aureli E, Smith P.
Modification of tooth size and shape in Down’s
syndrome. J Anat. 1996;188:167-72
Peretz B, Shapira J, Farbstein H, Aureli E, Smith
P. Modified cuspal relationships of mandibular
molar teeth in children with down’s syndrome.
J Anat. 1998;193:529-33
Pilcher ES. Treating the patient with Down syndrome. J Contemp Dent Pract. 2001;2:58
Roizen NJ, Patterson D. Down’s syndrome. Lancet. 2003;361:1281-9
Rubin SS, Rimmer JH, Chicoine B, Braddock
D, McGuire DE. Overweight prevalence in
persons with Down syndrome. Ment Retard.
1998;36:175-81
Russel BG, Kjaer I. Tooth agenesis in Down syndrome. Am J Med Genetics. 1995;55:466-71
Sato K, Shirakawa T, Niikuni N, Sakata H, Asanuma S. Effects of oral care in Down syndrome
children with obstructive sleep apnea. J Oral
Sci. 2010;52:145-7
Scully C, van Bruggen W, Diz Dios P, Casal B, Porter S, Davison MF. Down syndrome: lip lesions
(angular stomatitis and fissures) and Candida
www.buponline.com
52
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
albicans. Br J Dermatol. 2002;147:37-40
Sedaghat AR, Flax-Goldenberg RB, Gayler BW,
Capone GT, Ishman SL. A case-control comparison of lingual tonsillar size in children with
and without Down syndrome. Laryngoscope.
2012;122:1165-9
Shete MM, Stocks RM, Sebelik ME, Schoumacher
RA. Effects of adeno-tonsillectomy on polysomnography patterns in Down syndrome
children with obstructive sleep apnea: a comparative study with children without Down
syndrome. Int J Pediatr Otorhinolaryngol.
2010;74:241-4
Shin M, Besser LM, Kucik JE, Lu C, Siffel C, Correa A. Congenital Anomaly Multistate Prevalence and Survival Collaborative: Prevalence of
Down syndrome among children and adolescents in 10 regions of the United States. Pediatrics. 2009;124:1565-71
Shore S, Lightfoot T, Ansell P. Oral disease in children with Down syndrome: causes and prevention. Community Pract. 2010;83:18-21
Shott SR. Down syndrome: common otolaryngologic manifestations. Am J Med Genet C
Semin Med Genet. 2006;142:131-40
Stebbens VA, Dennis J, Samuels MP, Croft CB,
Southall DP. Sleep related upper airway obstruction in a cohort with Down’s syndrome.
Arch Dis Child. 1991;66:1333-8
Stoll C, Alembik Y, Dott B, Roth MP. Study of
Down syndrome in 238.942 consecutive
births. Ann Genet. 1998;41:44-51
Sureshbabu R, Kumari R, Ranugha S, Sathyamoorthy R, Udayashankar C, Oudeacoumar
P. Phenotipic and dermatological manifestations in Down Syndrome. Dermatol Online J.
2011;17:3
Suri S, Tompson BD, Cornfoot L. Cranial base,
maxillary and mandibular morphology in Down
syndrome. Angle Orthod. 2010;80:861-9
Trois MS, Capone GT, Lutz JA, Melendres MC,
Schwartz AR, Collop NA, Marcus CL. Obstructive sleep apnea in adults with Down syndrome. J Clin Sleep Med. 2009;5:317-23
Van Bysterveldt AK, Gillon G, Foster-Cohen S.
Integrated speech and phonological awareness intervention for pre-school children with
Down syndrome. Int J Lang Commun Disord.
2010;45:320-35
Waldman HB, Hasan FM, Perlman S. Down syndrome and sleep-disordered breathing: the dentist’s role. J Am Dent Assoc. 2009;140:307-12
Winter K, Baccaglini L, Tomar S. A review of
malocclusion among individuals with mental
and physical disabilities. Spec Care Dentist.
2008;28:19-26
Yang Q, Rasmussen SA, Friedman JM. Mortality
associated with Down’s syndrome in the USA
from 1983 to 1997: a population-based study.
Lancet. 2002;359:1019-25.
www.buponline.com
4. Sindrome di Klinefelter
Sinonimo 47,XXY Sindrome
Codici ICD 10: Q98.0 Q98.1 Q98.2 Q98.4
I. Cremonesi, M.G. Currà, T. Tagariello, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
GENETICA E DIAGNOSI
La Sindrome di Klinefelter (SK), descritta per la
prima volta da Harry Klinefelter nel 1942, definisce un gruppo di malattie cromosomiche rare
caratterizzate dalla presenza di almeno un cromosoma X in sovrannumero rispetto al normale cariotipo maschile. La SK, pur avendo una grande
varietà di espressione, è caratterizzata dalla presenza di manifestazioni comuni: testicoli piccoli, infertilità, alta statura, disturbi cognitivi e/o comportamentali (Klinefelter HF et al., 1942).
L’aneuploidia 47,XXY è l’anomalia cromosomica più frequente con una incidenza di 1 su 500
maschi nati vivi per anno (Hager K et al., 2012;
Paduch DA et al., 2008). Sono state descritte anche altre aneuploidie con incidenza estremamente
più bassa: le aneuploidie 48,XXYY e 48,XXXY
(1/17.000-50.000 maschi nati vivi per anno) e
49,XXXXY (1/85.000-100.000 maschi nati vivi per
anno). La SK non presenta predilezione razziale, etnica e geografica (Bojesen A et al., 2003). Circa il
40% dei feti portatori dell’aneuploidia nasce vivo.
L’indice di mortalità non è significativamente più
elevato rispetto alla popolazione generale (Bojesen
A et al., 2011).
La presenza di uno o più cromosomi X in sovrannumero di origine materna o paterna è causata
dalla mancata disgiunzione meiotica durante la
prima o la seconda divisione della gametogenesi o
da una mancata disgiunzione mitotica nello zigote in via di sviluppo (Jacobs PA et al., 1959). Sono
stati descritti anche maschi con cariotipo 46,XX,
causato dalla traslocazione di parte del cromosoma Y sul cromosoma X durante la meiosi paterna
(Vissotsak J et al., 2006; Linden MG et al., 1995;
Nielsen J et al., 1991).
I soggetti affetti da SK, pur presentando caratteristiche fenotipiche variabili, non hanno evidenti
dimorfismi, in particolare a livello facciale (Figura
1), e sono indistinguibili da soggetti con cariotipo normale (Caldwell PD et al., 1972). Per questo
motivo la diagnosi clinica è spesso casuale nel
corso di accertamenti resi necessari da una delle
manifestazioni cliniche. Nell’infanzia la SK può
essere ipotizzata nel corso di una valutazione per
ipospadia, pene ipoplasico, criptorchidismo, ritardo psicomotorio (Caldwell PD et al., 1972). In età
scolare possono indirizzare alla diagnosi il ritardo
del linguaggio, le difficoltà di apprendimento, i
www.buponline.com
54
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
disturbi comportamentali (Walzer S et al., 1978).
Nell’adolescenza può essere ipotizzata nel corso di
una visita endocrinologica per ritardo dello sviluppo
puberale, habitus eunucoide, ginecomastia, ipoplasia
dei testicoli (Robinson A et al., 1991). In età adulta
infertilità e tumori maligni del seno possono indirizzare alla diagnosi (Okada H et al., 1999). In caso di
sospetto diagnostico viene eseguita l’indagine genetica, indispensabile per confermare la diagnosi.
La diagnosi prenatale può essere effettuata mediante villocentesi o amniocentesi.
MANIFESTAZIONI CLINICHE
Figura 1: Ragazzo di 12 anni affetto da SK. Dimensioni
craniche leggermente diminuite in proporzione alle dimensioni corporee
Caratteristiche costanti nei soggetti con SK sono
testicoli piccoli, alta statura, infertilità, elevati
livelli di gonadotropine (LH, FSH) e bassi livelli di testosterone. Ginecomastia, scarsa peluria e
altre caratteristiche dell’habitus eunucoide hanno
espressività variabile (Wikström AM et al., 2011;
Caldwell PD et al., 1972).
Le manifestazioni cliniche della SK sono elencate
nella tabella 1.
Tabella 1: Manifestazioni cliniche caratteristiche della Sindrome di Klinefelter
Caratteristiche costanti (95-100%)
Alta statura
Testicoli piccoli
Azoospermia
Sterilità/ipofertilità
Elevati livelli sierici di gonadotropine (LH, FSH)
Bassi livelli sierici di testosterone
Caratteristiche frequenti (>80%)
Ritardo moderato del linguaggio, alterazioni della memoria, dislessia, deficit di attenzione
Ginecomastia/ghiandola mammaria iperplasica
www.buponline.com
55
Sindrome di Klinefelter
Caratteristiche associate (20-80%)
Vene varicose
Osteoporosi
Scoliosi
Brachicefalia/occipite piatto
Disturbi del comportamento
Obesità generalizzata
Iperglicemia/diabete mellito di tipo II
Ritardo mentale leggero/moderato
Carcinoma mammario
Neonati e bambini hanno altezza, peso e circonferenza cranica nella norma. L’aumento staturale è
significativo tra i 5 e gli 8 anni; l’altezza media a
fine crescita è di 179,2 +/- 6,2 cm; le braccia e le
gambe sono più lunghe della norma (Ratcliffe S,
1999; Schibler D et al., 1974).
Durante la pubertà, dopo un iniziale aumento,
le concentrazioni di testosterone raggiungono un
plateau in un range medio-basso che rimane costante; contemporaneamente i livelli di LH e FSH
aumentano gradualmente e, non essendo i caratteri sessuali secondari completamente sviluppati,
possono manifestarsi eunucoidismo e ginecomastia, con incidenza compresa tra il 56% e l’88%
(Wikström AM et al., 2011; Robinson A et al.,
1991).
I pazienti adulti affetti da SK sono caratterizzati
da ipogonadismo ipergonadotropico: le concentrazioni di LH e FSH sono elevate e le concentrazioni di testosterone nel 65-85% dei casi sono
inferiori ai valori normali (Wikström AM et al.,
2011). Il volume testicolare è in genere inferiore
a 10 ml nel periodo postpuberale. La maggior parte dei pazienti con SK non sono fertili anche se
sono descritti pazienti affetti da forme a mosaico
che hanno procreato in assenza di fecondazione
assistita (Smyth CM et al., 1998).
Il quoziente intellettivo è estremamente varia-
bile, da valori inferiori a valori superiori alla media. I risultati dei test Wechsler Intelligence evidenziano frequentemente un QI verbale inferiore
al QI di performance; tale discrepanza sembra essere causata da deficit nelle capacità verbali e nella
memoria uditiva (Rovet J et al., 1995). Deficit
nello sviluppo del linguaggio e nelle capacità
verbali possono evidenziarsi durante il periodo
scolare, in particolare nei casi 47,XX; a 7 anni il
bambino può avere problemi medio-gravi con la
lettura, l’articolazione delle parole, la narrazione,
la scrittura; problemi con la matematica si manifestano più tardi (Graham JM et al., 1988). La personalità dei soggetti con SK è variabile. Alcuni
sono timidi, immaturi, riservati, con difficoltà a
relazionarsi con i coetanei; altri amichevoli, gentili
ed in grado di avere buoni rapporti con gli altri; la
maggior parte si descrive come sensibile, apprensivo e insicuro. In adolescenti con SK è segnalata
un’incidenza di ansia e depressione maggiore rispetto a coetanei sani (Bender BG et al., 1995).
Il rischio di sviluppare il carcinoma mammario
nei soggetti adulti con SK è superiore di 20-50
volte rispetto alla popolazione generale (GómezRaposo C et al., 2010; Swerdlow AJ et al., 2005).
Patologie endocrine (diabete mellito, ipotiroidismo, ipoparatiroidismo) e patologie autoimmuni (lupus eritematoides sistemico, sindrome
www.buponline.com
56
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
di Sjogren, artrite reumatoide) sono di riscontro
relativamente frequente (Hsueh WA, 1978).
In età adulta sono frequenti vene varicose e ulcere alle gambe da stasi venosa (Campbell WA et
al., 1981).
La densità ossea è ridotta nel 25% dei pazienti
con SK (Campbell WA et al., 1981).
CARATTERISTICHE ORO-FACCIALI
Anomalie dentarie di dimensioni, di forma e di
eruzione sono descritte nei soggetti con SK.
Le dimensioni coronali sono mediamente superiori a quelle dei soggetti con cariotipo normale (Townsend GC et al., 1985; Alvesalo L et al.,
1980). In particolare, sono aumentati gli spessori
dello smalto degli incisivi centrali superiori e dei
canini permanenti; al contrario, gli spessori della dentina risultano inferiori rispetto a quelli di
maschi sani ma superiori rispetto a quelli delle
femmine (Sćepan I et al., 1993). Tali differenze
portano a ipotizzare che i cromosomi sessuali giochino un ruolo sulla crescita e sullo sviluppo dei
tessuti mineralizzati del dente: il cromosoma X
interverrebbe nella deposizione dello smalto e il
cromosoma Y nella deposizione sia dello smalto
che della dentina.
In soggetti con SK è descritta un’aumentata lunghezza delle radici dei denti permanenti (in particolare di molari e premolari) per incremento della crescita del terzo apicale (Lahdesmaki R et al.,
2007). È descritta un’alta prevalenza di taurodontismo (40%) (Figura 2) anche in molari decidui,
e di incisivi di forma “a pala” (shovel-shaped), in
particolare nelle varianti più severe della sindrome
(cariotipi 49,XXXXY e 48,XXXY) (Lia EN et al.,
2007; Hunter ML et al., 2003; Darbyshire PA et
al., 1989; Jaspers MT et al., 1980).
In pazienti con SK sono riportati casi di eruzione
ritardata degli elementi dentari della serie deci-
Figura 2: Bambino di 8 anni affetto da SK; ortopantomografia: detizione mista, primi molari permanenti di aspetto taurodontico, affollamento endosseo superiore e inferiore
di grado elevato con tendenza all’inclusione di 13 e 23
dua, confermando come i cromosomi sessuali giochino un ruolo nello sviluppo dentale (D’Alessandro et al., 2012).
Nei soggetti affetti da SK è riportata un’elevata
prevalenza di carie e di parodontopatie (PalinPalokas T et al., 1990; Vaisanen P et al., 1989).
Dal punto di vista ortopedico-ortodontico è
descritta un’elevata prevalenza di malformazioni a carico delle ossa craniche e dei mascellari:
dimensioni craniche diminuite, angolo della base
cranica diminuito, angolo goniaco aumentato
(Hata S et al., 2001; Ingerslev CH et al., 1978).
Nei pazienti affetti da SK il palato è caratterizzato
da aumentata dimensione anteroposteriore e da
ridotte dimensioni trasversale e verticale; la mandibola si presenta stretta, lunga e post-ruotata (Figura 3a, b, c). È riportata un’elevata prevalenza
di III classe dentale e di morso aperto anteriore
(Alvesalo L et al., 1993; Alvesalo L et al., 1992).
LINEE GUIDA DI TERAPIA
I pazienti con SK sono affetti da numerose alterazioni e patologie che rendono necessari interventi
multispecialistici. La diagnosi, le terapie e il follow-up richiedono la presenza di un team multi
www.buponline.com
57
Sindrome di Klinefelter
a
b
c
Figura 3 a, b, c: Morso crociato monolaterale sinistro,
morso coperto anteriore e affollamento inferiore in sede
incisiva. Bihelix inferiore in sede per espansione arcata
inferiore
e interdisciplinare adeguatamente formato sulla
sindrome.
La terapia sostitutiva ormonale con testosterone viene iniziata durante la pubertà (intorno ai 12
anni), aumentando progressivamente i dosaggi per
ottenere le concentrazioni sieriche adeguate all’età. La terapia sostitutiva porta ad aumento della
mascolinità, della forza, della libido, della densità
ossea, dei peli del corpo, determina una distribuzione più mascolina del tessuto adiposo e ha un
effetto positivo sull’umore e sul comportamento,
migliorando la capacità di concentrazione e le relazioni sociali; non ha tuttavia effetti sulla fertilità.
In caso di ginecomastia può essere indicata la terapia chirurgica.
In caso di deficit di acquisizione del linguaggio è
utile impostare un programma educativo dedicato.
Per quanto concerne le patologie di pertinenza
odontoiatrica, in relazione all’elevata prevalenza di patologie cariosa e parodontale descritta in
letteratura, è necessario, dal momento in cui è
formulata la diagnosi, promuovere la salute orale attraverso interventi di prevenzione primaria
mirati, con particolare riferimento all’alimentazione non cariogenica, all’igiene orale domiciliare, all’utilizzo di fluoro topico e quando indicato
sistemico, alle visite periodiche, alla sigillatura dei
solchi e delle fessure e dei fori ciechi. La sigillatura
è particolarmente indicata a livello dei taurodonti, caratterizzati da spessori dei tessuti duri diminuiti rispetto al normale e quindi da più precoce
coinvolgimento del tessuto pulpare da parte della
patologia cariosa (Joseph M, 2008; Yeh S, 1999).
Dalla letteratura si rileva la tendenza nei pazienti
con SK ad adottare soluzioni estrattive rispetto a
terapie conservative quando le lesioni cariose interessano elementi taurodonti, dal momento che
presentano una particolare anatomia del sistema
canalare che rende difficoltosa l’esecuzione di un
trattamento endodontico corretto (Palin-Palokas
T et al., 1990; Vaisanen P et al., 1989).
www.buponline.com
58
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
In presenza di malocclusioni è indicato il trattamento ortopedico-ortodontico se la collaborazione del paziente lo consente, in particolare per
quanto riguarda il mantenimento dell’igiene orale
domiciliare.
BIBLIOGRAFIA
Alvesalo L, Laine T. Palatal and alveolar arch dimensions in 47,XXY (Klinefelter syndrome)
men. Hum Biol. 1993;65:131-8
Alvesalo L, Laine T. Occlusion in 47,XXY (Klinefelter syndrome) men. Am J Phys Anthropol.
1992;87:161-5
Alvesalo L, Portin P. 47,XXY males: Sex chromosomes and tooth size. Am J of Hum Gen.
1980;32:955–9
Bender BG, Harmon RJ, Linden MG. Psychosocial adaptation of 39 adolescents with
sex chromosome abnormalities. Pediatrics.
1995;96:302-8
Bojsen A, Juul S, Højbjerg Gravholt C. Prenatal
and postnatal prevalence of Klinefelter syndrome: a national registry study. J Endocrinol
Metab. 2003;88:622-6
Bojesen A, Gravholt CH. Morbidity and mortality in Klinefelter syndrome (47,XXY). Acta
Paediatrica. 2011;100:807-13
Caldwell PD, Smith DW. The XXY (Klinefelter’s)
syndrome in childhood: detection and treatment. J Pediatr. 1972;80:250-8
Campbell WA, Price WH. Venous thromboembolic disease in Klinefelter’s syndrome. Clin
Genet. 1981;19:275-80
D’Alessandro G, Armuzzi L, Cocchi G, Piana
G. Eruption delay in a 47 XXY male: a case
report. Eur J Paediatr Dent. 2012;13:15960
Darbyshire PA, Witkop CJ Jr, Cervenka J. Prepubertal diagnosis of Klinefelter syndrome in a
patient with taurodontic teeth. Pediatr Dent.
1989;11:224-6
Gómez-Raposo C, Zambrana Tévar F, Sereno
Moyano M, López Gómez M, Casado E. Male
breast cancer ( Review ). Cancer Treatment
Reviews. 2010;36,451-7
Graham JM Jr, Bashir AS, Stark RE, Silbert A,
Walzer S. Oral and written language abilities
of XXY boys: implications for anticipatory
guidance. Pediatrics. 1988;81:795-806
Hager K, Jennings K, Hosono S, Howell S, Gruen
JR, Rivkees SA, Tartaglia NR, Rinder HM.
Molecular diagnostic testing for Klinefelter syndrome and other male sex chromosome aneuploidies. Int J Pediatr Endocrinol. 2012;2012:8
Hata S, Maruyama Y, Fujita Y, Mayanagi H. Dentofacial manifestation of XXXXY syndrome.
Int J Ped Dent. 2001;11:138-42
Hunter ML, Collard MM, Razavi T, Hunter B.
Increased primary tooth size in a 47,XXY
male: a first case report. Int J Paed Dent.
2003;13:271–3
Hsueh WA, Hsu TH, Federman DD. Endocrine
features of Klinefelter’s syndrome. Medicine.
1978;57:447-61
Ingerslev CH, Kreiborg S. Craniofacial morphology in Klinefelter syndrome: a roentgencephalometric investigation. Cleft Palate
Journal. 1978;15:100–8
Jacobs PA, Strong JA. A case of human intersexuality having possible XXY sex-determining mechanism. Nature. 1959;2:164-7
Jaspers MT, Witkop CJ. Taurodontism, an isolated trait associated with syndromes and XChromosome aneuploidy. American Journal
of Human Genetics. 1980;32:396-413
Joseph M. Endodontic treatment in three taurodontic teeth associated with 48,XXXY Klinefelter syndrome: a review and case report.
Oral Surg Oral Med Oral Pathol Oral Radiol
Endod. 2008;105:670-7
www.buponline.com
59
Sindrome di Klinefelter
Klinefelter HF, Reifenstein EC, Albright F.
Syndrome characterized by gynecomastia aspermatogenes without A-Leydigism
and increased excretion of follicle stimulating hormone. J Clin Endocrinol Metab.
1942;2:615-27
Lahdesmaki R, Alvesalo L. Root lengths in the
permanent teeth of Klinefelter (47XXY) men.
Archives of Oral Biology. 2007;52:822-7
Lia EN, Otero SA, Ferraz M, Gonalves LP: Oral
aspects of 49,XXXXY syndrome: a case report.
J Dent Child. 2007;74:136-9
Linden MG, Bender BG, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics.
1995;6:672-82
Nielsen J, Wohlert M. Chromosome abnormalities found among 34,910 newborn children:
results from a 13-year incidence study in
Arhus, Denmark. Hum Genet. 1991;87:81-3
Okada H, Fujioka H, Tatsumi N, Kanzaki M,
Okuda Y, Fujisawa M, Hazama M, Matsumoto O, Gohji K, Arakawa S, Kamidono S.
Klinefelter’s syndrome in the male infertility
clinic. Hum Reprod. 1999;14:946-52
Paduch DA, Fine RG, Bolyakov A, Kiper J. New
concepts in Klinefelter syndrome. Curr Opin
Urol. 2008;18:621-7
Palin-Palokas T, Alvesalo L, Takala I, Paunio K,
Suoranta K, Varrela J. Caries occurrence in
Klinefelter syndrome men (47,XXY males).
Proc Finn Dent Soc. 1990;86:143-7
Ratcliffe S. Long-term outcome in children of sex
chromosome abnormalities. Arch Dis Child.
1999;80:192-5
Robinson A, Bender BG, Linden MG. Summary of clinical findings in children and young
adults with sex chromosome anomalies. Birth
Defects. 1991;26:225-8
Rovet J, Netley C, Bailey J, Keenan M, Stewart D.
Intelligence and achievement in children with
extra X aneuploidy: a longitudinal perspective.
Am J Med Genet. 1995;60:356-63
Šćepan I, Babić M, Glišić B. The mesiodistal dimension of permanent teeth in men
with Klinefelter’s syndrome. Eur J Orthod.
1993;15:195-7
Schibler D, Brook CG, Kind HP, Zachmann M,
Prader A. Growth and body proportions in
53 boys and men with Klinefelter’s syndrome.
Helv Paediat Acta. 1974;29:325-33
Smyth CM, Bremner WJ. Klinefelter syndrome.
Arch Intern Med. 1998;158:1309-14
Swerdlow AJ, Schoemaker MJ, Higgins CD,
Wright AF, Jacobs PA; UK Clinical Cytogenetics Group. Cancer incidence and mortality in
men with Klinefelter syndrome: a cohort study. J Natl Cancer Inst. 2005;97:1204-10
Townsend GC, Alvesalo L. The size of permanent
teeth in Klinefelter (47,XXY) syndrome in
men. Archives of Oral Biology. 1985;30:83–4
Vaisanen P, Takala I, Alvesalo L, Markkanen H.
Periodontal health in 47,XXY men (Klinefelter syndrome). Proc Finn Dent Soc.
1989;85:441-4
Visootsak J, Graham JM. Klinefelter syndrome
and other sex chromosomal aneuploidies.
Orphanet Journal of Rare Diseases. 2006;1:42
Yeh S, Hsu T. Endodontic treatment in taurodontism with Klinefelter’s syndrome: a case report.
Oral Surg Oral Med Oral Pathol Oral Radiol
Endod. 1999;88:612-5
Walzer S, Wolff PH, Bowen D, Silbert AR, Bashir
AS, Gerald PS, Richmond JB. A method for
longitudinal study of behavioral development
in infants and children: the early development
of XXY children. J Child Psychol Psychiat.
1978; 9:213-22
Wikström AM, Dunkel L. Klinefelter syndrome.
Best Practice and Research. Clinical Endocrinology and Metabolism. 2011;25:239-50
www.buponline.com
www.buponline.com
5. Sindrome da delezione 22q11.2
Sinonimi: Sindrome di DiGeorge, Sindrome di Shprintzen,
Sindrome di Sedlackova, Sindrome di Cayler. Sindrome di
Takao, Sindrome Velo-Cardio-Facciale, CATCH 22, Anomalie
conotroncali e della faccia - Codice ICD 10: D82.1
S. Bagattoni, N. Al Khamis, T. Tagariello, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Sindrome da delezione 22q11.2 (Sd22q11.2)
è una malattia cromosomica rara caratterizzata
dall’associazione di cardiopatie congenite, ipoplasia del timo e delle paratiroidi, facies caratteristica e difficoltà di apprendimento (Goldmuntz
E, 2005).
La Sd22q11.2 è conosciuta anche con altri nomi:
sindrome di DiGeorge, sindrome Velo-CardioFacciale (VCF) e CATCH 22. Nel 1965 il pediatra Angelo DiGeorge ha descritto una sindrome
caratterizzata da immunodeficienza da alterato
sviluppo del timo, ipocalcemia da alterato funzionamento delle paratiroidi e cardiopatie congenite.
Nel 1978 il foniatra Robert Shprintzen ha descritto
una sindrome caratterizzata da anomalie del palato, voce nasale, cardiopatie congenite, difficoltà
di apprendimento e aspetto del volto caratteristico, definendola sindrome Velo-Cardio-Facciale.
Nei primi anni ’90 si è scoperto che la stessa alterazione cromosomica, la microdelezione a livello della banda 22q11 localizzata sul braccio lungo
del cromosoma 22, è responsabile di entrambe le
sindromi. Nel 1993 un gruppo di ricercatori inglesi ha proposto di denominare la sindrome con
l’acronimo CATCH 22 (C: Cardiac anomaly, A:
Anomalous face, T: Thymic hypoplasia, C: Cleft
palate, H: Hypocalcaemia, 22: chromosome number 22). Anche se la denominazione CATCH 22
è stata ampiamente utilizzata, sindrome da delezione 22q11.2 è attualmente quella più in uso
(Shprintzen RJ, 2008; Wilson DI et al., 1993).
L’incidenza è stimata di 1 su 4.000 nati vivi per
anno in assenza di predilezione razziale, etnica,
geografica e di genere. In pazienti con cardiopatie
congenite conotroncali la sua prevalenza è del 1550% e in persone con anomalie del palato è del
75% (Oskarsdottir S et al., 2004; Goodship J et
al., 1998; Tezenas Du Montcel ST et al., 1996).
Il decorso clinico è determinato dalla tipologia delle
malformazioni congenite e dalla gravità delle loro
manifestazioni cliniche, entrambe estremamente
eterogenee (Shprintzen RJ, 2008; Oskarsdottir S
et al., 2004; Goodship J et al., 1998; Tezenas Du
Montcel ST et al., 1996).
I pazienti affetti presentano un indice di mortalità
durante l’infanzia del 4% (McDonald-McGinn
DM et al., 2001) e in età adulta più elevato rispetto alla restante popolazione (Bassett AS et al.,
2009).
www.buponline.com
62
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
GENETICA E DIAGNOSI
La microdelezione a livello della banda q11.2
localizzata sul braccio lungo del cromosoma 22,
responsabile della mancata espressione del gene
TBX1 regolatore della adesione epiteliale e dello
sviluppo del palato, causa anomalie della morfogenesi, in particolare degli archi branchiali. Il difetto
embriogenetico, che si realizza tra la quarta e la sesta
settimana di gestazione, è responsabile di anomalie
delle strutture che derivano dal terzo e dal quarto
arco branchiale: cardiopatie, ipoplasia del timo,
ipoplasia/aplasia delle paratiroidi, dismorfismi facciali (Mesbah K et al., 2012; Kobrynski LJ et al.,
2007; Yagi H et al., 2003; Kelley RI et al., 1982; de
la Chapelle A et al., 1981). La penetranza genetica è
incompleta, comportando una variabilità di espressione fenotipica (Meinecke P et al., 1986).
Nel 90% dei casi la Sd22q11.2 è causata da una
mutazione ex novo e nel 10% è trasmessa secondo una modalità autosomica dominante da uno
dei genitori portatore della microdelezione (Bassett
AS et al., 2008; Shprintzen RJ, 2008; McDonaldMcGinn et al., 2001).
Per la diagnosi della microdelezione la tecnica
più affidabile ed utilizzata è l’ibridizzazione fluorescente in situ (FISH TEST); di più recente introduzione sono test di biologia molecolare quali
il Microarrays e il Multiplex Ligation Probe Assay
(Shprintzen RJ, 2008; Yakut T et al., 2006).
La diagnosi prenatale è possibile su villocentesi e
amniocentesi evidenziando la microdelezione; la
ricerca è indicata in presenza all’ecografia di malformazioni cardiache e del palato e di anomalie
del timo (Bataeva R et al., 2012; Bretelle F et al.,
2010).
MANIFESTAZIONI CLINICHE SISTEMICHE
Il quadro clinico della Sd22q11.2 può essere caratterizzato da numerosissime manifestazioni cliniche
e comportamentali elencate nella tabella 1 (Bassett
AS et al., 2011).
Le cardiopatie congenite, presenti nel 50%-70%
dei casi, si manifestano clinicamente nei primi
giorni di vita e spesso rappresentano la manifestazione clinica che porta alla diagnosi. Le più frequenti sono le anomalie conotroncali: interruzione
dell’arco aortico (>50%), tronco arterioso persistente (40%), difetti ventricolari settali (30%), tetralogia di Fallot (15%) (Bassett AS et al., 2011).
In pazienti con Sd22q11.2 sono riportate anche
severe anomalie delle arterie polmonari e dell’arco aortico (Momma K, 2010; Goldmuntz E et
al., 1998; Ryan AK et al., 1997; Wilson DI et al.,
1993).
Il timo può essere ipoplasico o aplasico, provocando un deficit immunitario da deficit e da alterata
funzionalità dei linfociti T, responsabile di infezioni ricorrenti che interessano prevalentemente
il tratto respiratorio superiore e l’orecchio medio;
bronchiti, bronchioliti, otiti, rinofaringiti e sinusiti
intercorrenti sono frequenti in età prescolare ma
generalmente scompaiono con l’età (Chaoui R et
al., 2002; Ford LC et al., 2000; Ryan AK et al.,
1997).
L’ipoparatiroidismo in un’elevata percentuale dei
casi (60%) è causa di ipocalcemia, responsabile
di convulsioni, che si manifestano soprattutto alla
nascita, e di difetti di mineralizzazione dei tessuti
duri, ossa e denti (Bassett AS et al., 2011; Hieronimus S et al., 2006; Choi JH et al., 2005; Ryan AK
et al., 1997).
L’ipotiroidismo (20%) è una delle possibili cause
di obesità in età adulta, presente nel 35% dei pazienti (Bassett AS et al., 2011).
Il deficit di secrezione dell’ormone della crescita
(GH) può essere responsabile durante l’infanzia e
l’adolescenza di una curva di crescita rallentata e di
un’altezza inferiore rispetto alla media (36%) e in
età adulta di bassa statura (20%) (Bassett AS et al.,
2011; Choi JH et al., 2005; Ryan AK et al., 1997).
www.buponline.com
63
Sindrome da delezione 22q11.2
Tabella 1: Manifestazioni cliniche in soggetti affetti da Sd22q11
Fenotipo clinico
Caratteri dismorfici
Anomalie congenite multiple
Difficoltà di apprendimento/ritardo dello sviluppo
Polidramnios
Cardiopatie Conotroncali:
interruzione dell’arco aortico
tronco arterioso persistente
difetti ventricolari settali
teratologia di Fallot
Naso lungo/grosso/a bulbo
Orecchie a impianto basso
Retrognazia/micrognazia
Insufficienza velofaringea ± schisi palatina sottomucosa
Voce nasale e/o rigurgito nasale
Schisi palatina
Anomalie dell’orecchio
Otite media cronica
Ipoacusia trasmissiva e/o neurosensoriale
Infezioni ricorrenti
Alterata funzionalità e deficit di linfociti T
Patologie autoimmuni
Ipotiroidismo
Ipertiroidismo
Ipocalcemia e/o ipoparatiroidismo
1. Caratteristiche genetiche
2. Malformazioni
cardiovascolari (50-70%).
Cardiopatie
congenite
3. Anomalie del
distretto cefalico
4. Anomalie del palato
e strutture adiacenti (75%)
Patologie
otorinolaringoiatriche
5. Immunodeficit
6. Patologie endocrine
Tiroide
Paratiroide
Ghiandola ipofisaria
7. Patologie
gastrointestinali
8. Anomalie del sistema
genito-urinario
9. Patologie oculari
10. Anomalie scheletriche
Obesità negli adulti
Bassa statura
Reflusso gastroesofageo
Disfagia
Colelitiasi
Anomalie strutturali del tratto urinario
Reflusso vescico-ureterale
Agenesia renale unilaterale
Displasia renale multicistica
Strabismo
Scoliosi
Anomalie spina cervicale e vertebre toraciche
www.buponline.com
64
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
11. Patologie ematologiche
12. Patologie neurologiche
13.Alterazioni della
crescita e dello sviluppo
14.Patologie
neuropsichiatriche
15. Altre
Respiratorie
Dermatologiche
Dentali
Caratteristiche sono le alterazioni dello sviluppo psicomotorio: ritardo dello sviluppo motorio
e della fonazione (>90%); difficoltà di apprendimento (>90%), ritardo mentale (35%) (Bassett AS
et al., 2011); il deficit cognitivo all’età di 5,5 anni
risulta essere maggiore nei maschi rispetto alle femmine (Duijff SN et al., 2012).
Alterazioni comportamentali sono molto frequenti. In particolare, nei bambini sindrome da
deficit di attenzione e iperattività (35-55%) e disordini dello spettro dell’autismo (14%); nell’adolescenza e in età adulta schizofrenia (>20%). Sono
descritte altre patologie psichiatriche: nei bambini
disturbo bipolare, depressione e ansietà; negli adulti disturbi ossessivo-compulsivi (Philip N et al.,
2011; Gothelf D et al., 2004).
CARATTERISTICHE ORO-FACCIALI
I soggetti affetti da Sd22q11 presentano una facies caratteristica con ipertelorismo, rime palpebrali strette e ruotate in basso, impianto delle
orecchie basso con padiglioni auricolari picco-
Trompocitopenia
Splenomegalia
Convulsioni ipocalcemiche ricorrenti
Crisi epilettiche
Ritardo dello sviluppo motorio e della fonazione
Difficoltà di apprendimento
Ritardo mentale
Sindrome da deficit di attenzione e iperattività
Disordini dello spettro dell’autismo
Ansia e depressione
Schizofrenia e altre psicosi
Patologie respiratorie non-infettive
Seborrea, dermatiti
Acne severa
Ipomineralizzazioni/ipoplasie dello smalto
Lesioni cariose
li, ponte nasale basso, naso prominente, bocca
piccola con labbra spesse (Figura 1 a, b). Altre
alterazioni craniofacciali caratteristiche sono ipoplasia malare, retrognazia dei mascellari, rotazione
posteriore della mandibola (Wentzel C et al., 2008;
Heliovaara A et al., 2006; Lipson AH et al., 1991;
Arvystas M et al., 1984).
L’insufficienza velofaringea (32%) è responsabile
nel neonato di difficoltà di suzione e di deglutizione e di fuoriuscita di latte dal naso e, in epoca successiva, di alterazioni della fonazione e di timbro di
voce nasale (Ryan AK et al., 1997).
Anomalie del palato, schisi palatina (9%) e sottomucosa (5%), sono responsabili di difficoltà di
suzione e di deglutizione, di rigurgito dal naso e
di disturbi del linguaggio e del sonno e predispongono all’insorgenza di otiti ricorrenti (Bassett AS
et al., 2011; Shprintzen RJ, 2008; Shprintzen RJ,
2000; Ryan AK et al., 1997; Shprintzen RJ, 1982;
Shprintzen RJ, 1978).
In pazienti con Sd22q11.2 sono descritte anomalie dentali di eruzione, numero, forma e struttura.
È descritta eruzione ritardata della dentatura permanente (Klingberg G et al., 2010; Klingberg G et
www.buponline.com
65
Sindrome da delezione 22q11.2
a
b
Figura 1 a, b: Bambina di 9 anni affetta da Sd22q11
al., 2002). Le alterazioni di numero più frequenti
sono le agenesie degli incisivi inferiori (9%); è presente una elevata correlazione tra ipodonzia e schisi
palatine (Heliovaara A et al., 2011). È descritta la
presenza di un unico incisivo centrale mascellare
o mandibolare (Oberoi S et al., 2005; Yang et al.,
2005). Per quanto concerne le anomalie di forma
sono descritti denti con cuspidi sovrannumerarie
(secondi premolari inferiori) e con cuspidi iposviluppate (primi premolari inferiori), denti conoidi,
denti con ridotto diametro mesio-distale (da Silva Dalben G et al., 2008). Per quanto concerne
le anomalie di struttura, sono descritte ipomineralizzazioni (11.2 % dei decidui e 24.6% dei permanenti) e ipoplasie (9.1% dei decidui e 6.2% dei
permanenti), fattori di rischio per la patologia cariosa (Klingberg G et al., 2005). Le potenziali cause delle anomalie dello smalto sono ipocalcemia,
prematurità, cardiopatie congenite e infezioni frequenti nell’infanzia (Nordgarden H et al., 2012).
Caratteristiche nei soggetti affetti da Sd22q11 sono
alterazioni della saliva sotto il profilo quantitativo e qualitativo. La secrezione salivare è inferiore
(0.71 ml/min) rispetto alla popolazione generale
(1.48 ml/min). La saliva presenta una concentrazione maggiore di proteine e di IgA e una minore di ioni calcio, fosfati e bicarbonato rispetto alla
popolazione generale. La ridotta quantità di calcio
e fosfati determina una ridotta capacità di rimineralizzazione delle lesioni cariose iniziali; la ridotta
quantità di bicarbonato determina un pH tendenzialmente acido, responsabile di un elevato rischio
di carie (Klingberg G et al., 2007).
Per quanto riguarda le patologie ortopedicoortodontiche, nei soggetti con Sd22q11.2 uno
dei riscontri più caratteristici è la faccia lunga
con profilo a convessità aumentata. L’elevata
prevalenza di respirazione orale è responsabile di
un pattern di crescita verticale, di un aumento
dell’altezza facciale anteriore e della post-rotazione
mandibolare. In particolare, nei pazienti con schi-
www.buponline.com
66
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
si palatina fattori predisponenti all’instaurarsi di
respirazione orale sono il ridotto diametro trasversale del mascellare superiore e la retrazione cicatriziale conseguente a pregressi interventi chirurgici di correzione dell’incompetenza velo-faringea
(Heliovaara A et al., 2006). Dal punto di vista
ortodontico sono descritti quadri di malocclusione scheletrica di II classe con morso aperto,
la cui eziopatogenesi è riconducibile a retrusione
mandibolare, riduzione della dimensione verticale posteriore e aumento dell’angolo goniaco, e di
malocclusione dentale di II classe molare e canina con angolo interincisivo aperto o con linguoinclinazione degli incisivi inferiori (da Silva Dalben
G et al., 2010) (Figura 2).
Figura 2: Malocclusione di II classe, morso profondo, linguo-inclinazione degli incisivi superiori
LINEE GUIDA DI TERAPIA
I pazienti con Sd22q11 sono affetti da numerose
alterazioni e patologie che rendono necessari interventi multispecialistici. La diagnosi, il follow-up e
le terapie richiedono la presenza di un team multi
e interdisciplinare adeguatamente formato sulla
sindrome.
L’approccio terapeutico varia in base alle manifestazioni cliniche dei singoli pazienti.
La diagnosi delle patologie cardiache viene fatta
in età pediatrica. Il tipo e i tempi degli interventi
cardiochirurgici, che spesso si effettuano in tempi
differiti, vengono decisi in base al tipo e alla gravità
della patologia; la prognosi è buona, ad eccezione
dei casi di stenosi/atresia dell’arteria polmonare; in
alcuni casi residua il rischio di endocardite batterica (Michielon G et al., 2006).
Le infezioni gravi sono rare nei soggetti affetti da
Sd22.q11.2; è comunque necessario il controllo
periodico del sistema immunitario dalla nascita,
in particolare prima delle vaccinazioni con vaccini
vivi (Bassett AS et al., 2011).
Le funzioni della tiroide e le ghiandole paratiroidi
devono essere monitorate. Il trattamento dell’ipocalcemia sintomatica grave, quasi sempre esclusiva
del periodo neonatale, richiede la somministrazione di calcio per via parenterale; l’ipocalcemia
asintomatica è corretta con una supplementazione
di calcio per via orale (Bassett AS et al., 2011).
Nei casi di documentato deficit di secrezione
dell’ormone della crescita (GH) deve essere instaurata la terapia sostitutiva (Bassett AS et al., 2011).
Nei casi di insufficienza velo-faringea particolare attenzione deve essere rivolta ai problemi di
alimentazione: il bambino deve essere tenuto in
posizione eretta durante l’allattamento e debbono
essere utilizzati biberon con tettarelle con fori ampi
per aumentare il flusso del latte; nei casi più gravi è
indicata l’alimentazione con sondino nasogastrico
(Shprintzen RJ, 2008).
Nei soggetti con schisi palatine, per facilitare l’alimentazione e la respirazione, è indicato nei primi
giorni di vita l’allestimento di otturatori palatini,
in attesa dell’intervento chirurgico, effettuato a circa 6 mesi (Tatum SA et al., 2002). Per la correzione
dei problemi di fonazione spesso è necessario un
intervento chirurgico di faringoplastica. L’ipoacusia sia trasmissiva che neurosensoriale necessita di
una diagnosi e terapia precoce al fine di prevenire
www.buponline.com
67
Sindrome da delezione 22q11.2
ritardo del linguaggio e difficoltà di apprendimento (Bassett AS et al., 2011).
La terapia delle patologie psichiatriche e del ritardo di sviluppo, caratteristici della Sd22q11.2,
sta ricevendo molta attenzione; per la sindrome da
deficit di attenzione e iperattività è utilizzato il metilfenidato (Gothelf D et al., 2003).
Per quanto concerne le patologie odontostomatologiche in un’ottica di interdisciplinarietà, è compito
del pediatra, dal momento in cui si effettua la diagnosi, inviare il paziente all’odontoiatra infantile perché informi la famiglia sull’importanza della salute
orale e sulla necessità di interventi di prevenzione.
Per quanto concerne la patologia cariosa, essendo
i pazienti affetti da Sd22q11 ad elevato rischio di
patologia cariosa e, quando affetti da cardiopatia
congenita, a rischio di Endocardite Batterica (EB), è
necessario attuare, dal momento in cui viene formulata la diagnosi, interventi di prevenzione primaria
mirati, con particolare riferimento all’alimentazione
non cariogenica, all’igiene orale domiciliare, all’utilizzo di fluoro topico e quando indicato sistemico,
alle visite periodiche trimestrali/semestrali, alla sigillatura di solchi, fessure e fori ciechi.
In relazione alla possibile presenza di agenesie,
all’età di 7-9 anni viene eseguita una ortopantomografia delle arcate dentali.
Nei pazienti a rischio di EB le terapie responsabili
di batteriemia devono essere precedute da profilassi antibiotica (vedi capitolo 2).
In letteratura non sono presenti dati relativi al trattamento ortopedico-ortodontico nei soggetti con
Sd22q11. La presenza di patologie ortopedicoortodontiche (retrusione mandibolare, morso
aperto) e una alterata crescita del mascellare superiore in presenza di schisi impongono l’intercettazione tempestiva e precoce delle malocclusioni
scheletriche e/o dentali. Le discrepanze trasversali, caratteristiche dei soggetti con palatoschisi, vengono corrette in dentizione mista o con espansore
rapido del palato tipo hi-rax, nei casi che necessita-
no di espansione parallela, o con espansore rapido
a ventaglio, soprattutto nelle lps bilaterali che richiedono un’espansione maggiore nei settori anteriori (Meazzini et al., 2001). Per la correzione delle
II Classi possono essere utilizzati apparecchi miofunzionali, quale il Twin-Bolck. In età adulta, al
fine di migliorare l’estetica facciale, per correggere
la retrusione mandibolare è possibile ricorrere alla
chirurgia ortognatica.
Per quanto concerne le lesioni dentali di origine
traumatica, pur non esistendo in letteratura alcun
riferimento specifico, considerando il deficit di attenzione e l’iperattività, è ipotizzabile una loro elevata prevalenza a livello degli elementi sia decidui
che permanenti; è quindi necessario fornire alla
famiglia linee guida per la loro gestione, in particolare per quanto concerne il reimpianto immediato
di elementi permanenti avulsi.
BIBLIOGRAFIA
Arvystas M, Shprintzen RJ. Craniofacial morphology in the velo-cardio-facial syndrome. J Craniofac Genet Dev Biol. 1984;4:39-45
Bassett AS, Marshall CR, Lionel AC, Chow EWC,
Scherer SW. Copy number variations and risk
for schizophrenia in 22q11.2 deletion syndrome. Hum Mol Genet. 2008;17:4045-53
Bassett AS, Chow EWC, Husted J, Hodgkinson
KA, Oechslin E, Harris L. Premature death in
adults with 22q11.2 deletion syndrome. J Med
Genet. 2009;46:324-30
Bassett AS et al. Practical guidelines for managing
patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159:332-442
Bataeva R, Bellsham-Revell H, Zidere V, Allan
LD. Reliability of fetal thymus measurement in
prediction of 22q11.2deletion: a retrospective
study using 4D STIC volumes. Ultrasound
Obstet Gynecol. 2013;41:172-6
www.buponline.com
68
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Bretelle F, Beyer L, Pellissier MC, Missirian C,
Sigaudy S, Gamerre M, D’Ercole C, Philip N. Prenatal and postnatal diagnosis of
22q11.2 deletion syndrome. Eur J Med Genet.
2010;53:367-70
Chaoui R, Kalache KD, Heling KS, Tennstedt C,
Bommer C, Körner H. Absent or hypoplastic
thymus on ultrasound: a marker for deletion
22q11.2 in fetal cardiac defects. Ultrasound
Obstet Gynecol. 2002;20:546-52
de la Chapelle A, Herva R, Koivisto M, Aula P.
A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet. 1981;57:2536
Choi JH, Shin YL, Kim GH, Seo EJ, Kim Y, Park
ES, Yoo HW. Endocrine manifestations of
chromosome 22q11.2 microdeletion. Horm
Res. 2005;63:294-9
da Silva Dalben G, Richieri-Costa A, de Assis Taveira LA. Craniofacial morphology in patients
with velocardiofacial syndrome. The Cleft Palate-Craniofacial J. 2010; 47:241-6
da Silva Dalben G, Richieri-Costa A, de Assis Taveira LA. Tooth abnormalities and soft tissue
changes in patients with velocardiofacial syndrome. Oral Surg Oral Med Oral Pathol Oral
Radiol Endod. 2008;106:46-51
Duijff SN, Klassen PWJ, Swanenburg de Veye
HFN, Beemer FA, Sinnema G, Vortsman
JAS. Cognitive development in children with
22q11.2 deletion syndrome. Br J Psychiatry.
2012;200:462-68
Ford LC, Sulprizio SL, Rasgon BM. Otolaryngological manifestations of velocardiofacial syndrome: a retrospective review of 35 patients.
Laryngoscope. 2000;110:362-7
Goldmuntz E. DiGeorge syndrome: new insights.
Clin Perinatol. 2005;32:963-78
Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in
infancy. Arch Dis Child. 1998;79:348-51.
Gothlef D, Gruber R, Presburger G, et al. Methylphenidate treatment for attention-deficit/hyperactivity disorder in chidren and adolescents
with velocardiofacial syndrome: an open-label
study. J Clin Psychiatry. 2003; 64:1163-9
Gothelf D, Presburger G, Levy D, Nahmani A,
Birg M, Berant M, Blieden LC, Finkelstein
Y, Frisch A, Apler A, Weizman A. genetic, developmental and physical factors associated
with attention deficit hyperactivity disorder
in patients with velocardiofacial syndrome.
Am J Med Genet Neuropsychiatr Genet.
2004;126:116-21
Heliovaara A, Hurmertina K. Craniofacial
cephalometric morphology in children with
CATCH 22 syndrome. Orthod Craniofac Res.
2006;9:186-92
Hieronimus S, Bec-Roche M, Pedeutour F, et al.
The spectrum of parathyroid gland dysfunction
associated with the microdeletion 22q11. Eur J
Endocrinol. 2006;155:47-52
Kelley RI, Zackai EH, Emanuel BS, Kistenmacher M, Greenberg F, Punnett HH. The association of the DiGeorge anomalad with partial
monosomy of chromosome 22. J Pediatrics.
1982;101:197-200.
Klingberg G, Oskarsdottir S, Johannesson LE Noren JG. Oral manifestation in 22q11 deletion
syndrome. Inter J Pediatr Dent. 2002;12:1423
Klingberg G, Dietz W, Oskarstottir S, Odelius H,
Gelander L, Noren JG. Morphological appearance and chemical composition of enamel in
primary teeth from patient with 22q11 deletion syndrome. Eur J Oral Sci. 2005;113:30311
Klingberg G, Lingstrom P, Oskarsdottir S, Friman
V, Bohman E, Carlen A. Caries-related saliva
properties in individuals with 22q11 deletion
syndrome. Oral Surg Oral Med Oral Pathol
Oral Radiol Endod. 2007; 103:497-504
www.buponline.com
69
Sindrome da delezione 22q11.2
Klingberg G, Hallberg U, Oskarsdóttir S. Oral
health and 22q11 deletion syndrome: thoughts
and experiences from the parents’ perspectives.
Int J Paediatr Dent. 2010;20:283-92
Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeroge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet.
2007;370:1443-52
Lipson AH, Yuille D, Angel M, Thompson PG,
Vandervoord JG, Beckenham EJ. Velocardiofacial (Shprintzen) syndrome: an important syndrome for the dysmorphologist to recognise. J
Med Genet. 1991;28:596-604
McDonald-McGinn DM, Zackai EH. Genetic
counseling for the 22q11.2 deletion. Dev Disabil Res Rev. 2008;14:69-74.
McDonald-McGinn DM, Tonnesen MK,
Laufer-Cahana A, Finucane B, Driscoll DA,
Emanuel BS, et al. Phenotype of the 22q11.2
deletion in individuals identified through an
affected relative: cast a wide FISHing net! Genet Med. 2001;3:23–9
Meazzini M.C., Brusati R., Bozzetti A., Mazzoleni
F., Felisati G., Garattini G., Lalatta F., Rezzonico A. Malformazioni craniofacciali: Coordinamento ortodontico-chirurgico - 2011 - Ed.
Martina Sel.
Meinecke P, Beemer FA, Schinzel A, Kushnick
T. The Velo-Cardio-Facial (Shprintzen) syndrome. Clinical variability in eight patients.
Eur J Pediatr. 1986;145:538-44
Mesbah K, Rana MS, Francou A, van Duijvenboden K, Papaioannou VE, Moorman AF,
Kelly RG, Christoffels VM. Identification of a
Tbx1/Tbx2/Tbx3 genetic pathway governing
pharyngeal and arterial pole morphogenesis.
Hum Mol Genet. 2012;21:1217-29
Michielon G, Marino B, Formigari R, Gargiulo G,
Picchio F, Digilio MC. Genetic syndromes and
outcome after surgical correction of teratology
of Fallot. Ann Thorac Surg. 2006; 81:968-75
Momma K, Cardiovascular anomalies associated
with chromosome 22q11.2 deletion syndrome.
Am J Cardiol. 2010;105:1617-24.
Nordgarden H, Lima K, Skogedal N, Følling I,
Storhaug K, Abrahamsen TG. Dental developmental disturbances in 50 individuals with
the 22q11.2 deletion syndrome; relation to
medical conditions? Acta Odontol Scand.
2012;70:194-201.
Oberoi S, Vargervik K: Velocardiofacial syndrome
with single central incisor. Am J Med Genet.
2005;132:194-7
Oskarsdottir S, Vujic M, Fasth A. Incidence and
prevalence of the 22q11 deletion syndrome:
a population-based study in Western Sweden.
Arch Dis Child. 2004;89:148-51
Philip N, Bassett A. Cognitive, behavioral and psychiatric phenotype in 22q11.2 deletion syndrome. Behav Genet. 2011;41:403-12.
Ryan AK, Goodship JA, Wilson DI, et al. Spectrum
of clinical features associated with interstitial
chromosome 22 deletions: a European collaborative study. J Med Genet. 1997;34:798-804
Shprintzen RJ, Goldberg RB, Lewin ML, et al. A
new syndrome involving cleft palate, cardiac
anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J.
1978;15:56-62
Shprintzen RJ. Palatal and pharyngeal anomalies in craniofacial syndromes. Birth Defects.
1982;18:53-78
Shprintzen RJ. Velocardiofacial syndrome. Otolaryngol Clin North Am. 2000; 33:1217-40
Shprintzen RJ, Velo-Cardio-Facial Syndrome:
30 Years of Study. Dev Disabil Res Rev.
2008;14:3-10
Tatum SA 3rd, Chang J, Havkin N, Shprintzen RJ.
Pharyngeal flap and the internal carotid in the
velo-cardio-facial sindrome. Arch Facial Plast
Surg. 2002;4:73-80
Tezenas Du Montcel ST, Mendizabal H, Ayme S,
www.buponline.com
70
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Levy A, Philip N. Prevalence of 22q11 microdeletion. J Med Genet. 1996;33:719
Wentzel C, Fernström M, Ohrner Y, Annerén
G, Thuresson AC. Clinical variability of the
22q11.2 duplication syndrome. Eur J Med
Genet. 2008;51:501-10
Wilson DI, Burn J, Scambler P, Goodship J. DiGeorge syndrome: part of CATCH 22. J Med
Genet. 1993;30:852-6
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa
S, Minoshima S, Ichida F, Joo K, Kimura M,
Imamura S, Kamatani N, Momma K, Takao A,
Nakazawa M, Shimizu N, Matsuoka R. Role of
TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366-73.
Yakut T, Kilic SS, Cil E, Yapici E, Egeli U.FISH
investigation of 22q11.2 deletion in patients
with immunodeficiency and/or cardiac abnormalities. Pediatr Surg Int. 2006;22:380-3
Yang HC, Shyur SD, Huang LH, Chang YC, Wen
DC, Liang PH, Lin MT. DiGeorge syndrome
associated with solitary median maxillary
central incisor. Asian Pac J Allergy Immunol.
2005;23:159-63
www.buponline.com
6. Sindrome di Turner
Sinonimo: Sindrome da parziale o completa assenza
di un cromosoma X - Codice ICD 10: Q96
I. Cremonesi, S. Bagattoni, T. Tagariello, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Sindrome di Turner (ST), descritta per al prima volta da Henry Turner nel 1938, è una malattia cromosomica rara causata dall’assenza di un
cromosoma X, caratterizzata da iposomia e da insufficienza ovarica con infertilità. Malformazioni
e aumentato rischio di patologie sistemiche sono
incostanti (Turner HH, 1938).
L’incidenza è stimata di 1 su 2500 femmine nate
vive in assenza di predilezione razziale, etnica e
geografica(National Institutes of Health, 2004). Il
99% dei concepimenti affetti da ST non sopravvive oltre la 28a settimana di vita intrauterina, rappresentando la ST la causa del 15% degli aborti
spontanei (Menasha J et al., 2005).
L’indice di mortalità sembra essere sovrapponibile
a quello della popolazione generale.
GENETICA E DIAGNOSI
In circa il 50% dei casi la ST è caratterizzata da
monosomia del cromosoma X; la causa principale è la non disgiunzione del cromosoma sessuale
durante la meiosi, solitamente per un errore nella
spermatogenesi che provoca la perdita del cromosoma sessuale paterno (Kesler SR, 2007; Loscalzo
ML, 2008).
Le restanti forme sono a mosaico (con una linea cellulare 45,X e una linea cellulare normale
46,XX), causate da anomalie strutturali di un
cromosoma X (isocromosoma [X i(Xq)], ad anello
[r(X)], delezione del braccio lungo o del braccio
corto [del(X)]) (Kesler SR, 2007).
La diagnosi prenatale può essere effettuata su villocentesi o amniocentesi; le forme tipiche possono
essere identificate con l’ecografia (Cabrol S, 2007).
MANIFESTAZIONI CLINICHE SISTEMICHE
Le numerose manifestazioni cliniche caratteristiche della ST, eterogenee e non sempre presenti contemporaneamente in relazione ai diversi
cariotipi, sono descritte nella Tabella 1 (Lleo A
et al., 2012; Conway GS et al., 2010; Bondy C,
2007;Moreno-Garcia M et al., 2005; Conway GS
et al., 2004; Savendahl L et al., 2000; Rao E et al.,
1997).
www.buponline.com
72
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Tabella 1: Manifestazioni cliniche caratteristiche della Sindrome di Turner
APPARATO
Apparato scheletrico
Apparato riproduttore
Apparato cutaneo
Apparato
odontostomatognatico
Apparato otorinolaringoiatrico
Apparato endocrino
Apparato cardiovascolare
MANIFESTAZIONI CLINICHE
Statura bassa
FREQUENZA DI
MANIFESTAZIONE
100%
Torace largo (“a scudo”)
70%
Osteoporosi
50%
Cubito valgo
50%
Scoliosi
10%
Discinesia gonadica
90%
Teletelia con ipoplasia dei capezzoli
80%
Linfedema del dorso della mani e dei piedi
70%
Displasia delle unghie
70%
Pterigium colli
70%
Nevi pigmentati multipli
27%
Impianto basso dei capelli nella nuca
40%
Micrognanzia
70%
Agenesie dentali e alterazioni dell’odontogenesi
70%
Palato ogivale
35%
Anomalie auricolari
Ipoacusia
50%
Ipotiroidismo
50%
Malattia celiaca
2-8%
Diabete Mellito tipo 1
2,5-6%
Ipertensione
25-40%
Cardiopatie congenite
17-45%
Le pazienti affette da ST presentano un fenotipo
estremamente eterogeneo in relazione al cariotipo: da quadri severi legati a monosomia, caratterizzati da bassa statura, disgenesia gonadica,
linfedema, malformazioni a livello di numerosi
apparati, a quadri sfumati legati a mosaicismo,
caratterizzati da morfotipo normale, bassa statura
e insufficienza ovarica (Morgan T, 2007).
Circa la metà delle pazienti con ST presenta un
ritardo di crescita intrauterino, più pronunciato
www.buponline.com
73
Sindrome di Turner
per l’altezza che per il peso. Fino ai 3 anni la crescita si mantiene sul 50° percentile ed è presente
un ritardo di maturazione ossea. Tra i 3 e i 12 anni
la velocità di crescita diminuisce progressivamente fino a raggiungere il 10° percentile, mentre l’età
ossea ha una progressione normale. Dai 12 anni
la velocità di crescita si discosta sempre più dal
range di normalità. A 20 anni viene raggiunta la
statura finale, di tre deviazioni standard sotto la
media della popolazione femminile normale (Moreno-Garcia M et al., 2005; Rao E et al., 1997).
La causa del ritardo di crescita, ancor oggi in parte
sconosciuta, è probabilmente legata ad alterazioni
di secrezione o di meccanismo d’azione dell’ormone della crescita (GH) (Bannink EM et al., 2009;
Mazzanti L et al., 2009).
Elemento patognomonico della ST è la disgenesia
ovarica. Secondo alcuni autori il ridotto numero
di cellule germinali è imputabile ad una difettosa
migrazione di cellule dalle creste genitali durante
la vita fetale (Bondy C, 2007). Secondo altri le
cellule germinali dell’ovaio, normali fino al terzo
mese di gestazione, dopo tale periodo subiscono
un arresto del processo di maturazione e gli ovociti
vengono sostituiti da tessuto connettivo, che residua in strutture definite “streaks” (Siddiqui MN
et al., 2002). Nelle pazienti affette da monosomia
l’ipogonadismo si manifesta alla pubertà con mancata comparsa dei caratteri sessuali secondari
ed amenorrea primaria (Morgan T, 2007; De La
Chapelle, 1962; Acheson et al., 1961). L’anomala
funzione ovarica è responsabile di alterazioni ormonali, in particolare della secrezione di GH (Quigley CA et al., 2002), androgeni (Rosenfield RL et
al, 2005; Hanton L et al., 2003) e ormoni tiroidei
(Larizza D et al., 2009; Medeiros CC et al., 2009).
Al contrario, le pazienti affette da mosaicismo nel
40-75% dei casi hanno una pubertà spontanea.
Di frequente riscontro sono anomalie ossee: collo
corto, vertebre cervicali ipoplasiche, gambe corte
con sproporzione tra segmento superiore e infe-
riore, torace largo (“a scudo”) con capezzoli iperdistanziati e ipoplasici. Il cubito valgo (aumento
della deviazione dell’avambraccio sul braccio nella
direzione del radio, ad arto completamente esteso)
è uno dei segni patognomonici descritti da Turner.
Spesso sono presenti una riduzione di lunghezza del
quarto osso metacarpale (segno di Archibald) e una
deformità “a baionetta” del polso (segno di Madelung), risultato di una curvatura laterale o dorsale
del radio e di una sublussazione dell’ulna. In alcuni
casi è rilevabile agenesia del condilo mediale della
tibia (segno di Kosowicz). Le anomalie scheletriche sono diagnosticabili all’esame radiografico, che
in alcuni casi evidenzia anche segni di osteoporosi
(Bondy CA et al., 2007; Moreno-Garcia M et al.,
2005; Sutton EJ et al., 2005; Savendahl L et al.,
2000).
Le patologie cardiovascolari congenite nelle pazienti con ST hanno una prevalenza significativamente più elevata rispetto alla restante popolazione
femminile, essendo presenti in circa il 30% dei
casi, in particolare nelle monosomie (Mazzanti L.
et al., 1998). Le malformazioni più frequenti sono
l’aorta bicuspide (12,5%), la coartazione aortica
(6,9%), la valvulopatia aortica (3,2%) (Sharma J
et al., 2009; Elsheikh M et al., 2001). Patologie
cardiovascolari, in particolare la dissezione aortica
e la malattia ischemica, rappresentano la prima
causa di morte nelle pazienti con ST (Poprawski
K et al., 2009).
Patologie endocrine come ipotiroidismo e diabete possono fare parte del quadro clinico della ST.
A fronte di una positività agli anticorpi antitiroidei
nel 50% delle pazienti con ST, solo nel 24% è presente ipotiroidismo e nel 2,5% ipertiroidismo. Le
patologie tiroidee possono insorgere durante l’infanzia; la loro frequenza aumenta con l’età (Savendahl L et al., 2000). Il rischio di sviluppare diabete
mellito di tipo 1 risulta aumentato nelle pazienti
affette da ST (Jorgensen KT et al., 2010; Mortesen
KH et al., 2009).
www.buponline.com
74
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Anomalie renali sono presenti in circa il 7-8%
delle pazienti con ST: malposizioni, rene a ferro di
cavallo, idronefrosi asintomatica. L’ecografia permette la diagnosi precoce.
Le patologie otorinolaringoiatriche sono molto
frequenti nelle pazienti affette da ST. Anomalie
funzionali e anatomiche delle tube di Eustachio
sono spesso causa di otiti medie acute ricorrenti
(in età compresa tra 1 e 6 anni, con un picco di incidenza a 3 anni), che rappresentano un fattore di
rischio per una ipoacusia neurosensoriale (il 70%
delle pazienti tra i 15 e 35 anni ha un deficit uditivo che peggiora con l’età e a 35 anni il 61% delle
pazienti è affetta da sordità (Conway GS, 2004).
Nella ST sono descritte patologie dell’apparato
digerente: la prevalenza di rettocolite ulceroemorragica e di morbo di Crohn è doppia rispetto alla
popolazione generale (Conway GS, 2004). Nelle
pazienti affette da ST il rischio di sviluppare malattia celiaca risulta undici volte più elevato rispetto
alla popolazione generale (Lleo A et al., 2012; Mortesen KH et al., 2009; Elsheikh M et al., 2002).
La prevalenza delle patologie epatiche risulta elevata nelle ST: l’80% delle pazienti con età superiore ai 35 anni presenta alterazioni delle funzionalità
epatica, generalmente legate ad anomalie vascolari
(Roulot D et al., 2004).
Di riscontro relativamente frequente sono il linfedema del dorso delle mani e dei piedi e lo pterigium
colli che, in epoca neonatale, sono segni orientativi
per la diagnosi di ST (Szilágyi et al., 2000).
Di riscontro relativamente frequente sono anche i
nevi pigmentati, solitamente benigni ma che tendono ad accrescersi dimensionalmente durante la
pubertà (Zvulunov A et al., 1998).
CARATTERISTICHE ORO-FACCIALI
L’eterogeneità cariotipica delle ST giustifica le significative variazioni fenotipiche che si evidenzia-
a
b
Figura 1 a, b: Bambina di 9 anni affetta da ST. Facies
caratteristica: ipertelorismo, zigomi iposviluppati, impianto
basso delle orecchie, collo corto e tozzo
no in queste pazienti a livello orofacciale. Esistono
tuttavia tratti di frequente riscontro: ipertelorismo (occhi molto distanziati), epicanto (palpebre
superiori dirette verso il basso e l’esterno), ptosi
www.buponline.com
75
Sindrome di Turner
palpebrale, zigomi iposviluppati, orecchie con
impianto basso e retro-posto e lobi rivolti verso
l’esterno, collo corto e tozzo con pterigio. Caratteristica è la facies adenoidea (viso allungato con
colorito pallido, occhiaie, bocca semiaperta, narici
piccole, labbra screpolate), associata alla respirazione prevalentemente orale e a frequenti e intercorrenti infezioni respiratorie (Figura 1 a, b).
Di frequente riscontro sono anomalie dentali di
forma e di struttura, nella cui patogenesi la genetica riveste un ruolo chiave (Rizell S et al., 2010;
Vandewalle KS et al., 1993). In corrispondenza
del braccio corto (locus p 22) del cromosoma X
sono localizzati geni deputati alla modulazione
dell’odontogenesi, in particolare il gene AMGX
che codifica l’amelogenina, proteina secreta dagli
ameloblasti. Nelle pazienti affette da ST l’assenza o
le anomalie strutturali di un cromosoma X possono estrinsecarsi in alterazioni nella produzione di
smalto, in senso quantitativo, responsabili di dimensioni ridotte del dente sia deciduo che permanente, e qualitativo, responsabili di ipoplasie dello
smalto, fattore di rischio per la patologia cariosa
(Rizell S et al., 2010).
La patologia cariosa è di frequente riscontro nelle
pazienti con ST, come evidenziano gli elevati valori
di DMFT descritti in queste pazienti (Faggella A
et al., 2006).
Per quanto riguarda la patologia parodontale nelle pazienti con ST sono riportati elevati indici di
placca e gengivali, fattori di rischio per parodontopatie superficiali e profonde (Szilágyi A et al.,
2000) (Figura 2).
L’elevata prevalenza di patologie di pertinenza
ortopedico-ortodontica è causata dalle caratteristiche geneticamente determinate dalla sindrome. Alcuni autori ipotizzano che le alterazioni
del distretto cefalico siano legate ad un disturbo
dell’ossificazione encondrale, dal momento che
colpiscono principalmente le strutture derivate
dal condrocranio, in particolare quelle della base
Figura 2: Adolescente di 13 anni affetto da ST. Morso aperto anteriore, presenza di diastemi, gengivite marginale
cranica (Rongen-Westerlaken C et al., 1993). Sono
tuttavia necessarie ulteriori ricerche per valutare le
modalità attraverso le quali un’anomalia del cromosoma X possa interferire con la sintesi del collagene ed essere responsabile di alterazioni dei tessuti
connettivi.
Nelle pazienti affette da ST è descritta una tendenza alla II classe scheletrica con crescita mandibolare in senso orario, riduzione dell’altezza facciale posteriore e allungamento del terzo inferiore
del volto (Andersen E et al., 2000; Szilagyi A et al.,
2000) (Figura 3 a, b, c).
Indagini cliniche e radiografiche hanno inoltre
evidenziato che, indipendentemente dalla crescita
della mandibola (che è prevalentemente sotto controllo genetico) e dalla conseguente classe molare,
la totalità delle pazienti con ST presenta palato
ogivale con incompetenza trasversale tra le arcate, correlabile a fattori funzionali quali la respirazione orale, complicanza di infezioni respiratorie
frequenti e intercorrenti causate da patologie otorinolaringoiatriche (Hederstierna C et al., 2009).
LINEE GUIDA DI TERAPIA
Per il trattamento dell’iposomia è utilizzata la terapia con GH, che permette di accelerare la velo-
www.buponline.com
76
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
a
b
c
Figura 3 a, b, c: Bambina di 6 anni affetta da ST. Modelli
in gesso delle arcate dentarie. Si evidenziano: palato ogivale, seconda classe molare, morso aperto anteriore, overjet
aumentato
cità di crescita e di aumentare la statura finale, che
risulta più elevata nelle pazienti in cui il trattamento inizia prima degli 8-9 anni di età e dura più di
4 anni (Stephure DK, 2005). Gli effetti collaterali
della terapia con GH sono scarsi; è frequente un’insulinoresistenza che regredisce dopo la sospensione
della terapia. Per le pazienti affette da ipertensione
e da anomalie cardiovascolari è comunque richiesta
una stretta sorveglianza in corso di terapia.
Per le pazienti con insufficienza ovarica è indispensabile la terapia con estrogeni che, senza accelerare il processo di sinostosi delle suture di accrescimento, induce lo sviluppo dei caratteri sessuali
secondari migliorando l’autostima e l’inserimento
sociale ed esercita una azione preventiva sull’osteoporosi e sulle malattie cardiovascolari (Conway
GS, 2004).
Le patologie otorinolaringoiatriche, molto frequenti nell’infanzia nelle pazienti affette da ST,
spesso necessitano di interventi chirurgici di adenoidectomia e/o di posizionamento di drenaggi
timpanici.
Per quanto concerne le patologie di pertinenza
odontostomatologica, nella consapevolezza che
una paziente con ST è ad alto rischio di patologia cariosa, in un’ottica di interdisciplinarietà, è
compito del pediatra, dal momento in cui si effettua la diagnosi, inviare la paziente all’odontoiatra
infantile perché informi la famiglia sull’importanza della salute orale e sulla necessità di interventi
di promozione della salute orale (igiene orale
domiciliare, alimentazione non cariogenica, utilizzo di fluoro topico e, quando indicato, sistemico, visite periodiche, sigillature dei solchi e delle
fessure).
Una grave insufficienza renale rappresenta una controindicazione all’assunzione sistemica di fluoro.
Nei pazienti con ST con cardiopatie congenite a
rischio di EB, gli interventi odontoiatrici a rischio
di batteriemia debbono essere eseguiti in un regime
di profilassi antibiotica (vedi cap. 2).
Per quanto concerne la terapia ortopedico-ortodontica, è garanzia di miglior risultato la precocità
di intervento, in dentizione decidua o mista, non
appena il livello di collaborazione della paziente lo
consenta. L’ottimizzazione dell’intervento necessita di un approccio interdisciplinare ortodontista/
otorinolaringoiatra, dal momento che il quadro
clinico è caratterizzato da ipoplasia del mascellare superiore e da disturbi respiratori. L’espansio-
www.buponline.com
77
Sindrome di Turner
ne del palato, determinando l’abbassamento della
volta con aumento volumetrico delle fosse nasali,
può contribuire a migliorare la respirazione nasale. L’utilizzo di apparecchiature di tipo ortopedico
sfrutta l’immaturità suturale per indurre la crescita del mascellare superiore sia sul piano trasversale che, quando necessario, sul piano sagittale.
Il dispositivo più frequentemente utilizzato è il
disgiuntore rapido del palato per un tempo minimo di 8-9 mesi, che trova applicazione nei casi
di morso crociato posteriore, di discrepanza dentoalveolare da affollamento e di openbite da incompetenza trasversale.
Nelle II classi da retrusione/ipoplasia mandibolare, nell’obiettivo di stimolare la crescita mandibolare possono essere utilizzati attivatori funzionali
per un tempo variabile in base alla collaborazione e all’epoca di applicazione. Quando la crescita
viene programmata e farmacologicamente indotta
con GH, è necessaria una stretta collaborazione
tra ortodontista ed auxologo, poiché i risultati
migliori e più rapidi si ottengono nel periodo
immediatamente precedente il picco di crescita
puberale.
In dentizione permanente, quando ve ne sia la
necessità funzionale e/o estetica, sono indicate le
apparecchiature fisse multibande ad azione ortodontica (Andersen et al., 2000; Szilagyi et al.,
2000). In caso di terapia ortodontica fissa, la presenza di ipoplasie dello smalto e l’elevato rischio di
carie rendono necessari interventi di prevenzione
mirati (igiene orale domiciliare molto accurata,
utilizzo di fluoro topico domiciliare, applicazione
ambulatoriale di gel o vernici al fluoro, sigillatura
dei solchi e delle fessure).
I vantaggi che si ottengono dall’ortopedia e dall’ortodonzia intercettiva sono sia funzionali che estetici. Masticazione, respirazione e fonazione efficaci e
un viso armonico contribuiscono al miglioramento
della qualità della vita e dell’autostima di queste
pazienti, facilitandone l’inserimento sociale.
BIBLIOGRAFIA
Acheson RM, Zampa GA: Skeletal maturation
in ovarian dysgenesis and Turner’s syndrome.
Lancet. 1961;1:917-20
Andersen E, Sonnesen L, Kjaer MS, Fischer
Hansen B, Kjaer I. The prenatal cranial base
complex and hand in Turner syndrome. Eur J
Orthod. 2000;22:185-94
Bannink EM, van der Palen RL, Mulder PG,
de Muinck Keizer-Schrama SM. Long-term
follow-up of GH-treated girls with Turner
syndrome: BMI, blood pressure, body proportions. Horm Res. 2009;71:336-42
Bondy C. Clinical practice guidelines: Care of girls
and women with Turner syndrome: A guideline of the TS Study Group. J Clin Endocrinol
Metab. 2007; 92:10-25
Cabrol S. Turner syndrome. Ann Endocrinol (Paris). 2007;68:2-9
Conway GS: Considerations for transition from
paediatric to adult endocrinology: women with
Turner’s syndrome. Growth Horm IGF Res.
20040;14:77-84
De La Chapelle A, Hortling H: XY/XO mosaicism. Lancet. 1962;13;2:783-4
Elsheikh M, Casadei B, Conway GS, Wass JA: Hypertension is a major risk factor for aortic root
dilatation in women with Turner syndrome.
Clin Endocrinol. 2001;54:69-73
Faggella A, Guadagni MG, Cocchi S, Tagariello T,
Piana G: Dental features in patients with Turner sindrome. Eur J Paediatr Dent. 2006;7:1658
Hanton L, Axelrod L, Bakalov V, Bondy CA: The
importance of estrogen replacement in young
women with Turner syndrome. J Womens
Health 2003;12:971-7
Hederstierna C, Hultcrantz M, Rosenhall U: A
longitudinal study of hearing decline in women with Turner syndrome. Acta Otolaryngol.
2009;129:1434-41
www.buponline.com
78
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Kesler SR: Turner syndrome. Child Adolesc Psychiatr Clin N Am. 2007;6:709-22
Jørgensen KT, Rostgaard K, Bache I, Biggar RJ,
Nielsen NM, Tommerup N, Frisch M. Autoimmune diseases in women with Turner’s
syndrome. Arthritis Rheum. 2010;62:658-66
Larizza D, Calcaterra V, Martinetti M: Autoimmune stigmata in Turner syndrome: when lacks an
X chromosome. J Autoimmun. 2009;33:25-30
Lleo A, Moro00ni L, Caliari L, Invernizzi P. Autoimmunity and Turner’s syndrome. Autoimmun Rev. 2012;11:538-43
Loscalzo ML: Turner syndrome. Pediatr.
2008;29:219-27
Mazzanti L, Cacciari E, for the Italian Study
Group for Turner Syndrome (ISGTS): Congenital heart disease in patients with Turner
syndrome. J Pediatr. 1998;133:688-92
Mazzanti L, Tamburrino F, Bergamaschi R, Scarano E, Montanari F, Torella M, Ballarini E, Cicognani A: Developmental syndromes: growth
hormone deficiency and treatment. Endocr
Dev. 2009;14:114-34
Medeiros CC, de Lemos-Marini SH, Filho MB,
Camargo EE, Santos AO, Magna LA, GuerraJúnior G, Baptista MT, Maciel-Guerra AT:
Turner’s syndrome and subclinical autoimmune thyroid disease: a two-year follow-up study.
J Pediatr Endocrinol Metab. 2009;22:109-18
Menasha J, Levy B, Hirschhorn K, Kardon NB:
Incidence and spectrum of chromosome abnormalities in spontaneous abortions: new
insights from a 12-year study. Genet Med.
2005;7:251-63
Moreno-Garcia M, Fernandez-Martinez FJ, Barreiro Miranda E: Chromosomal anomalies
in patients with short stature. Pediatr Int.
2005;47:546-9
Morgan T: Turner syndrome: diagnosis and management. Am Fam Physician. 2007;76:405-10
Mortensen KH, Cleemann L, Hjerrild BE, Nexo
E, Locht H, Jeppesen EM, Gravholt CH. Increased prevalence of autoimmunity in Turner
syndrome-influence of age. Clin Exp Immunol. 2009;156:205-10
National Institutes of Health: Clinical Features of
Turner syndrome. 2004
Poprawski K, Michalski M, Ławniczak M, Łacka
K: Cardiovascular abnormalities in patients
with Turner syndrome according to karyotype:
own experience and literature review. Pol Arch
Med Wewn. 2009;119:453-60
Quigley CA, Crowe BJ, Anglin DG, Chipman
JJ: Growth hormone and low dose estrogen
in Turner syndrome: results of a United States
multi-center trial to near-final height. J Clin
Endocrinol Metab. 2002;87:2033-41
Rao E, Weiss B, Fukami M, Rump A, Niesler B,
Mertz A: Pseudoautosomal deletions encompassing a novel homeobox gene cause growth
failure in idiopathic short stature and Turner
syndrome. Nat Genet. 1997;16:54-63
Rizell S, Kjellberg H, Dietz W, Norén JG, Lundgren T. Altered inorganic composition of dental enamel and dentin in primary teeth from
girls with Turner syndrome. Eur J Oral Sci.
2010;118:183-90
Rongen-Westerlaken C, vd Born E, Prahl-Andersen B, von Teunenbroek A, Manesse P,
Otten BJ, vd Tweel I, Kuijpers-Jagtman AM,
Delemarre vd Waal HA, Drayer NM: Effect
of growth hormone treatment on craniofacial
growth in Turner’s syndrome. Acta Paediatr.
1993;82:364-8
Rosenfield RL, Devine N, Hunold JJ, Mauras N,
Moshang T Jr, Root AW: Salutary effects of
combining early very low-dose systemic estradiol with growth hormone therapy in girls with
Turner syndrome. J Clin Endocrinol Metab.
2005;90:6424-30
Roulot D, Degott C, Chazouillères O, Oberti F,
Calès P, Carbonell N, Benferhat S, Bresson-
www.buponline.com
79
Introduzione
Hadni S, Valla D: Vascular involvement of
the liver in Turner’s syndrome. Hepatology.
2004;39:239-47
Savendahl L, Davenport ML: Delayed diagnoses
of Turner syndrome: proposed guidelines for
change. J Pediatr. 2000;137:455-9
Sharma J, Friedman D, Dave-Sharma S, Harbison
M: Aortic distensibility and dilation in Turner’s
syndrome. Cardiol Young. 2009;19:568-72
Siddiqui MN, Mia MA, Perveen R, Uddin MM,
Chowdhury KS: Turner syndrome: a case
of gonadal dysgenesis. Mymensingh Med J.
2002;11:122-4
Stephure DK; Canadian Growth Hormone Advisory Committee: Impact of growth hormone
supplementation on adult height in turner
syndrome: results of the Canadian randomized
controlled trial. J Clin Endocrinol Metab.
2005;90:3360-6
Sutton EJ, McInerney-Leo A, Bondy CA, Gollust
SE, King D, Biesecker B: Turner syndrome:
four challenges across the lifespan. Am J Med
Genet A. 2005; 39:57-66
Szilágyi A, Keszthelyi G, Nagy G, Madléna M:
Oral manifestations of patients with Turner
syndrome. Oral Surg Oral Med Oral Pathol
Oral Radiol Endod. 2000;89:577-84
Turner HH. Classic pages in obstetrics and gynecology by. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology. 1938;23:566-74
Vandewalle KS, Castro GW, Camm JH: Dental
management of a patient with Turner syndrome. J Clin Pediatr Dent. 1993;18:26-30
Zvulunov A, Wyatt DT, Laud PW, Esterly NB:
Influence of genetic and environmental factors
on melanocytic naevi: a lesson from Turner’s
syndrome. Br J Dermatol. 1998;138:993-7
www.buponline.com
www.buponline.com
7. Sindrome dell’X fragile
Sinonimi: Sindrome di Martin Bell, Sindrome FRA-X,
Sindrome di Escalante - Codice ICD 10: Q99.2
S. Bagattoni, N. Al Kamhis, T. Tagariello, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Sindrome dell’X fragile (SXF), descritta per
la prima volta da Martin e Bell nel 1943, è una
malattia cromosomica rara legata al cromosoma
X caratterizzata da ritardo mentale, disturbi del
comportamento, dismorfia facciale, iperlassità
ligamentosa, alterazioni fisiche (Terracciano A
et al., 2005). È la causa più frequente di ritardo mentale ereditario. Le manifestazioni cliniche
sono molto variabili da un individuo all’altro e
sono meno evidenti nel genere femminile.
L’incidenza è stimata di 1 su 4.000-6.000 maschi
nati vivi e di 1 su 7.000-10.000 femmine nate
vive, in assenza di predilezione razziale, etnica e
geografica (Crawford DC et al., 2001). La SXF è
ancora oggi sottodiagnosticata, in particolare nel
genere femminile in cui le manifestazioni possono
essere scarse o assenti.
GENETICA E DIAGNOSI
La SXF è causata da un incremento del numero
di ripetizioni del trinucleotide citosina-guaninaguanina (CGG) in una zona in prossimità del gene
FMR-1 localizzato sul cromosoma X. I soggetti
sani presentano un numero di ripetizioni della tripletta CGG non superiore a 50. In relazione al numero di triplette CGG ripetute, le alterazioni della
sequenza genica associata alla SXF si presentano in
due forme definite premutazione (PM) e mutazione completa (MC). Si parla di PM quando il
numero di ripetizioni è compreso tra 50 e 200 e
di MC quando è superiore a 200 (Pieretti M et
al., 1991). La premutazione è generalmente asintomatica. La mutazione completa è responsabile
del mancato funzionamento del gene FMR1, che
causa assenza o carenza della proteina FMRP,
implicata nei processi di connessione neuronale
(Wattendorf DJ et al., 2005).
La sindrome è ereditata da uno dei genitori portatore di una premutazione, in genere la madre.
La diagnosi prenatale, effettuata tramite Southern blot su villocentesi o amniocentesi, permette
l’identificazione della PM e della MC (Willemsen
R et al., 1996).
Si deve far diagnosi di sospetto di SXF quando un
bambino presenta ritardo psicomotorio e del linguaggio, in particolare se esistono individui affetti
nello stesso ambito familiare. Nei casi di sospetto
clinico, per la diagnosi citogenetica si utilizzano i
www.buponline.com
82
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
test Southern blot e PCR. Nel 1999 è stata sviluppata una tecnica di screening di tipo immunocitochimico basata sulla individuazione diretta della
proteina FMRP nella radice dei capelli o nei linfociti prelevati da striscio ematico. Questa tecnica,
non invasiva e di estrema rapidità, non è però in
grado di individuare i portatori di PM (Iwahashi
C et al., 2009).
La consulenza genetica è sempre indicata a livello
familiare in caso di parente affetto dalla sindrome
per individuare eventuali portatori e per informare
sul rischio di ricorrenza della malattia.
MANIFESTAZIONI CLINICHE SISTEMICHE
Il quadro clinico della sindrome è estremamente
variabile (Tabella 1). Le femmine sono meno colpite rispetto ai maschi: nei casi con MC il 100%
dei maschi presenta il quadro completo della malattia mentre le femmine solo nel 50% dei casi
(Nunn JH et al., 1990).
I maschi con MC presentano un fenotipo caratterizzato da anomalie del distretto cranio-cefalico,
iperestensibilità delle articolazioni, scoliosi,
piedi piatti, ginocchio valgo, macrorchidismo;
solitamente il grado di gravità delle alterazioni fisiche e del ritardo mentale sono associati.
(Escalante JA et al., 1971). Le femmine con MC
possono presentare, in forma attenuata, le stesse alterazioni fisiche dei maschi; solo alcuni casi
presentano deficit psichici e cognitivi in assenza di
evidenti anomalie fisiche.
Il primo segno della malattia è generalmente il ritardo dello sviluppo psicomotorio, in particolare
dell’apprendimento del linguaggio. Sono caratteristici problemi di comprensione e di memoria a
breve termine e di lavoro, mentre la memoria visiva
è buona. Il ritardo mentale è di grado variabile,
da lieve a grave: la maggior parte dei maschi ha un
QI inferiore a 50, mentre le femmine hanno un QI
compreso tra 70 e 85 (Tejada-Minguez MI, 2006).
Di norma il ritardo mentale non interferisce con la
capacità di rispondere a semplici istruzioni verbali e non verbali ma ha un impatto sfavorevole sui
compiti che richiedono l’acquisizione di informazioni astratte non sequenziali o correlate alla valutazione degli stati interiori.
Sono presenti disturbi del comportamento: deficit dell’attenzione e iperattività, ansietà, disturbi
del sonno, instabilità psicomotoria, incapacità a
fissare l’attenzione, comportamenti di tipo autistico (scarso contatto oculare, battere le mani, difesa
tattile, inclusa la difesa orale), comportamenti autolesionisti (morsicature). Sono descritti casi di disturbi ossessivo-compulsivi caratterizzati da rituali
per limitare l’ansia legata a un’ossessione. Sono
frequenti altri comportamenti atipici quali mordicchiamento delle dita, movimenti stereotipati
delle mani, eloquio veloce, imitazione del linguaggio altrui, instabilità emotiva, resistenza ai cambiamenti ambientali, fobia sociale accompagnata da
comportamenti impulsivo-aggressivi o da mutismo
(Levitas A, 1996).
La curva della crescita staturale è caratterizzata
da aumentato accrescimento nell’età prepuberale
compensato da riduzione in velocità e quantità di
crescita in età puberale, con possibile esito di iposomia in età adulta.
Otiti medie e sinusiti ricorrenti sono frequenti
nei primi anni di vita.
Lassità ligamentosa, iperestensibilità articolare
(in particolare delle dita) e piedi piatti possono
essere presenti e aggravarsi con l’età (Kulkarni GV
et al., 1994).
Il prolasso della valvola mitrale, presente nel
50% dei pazienti con SXF, generalmente non dà
segni clinici e non comporta limitazioni dell’attività fisica (Murray J et al., 1997).
Possono essere presenti problemi visivi (strabismo, miopia e ipermetropia).
Reflusso gastro-esofageo (RGE) si manifesta in
www.buponline.com
83
Sindrome dell’X fragile
circa 1/3 dei bambini con SXF ed è responsabile di
irritabilità e vomito ricorrente.
Epilessie e convulsioni sono relativamente frequenti durante i primi 6 anni di vita, in particolare nei
maschi (13-18% dei maschi, 5% delle femmine).
Manifestazione clinica caratteristica della sindrome nei maschi post-puberi è il macrorchidismo
(nell’80-92%), mai riscontrato nel neonato.
In soggetti portatori di PM in età adulta sono stati
descritti abuso di alcool e stupefacenti, quadri di
depressione maggiore associati ad ansia e ad attacchi di panico.
I soggetti portatori di PM sono inoltre a rischio di
Sindrome del Tremore e Atassia associata all’X
Fragile (FXTAS) e le femmine di Insufficienza
Ovarica Primaria (IOP).
La FXTAS, che compare nel 40-45% dei maschi e
nell’8-16% delle femmine tipicamente dopo i 50
anni, è una patologia neurodegenerativa caratterizzata da atassia cerebellare progressiva, tremori,
declino delle funzioni cognitive con perdita della
memoria a breve termine, demenza, neuropatie
periferiche, debolezza muscolare a carico degli arti
inferiori (Leehey MA et al., 2003; Hagerman RJ
et al., 2001).
La IOP è caratterizzata dall’interruzione precoce,
in età < a 40 anni, del ciclo ovarico e mestruale;
colpisce il 20% delle donne con premutazione e
non si presenta mai nei casi di mutazione completa
(Allingham-Hawkins DJ et al., 1999; Conway GS
et al., 1995).
Tabella 1: Manifestazioni cliniche nella Sindrome dell’X Fragile
Manifestazioni cliniche
Ritardo mentale
Disturbi del linguaggio
Infezioni dell’orecchio
Iperestensibilità articolare
Deficit dell’attenzione ed iperattività
Ansietà
Depressione
Macrorchidismo
Piedi piatti e ginocchia iperestensibili
Prolasso della mitrale
Miopia ed ipermetropia
Strabismo
Reflusso gastro-esofageo
Scoliosi
Epilessie e convulsioni
Menopausa precoce
Ernie
Dislocazioni articolari
www.buponline.com
%
100
molto frequenti
>85
73
80 maschi
30 femmine
64-79 maschi
36-40 femmine
65 femmine
80-92 maschi
>50
50
17-57
8-44
30
20
<20
15-20 femmine
15
3
84
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
CARATTERISTICHE ORO-FACCIALI
I pazienti affetti da SXF presentano una facies
caratteristica (Figure 1-2-3): dolicocefalia,
fronte alta, orecchie grandi e prominenti, lieve ipertelorismo, naso lungo e a bulbo, labbra
sottili e retratte, palato ogivale (Figura 1 a, b, c)
(Murray J et al., 1997). Lassità ligamentosa a livello dell’articolazione temporo-mandibolare può
essere responsabile di lussazione mandibolare
ricorrente.
In letteratura sono riportate anomalie dentali di
eruzione (eruzione anticipata dei denti decidui e
permanenti), di numero (denti sovrannumerari),
di dimensioni (macrodonzia) e di forma (taurodontismo, anomalie radicolari) (Ridaura-Ruiz L et
al., 2009; Kotilainen J et al., 1999; Peretz B et al.,
1988).
In letteratura non sono presenti dati relativi alla
prevalenza della patologia cariosa. Tuttavia la presenza di RGE, le difficoltà nelle manovre di igiene
orale domiciliare e le abitudini alimentari non corrette rappresentano fattori di rischio per la patologia cariosa.
La prevalenza della patologia parodontale è sovrapponibile a quella dei soggetti sani. Nei casi in
cui siano presenti crisi epilettiche, l’assunzione di
antiepilettici (Fenitoina nel 50-60%, Valproato,
b
a
c
Figura 1 a, b, c: Paziente affetto da SXF, 16 anni; facies caratteristica: dolicocefalia, fronte alta, orecchie grandi e prominenti, lieve ipertelorismo, naso lungo e a bulbo, labbra sottili e retratte, prognatismo mandibolare; II classe dentale e scheletrica,
biprotrusione, morso aperto, affollamento in arcata superiore ed inferiore, palato ogivale
www.buponline.com
85
Sindrome dell’X fragile
Fenobarbital e Etosuccimide con frequenza significativamente inferiore) può causare ipertrofia
gengivale, che si manifesta con un aumento del
volume gengivale e formazione di pseudotasche in
assenza di riassorbimento osseo.
Per quanto concerne le patologie di pertinenza
ortopedico-ortodontica sono di frequente riscontro palato ogivale, cross-bite, open bite anteriore
e malocclusione di III classe (Tejada-Minguez MI,
2006).
In letteratura si evidenzia un’elevata prevalenza di
lesioni dentali di origine traumatica, in particolare a livello del settore frontale superiore, attribuibile a fattori predisponenti legati alla patologia di
base quali ritardo mentale, iperattività, crisi epilettiche (Nunn JH et al., 1990; Shellhart WC et al.,
1986).
LINEE GUIDA DI TERAPIA
I pazienti con SXF sono affetti da numerose alterazioni e patologie; la diagnosi, il follow-up e le
terapie richiedono la presenza di un team multi
e interspecialistico adeguatamente formato sulla
sindrome.
Non esiste attualmente un trattamento farmacologico specifico per questa sindrome. La terapia è
di tipo riabilitativo, motorio e psicopedagogico; in
particolare l’assistenza psicopedagogica da parte
di educatori specializzati può migliorare sensibilmente le potenzialità del bambino ed aiutarlo a vivere i rapporti con gli altri in modo armonico (Hall
DA et al., 2006).
Nei bambini con SXF sono di riscontro molto
frequente disturbi del sonno (difficoltà di addormentamento, risvegli frequenti nella notte). Di qui
la necessità di informare i genitori sui rischi che
derivano dall’utilizzo di soluzioni zuccherate attraverso il biberon per favorire l’addormentamento.
Creare una ritualità dell’addormentamento (met-
tere il bambino a letto alla stessa ora, camera buia,
musica rilassante, racconti di favole/storie, pigiama
morbido, succhiotto anatomico), fornendo segnali
che è tempo di dormire e creando un’atmosfera di
serenità, può aiutare ad ottimizzare il sonno.
Molti bambini con SXF hanno difficoltà ad alimentarsi nei primi mesi di vita e molti danno la
preferenza a cibi morbidi, spesso cariogenici. In
questi casi può rendersi necessario un intervento
fisioterapico per migliorare la funzionalità masticatoria.
Il RGE richiede la posizione eretta del bambino
durante e dopo i pasti e, nei casi più gravi, l’utilizzo
di farmaci anti-reflusso (Gaviscon, Ranitidina).
Per i disturbi d’ansia e ossessivi-compulsivi, per
l’autolesionismo, per il comportamento aggressivo
è indicato un trattamento farmacologico. È in
fase di studio l’utilizzo di antagonisti dei recettori mGluR5, di agonisti dei recettori GABA-A e
GABA-B e della minociclina; i primi risultati sono
promettenti e potrebbero modificare il decorso e
migliorare la prognosi (Thomas AM et al., 2012;
Heulens I et al., 2011; Rooms L et al., 2011).
Per promuovere l’integrazione sociale sono utilizzati piani educativi individualizzati, interventi cognitivo-comportamentali e terapia occupazionale.
Per quanto concerne le patologie odontostomatologiche, in relazione alla presenza di fattori di
rischio per la patologia cariosa, in un’ottica di
interdisciplinarietà, è compito del pediatra, dal
momento in cui si effettua la diagnosi, inviare il
paziente all’odontoiatra infantile perché informi la
famiglia sull’importanza della salute orale e sulla
necessità di interventi di prevenzione (igiene orale domiciliare, alimentazione non cariogenica, utilizzo di fluoro topico e quando indicato sistemico,
visite periodiche, sigillatura dei solchi, delle fessure
e dei fori ciechi dei molari permanenti).
Molti soggetti con SXF incontrano difficoltà nelle manovre dell’igiene personale, inclusa l’igiene
orale. Per migliorare la loro collaborazione possono
www.buponline.com
86
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
essere utilizzate illustrazioni relative alle varie manovre da eseguire da proporre al bambino in modo
interattivo. È consigliabile l’utilizzo di uno spazzolino elettrico.
Nei pazienti con ipertrofia gengivale e pseudotasche può essere indicato il trattamento chirurgico e
l’inserimento del paziente in un ciclo di follow-up
per il controllo igienico e il monitoraggio di eventuali recidive e/o complicanze.
In considerazione delle caratteristiche psicocomportamentali della SXF, il trattamento odontoiatrico è spesso difficoltoso a causa dello scarso
livello di collaborazione. Nei pazienti con SXF è
importante utilizzare un approccio psicologico e
comportamentale mirato ad individuare i canali
comunicativi più efficaci per conquistare la fiducia
e la collaborazione del paziente; per raggiungere
questo obiettivo, di grande utilità è il colloquio
preliminare con la famiglia.
Per ottenere la collaborazione si possono utilizzare
varie tecniche.
Il role-play: far partecipare il bambino ad una rappresentazione gioco-favola in cui l’educatore gioca
il ruolo del dentista utilizzando un abbigliamento
realistico e simulando l’ambiente odontoiatrico.
Il modeling: far vedere al bambino un filmato
in cui è ripresa una seduta odontoiatrica con un
bambino molto collaborante o farlo partecipare di
persona ad una seduta con un bambino molto collaborante.
La “pedagogia delle immagini”: proporre al
bimbo, prima dell’appuntamento odontoiatrico,
un album descrittivo dei luoghi, delle persone
e degli oggetti relativi alla seduta odontoiatrica,
in modo che possa imparare cosa deve fare, dove,
quando, come, con chi e quale sarà l’esatta successione delle azioni che avverranno e che lui dovrà
compiere. Con questo strumento viene data al
bambino la possibilità di prevedere gli eventi che
lo coinvolgeranno, creando uno stato di fiducia e
valorizzando le sue capacità di comprensione. L’al-
bum viene consegnato due settimane prima della
seduta ai genitori o agli educatori perché lo propongano al bambino; una copia dell’album viene
allegata alla cartella odontoiatrica del paziente. Il
significato clinico ed educativo delle immagini
associate alla verbalizzazione delle varie sequenze,
mediante semplici didascalie, permette di tranquillizzare il bambino sulle novità del contesto e
di renderlo consapevole del significato delle cure
odontoiatriche. Le immagini vanno proposte una
alla volta. Durante la seduta gli operatori utilizzano le stesse parole delle didascalie illustrative. Le
immagini possono essere anche un utile strumento
per far accettare al paziente le manovre di igiene
orale domiciliare.
Nei casi in cui il livello di collaborazione risulti
nullo le terapie odontoiatriche debbono essere eseguite in regime di anestesia generale. L’intervento
in AG deve essere considerato come il punto di
partenza per un programma di controlli periodici
ravvicinati, nel corso dei quali attuare igiene orale professionale e intercettare le lesioni allo stadio
iniziali, quando le terapie sono più semplici quindi
più accettate dal paziente.
In presenza di prolasso della valvola mitrale, in
caso di interventi odontoiatrici a rischio deve essere valutata la necessità di profilassi antibiotica
dell’endocardite batterica (vedi cap. 2).
Spesso è necessaria una terapia ortopedico-ortodontica per intercettare una malocclusione scheletrica di III classe. Quando il livello di collaborazione del bambino alla terapia e alle manovre di
igiene orale domiciliare lo consenta, si utilizzano
due dispositivi ad azione ortopedica: l’espansore
rapido del palato (REP), per correggere il deficit
trasversale derivante dall’ipoplasia palatina, e la
maschera di Delaire che, stimolando la protrazione ed anterorotazione del mascellare superiore,
attenua il deficit sagittale e verticale.
In relazione all’elevata prevalenza di lesioni dentali di origine traumatica è importante fornire
www.buponline.com
87
Sindrome dell’X fragile
alla famiglia linee guida per la loro gestione, in
particolare sull’opportunità del reimpianto immediato in caso di avulsione di denti permanenti, e
attuare misure preventive come la realizzazione di
paradenti.
BIBLIOGRAFIA
Allingham-Hawkins DJ, Babul-Hirji , Chitayat
HD, Holden JJ, Yang KT, Lee C, Hudson R,
Gorwill H, Nolin SL, Glicksman A, Jenkins
EC, Brown WT, Howard-Peebles PN, Becchi C, Cummings E, Fallon L, Seitz S, Black
SH, Vianna-Morgante AM, Costa SS, Otto
PA, Mingroni-Netto RC, Murray A, Webb J,
Vieri F Fragile X premutation is a significant
risk factor for premature ovarian failure: the
International Collaborative POF in Fragile
X study-preliminary data. Am J Med Genet.
1999;83:322-5
Conway GS, Hettitarachein S, Murray A, Jacobs
PA. Fragile X premutations in familial premature ovarian failure. Lancet. 1995;346:309-310
Crawford DC, Acuña JM, Sherman SL. FMR1
and the fragile X syndrome: human genome
epidemiology review. Genet Med. 2001;3:
359-71
Escalante JA, Grunspun H, Frota-Pessoa O. Severe
sexlinkedmental retardation. J Genet Hum.
1971;19:137-40
Hagerman RJ, Leehey M, Heinrichs W, Tassone F,
Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ. Intention tremor, parkinsonism, and
generalized brain atrophy in male carriers of
fragile X. Neurology. 2001;57:127-30
Hall DA, Berry-Kravis E, Hagerman RJ, Hagerman
PJ, Rice CD, Leehey MA. Symptomatic treatment in the fragile X-associated tremor/ataxia
syndrome. Mov Disord. 2006;21:1741-4
Heulens I, Kooy F. Fragile X syndrome: From gene
discovery to therapy. Frontiers in Bioscience.
2011;16:1211-32
Iwahashi C, Tassone F, Hagerman RJ, Yasui D,
Parrott G, Nguyen D, Mayeur G, Hagerman
PJ. A quantitative ELISA assay for the fragile
x mental retardation 1 protein. J Mol Diagn.
2009;11:281-9
Kotilainen J, Pirinen S. Dental maturity is advanced in Fragile X syndrome. Am J Med Genet. 1999;83:298-301
Kulkarni GV, Levine N. Fragile X (Martin Bell)
syndrome. Spec Care Dentist. 1994;14:21-5
Leehey MA, Munhoz RP, Lang AE, Brunberg JA,
Grigsby J, Greco C, Jacquemont S, Tassone F,
Lozano AM, Hagerman PJ, Hagerman RJ. The
fragile X premutation presenting as essential
tremor. Arch Neurol. 2003;60:117-21
Levitas A. Neuropsychiatric aspects of fragile
X syndrome. Semin Clin Neuropsychiatry.
1996;1:154-67
Murray J, Cuckle H, Taylor G, Hewison J. Screening for fragile X syndrome. Health Technol Assess. 1997;1:1-71
Nunn JH, Durning P. Fragile X (Martin Bell)
syndrome and dental care. Br Dent J.
1990;168:160-2
Peretz B, Ever-Hadani P, Casamassimo P, Eidelman E, Shellhart C, Hagerman R. Crown size
asymmetry in males with fra (X) or Martin-Bell
syndrome. Am J Med Genet. 1988;30:185-90
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra
BA, Caskey CT and Nelson DL. Absence of
expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817-22
Ridaura-Ruiz L, Quinteros-Borgarello M, BeriniAytés L, Gay-Escoda C. Fragile X-syndrome:
Literature review and report of two cases. Med
Oral Patol Oral Cir Bucal. 2009;14:434-9
Rooms L, Kooy RF. Advances in understanding fragile X syndrome and related disorders (Review). Current Opinion in Pediatrics.
www.buponline.com
88
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
2011;23:601-6
Shellart WC, Casamassimo P, Hagerman RJ,
Belanger GK. Oral findings in fragile X syndrome. American Journal of Medical Genetics.
1986;23:179-87
Tejada-Minguez MI. Síndrome X frágil. Libro de
consulta para familiares y profesionales. Madrid: Artegraf; 2006
Terracciano A, Chiurazzi P, Neri G. Fragile X syndrome. Am J Med Genet. C Semin Med Genet. 2005;137:32-7
Thomas AM, Bui N, Perkins JR, Yuva-Paylor
LA, Paylor R. Group I metabotropic glutamate
receptor antagonists alter select behaviors in a
mouse model for fragile X syndrome. Psychopharmacology. 2012;219:47-58
Wattendorf DJ, Muenke M. Diagnosis and management of fragile X syndrome. Am Fam Physician. 2005;72:111-3
Willemsen R, Oosterwijk JC, Los FJ, Galjaard H,
Ostra BA. Prenatal diagnosis of fragile X syndrome. Lancet. 1996;348:967-8
www.buponline.com
8. Sindrome di Williams
Sinonimi: Delezione 7q11.23; Monosomia 7q11.23;
Sndrome di Williams-Beuren - Codice ICD 10: Q87.8
I. Cremonesi, S. Bagattoni, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Sindrome di Williams (SW), conosciuta anche come Sindrome di Williams-Beuren, descritta in modo indipendente nel 1961 da Williams e
nel 1962 da Beuren, è una malattia cromosomica rara caratterizzata da disturbi dello sviluppo,
cardiopatie congenite, dismorfismo facciale con
aspetto da elfo, ritardo psicomotorio e profilo
cognitivo e comportamentale specifici (Berdon
WE et al., 2011; Beuren AJ et al., 1962; Williams
JC et al., 1961).
L’incidenza è stimata di 1 su 8000 nati vivi per
anno, in assenza di predilezione razziale, etnica, geografica e di genere (Haas WB et al., 2012); considerando che alcune forme rimangono non diagnosticate, questi valori rappresentano una sottostima
(Strømme P et al., 2002).
L’indice di mortalità, in assenza di patologie cardiovascolari responsabili di morte precoce, è simile
alla popolazione generale.
GENETICA E DIAGNOSI
delezione nella regione q.11.23 del cromosoma
7 che determina la perdita di 26-28 geni, tra cui
quello dell’elastina. La delezione si manifesta quasi
sempre “ex novo”, da cui il carattere sporadico della sindrome (Kaplan P et al., 2001). Sono riportati
rari casi ad ereditarietà autosomica dominante.
La diagnosi è clinica e viene per lo più formulata durante l’infanzia sulla base delle caratteristiche facciali, del profilo cognitivo e delle patologie
cardiache. La diagnosi è confermata nel 95% dei
casi dall’indagine genetica: la microdelezione del
cromosoma 7, non visibile sul cariotipo standard,
può essere messa in evidenza mediante ibridazione
fluorescente in situ (FISH); in alcuni casi la delezione è così piccola da non essere registrata e l’esame risulta negativo.
Non vi è un test di screening per la diagnosi prenatale nella popolazione generale; solo nei rari casi
di rischio per familiarità può essere effettuata su
villocentesi o amniocentesi (Kaplan P et al., 2001;
Lashkari A et al., 1999). Recentemente sono stati
descritti casi di diagnosi prenatale di SW effettuata
utilizzando la tecnica BACs-on-Beads, una nuova
tecnologia in grado di individuare nel feto aneuploidie e microdelezioni (Popowski T et al., 2011).
La SW è causata nel 95% dei casi da una microwww.buponline.com
90
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
MANIFESTAZIONI CLINICHE SISTEMICHE
Molte sono le caratteristiche comuni ai pazienti affetti da SW, nonostante l’estrema variabilità
fenotipica. Le principali manifestazioni cliniche
sono descritte nella Tabella 1 (Pober BR, 2010).
Alla nascita sono spesso presenti difficoltà di alimentazione, episodi di vomito, reflusso gastroesofageo (RGE) e crescita rallentata. Possono essere presenti anche disturbi del sonno, stipsi, ernie
inguinali (in 1/3 dei casi), ipercalcemia idiopatica
(che frequentemente scompare tra i 18 e i 24 mesi)
Tabella 1: Manifestazioni cliniche della Sindrome di Williams
MANIFESTAZIONI CLINICHE
Apparato cardiovascolare
Stenosi aortica sopravalvolare (SASV)
Stenosi polmonare periferica (SPP)
Prolasso della valvola mitrale (PVM)
Difetto del setto ventricolare (DSV)
Ipertensione
Ictus
Morte improvvisa
Sviluppo cognitivo
Compromissione cognitiva globale (QI circa 55)
Caratteristico pattern di forza e debolezza cognitiva
Distretto cefalico e apparato stomatognatico
Microcefalia
Dismorfismo facciale
Anomalie dentali di eruzione, numero, forma e struttura
Malocclusioni
Apparato endocrino
Pubertà anticipata
Intolleranza al glucosio/diabete mellito
Osteopenia
Ipotiroidismo (subclinico)
Ipercalcemia
Apparato gastrointestinale
Coliche
Difficoltà di alimentazione
Intolleranze alimentari
Stitichezza
Reflusso gastroesofageo (RGE)
Diverticoli
Prolasso rettale
Celiachia
www.buponline.com
91
Sindrome dell’X fragile
Apparato genitourinario
Anomalie renali
Infezioni del tratto urinario frequenti
Calcoli renali
Apparato muscolo-scheletrico
Ipotonia muscolare
Scoliosi
Anchilosi articolari
Sinostosi radio cubitale
Lussazione recidivante delle rotule
Sistema nervoso
Ipotonia centrale
Iperreflessia
Sintomi cerebellari
Apparato oculare
Strabismo
Alterata acuità visiva
Ridotta stereopsi
Restringimento dei dotti lacrimali
Apparato otorinolaringoiatrico
Iperacusia
Perdita dell’udito lieve-moderata per i toni alti
Otiti medie ricorrenti
Cute e annessi cutanei
Invecchiamento prematuro della cute
Capelli precocemente grigi
Ernie (inguinali e altre)
Personalità, comportamento e benessere emozionale
Personalità amichevole
Iperattività, scarsa attenzione (ADHD)
Ansia, fobie, tratti ossessivi-compulsivi
Distimia
Varie
Bassa statura
Disturbi del sonno
Anomalo aumento di peso
(Gilbert-Dussardier B, 2006; Metcalfe K, 1999).
Le malformazioni cardiache sono presenti nel
75% dei casi. La più frequente è la stenosi aortica sopravalvolare (SASV), la cui severità varia da
lieve a grave. La stenosi delle arterie medie e lar-
ghe, causata dall’ispessimento della tonaca media,
costituisce l’anomalia vascolare tipica della SW;
il restringimento arterioso può essere o isolato o
presente contemporaneamente in più siti: arco aortico, aorta discendente, arterie polmonari, corona-
www.buponline.com
92
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
riche, renali, mesenteriche ed intracraniche (Pober
BR, 2010; Gilbert-Dussardier B, 2006).
L’ipertensione si sviluppa circa nel 50% dei pazienti adulti e occasionalmente può comparire
durante l’infanzia; può essere a eziopatogenesi sconosciuta o secondaria a stenosi delle arterie renali
(Pober BR, 2010; Morris CA, 2006).
La crescita postnatale è rallentata nei primi anni
di vita; l’altezza media in età adulta è 159 cm nei
maschi e 147 cm nelle femmine. Nel 75% dei casi
è presente microcefalia (Metcalfe K, 1999).
Il ritardo nelle acquisizioni motorie si manifesta
con alterazioni della motricità fine e della coordinazione; le azioni che necessitano di una buona
capacità motoria e della programmazione dei movimenti (svestirsi, spogliarsi, lavarsi) spesso sono di
difficile esecuzione.
Per quanto concerne l’apparato muscolo-scheletrico, è di riscontro frequente l’ipotonia muscolare, che porta i pazienti ad assumere posture compensatorie per raggiungere un equilibro corporeo
(spalle spioventi, flessione delle anche e delle ginocchia, iperlordosi lombare) (Gilbert-Dussardier
B, 2006). Di riscontro relativamente frequente
sono anche scoliosi (17%), anchilosi delle grandi
articolazioni (15%), sinostosi radiocubitale (10%),
lussazione recidivante delle rotule (5%) (Morris
CA, 2006; Metcalfe K, 1999).
La presenza di anomalie renouretrali necessita del
monitoraggio della funzionalità renale.
Per quanto riguarda le patologie oculari, il 40%
dei bambini affetti da SW presenta strabismo e/o
difetti di rifrazione (Gilbert-Dussardier B, 2006).
In pazienti affetti da SW sono descritti casi di ipotiroidismo e di pubertà precoce (Pober BR, 2010;
Metcalfe K, 1999; Gorlin RJ et al., 1990).
I capelli diventano grigi e la cute invecchia precocemente ma non ci sono evidenze sufficienti per
definire la SW una sindrome ad invecchiamento
precoce (Pober BR, 2010).
L’incidenza di malattia celiaca risulta aumentata
(9,6%) rispetto alla popolazione generale (0,5%)
(Giannotti A et al., 2001).
Per quanto riguarda il linguaggio, dopo un iniziale
ritardo nell’acquisizione, la maggior parte dei bambini diventa molto loquace e con un timbro di voce
caratteristico (Gilbert-Dussardier B, 2006).
Il profilo cognitivo è caratterizzato da buone
competenze verbali e da una buona memoria ma
anche da gravi difficoltà a orientarsi nello spazio e
da scarsa capacità di concentrazione. Il quoziente
intellettivo (QI) nei bambini ha una media di 55
con discordanza tra un QI verbale buono e un QI
nelle performance basso.
Negli individui con SW si evidenzia un fenotipo
psicologico molto particolare, caratterizzato da
punti di forza e di debolezza su più ambiti cognitivi
e da un modello definito di comportamento sociale. Uno degli aspetti più interessanti è l’aumentata
propensione verso l’interazione sociale e la tendenza ad assumere atteggiamenti eccessivamente confidenziali nei confronti anche di sconosciuti come
avvicinarsi molto alle persone e fissarle negli occhi
(“face processing”) (Jarvinen-Pasley A. et al., 2008).
A differenza degli individui affetti da Autismo e
da Sindrome della X fragile, che tendono ad assumere un comportamento “socialmente distante”, i
soggetti con SW sono considerati “iper-sociali”.
Presentano inoltre una atipica capacità di elaborare le emozioni: hanno scarsa capacità di percepire
le emozioni negative veicolate attraverso la tonalità
della voce e le espressioni facciali (volti arrabbiati)
e polarizzano l’attenzione sulle emozioni positive
(volti felici) (Frigerio E et al., 2006). I bambini presentano solitamente un comportamento iperattivo,
a volte aggressivo nei confronti dei coetanei (Pober
BR, 2010; Gilbert-Dussardier B, 2006).
Nel passaggio dall’adolescenza all’età adulta si può
manifestare una sindrome depressiva.
Molto frequente è l’ipersensibilità al rumore, che
tende ad attenuarsi negli anni (Pober BR, 2010;
Mervis CB et al., 2000).
www.buponline.com
93
Sindrome dell’X fragile
CARATTERISTICHE ORO-FACCIALI
I pazienti affetti da SW presentano una facies, definita da elfo, con caratteristiche peculiari che si
accentuano negli anni: naso con radice appiattita
e punta globosa, bocca grande con labbro inferiore ampio ed everso, guance paffute, edema
periorbitale, epicanto, iride “a stella” (Figura
1 a, b) (Axelsson S., 2005; American Academy of
Pediatrics Committee on Genetics, 2001; Winter
M et al., 1996). Con l’età, il viso diventa più stretto e assume lineamenti grossolani.
Sono frequenti anomalie dentali di eruzione, di
numero, di dimensione, di forma e di struttura
(Ohazama A et al., 2007; Kashyap AS et al., 2000).
L’eruzione e la permuta dentaria sono ritardate,
in relazione al ritardo di crescita e di sviluppo (Moskovitz M et al., 2005; Kashyap AS et al., 2000).
Sono descritti casi di agenesie dentali multiple
della dentatura sia decidua che permanente (nel
40,5% agenesia di più di 1 elemento permanen-
te, nell’11,9% agenesia di 6 o più elementi permanenti) (Axelsson S et al., 2003). Caratteristica
della SW è la microdonzia, con formazione di
ampi diastemi. Per quanto riguarda le anomalie
di forma sono descritte anomalie coronali (denti
conoidi, denti a gemma) e radicolari (radici corte
e sottili) nel 12,5% dei denti decidui e nel 40,7%
dei permanenti (Ohazama A et al., 2007; Kashyap
AS et al., 2000).
Sono di riscontro relativamente frequente ipoplasie dello smalto (Figura 2) di elementi sia decidui
che permanenti (Ohazama A et al., 2007; American Academy of Pediatrics Committee on Genetics, 2001).
Per quanto riguarda la patologia cariosa, la prevalenza è superiore rispetto alla popolazione generale. La presenza di RGE e/o ipoplasie dello smalto
rappresentano fattori di rischio per erosioni e carie
(Ohazama A et al., 2007; American Academy of
Pediatrics Committee on Genetics, 2001).
Sono descritte patologie mucogengivali: ispes-
b
Figura 1 a, b: Bambina di 6 anni affetta da SW. Facies
caratteristica: edema periorbitale, guance paffute, naso con
radice appiattita e punta globosa, bocca grande con labbro
inferiore ampio
a
www.buponline.com
94
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
mandibolare in post-rotazione, con dolicocefalia
e deficit di sviluppo del mascellare superiore. Di
riscontro frequente nei pazienti con SW è il succhiamento della lingua. Nell’85% dei pazienti con
SW sono descritte malocclusioni: II e III classi,
morso profondo, morso aperto (Habersack K et
al., 2007, Axelsson S et al., 2003, American Academy of Pediatrics Committee on Genetics, 2001).
Figura 2: Ipoplasie dello smalto a livello degli incisivi centrali superiori e inferiori
simenti della mucosa orale e presenza di frenuli
labiali accessori e, in pazienti con malattia celiaca, stomatiti aftose ricorrenti (Giannotti A et al.,
2001).
Per quanto riguarda la malattia parodontale, i dati
presenti in letteratura sono scarsi. La SW è tuttavia
caratterizzata da alterazioni a carico dell’elastina,
macromolecola strutturale della gengiva che svolge un ruolo importante nella resistenza del tessuto
parodontale. Secondo un case series la perdita del
gene che codifica per l’elastina non si assocerebbe
all’insorgenza di malattia parodontale: in pazienti
affetti da SW, pur in presenza di elevati valori degli
indici di placca causati da scarsa igiene domiciliare,
non sono descritti elevati livelli di infiammazione
gengivale e perdita di attacco (Joseph et al., 2008).
In pazienti con SW sono anche descritti quadri di
ipertrofia gengivale associata a presenza di pseudotasche e conseguente accumulo di placca batterica
per difficile mantenimento di un adeguato standard di igiene orale (Canargiu et al., 2009; Joseph
C et al., 2008; Ohazama A et al., 2007; Axelsson
S, 2005; Moskovitz M et al., 2005; Tarjan I et al.,
2003; Axelsson S et al., 2003; Kashyap AS et al.,
2000; Hertzberg et al., 1994).
Per quanto riguarda le patologie ortopedicoortodontiche, l’ipotonia muscolare causa incompetenza labiale, respirazione orale e postura bassa
della lingua, condizioni che inducono una crescita
LINEE GUIDA DI TERAPIA
I pazienti con SW sono affetti da numerose alterazioni e patologie che rendono necessari interventi multispecialistici. La diagnosi, le terapie e il
follow-up richiedono la presenza di un team multi
e interspecialistico adeguatamente formato sulla
sindrome.
Le problematiche cardiovascolari sono le più
gravi per i pazienti Williams e richiedono terapie
dedicate e follow-up periodici. Nei casi di SASV
moderata o grave è richiesto l’intervento chirurgico. Il trattamento dell’ipertensione richiede terapia farmacologica e stili di vita corretti (Pober BR,
2010).
Il RGE richiede la posizione eretta del bambino
durante e dopo i pasti e, nei casi più gravi, l’utilizzo di farmaci anti-reflusso (Gaviscon, Ranitidina).
Per quanto concerne le patologie endocrine è necessario un attento monitoraggio e, se necessario,
terapie dedicate: dieta povera di calcio in caso di
ipercalcemia, assunzione di ormone tiroideo in
caso di ipotiroidismo, assunzione di insulina in
caso di diabete (Metcalfe K, 1999).
Per adattare il paziente alla propria situazione e favorire il suo inserimento nella società è utile l’intervento di psicologi, logopedisti e fisioterapisti
(Pober BR, 2010).
Per quanto concerne le patologie di pertinenza
odontoiatrica, essendo i pazienti affetti da SW
ad elevato rischio di patologia cariosa e, quando
www.buponline.com
95
Sindrome dell’X fragile
affetti da cardiopatia congenita, a rischio di Endocardite Batterica (EB), è necessario attuare, dal
momento in cui viene formulata la diagnosi, interventi di prevenzione primaria mirati e intensivi,
con particolare riferimento all’alimentazione non
cariogenica, all’igiene orale domiciliare, all’utilizzo
di fluoro topico e quando indicato sistemico, alle
visite periodiche trimestrali/semestrali, alla sigillatura di solchi, fessure e fori ciechi.
Essendo compromessa la motricità fine, le manovre di igiene orale risultano difficili; è quindi consigliabile l’utilizzo di uno spazzolino elettrico. Gli
interventi di educazione motoria, che garantiscono
buoni risultati, possono prevedere l’insegnamento
dello spazzolamento dei denti.
Nei pazienti con ipertrofia gengivale e pseudotasche può essere indicato il trattamento chirurgico
e l’inserimento del paziente in un ciclo di followup per il controllo igienico e il monitoraggio di
eventuali recidive e/o complicanze (Canargiu et
al., 2009).
Il trattamento odontoiatrico dei pazienti con
SW necessita di un approccio psicologico e
comportamentale mirato in relazione ai comportamenti ansiosi e iperattivi e alla scarsa capacità di concentrazione caratteristici della sindrome
(Holinger DP et al., 2005; Moskovitz M et al.,
2005). Di fondamentale importanza è individuare i canali comunicativi più efficaci per conquistare la fiducia e la collaborazione del paziente; per
raggiungere questo obiettivo, di grande utilità è il
colloquio preliminare con la famiglia. Nei casi di
elevata sensibilità al rumore è importante ridurre
al minimo la rumorosità dell’ambiente, utilizzare,
quando possibile, strumentazione odontoiatrica
manuale e consigliare l’uso di tappi auricolari o
l’ascolto di musica con cuffie.
In presenza di patologie cardiovascolari a rischio
di EB, prima di effettuare procedure odontoiatriche che provocano sanguinamento è necessaria la
profilassi antibiotica (vedi capitolo 2).
Considerando l’elevata prevalenza di anomalie dentarie e di patologie ortopedico-ortodontiche, è importante effettuare una diagnosi precoce ed intercettare abitudini viziate nell’obiettivo di attuare il piano
di trattamento più idoneo e più efficace (Axelsson S,
2005; Axelsson S et al., 2003; American Academy of
Pediatrics Committee on Genetics, 2001)
Le alterazioni della sfera oro-facciale associate alla
sindrome sono variabili, spesso su base disfunzionale a causa dell’ipotonia muscolare e debbono essere affrontate in un’ottica interdisciplinare di collaborazione (foniatra, logopedista, ortodontista). Il
trattamento ortopedico-ortodontico intercettivo mediante apparecchiature ortopedico-ortodontico-funzionali permette di ridurre o prevenire lo
sviluppo di malocclusioni più importanti (Foto
3 a, b, c). La microdonzia spesso presente, la più
alta frequenza di agenesie e le caratteristiche comportamentali che influiscono sulla collaborazione
rendono particolarmente complesso l’approccio
ortopedico-ortodontico. Quando la terapia non
riesce a compensare i casi di gravi malocclusioni,
può rendersi necessario il ricorso alla chirurgia
ortognatica in età adulta per il corretto posizionamento dei mascellari (Habersack K et al., 2007).
Per quanto riguarda le lesioni dentali di origine
traumatica, pur non esistendo in letteratura alcun riferimento specifico, considerando il ritardo
nello sviluppo motorio e i conseguenti problemi
nell’equilibrio e nella coordinazione dei movimenti, è ipotizzabile una loro elevata prevalenza a
livello degli elementi sia decidui che permanenti;
è quindi necessario fornire alla famiglia linee guida per la loro gestione e attuare misure preventive
come la realizzazione di paradenti.
BIBLIOGRAFIA
American Academy of Pediatrics Committee on
Genetics. Health care supervision for chil-
www.buponline.com
96
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
dren with William syndrome. Pediatrics.
2001;107:1192-204
Axelsson S, Bjornland T, et al. Dental characteristics in Williams syndrome: a clinical and radiographic evaluation. Acta Odontol Scand.
2003;61:129-36
Axelsson S. Variability of the cranial and dental
phenotype in Williams syndrome. Swed Dent J
Suppl. 2005;170:3-67
Berdon WE, Clarkson PM, Teele RL. Williams-Beuren syndrome: Historical aspects;
Pediatric Radiology. 2011;41-2:262-6
Beuren AJ, Apitz J, Harmjanz D. Supravalvular
aortic stenosis in association with mental retardation and a certain facial appearance. Circulation. 1962;26:1235-40
Canargiu F, Erriu M, Piras A, Dibart SN. Modifications of periodontal tissues associated with
Williams syndrome. A case report. Minerva
Stomatol. 2009;58:375-81.
Frigerio, E., Burt, D. M., Gagliardi, C., Cioffi, G.,
Martelli, S., Perrett, D. I., and Borgatti, R. Is
everybody always my friend? Perception of approachability in Williams syndrome. Neuropsychologia. 2006;44:254-9
Giannotti A, Tiberio G, Castro M, Virgilii F, Colistro F, Ferretti F, Digilio MC, Gambarara M,
Dallapiccola B. Coeliac disease in Williams
syndrome. J Med Genet. 2001;38:767-8
Gilbert-Dussardier B. Le syndrome de Williams-Beuren. Encyclopédie Orphanet. Décembre 2006
Gorlin RJ, Cohen MM, Levin LS. Syndromes of
the head and neck. Monographs on medical
genetics. New York: Oxford University Press.
1990
Habersack K, Grimaldi B, Paulus GW. Orthodontic orthognathic surgical treatment of a subject
with Williams Beuren syndrome a follow-up
from 8 to 25 years of age. Eur J Orthod.
2007;29:332-7
Haas BW, Reiss AL. Social brain development in
williams syndrome: the current status and directions for the future research. Front Psychol.
2012;3:186
Hertzberg J, Nakisbendi L et al. Williams syndrome oral presentation of 45 cases. Pediatr
Dent. 1994;16:262-7
Holinger DP, Bellugi U, Mills DL, Korenberg JR,
Reiss AL, Sherman GF, Galaburda AM. Relative sparing of primary auditory cortex in Williams Syndrome. Brain Res. 2005;1037:35-42
Jarvinen-Pasley A, Bellugi U, Reilly J, Mills DL,
Galaburda A, Reiss AL, Korenberg JR. Defining the social fenotype in Williams syndrome:
a model for linking gene, the brain and behaviour. Dev Psychopathol. 2008;20:1-35
Joseph C, Landru MM et al. Periodontal conditions in Williams Beuren syndrome: a series of
8 cases. Eur Arch Paediatr Dent. 2008;9:142-7
Kaplan P, Wang PP, Francke U. Williams (Williams
Beuren) syndrome: a distinct neurobehavioral
disorder. J Child Neurol. 2001;16:177-90
Kashyap AS, Sharma HS et al. Dental anomalies in Williams syndrome. Postgrad Med J.
2000;76:712
Lashkari A, Smith AK, Graham JM Jr. Williams-Beuren syndrome: an update and review for the primary physician. Clin Pediatr.
1999;38:189-208
Mervis CB, Robinson BF, Bertrand J, Morris CA,
Klein-Tasman BP, Armstrong SC. The Williams syndrome cognitive profile. Brain Cogn.
2000;44:604-28
Metcalfe K: Williams sindrome: an update on clinical and molecular aspects. Arch Dis Child.
1999;81:198-200
Morris CA. Williams Syndrome. In: Pagon RA,
Bird TD, Dolan CR, Stephens K, Adam MP;
GeneReviews™. Seattle (WA): University of
Washington, Seattle; 1993
Moskovitz M, Brener D, Faibis S, Peretz B. Med-
www.buponline.com
97
Sindrome dell’X fragile
ical considerations in dental treatment of
children with Williams syndrome. Oral Surg
Oral Med Oral Pathol Oral Radiol Endod.
2005;99:573-80
Ohazama A, Sharpe PT. TFII-I gene family during
tooth development: candidate genes for tooth
anomalies in Williams syndrome. Dev Dyn.
2007;236:2884-8
Pober BR. Williams-Beuren syndrome. N Engl J
Med. 2010;362:239-52
Popowski T, Vialard F, Leroy B, Bault JP, MolinaGomes D. Williams-Beuren syndrome: the
prenatal phenotype. Am J Obstet Gynecol.
2011;205:6-8
Strømme P, Bjørnstad PG, Ramstad KJ. Prevalence
estimation of Williams syndrome. Child Neurol. 2002;17:269-71
Tarjan I, G Balaton et al. Facial and dental appearance of Williams syndrome. Postgrad Med J.
2003;79:241
Williams JC, Barratt-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation.
1961;4:1311-8
Winter M, Pankau R, Amm M, Gosch A, Wessel A. The spectrum of ocular features in the
Williams-Beuren syndrome. Clin Genet.
1996;49:28-31
www.buponline.com
www.buponline.com
9. Sindrome di Angelman
Sinonimo: Happy puppet syndrome
Codice ICD 10: Q93.5
T. Tagariello, N. AlKhamis, L. Armuzzi, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Sindrome di Angelman (SA), descritta per la
prima volta nel 1965 dal pediatra inglese Harry
Angelman che la definì “happy puppet syndrome”
per la tipica espressione del volto, è una malattia
cromosomica rara caratterizzata da facies caratteristica, ritardo mentale, assenza di linguaggio,
comportamenti specifici (ipereccitabilità, iperattività, scarsa attenzione, episodi di riso eccessivo e
immotivato), alterazioni dello sviluppo motorio
e crisi epilettiche (Angelman H, 1965).
L’incidenza è stimata fra 1 su 12.000 e 1 su 20.000
nati vivi per anno, in assenza di predilezione razziale, etnica, geografica e di genere (Campos-Castellò
J, 2004; Clayton-Smith J et al., 2003; Kyllerman
M, 1995).
Non sono presenti in letteratura dati relativi all’aspettativa di vita dei pazienti con SA.
GENETICA E DIAGNOSI
La SA è causata da mutazione o delezione di uno
o più geni localizzati sul cromosoma 15 a livello della regione q11-q13, che contiene il gene
UBE3A. Nel 70% dei casi è presente delezione
della regione q11-q13 del cromosoma 15 materno e il gene UBE3A di origine materna è assente. Nel 10% dei casi sono presenti mutazioni
nel gene UBE3A del cromosoma 15 materno.
Nel 2-5% dei casi la sindrome è causata da disomia uniparentale paterna (UPD); in questi
pazienti il quadro clinico è meno severo rispetto
a quelli con delezione; in particolare microcefalia, crisi epilettiche e alterazioni dello sviluppo
motorio sono meno frequenti (Clayton-Smith
J et al., 2003). Nel 2-5% dei casi sono presenti
mutazioni a livello del cromosoma 15 materno
che rendono inattivo il gene UBE3A; in questi
pazienti microcefalia, crisi epilettiche e alterazioni
dello sviluppo motorio sono meno gravi e in alcuni le capacità di linguaggio sono buone (Mabb
AM et al., 2011; Van Buggenhout G et al., 2009).
Nel 5-20% dei pazienti con diagnosi clinica di
SA intervengono meccanismi genetici ad oggi
sconosciuti.
I meccanismi genetici differenti determinano fenotipi simili ma con manifestazioni cliniche
differenti (Tan WH et al., 2011; Jiang Y et al.,
1999).
Il rischio che ulteriori figli abbiano la sindrome è
www.buponline.com
100
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
elevato (fino al 50%) in presenza di mutazioni che
rendono inattivo il gene UBE3A mentre è inferiore
all’1% nei casi di delezione 15q11-q13 e di disomia uniparentale paterna (Mabb AM et al., 2011;
Van Buggenhout G et al., 2009).
La diagnosi prenatale in presenza di un figlio
affetto è possibile per i meccanismi genetici conosciuti su villocentesi o amniocentesi (ClaytonSmith J et al., 2003).
MANIFESTAZIONI CLINICHE SISTEMICHE
Nonostante la variabilità fenotipica correlata
all’eterogeneità genetica, molte sono le caratteristiche comuni ai pazienti affetti da SA.
Nel 2006 la Scientific Advisory Committee of the
US Angelman Syndrome Foundation ha definito
i criteri clinico-diagnostici della SA, suddividendoli in tre categorie a seconda della frequenza con
cui si manifestano (Tabella 1) (Williams CA, 2010;
Williams CA et al., 2006).
Tabella 1: Criteri clinico-diagnostici della SA
Caratteristiche costanti (100%)
-
-
-
-
grave ritardo nello sviluppo psico-motorio
deficit motori e disturbi dell’equilibrio (atassia/tremori)
comportamenti specifici (frequenti risate, eccitabilità, iperattività)
difficoltà di comunicazione (linguaggio verbale minimo o assente)
Caratteristiche frequenti (>80%)
-
-
-
alterata crescita del cranio (microcefalia/circonferenza cranica non proporzionata)
crisi epilettiche (esordio in età < 3 anni)
EEG caratteristico (spesso non correlato a crisi epilettiche)
Caratteristiche associate (20-80%)
-
-
-
-
-
-
-
-
-
-
-
-
-
osso occipitale piatto
solco occipitale
lingua protrusa
spinta linguale
difficoltà di succhiamento e di deglutizione
difficoltà di alimentazione nella prima
infanzia / ipotonia del tronco
mento prominente
bocca ampia, denti diastemati
scialorrea
eccessivi movimenti di masticazione e della
bocca
strabismo
cute ipopigmentata
aumento dei riflessi tendinei profondi
-
-
-
-
-
-
-
-
-
arti superiori in posizione flessa nella
deambulazione
andatura con piedi larghi, in posizione prona
o valga
sensibilità al calore aumentata
disturbi del sonno
fascino per l’acqua
comportamenti anomali legati
all’alimentazione (mangiare prodotti non
alimentari/apparente aumento dell’appetito)
obesità
scoliosi
stipsi
www.buponline.com
101
Sindrome di Angelman
Caratteristici sono i comportamenti specifici, in
particolare le frequenti risate con atteggiamento di
apparente felicità, l’iperattività, il deficit dell’attenzione, la personalità facilmente eccitabile. Le
risate, provocabili fin dalle prime settimane di vita,
possono essere inappropriate rispetto agli stimoli;
l’atteggiamento “felice” sembra essere espressione
di un evento motorio.
La difficoltà di comunicazione è uno degli aspetti
predominanti nelle SA (Jolleff N et al., 1993). Il
linguaggio non si sviluppa e la maggior parte dei
pazienti ha un vocabolario di sole 2 o 3 parole
(Andersen WH et al., 2001). L’abilità di comunicazione non verbale è superiore rispetto a quella
verbale. Molti pazienti sono in grado di comprendere semplici comandi nel contesto della routine
quotidiana. In età adulta possono svilupparsi comportamenti aggressivi.
Nella SA sono caratteristici l’assenza di coordinazione motoria, i difetti di movimento e i disturbi
dell’equilibrio con atassia, movimenti traballanti
(tipo burattino) e ipercinetici degli arti inferiori e
del tronco (Clayton-Smith J, 1993).
Molto frequenti sono i problemi del sonno con
ridotta necessità di dormire (Pelc K et al., 2008).
Caratteristica peculiare della SA è il reflusso gastroesofageo (RGE), presente in tutte le età ma più
frequente nei bambini (Clayton-Smith J, 1993).
Le crisi epilettiche iniziano in genere nei primi 3
anni di vita, più spesso a 18-24 mesi (Laan LA et
al., 1996; Viani F et al., 1995). Sono riportati diversi tipi di crisi: assenze atipiche, crisi mio-cloniche, stato epilettico non convulsivo. L’evoluzione è
relativamente benigna e con l’età adulta si ha una
diminuzione della frequenza di attacchi epilettici (Clayton-Smith J, 1993; Matsumoto A et al.,
1992).
Sono descritti casi di ipertono vagale con disturbi
del ritmo cardiaco.
La scoliosi, presente nel 10% dei bambini e nella totalità degli adulti, si aggrava con l’età deter-
minando una diminuzione delle capacità motorie
(Laan LA et al., 1996).
CARATTERISTICHE ORO-FACCIALI
La caratteristica più frequente a livello del distretto
oro-facciale è la microcefalia da ridotta circonferenza cranica, più pronunciata nei soggetti con delezione 15q11-q13.
Altri aspetti che contribuiscono a delineare la facies caratteristica sono gli occhi infossati, la lingua protrusa, la macrostomia, il prognatismo
mandibolare con mento prominente, gli ampi
diastemi interdentali (Campos-Castellò J, 2004)
(Figure 1 a, b, 2).
Sono di frequente riscontro difficoltà di deglutizione/suzione, iperattività masticatoria, movimenti afinalistici delle labbra e digrignamento.
La scialorrea, causata dalla postura della bocca
aperta in assenza di un corretto controllo della muscolatura periorale, è comune nel bambino piccolo
e tende a diminuire con l’età, anche se in alcuni
casi persiste nell’adulto (Boyce HW et al., 2005;
Zori RT et al., 1992).
In pazienti con SA sono di frequente riscontro erosioni dello smalto di elementi decidui e permanenti, correlate alla presenza di RGE.
Per quanto riguarda la prevalenza delle patologie
cariosa e parodontale, in letteratura non sono presenti studi epidemiologici in pazienti affetti da SA.
Un solo case report evidenzia una elevata prevalenza di carie a livello della dentatura sia decidua che
permanente (Murakami C et al., 2008). La nostra
esperienza clinica condotta su 5 pazienti di età
compresa tra 10 e 14 anni conferma tale dato. L’elevato rischio di patologia cariosa è correlato ad
abitudini alimentari errate (ad esempio in pazienti
con problemi di sonno l’utilizzo di soluzioni zuccherate per favorire l’addormentamento), all’utilizzo di farmaci sotto forma di sciroppi (edulcorati
www.buponline.com
102
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Figura 2: Adolescente di 12 anni affetta da SA. Abbondanti
accumuli di placca batterica e tartaro, gengivite marginale, ampi diastemi, morso aperto anteriore, elemento 21 con
frattura smalto/dentina di origine traumatica, elemento 22
con discromia grigia e lesione cariosa cervicale vestibolare
a
b
Figura 1 a, b: Adolescente di 14 anni affetta da SA. Facies
caratteristica: ipotonia dei muscoli del volto, macrostomia
con zuccheri cariogenici e spesso a pH acido), alle
erosioni dentali, alla scarsa igiene orale domiciliare,
resa problematica dalle difficoltà motorie e comportamentali insite nella SA.
L’assunzione di farmaci antiepilettici (Fenitoina nel 50-60% dei casi, Valproato, Fenobarbital
e Etosuccimide con frequenza significativamente
inferiore) può causare ipertrofia gengivale, che si
manifesta con un aumento del volume gengivale e
formazione di pseudotasche in assenza di riassorbimento osseo.
Per quanto concerne le patologie ortopedicoortodontiche i dati presenti in letteratura sono
scarsi. Sono descritti pazienti affetti da SA con
malocclusione scheletrica di III classe riconducibile a protrusione della mandibola e del mento
(Campos-Castellò J, 2004) e pazienti con pattern
di crescita di tipo verticale e malocclusione dentale caratterizzata da ampi diastemi e morso aperto
anteriore correlati alla spinta linguale (Van Buggenhout G et al., 2009; Maciel CT et al., 2005). In
letteratura è descritto un solo caso di trattamento
ortodontico in un paziente di 11 anni finalizzato a
ridurre un ampio diastema tra gli incisivi centrali
per esigenze estetiche dei genitori (Murakami C et
al., 2008).
www.buponline.com
103
Sindrome di Angelman
LINEE GUIDA DI TERAPIA
I pazienti con SA sono affetti da numerose alterazioni e patologie; la diagnosi, il follow-up e le terapie richiedono la presenza di un team multispecialistico adeguatamente formato sulla sindrome.
La difficoltà nell’alimentazione nei neonati può
essere migliorata con l’utilizzo di tettarelle che facilitino la suzione dal biberon.
Il RGE richiede la posizione eretta del bambino
durante e dopo i pasti e, nei casi più gravi, l’utilizzo di farmaci anti-reflusso (Gaviscon, Ranitidina). Nei casi in cui la terapia farmacologica non
abbia effetto, può rendersi necessario l’intervento
chirurgico (fundoplicatio). In relazione al rischio
di erosioni e di patologia cariosa che il RGE comporta, è necessario fornire ai genitori, dal momento
in cui è formulata la diagnosi, informazioni relative
all’alimentazione non cariogenica, all’igiene orale
domiciliare, all’utilizzo di fluoro topico e sistemico.
Nei pazienti con stipsi è consigliabile una dieta
ricca di fibre, utile anche per favorire una migliore
masticazione, e l’utilizzo di farmaci lassativi.
In caso di crisi epilettiche, la terapia prevede l’assunzione di farmaci anticonvulsivanti. In relazione al rischio di ipertrofia gengivale che questi farmaci comportano, è necessario fornire ai genitori
informazioni relative alla necessità di una corretta
igiene orale domiciliare e di sedute di igiene orale
professionale periodiche (Pelc K et al., 2008; Bjerre
I et al., 1984).
Lo strabismo viene corretto con la chirurgia (Mah
ML et al., 2000; King RA, et al., 1993; Dickinson
AJ et al., 1990).
Per i bambini con instabilità motoria, la fisioterapia è necessaria per migliorare la deambulazione
(Clayton-Smith J et al., 2003). Per stimolare competenze motorie fini degli arti è necessaria una terapia occupazionale. In questa ottica l’insegnamento
delle manovre di igiene orale può essere utilizzato
per migliorare le competenze motorie delle mani.
La logopedia, compresi i metodi non verbali di
comunicazione, permette di migliorare le scarse
capacità di comunicazione (Andersen WH et al.,
2001).
Nei bambini con SA sono di riscontro molto frequente disturbi del sonno (difficoltà di addormentamento, risvegli frequenti nella notte). Di qui
la necessità di informare i genitori sui rischi che
derivano dall’utilizzo di soluzioni zuccherate attraverso il biberon per favorire l’addormentamento.
Creare una ritualità dell’addormentamento (mettere il bambino a letto alla stessa ora, camera buia,
musica rilassante, racconti di favole/storie, pigiama
morbido, succhiotto anatomico), fornendo segnali
che è tempo di dormire e creando un’atmosfera di
serenità, può aiutare ad ottimizzare il sonno. Nei
casi gravi possono essere somministrati farmaci
sedativi o, in alternativa, la melatonina, regolatore
del ciclo sonno-veglia (Miano S et al., 2005; Bruni
O et al., 2004).
Per l’iperattività masticatoria e i movimenti afinalistici delle labbra, il più delle volte dovuti a
stress o a noia, è molto utile un approccio comportamentale di distrazione (Boyce HW et al.,
2005).
La scialorrea è molto difficile da trattare. I genitori
debbono incoraggiare la chiusura della bocca sia
con messaggi verbali sia chiudendo delicatamente
la bocca del bambino. Interventi fisioterapici e
logopedici sono utilizzabili per il controllo della
muscolatura periorale. La terapia farmacologica
della scialorrea si può attuare mediante farmaci
anticolinergici, quali scopolamina e glicopirrolato
sotto forma di cerotti (iniziando con bassi dosaggi,
un quarto o mezzo cerotto, da aumentare alla necessità), che possono comportare effetti collaterali
quali sonnolenza, secchezza oculare, stipsi e riduzione delle secrezioni bronchiali. È utilizzata anche
l’iniezione di tossina botulinica all’interno delle
ghiandole salivari, che riduce il flusso salivare per
un periodo massimo di quattro mesi. L’ultima al-
www.buponline.com
104
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
ternativa è rappresentata da tecniche chirurgiche
(deviazione o legatura dei dotti salivari). Si evidenzia che le terapie utilizzate per diminuire la scialorrea possono comportare una significativa riduzione
del flusso salivare, tale da rappresentare un ulteriore fattore di rischio di patologia cariosa.
Per quanto concerne le patologie di pertinenza
odontostomatologica, in un’ottica di interdisciplinarietà, è compito del pediatra, dal momento in cui
si effettua la diagnosi, inviare il paziente all’odontoiatra infantile perché informi la famiglia sull’importanza della salute orale e sulla necessità di interventi
di prevenzione (igiene orale domiciliare, alimentazione non cariogenica, utilizzo di fluoro topico e,
quando indicato, sistemico, visite periodiche trimestrali/semestrali, sigillatura dei solchi, delle fessure e
dei fori ciechi dei molari permanenti).
Per quanto riguarda l’igiene orale domiciliare,
il paziente con SA, a causa dello scarso livello di
collaborazione, dei movimenti involontari e dello
scarso coordinamento motorio, nella maggior parte dei casi non è in grado di acquisire l’autonomia
nello spazzolamento. È quindi necessario insegnare
ai genitori e ai tutori tecniche “speciali” e mezzi
di “contenzione dolce”. Ad esempio, si può consigliare di sdraiare il bambino, mantenendo aperto il
cavo orale con un dito protetto da uno spessore di
gomma o con un apribocca di gomma (evidenziando che il continuo masticare può rendere difficile
il posizionamento e il mantenimento in situ dell’apribocca); una seconda persona può essere necessaria per contenere braccia e gambe. Lo spazzolino
elettrico può essere utile in molti casi. Sono da
consigliare la pulizia dei denti con garza bagnata su
dito dopo la somministrazione di sciroppi e l’utilizzo settimanale di gel al fluoro su spazzolino o garza.
Il programma di prevenzione prevede visite di
controllo periodiche a intervalli stabiliti in funzione del controllo domiciliare della placca batterica, in occasione delle quali eseguire igiene orale
professionale, applicazione topica di fluoro e sigil-
latura dei solchi, delle fessure e dei fori ciechi dei
molari permanenti con sigillanti resinosi, appena
l’eruzione ne permette l’isolamento con diga di
gomma. Qualora i solchi e le fessure fossero ad elevato rischio di carie in denti neoerotti non isolabili
con la diga di gomma, è consigliabile la sigillatura
provvisoria con sigillanti vetroionomerici.
Le visite di controllo rappresentano l’occasione
per creare familiarità con gli operatori e l’ambiente odontoiatrico. Per ottenere la collaborazione, gli
operatori mettono in atto tecniche di comunicazione: tell-show-do, modeling, rinforzo positivo e
comunicazione non verbale. Gli appuntamenti devono essere fissati nella prima parte della mattinata,
momento della giornata in cui il livello di collaborazione sembra essere migliore anche per l’effetto dei
farmaci sedativi utilizzati per i disturbi del sonno.
Poiché i bambini con SA sono molto attratti dalle
superfici riflettenti e dall’acqua, queste possono essere utilizzate per gestire il comportamento del bambino e per facilitare la comunicazione (Clayton-Smith
J et al., 2003; Williams CA et al., 2006).
In alcuni casi la collaborazione del paziente con
SA è estremamente scarsa, tale da non consentire
l’attuazione del piano di trattamento, in particolare quando complesso, in anestesia locale. In questi casi le terapie odontoiatriche vengono eseguite
in anestesia generale (AG). Nel corso della visita
anestesiologica, nell’obiettivo di evitare complicanze intra e/o postoperatorie, vengono considerate
condizioni di rischio quali reflusso gastroesofageo,
epilessia, bradicardia (per l’elevato tono vagale),
anomalie cranio-facciali e atrofia muscolare periferica. Va evidenziato che molti farmaci utilizzati
nell’AG esercitano i loro effetti attraverso i recettori del GABA (acido gamma aminobutirrico A);
essendo la SA causata da alterazioni degli stessi
geni che controllano la produzione dei recettori del
GABA sono ipotizzabili interazioni di cui non sono
attualmente noti gli effetti. In letteratura sono descritti casi trattati con esito positivo mediante uso
www.buponline.com
105
Sindrome di Angelman
Figura 3: Rx endorale post-traumatica degli elementi 21 e
22. Elemento 21: frattura smalto-dentinale, lesione periapicale in corrispondenza di apice aperto in fase di formazione. Elemento 22: riassorbimento interno, canale pulpare
in comunicazione con lesione cariosa cervicale vestibolare
combinato di propofol, ketamina e sevoflurano
(Landsman IS et al., 2012; Witte W et al., 2011;
Kim BS et al., 2010). L’intervento in AG deve essere considerato come il punto di partenza per un
programma di controlli periodici ravvicinati, nel
corso dei quali attuare igiene orale professionale e
intercettare le lesioni allo stadio iniziale, quando le
terapie sono più semplici e di rapida esecuzione,
quindi più accettate dal paziente.
Per quanto concerne le terapie ortopedico-ortodontiche, le difficoltà al mantenimento di una
corretta igiene orale domiciliare, lo scarso livello di
collaborazione e i movimenti afinalistici a livello
Figura 4: Rx endorale degli elementi 21 e 22 dopo trattamento. Elemento 21: apertura del canale mostra polpa
necrotica, eseguiti lavaggi con ipoclorito, si attende la risoluzione della lesione periapicale per procedere con il posizionamento di idrossido di calcio per promuovere l’apicogenesi.
Elemento 22: apertura del canale pulpare, posizionamento
di idrossido di calcio nel canale e MTA a livello della comunicazione vestibolare
del cavo orale, le rendono di difficile attuazione
nei pazienti con SA.
Per quanto concerne le lesioni dentali di origine
traumatica, pur non esistendo in letteratura alcun riferimento specifico, considerando l’assenza
di coordinazione motoria, i difetti di movimento,
i disturbi dell’equilibrio e la possibile presenza di
crisi epilettiche è ipotizzabile una loro elevata prevalenza a livello degli elementi sia decidui che permanenti (Figure 3, 4); è quindi necessario fornire
alla famiglia linee guida per la loro gestione, in particolare sull’opportunità del reimpianto immediato
in caso di avulsione di denti permanenti.
www.buponline.com
106
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
BIBLIOGRAFIA
Andersen WH, Rasmussen RK, Stromme P. Levels
of cognitive and linguistic development in Angelman syndrome: a study of 20 children. Logoped Phoniatr Vocol. 2001;26:2-9
Angelman H. Puppet children: a report on three
cases. Dev Med Child Neurol. 1965;7:681-8
Bjerre I, Fagher B, Ryding E, Rosén I. The Angelman or “happy puppet” syndrome. Clinical and electroencephalographic features
and cerebral blood flow. Acta Paediatr Scand.
1984;73:398-402
Boyce HW and Bakheet MR. Sialorrhea: a review
of a vexing, often unrecognized sign of oropharyngeal and esophageal disease. J Clin Gastroenterol. 2005;39:89-97
Bruni O, Ferri R, D’Agostino G, Miano S, Roccella M, Elia M. Sleep disturbances in Angelman
syndrome: a questionnaire study. Brain Dev.
2004;26:233-40
Campos-Castellò J. Angelman syndrome. Orphanet Encyclopedia. 2004
Clayton-Smith J, Laan L. Angelman syndrome:
a review of the clinical and genetic aspects. J
Med Genet. 2003;40:87-95
Clayton-Smith J. Clinical research on Angelman
syndrome in the United Kingdom: observations on 82 affected individuals. Am J Med
Genet. 1993;46:12-5
Dickinson AJ, Fielder AR, Young ID, Duckett DP.
Ocular findings in Angelman’s (happy puppet) syndrome. Ophthalmic Paediatr Genet.
1990;11:1-6
Jiang Y, Lev-Lehman E, Bressler J, Tsai TF, Beaudet AL. Genetics of Angelman syndrome. Am J
Hum Genet. 1999;65:1-6
Jolleff N, Ryan MM. Communication development in Angelman’s syndrome. Arch Dis
Child. 1993;69:148-50
Kim BS, Yeo JS, Kim SO. Anesthesia of a dental
patient with Angelman syndrome -A case report-. Korean J Anesthesiol. 2010;58:207-10
King RA, Wiesner GL, Townsend D, White JG.
Hypopigmentation in Angelman syndrome.
Am J Med Genet. 1993;46:40-4
Kyllerman M. On the prevalence of Angelman
syndrome. Am J Med Genet. 1995;59:405
Laan LA, den Boer AT, Hennekam RC, Renier
WO, Brouwer OF. Angelman syndrome in
adulthood. Am J Med Genet. 1996;66:356-60
Landsman IS, Mitzel HM, Peters SU, Bichell TJ.
Are children with Angelman syndrome at high
risk for anesthetic complications? Paediatr
Anaesth. 2012;22:263-7
Mabb AM, Judson MC, Zylka MJ, Philpot BD.
Angelman syndrome: Insights into genomic
imprinting and neurodevelopmental phenotypes. Trends in Neurosciences. 2011;34:293303
Maciel CT, Leite IC. Etiological aspects of anterior
open bite and its implications to the oral functions. Pro Fono. 2005;17:293-302
Mah ML, Wallace DK, Powell CM. Ophthalmic
manifestations of Angelman syndrome. JAAPOS. 2000;4:248-9
Matsumoto A, Kumagai T, Miura K, Miyazaki S,
Hayakawa C, Yamanaka T. Epilepsy in Angelman syndrome associated with chromosome
15q deletion. Epilepsia. 1992;33:1083-90
Miano S, Bruni O, Elia M, Musumeci S. A, Verrillo E, Ferri R. Sleep breathing and periodic
leg movement pattern in Angelman Sindrome:
A polysomnographic study. Clinical Neurophisiology. 2005;116:2685-92
Murakami C, Nahás Pires Corrêa MS, Nahás Pires
Corrêa F, Nahás Pires Corrêa JP. Dental treatment of children with Angelman syndrome:
a case report. Spec Care Dentist. 2008;28:8-11
Pelc K, Cheron G, Boyd SG, Dan B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008;9:434-41
www.buponline.com
107
Sindrome di Angelman
Pelc K, Boyd SG, Cheron G, Dan B. Epilepsy in
Angelman syndrome. Seizure. 2008;17:211-7
Tan WH, Bacino CA, Skinner SA, Anselm I, Barbieri-Welge R, Bauer-Carlin A, Beaudet AL,
Bichell TJ, Gentile JK, Glaze DG, Horowitz
LT, Kothare SV, Lee HS, Nespeca MP, Peters
SU, Sahoo T, Sarco D, Waisbren, SE, Bird
LM. Angelman syndrome: mutations influence
features in early childhood. Am Journal Med
Genet. 2011;155:81-90
Van Buggenhout G, Fryns JP. Angelman syndrome (AS, MIM 105830). Eur J Hum Genet.
2009;17:1367-73
Viani F, Romeo A, Viri M, Mastrangelo M, Lalatta F, Selicorni A, Gobbi G, Lanzi G, Bettio D, Briscioli V. Seizure and EEG patterns
in Angelman’s syndrome. J Child Neurol.
1995;10:467-1
Williams CA, Beaudet AL, Clayton-Smith J, Knoll
JH, Kyllerman M, Laan LA, Magenis RE,
Moncla A, Schinzel AA, Summers JA, Wagstaff
J. Angelman syndrome. Am J Med Genet A.
2006;140:413-8
Williams CA.The behavioral phenotype of the
Angelman syndrome. Am J Med Genet Part C
Semin Med Genet. 2010;154:432-7
Witte W, Nobel C, Hilpert J. Anesthesia and Angelman syndrome. Anaesthesist. 2011;60:63340
Zori RT, Hendrickson J, Woolven S, Whidden EM,
Gray B, Williams CA. Angelman syndrome:
clinical profile. J Child Neurol. 1992;7:270-80
www.buponline.com
www.buponline.com
10. Sindrome di Prader Willi
Sinonimi: Sindrome di Prader-Labhar-Willi
Codice ICD 10: Q87.1
I. Cremonesi, S. Bagattoni, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
GENETICA E DIAGNOSI
La Sindrome di Prader-Willi (SPW), descritta nel 1956 da Andrea Prader, Heinrich Willi
e Alexis Labhart, è una malattia cromosomica
rara caratterizzata da disfunzioni del sistema
ipotalamo-ipofisario, dismorfia facciale caratteristica, mani e piedi piccoli, grave ipotonia nel periodo neonatale e nei primi due anni
di vita, iperfagia dopo i 3 anni, bassa statura,
ipogonadismo, difficoltà di apprendimento e
disturbi psico-comportamentali anche gravi
(Prader A et al., 1956). La SPW è la causa più
frequente di obesità di origine sindromica (Jin
DK, 2011).
L’incidenza è stimata di 1 caso su 25.000 nati
vivi per anno, in assenza di predilezione razziale,
etnica, geografica e di genere (Jin DK, 2011).
I pazienti affetti presentano un indice di mortalità del 3% indipendentemente dall’età; le cause
di decesso più frequenti sono insufficienza respiratoria nell’infanzia e patologie correlate all’obesità in età adulta (Jin DK, 2011; Butler JV et
al., 2002).
La SPW è ascrivibile ad alterazioni della regione
15q11-q13 del braccio lungo del cromosoma 15.
Nel 70% dei casi è causata dalla delezione della
porzione 15q11-q13 del cromosoma 15 di origine
paterna e nel 25-30% da disomia uniparenterale materna. In meno del 2% dei casi si riscontra
una microdelezione, non identificabile con la citogenetica, che interessa un gene di controllo della
regione 15q11-q13 del cromosoma 15 paterno.
La maggior parte dei casi sono causati da mutazioni accidentali e non comportano il rischio di
ricorrenza in gravidanze successive (Goldstone AP,
2004; Nicholls RD et al., 1989).
La diagnosi, primariamente clinica e basata sulle
caratteristiche fenotipiche, viene confermata dai
test genetici.
La diagnosi prenatale eseguita su villocentesi o
amniocentesi con test di biologia molecolare (Test
di Metilazione, Metylation Specific PCR, Fluorescence In Situ Hibridization) è indicata in caso di
posizione anomala di mani e piedi del feto, di
diminuzione dei movimenti fetali e di polidramnios (Bigi N et al., 2008).
www.buponline.com
110
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
MANIFESTAZIONI CLINICHE SISTEMICHE
Le caratteristiche somatiche comuni ai pazienti affetti da SPW sono molte, nonostante
la variabilità presente tra i vari individui. Nella
Tabella 1 sono descritti i criteri clinici e anamnestici utilizzati per la diagnosi (Gunay-Aygun
M et al., 2001; Holm VA et al., 1993).
Alla nascita, spesso pretermine, il quadro clinico è caratterizzato da marcata ipotonia muscolare responsabile, nei primi anni di vita, di problemi di suzione e deglutizione e di importanti
difficoltà nell’alimentazione, che necessitano
di periodi prolungati di ospedalizzazione. Nonostante l’ipotonia muscolare tenda ad attenuarsi,
l’acquisizione della deambulazione è ritardata
(in media a 24 mesi) e persistono ridotta massa muscolare, affaticamento muscolare, goffaggine nei movimenti, disartrie, difficoltà di
masticazione e di respirazione, responsabili di
infezioni respiratorie ricorrenti durante l’infanzia (Diene G et al., 2007; Gunay-Aygun M et
al., 2001).
Verso i 3 anni si manifesta iperfagia da appetito
insaziabile, probabilmente causata da alterato
funzionamento del centro ipotalamico di regolazione della sazietà. L’aumento di appetito incontrollato e continuo, associato a ridotta sensazione di sazietà, spinge il paziente ad ingerire in
modo ossessivo-compulsivo tutto quanto possa
reperire. L’iperfagia e lo scarso consumo energetico causato dall’ipotonia sono potenziali cause
di obesità di grado elevato, particolarmente resistente al trattamento dietetico e farmacologico.
L’obesità è spesso responsabile di gravi complicanze cardiovascolari, respiratorie e metaboliche, in particolare diabete mellito di tipo 2 e
dislipidemie (Dykens EM, 2000).
Anomalie endocrine correlate al deficit ipotalamo-ipofisario possono essere alla base di sviluppo puberale lento ed incompleto, in particola-
re nei maschi (Eiholzer U et al., 2006) e di bassa
statura da deficit dell’ormone della crescita
(GH), descritta in circa il 50% dei casi (Wattendorf DJ et al., 2005; Burman PE et al., 2001).
Il deficit cognitivo è estremamente variabile e
si associa a difficoltà nell’apprendimento e nel
linguaggio, in particolare nei soggetti colpiti da
delezione (Whittington J et al., 2004).
I bambini con SPW hanno un carattere gioviale
ed allegro. Quadri di nevrosi e/o psicosi sono
di raro riscontro. Sono invece frequenti, in particolare nei casi di disomia di origine materna,
disturbi psico-comportamentali non precisamente tipizzabili ma caratteristici per intensità
e modalità di espressione, quali crisi di collera
incontrollata in seguito ad episodi banali come
restrizioni alimentari, che evolvono in atti di
violenza contro cose e persone, comportamenti
ossessivo-compulsivi con ripetizione di parole,
tendenza alla depressione, comportamenti autolesionisti come procurarsi escoriazioni cutanee (Diene G et al., 2007).
Lesioni cutanee da grattamento, che compaiono generalmente a partire dai sei anni di età
e tendono a cronicizzare e ad aggravarsi per la
fragilità vascolare e per l’alta soglia del dolore
caratteristiche della SPW, possono essere particolarmente invalidanti (Diene G et al., 2007).
Nei pazienti con SPW sono frequenti apnee
ostruttive notturne (OSAS) di origine centrale
e periferica (grave obesità e ipotonia dei muscoli della gabbia toracica), responsabili di insufficiente ventilazione polmonare, con disturbi del
sonno e sonnolenza durante il giorno (Nixon
GM et al., 2002).
I pazienti affetti da SPW presentano alterazioni
posturali: la scoliosi può insorgere anche molto precocemente mentre la cifosi, che spesso
accompagna la scoliosi, compare più frequentemente nell’adolescenza o nell’età adulta. La prevalenza di scoliosi e di lussazione delle anche è
www.buponline.com
111
Sindrome di Prader Willi
Tabella 1: Criteri clinici e anamnestici per la diagnosi di SPW
1.
Criteri maggiori
Ipotonia neonatale (di origine centrale)
Problemi di alimentazione nella prima infanzia
Iperfagia, ricerca ossessiva di cibo, obesità dopo i 3 anni
Tratti somatici caratteristici
Ipogonadismo
Deficit mentale e ritardo dello sviluppo psicomotorio
2.
Criteri minori
Riduzione dei movimenti, letargia, pianto debole nel neonato
Caratteristiche comportamentali
Disturbi del sonno
Bassa statura
Ipopigmentazioni
Acromicria (mani e piedi piccoli)
Mani affusolate con margine ulnare rettilineo
Anomalie oculari
Saliva densa e viscosa, cheilite angolare
Difetti nell’articolazione delle parole
Lesioni cutanee da grattamento
Criteri aggiuntivi
Elevata soglia del dolore
Diminuito senso del vomito
Alterazioni della termoregolazione
Scoliosi
Cifosi
Adrenarca precoce
Osteoporosi
Abilità nei giochi di pazienza (tipo puzzle)
www.buponline.com
112
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
cinque volte superiore rispetto alla popolazione
generale (Diene G et al., 2007).
Sul piano oftalmologico sono frequenti strabismo, miopia, astigmatismo e ipermetropia
(Diene G et al., 2007).
CARATTERISTICHE ORO-FACCIALI
I pazienti affetti da SPW presentano una facies
caratteristica con fronte piatta e stretta, rima
palpebrale rivolta verso l’alto, occhi a mandorla, strabismo, ponte nasale stretto, micrognazia,
labbro superiore sottile con angoli rivolti verso il
basso (bocca a cappello di gendarme) (Figura 1)
(Jin DK, 2011; Wattendorf DJ et al., 2005).
Per quanto concerne le anomalie dentali, in letteratura sono descritte anomalie di numero (elementi soprannumerari), di dimensione (microdonzia), di forma (taurodonzia), di struttura
(ipoplasie dello smalto) e di eruzione (ritardo
nelle permute) (Atar M et al., 2010; Scardina GA
et al., 2007; Banks et al., 1996; Bassarelli V et al.,
1991).
Molti pazienti affetti da SPW presentano xerostomia, causata da diminuito flusso salivare da disfunzione delle ghiandole salivari maggiori e aggravata
dalla respirazione orale. È alterata anche la composizione della saliva, caratterizzata da alti livelli
di ioni e proteine, in particolare mucine, che ne
determinano una consistenza molto viscosa (Scardina GA et al., 2007; Butler MG et al., 2004; Boer
H et al., 2002; Cummings DE et al., 2002; Young
W et al., 2001).
L’alterazione quantitativa e qualitativa della saliva, la scarsa igiene orale causata dalla ridotta
abilità motoria e le abitudini alimentari scorrette
sono le principali cause dell’elevata prevalenza di
lesioni cariose e di erosioni dello smalto (Figura
2) (Scardina GA et al., 2007; Bots CP et al., 2004;
Young W et al., 2001; Salako NO et al., 1995).
Figura 1: Bambino di 2 anni affetto da SPW; marcata ipotonia muscolare a livello del volto con evidenti screpolature
agli angoli della bocca
Tuttavia, due studi recenti, condotti rispettivamente su cinquanta e su quindici pazienti affetti da
SPW di età compresa tra 3 e 40 anni, evidenziano
bassi livelli di carie correlabili all’attuazione di un
programma di promozione della salute orale mirato (Saeves R et al., 2011; Bailleul-Forestier I et
al., 2008).
In età adulta sono descritte erosioni dello smalto
correlate ad abbassamento del pH salivare causato
da assunzione frequente e ripetuta di cibo (Saeves
R et al., 2012; Bots CP et al., 2004).
Per quanto riguarda le patologie parodontali sono
www.buponline.com
113
Sindrome di Prader Willi
Figura 2: Erosioni e ipoplasie dello smalto; quadro di ECC
descritti quadri di parodontite giovanile aggressiva
in due case report, in un ragazzo di 20 anni e in
una ragazza di 12 anni (Yanagita M et al., 2011;
Greenwood RE et al., 1990). Nel case report del
paziente di 20 anni gli autori correlano la parodontopatia con la presenza di trauma occlusale e di
severa malocclusione.
Per quanto riguarda le patologie di pertinenza
ortopedico-ortodontica, la respirazione orale,
l’ipotonia muscolare e le alterazioni posturali possono influenzare negativamente lo sviluppo craniofacciale. In letteratura non sono tuttavia presenti
dati relativi a patologie ortopedico/ortodontiche
associate alla SPW. L’esperienza clinica evidenzia
che nei pazienti affetti da SPW sono di frequente riscontro patologie ortopedico-ortodontiche,
in particolare deficit trasversale del mascellare
superiore con presenza di palato ogivale, correlabile alle intercorrenti infezioni respiratorie durante
l’infanzia.
LINEE GUIDA DI TERAPIA
I pazienti con SPW sono affetti da numerose alterazioni e patologie che rendono necessari interventi interdisciplinari. La diagnosi precoce, il follow-
up e le terapie hanno migliorato sensibilmente la
qualità della vita di questi bambini, in particolare
prevenendo i rischi legati all’obesità e riducendo i
disturbi dell’apprendimento e del linguaggio.
Nei primi mesi di vita l’ipotonia muscolare può
rendere impossibile l’allattamento al seno; qualora
non si riesca ad attuare un’alimentazione per os si
ricorre al sondino nasogastrico.
Durante tutta la vita del paziente è indispensabile
uno stretto controllo del regime alimentare. Nei
primi anni di vita è necessario evitare carenze di
apporto calorico a causa delle difficoltà di alimentazione. A partire dai tre anni è necessaria una dieta
bilanciata e corretta per evitare le problematiche
legate all’obesità causata dagli eccessi alimentari da
appetito insaziabile.
Nei maschi l’ipogonadismo può essere trattato
nella prima infanzia chirurgicamente.
In entrambi i sessi la persistenza di bassi tassi di
ormoni sessuali durante la pubertà richiede una
terapia sostitutiva (Eiholzer U et al., 2006).
Per quanto concerne il deficit di GH, il trattamento sostitutivo permette un’accelerazione della velocità di crescita nell’adolescente, una diminuzione della massa grassa e una statura più alta
nell’adulto (Jin DK, 2011; Butler MG et al., 2004;
Carrel AL et al., 2002; Burman PE et al., 2001).
In letteratura sono riportati casi di decesso di bambini con SPW trattati con GH che presentavano
all’anamnesi medica uno o più fattori di rischio:
obesità severa, pregressi episodi di insufficienza respiratoria, apnee ostruttive del sonno, infezioni respiratorie non specificate. I pochi dati recenti della
letteratura sembrano evidenziare che il trattamento
con GH non sia causa di aggravamento delle apnee ostruttive ma che ne determini una diminuzione del numero di episodi. Tuttavia, in attesa di
dati più precisi, l’indicazione attuale è improntata
alla cautela: la terapia con GH è controindicata
in presenza di apnee severe non trattate, diabete
mellito non controllato, obesità severa, patologie
www.buponline.com
114
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
neoplastiche e psicosi. In corso di terapia con GH
la funzione respiratoria deve essere sottoposta a
monitoraggio costante; qualora insorgano alterazioni della funzione respiratoria, la terapia deve
essere sospesa (Deal CL et al., 2013; Haqq AM
et al., 2003).
In relazione alle ridotte abilità motorie orali, è
necessario un intervento logopedico precoce
anche prima dell’età dell’acquisizione del linguaggio, spesso ritardata, nell’obiettivo di stimolare la
motricità buccofacciale e di migliorare la deglutizione.
Per la terapia dei disturbi psico-comportamentali, in particolare dei disturbi ossessivo-compulsivi, vengono utilizzati farmaci che hanno come
effetto collaterale xerostomia.
Per quanto concerne la patologia cariosa, in
un’ottica di interdisciplinarietà, è compito del
pediatra, dal momento in cui si effettua la diagnosi, inviare il paziente all’odontoiatra infantile
perché informi la famiglia sull’importanza della
salute orale e sulla necessità di interventi di promozione della salute orale (igiene orale domiciliare, alimentazione non cariogenica, utilizzo di
fluoro topico e, quando indicato, sistemico, visite
periodiche trimestrali/semestrali, sigillatura dei
solchi, delle fessure e dei fori ciechi dei molari
permanenti).
I deficit di tono muscolare e di coordinazione
motoria rendono difficile una gestione autonoma dell’igiene orale, che deve essere sempre
assistita dai familiari. La forte propensione alla
percezione visiva caratteristica di molti pazienti
affetti da SPW può essere utilizzata come via di
comunicazione preferenziale per impartire istruzioni di igiene orale domiciliare.
In considerazione delle caratteristiche psicocomportamentali della SPW il trattamento
odontoiatrico è spesso difficoltoso a causa dello scarso livello di collaborazione. Nei pazienti
con SPW è importante utilizzare un approccio
psicologico e comportamentale mirato ad individuare i canali comunicativi più efficaci per conquistare la fiducia e la collaborazione del paziente;
per raggiungere questo obiettivo, di grande utilità
è il colloquio preliminare con la famiglia.
Qualora nel corso della seduta odontoiatrica subentri una crisi di collera nessun intervento né
verbale, né di negoziazione, né di punizione, permette di interrompere la crisi; il paziente va fatto
alzare dalla poltrona, isolato in un ambiente tranquillo e sorvegliato perché non si procuri lesioni.
Le crisi sono solitamente di breve durata; quando
il paziente è ritornato calmo è necessario ricercare
con lui la causa scatenante, per evitare che la crisi
si ripresenti.
Solo in caso di situazioni particolarmente complesse è necessario ricorrere all’anestesia generale
(AG). L’intervento in AG deve essere considerato
come il punto di partenza per un programma di
controlli periodici ravvicinati, nel corso dei quali
attuare igiene orale professionale e intercettare le
lesioni allo stadio iniziali, quando le terapie sono
più semplici quindi più accettate.
Per quanto riguarda le patologie di tipo ortopedico-ortodontico, è importante stabilire un
protocollo diagnostico precoce nell’obiettivo di
attuare un piano di trattamento efficace, in particolare nei casi di OSAS, che i bambini affetti da
SPW presentano con elevata frequenza. In questi
casi va valutato, in collaborazione con l’otorinolaringoiatra, il ruolo etiopatogenetico del deficit
dei diametri palatali, dell’ipotonia, dell’obesità.
Stabilita la necessità di espansione palatale si utilizza un’apparecchiatura ortopedica, l’espansore
rapido del palato, che sfrutta l’immaturità suturale per indurre la crescita trasversale del mascellare superiore sul piano trasversale, determinando
l’abbassamento della volta palatina e l’aumento
volumetrico delle fosse nasali, situazioni anatomiche che favoriscono la respirazione nasale.
Il tipo di malocclusione più frequente è la II clas-
www.buponline.com
115
Sindrome di Prader Willi
se da retrusione mandibolare, spesso correlata a
respirazione orale nei casi in cui trovi indicazione
un approccio di tipo funzionale, nel paziente
in terapia con GH, quindi con crescita indotta,
l’ortodontista deve razionalizzare il timing d’intervento, operando in sinergia con l’auxologo per
sfruttare l’epoca del picco di crescita ossea.
La finalizzazione ortodontica a permuta completata, che fa seguito alla fase ortopedico-funzionale, in questi pazienti è spesso difficoltosa a causa
della scarsa collaborazione, in particolare all’igiene orale domiciliare.
Per quanto concerne le lesioni dentali di origine
traumatica, pur non esistendo in letteratura alcun riferimento specifico, considerando il deficit
di tono muscolare e di coordinazione motoria, è
ipotizzabile una loro elevata prevalenza a livello
degli elementi sia decidui che permanenti; è quindi necessario fornire alla famiglia linee guida per
la loro gestione, in particolare sull’opportunità
del reimpianto immediato in caso di avulsione di
denti permanenti.
BIBLIOGRAFIA
Atar M, Körperich EJ. Systemic disorders and
their influence on the development of dental hard tissues: a literature review. J Dent.
2010;38:296-306
Bailleul-Forestier I, Verhaeghe V, Fryns JP, Vinckier
F, Declerck D, Vogels A. The oro-dental phenotype in Prader-Willi syndrome: a survey of
15 patients. Int J Paediatr Dent. 2008;18:40-7
Banks PA, Bradley JC, Smith A. Prader–Willi
syndrome–a case report of the multidisciplinary management of the orofacial problems. Br J
Orthod. 1996;23:299-304
Bassarelli V, Baccetti T, Bassarelli T, Franchi L. The
dentomaxillofacial characteristics of the Prader-Labhart-Willi syndrome. A clinical case re-
port. Minerva Stomatol. 1991;40:811-9
Bigi N, Faure JM, Coubes C, Puechberty J, Lefort
G, Sarda P, Blanchet P. Prader-Willi syndrome:
is there a recognizable fetal phenotype? Prenat
Diagn. 2008;28:796-9
Boer H, Holland A, Whittington J, Butler J, Webb
T, Clarke D. Psychotic illness in people with
Prader Willi syndrome due to chromosome
15 maternal uniparental disomy. Lancet.
2002;359:135-6
Bots CP, Schueler YT, Brand HS, van Nieuw Amerongen A. A patient with Prader-Willi syndrome. Characteristics, oral consequences and
treatment options. Ned Tijdschr Tandheelkd.
2004;111:55-8
Burman PE, Ritzén M, Lindgren AC. Endocrine
dysfunction in Prader- Willi syndrome: a review with special reference to GH. Endocr Rev.
2001;22:787-99
Butler JV, Whittington JE, Holland AJ, Boer H,
Clarke D, Webb T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study.
Dev MedChild Neurol. 2002;44:248-55
Butler MG, Bittel DC, Kibiryeva N, Talebizadeh
Z, Thompson T. Behavioral differences among
subjects with Prader-Willi syndrome and type I
or type II deletion and maternal disomy. Pediatrics. 2004;113:565-73
Carrel AL, Myers SE, Whitman BY, Allen DB.
Benefits of long-term GH therapy in Prader-Willi syndrome: a 4-year study. J Clin Endocrinol Metab. 2002;87:1581-5
Cummings DE, Clement K, Purnell JQ, et al. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med. 2002;8:643-4
Deal CL, Tony M, Höybye C, Allen DB, Tauber M, Christiansen JS; the 2011 GH in PWS
Clinical Care Guidelines Workshop Participants. Growth Hormone Research Society
Workshop Summary: Consensus Guidelines
www.buponline.com
116
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
for Recombinant Human Growth Hormone
Therapy in Prader-Willi Syndrome. J Clin Endocrinol Metab. 2013 first published ahead of
print March 29, 2013 as doi:10.1210/jc.20123888
Diene G, Postel-Vinay A, Pinto G, Polak M, Tauber M. The Prader-Willi syndrome. Ann Endocrinol. 2007;68:129-37
Dykens EM. Contaminated and unusual food
combinations: what do people with Prader-Willi syndrome choose? Ment Retard.
2000;38:163-71
Eiholzer U, l’Allemand DE, Rousson V, et al.
Hypothalamic and gonadal components of
hypogonadism in boys with Prader-Labhart-Willi syndrome. J Clin Endocrinol Metab.
2006;91:892-8
Goldstone AP. Prader-Willi syndrome: advances
in genetics, pathophysiology and treatment.
Trends Endocrinol Metab. 2004;15:12-20
Greenwood RE, Small IC. Case report of the
Prader-Willi syndrome. J Clin Periodontol.
1990;17:61-3
Gunay-Aygun M, Schwartz S, Heeger S, O’Riordan MA, Cassidy SB. The changing purpose
of Prader-Willi syndrome clinical diagnosis criteria and proposed revised criteria. Pediatrics.
2001;108:92
Haqq AM, Stadler DD, Jackson RH, Rosenfeld RG, Purnell JQ, LaFranchi SH. Effects
of growth hormone on pulmonary function,
sleep quality, behavior, cognition, growth velocity, body composition, and resting energy
expenditure in Prader-Willi syndrome. J Clin
Endocrinol Metab. 2003;88:2206-12
Holm VA, Cassidy SB, Butler MG, Hanchett JM,
Greenswag LR, Whitman BY, Greenberg F:
Prader-Willi syndrome: consensus diagnostic
criteria. Pediatrics. 1993;91:398-402
Jin DK. Systematic review of the clinical and genetic aspects of Prader-Willi syndrome. Korean
J Pediatr. 2011;54:55-63
Nicholls RD, Knoll JH, Butler MG, Karam S,
Lalande M. Genetic imprinting suggested by
maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature.1989;342:281-5
Nixon GM, Brouilette RT. Sleep and breathing
in Prader-Willi syndrome. Pediatr Pulmonol.
2002;34:209-17
Prader A, Labhart A, Willi H. Ein syndrom von
adipositas, klein-wuchs, krytorchidismus und
oligophrenie nach myotonieartigem zustand
im neugeborenenalter. Schweiz Med Wochenshr. 1956;86:1260-1
Saeves R, Espelid I, Storhaug K, Sandvik L,
Nordgarden H. Severe tooth wear in Prader-Willi syndrome. A case-control study.
BMC Oral Health. 2012;12:12
Saeves R, Nordgarden H, Storhaug K, Sandvik L,
Espelid I. Salivary flow rate and oral findings
in Prader-Willi syndrome: a case-control study.
Int J Paediatr Dent. 2011;22:27-36
Salako NO, Ghafouri HM. Oral findings in a child
with Prader-Labhart-Willi syndrome. Quintessence Int. 1995;26:339-41
Scardina GA, Fucà G, Messina P. Oral diseases in
a patient affected with Prader-Willi syndrome.
Eur J Paediatr Dent. 2007;8:96-9
Wattendorf DJ, Muenke M. Prader-Willi syndrome. Am Fam Physician. 2005;72:827-30
Whittington J, Holland A, Webb T, Butler J,
Clarke D, Boer H. Cognitive abilities and
genotype in a population-based sample people
with Prader-Willi syndrome. J Intellect Disabil
Res. 2004;48:172-87
Yanagita, M., Hirano, H., Kobashi, M., Nozaki,
T., Yamada, S., Kitamura, M. Periodontal disease in a patient with Prader-Willi syndrome: a
case report. Journal of Medical Case Reports.
2011;5:329
Young W, Khan F, Brandt R, Savage N, Razek
AA, Huang Q. Syndromes with salivary dys-
www.buponline.com
117
Sindrome di Prader Willi
function predispose to tooth wear. Case reports
of congenital dysfunction of major salivary
glands, Prader-Willi, congenital rubella, and
Sjogren’s syndromes. Oral Surg Oral Med Oral
Pathol Oral Radiol Endod. 2001;92:38-48
www.buponline.com
www.buponline.com
11. Sindrome di Silver Russel
Sinonimo: nanismo Silver-Russell
Codice ICD 10: Q87.1
N. Alkhamis, F. Skendo, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Sindrome di Silver-Russel (SSR), descritta da
Silver nel 1953 e da Russel nel 1954, è una malattia
cromosomica rara caratterizzata da ritardo di crescita ad esordio prenatale, asimmetria degli arti, macrocefalia e facies caratteristica (Saal HM, 2002).
L’incidenza è stimata di 1 su 100.000 nati vivi per
anno, in assenza di predilezione razziale, etnica,
geografica e di genere (Eggermann T et al., 2010;
Christoforidis A et al., 2005).
La prognosi a lungo termine è buona.
GENETICA E DIAGNOSI
La SSR è una condizione geneticamente eterogenea con un fenotipo tipico ma variabile.
In circa il 50% dei pazienti che presentano le caratteristiche della SSR sono presenti anomalie cromosomiche: nel 35-50% ipometilazione a livello
della regione 11p15.5 del cromosoma 11 paterno e
nel 10% disomia uniparentale materna del cromosoma 7 (UPD7).
Sono descritti modelli ereditari autosomici dominanti e autosomici recessivi ma si tratta prevalen-
temente di casi sporadici la cui eziologia è sconosciuta. (Eggermann T et al., 2010; Abu-Amero S et
al., 2008; Saal HM, 2002).
La diagnosi prenatale non è di solito possibile.
Nelle gravidanze in cui venga identificato mediante
ecografia un ritardo di crescita intrauterino del feto
(ma il ritardo di crescita intrauterino spesso non
può essere identificato in maniera soddisfacente
fino al terzo trimestre) possono essere eseguiti test
genetici (Saal HM, 2011).
La diagnosi è prevalentemente clinica e si basa
sulle manifestazioni fenotipiche, in primo luogo il
ritardo di crescita valutato in rapporto alla circonferenza cranica nella norma. Test genetici di metilazione e delezione/duplicazione possono fornire
una conferma alla diagnosi di SSR (Eggermann
T et al., 2010). La diagnosi differenziale si pone
con il ritardo di crescita prenatale da disfunzione
placentare, con la progeria neonatale (sindrome di
Wiedemann-Rautenstrauch), con la sindrome 3M,
con il nanismo muliebre (Saal HM, 2002).
I criteri maggiori, minori e di supporto che guidano il clinico nella diagnosi di SSR sono indicati
nella Tabella n. 1 (Wakeling EL et al., 2010; Bartholdi D et al., 2009; Eggermann T et al. 2009;
Netchine I et al., 2007).
www.buponline.com
120
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Tabella 1: Criteri diagnostici della SSR
Criteri maggiori
Ritardo nella crescita intrauterina / Riduzione della crescita intrauterina < 10° percentile
Riduzione della crescita postatale: peso / lunghezza del neonato < 3° percentile
Circonferenza del cranio nella norma
Asimmetrie degli arti, del corpo e/o del viso
Criteri minori
Braccia corte con rapporto braccio-avambraccio normale
Clinodattilia del 5° dito
Forma del viso triangolare
Bozze frontali / Fronte prominente
Criteri di supporto
Macchie caffelatte o alterazioni della pigmentazione della pelle
Anomalie genito-urinarie (criptorchidismo, ipospadia)
Ritardo nelle funzioni motorie, cognitive e/o del linguaggio
Disturbi dell’alimentazione
Ipoglicemia
MANIFESTAZIONI CLINICHE SISTEMICHE
Le manifestazioni sistemiche più frequenti nei
soggetti con SSR sono riportate nella Tabella 2
(Abu-Amero S et al., 2008).
La SSR è caratterizzata da severo ritardo della
crescita intrauterino, da scarsa crescita postnatale e da ritardata maturazione ossea; sia il
peso alla nascita che la crescita postnatale sono
significativamente inferiori rispetto alla media
(< 2 deviazioni standard). Gli individui affetti da SSR hanno bassa statura in età adulta:
se non trattati con ormone della crescita (GH)
i maschi raggiungono in media un’altezza di
151,2±7,8 cm e le femmine di 139,9±9 cm (Saal
HM, 2002; Price SM et al., 1999). Tuttavia solo
in alcuni pazienti è descritto un deficit di GH.
Il peso è maggiormente compromesso rispetto
alla statura per scarsezza del pannicolo adiposo
sottocutaneo. Spesso è presente scarso appetito, per cui la crescita è ulteriormente rallentata
e i soggetti sono a rischio di ipoglicemia.
Nel 60-80% dei casi è descritta asimmetria
nella lunghezza degli arti, di solito parziale e
non progressiva. Displasia dell’anca, scoliosi e
dislocazioni a carico delle articolazioni del ginocchio e del gomito, anomalie scheletriche a
carico delle mani e dei piedi (brachidattilia e/o
clinodattilia del quinto dito) sono manifestazioni frequenti (Lahiri A et al., 2009; Abrahm E et
al., 2004; Price SM et al., 1999).
Patologie gastrointestinali sono frequenti (nel
77% dei casi), in particolare reflusso gastroesofageo (RGE) ed esofagiti (Saal HM et al., 2002;
Anoderson J et al., 2002).
Nella maggior parte degli individui affetti lo
sviluppo sessuale secondario e la pubertà
sono nella norma. I maschi possono presentare
www.buponline.com
121
Sindrome di Silver Russel
Tabella 2: Manifestazioni cliniche della sindrome di Silver-Russel
Caratteristiche fisiche
Facies caratteristica con viso triangolare e piccolo, fronte alta, mento appuntito, angoli della bocca rivolti
verso il basso
Bassa statura
Dismorfismi scheletrici: asimmetrie scheletriche, displasia dell’anca, scoliosi, dislocazioni a carico delle
articolazioni del ginocchio e del gomito, clinodattilia, campodattilia con artrogriposi distale, sindattilia tra
secondo e terzo dito
Dismorfismi urogenitali: ipospadia, valvole uretrali posteriori, ernia inguinale
Caratteristiche cliniche
Peso alla nascita inferiore di 2 deviazioni standard rispetto alla media
Scarsa crescita
Scarso controllo della postura della testa durante l’infanzia, dovuto alla relativa grandezza del cranio
rispetto al corpo
Disfunzioni motorie da mancanza di forza e di massa muscolare
Difficoltà di alimentazione durante l’infanzia responsabili di ipoglicemia a digiuno
Cospicua sudorazione durante l’infanzia
Patologie gastrointestinali: reflusso gastroesofageo, esofagiti, avversione verso il cibo
Sviluppo psico-motorio ritardato spesso associato a difficoltà nell’apprendimento
Ritardata età ossea
Neoplasie
anomalie genitali, in particolare criptorchidismo e ipospadia.
Sono descritti rari casi di pazienti con SSR affetti da patologie renali: idronefrosi, acidosi
renale tubulare, valvole uretrali posteriori (Saal
HM et al., 2002; Price SM et al., 1999).
È frequente un ritardo dello sviluppo motorio
e cognitivo e, pur risultando il QI normale o
non significativamente più basso rispetto alla
media, sono frequenti difficoltà nell’apprendimento (Noeker M et al., 2004). Circa la metà
dei pazienti affetti da SSR necessita di terapie
logopediche e circa 1/3 di supporto scolastico
(Lai K et al., 1994).
Il rischio di insorgenza di neoplasie non risulta
aumentato, tuttavia in letteratura sono riportati
casi di tumore di Wilms, di carcinoma epato-
cellulare e di craniofaringioma associati a SSR
(Brukheimer E et al., 1993; Chitayat D et al.,
1988; Draznin MB et al., 1980).
CARATTERISTICHE ORO-FACCIALI
Alla nascita la circonferenza cranica è nella
norma, mentre altezza e peso sono inferiori
alla norma, conferendo al neonato un aspetto
pseudoidrocefalico. (Perkins RM et al., 2002).
I bambini affetti da SSR hanno una facies caratteristica con viso triangolare e piccolo, fronte
larga e prominente, mento piccolo e appuntito, bocca larga, labbra sottili con angoli rivolti verso il basso, occhi grandi e sclere blu.
Tali caratteristiche tendono a diventare sfumate
www.buponline.com
122
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
in età adulta. Sono descritte asimmetrie a livello della faccia (Bergman A et al., 2003), delle
labbra e dei denti (Kotilainen J et al., 1995)
(Figura 1 a, b, c).
Il 7% dei pazienti affetti da SSR presenta palatoschisi e ugola bifida (Wakeling EL et al.,
2010).
Per quanto concerne le anomalie dentarie, in
letteratura è reperibile solo un esiguo numero di
studi su pazienti affetti da SSR che evidenziano
la presenza di anomalie di eruzione, quali eruzione ritardata (Bergman A et al., 2003; Kotilainen J et al., 1995) e anticipata (Rubenstein LK
et al., 1988), di numero, quali molari decidui
soprannumerari (Bedi R et al., 1991) e ageneb
a
c
Figura 1 a, ,b, c: Ragazza di 11anni affetta da SSR. Facies caratteristica: viso triangolare, piccolo e asimmetrico, fronte larga
e prominente, labbra sottili, mento piccolo e appuntito
www.buponline.com
123
Sindrome di Silver Russel
sia dei secondi premolari permanenti (Figura 2)
(Cullen CL et al., 1987); di forma quali microdonzia (Kulkarni ML et al., 1995; Cullen CL et
al., 1987); di struttura quali ipoplasie dello smalto della dentatura decidua e permanente (Bergman A et al., 2003; Kotilainen J et al., 1995).
Nei pazienti affetti da SSR è riportata un’elevata
prevalenza di patologie cariosa e parodontale,
correlate ad una scarsa igiene orale domiciliare
(Rubenstein LK et al., 1988; Cullen CL et al.,
1987). L’esperienza clinica evidenzia che all’elevato rischio di patologia cariosa contribuiscono
la frequente assunzione di zuccheri per contrastare gli episodi di ipoglicemia, le ipoplasie dello
smalto e la presenza di RGE.
Per quanto concerne le patologie ortopedicoortodontiche (Figure 3, 4, 5, 6 a, b, c, d) in
letteratura sono riportati:
• affollamento dentale in mandibola piccola
(Bergman A et al., 2003; Kisnisci RS et al.,
1999; Kotilainen J et al., 1995; Cullen CL
et al., 1987);
• II classe scheletrica, aumentati valori di
overjet e overbite, ipodivergenza (Bergman
A et al., 2003);
Figura 2: Ortopantomografia: asimmetria dei rami orizzontali e verticali della mandibola, affollamento endosseo
superiore con tendenza all’inclusione di 13 e 23; agenesia
del secondo premolare inferiore di sinistra
•
•
•
altezza facciale, ampiezza cranica e facciale
e lunghezza della base cranica ridotte (Kotilainen J et al., 1995; Cullen CL et al.,
1987);
palato ogivale (Bergman A et al., 2003; Cullen CL et al., 1987);
condili smussati (Cullen CL et al., 1987).
LINEE GUIDA DI TERAPIA
I pazienti con SSR sono affetti da numerose patologie che rendono necessari interventi multispecialistici. La diagnosi, le terapie e il follow-up
richiedono la presenza di un team multi e interdisciplinare adeguatamente formato sulla sindrome.
Per quanto riguarda lo sviluppo neuromotorio,
i problemi connessi all’ipotonia debbono essere
trattati dai primi mesi di vita con interventi fisioterapici.
Figura 3: Teleradiografia latero-laterale: seconda classe scheletrica da ipoplasia mandibolare, note dismorfiche
www.buponline.com
124
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Figura 4: Teleradiografia antero-posteriore: sul piano frontale asimmetria cranio-facciale che origina dalla base cranica con deviazione mandibolare verso il lato sinistro
Figura 5: Visione frontale intraorale. Gengivite marginale
diffusa
Problemi dello sviluppo cognitivo, deficit di
linguaggio e difficoltà di apprendimento debbono essere intercettati e trattati tempestivamente da psicologi, logopedisti e insegnanti
di sostegno per migliorare l’inserimento socia-
le del bambino (Saal HM, 2002; Lai K et al.,
1994).
Il RGE richiede la posizione eretta del bambino
durante e dopo i pasti e, nei casi più gravi, l’utilizzo di farmaci anti-reflusso (Gaviscon, Ranitidina).
Nei casi in cui la terapia farmacologia non abbia
effetto può rendersi necessario l’intervento chirurgico (fundoplicatio).
L’asimmetria delle estremità inferiori richiede un intervento quando la differenza nella
lunghezza degli arti è superiore a 3 centimetri.
Durante la crescita può essere usato uno spessore nella scarpa dell’arto più corto; a crescita
terminata devono essere prese in considerazione terapie chirurgiche (epifisiodesi, distrazione
osteogenetica). Anche le anomalie delle mani e
dei piedi possono essere risolte chirurgicamente.
(Lahiri A et al., 2009; Saal HM, 2002; Abrahm
E et al., 2004).
In relazione alle anomalie genitali, è importante
un consulto urologico per il trattamento chirurgico
del criptorchidismo, quando non risolto spontaneamente entro il primo anno di età.
La terapia con ormone della crescita (GH), frequentemente prescritta come parte del trattamento
della SSR, viene effettuata tramite iniezione giornaliera dall’età di 2 anni fino all’adolescenza; sembra dare risultati positivi anche nei soggetti senza
deficit di GH determinando un aumento della crescita staturale (Saal HM, 2002). Consentendo al
bambino di avere un’altezza normale al momento
dell’inserimento scolastico, ne rafforza l’autostima e favorisce le relazioni interpersonali; inoltre,
determinando un aumento del metabolismo, aumenta l’appetito e induce il bambino a mangiare
cibo ad intervalli regolari. L’assunzione di GH non
corregge le asimmetrie del corpo e degli arti (Rizzo
V. et al., 2001).
L’ipoglicemia, soprattutto notturna, viene contrastata con frequenti merende a base di zuccheri,
fattori di rischio per lo sviluppo di carie (Mazzanti
www.buponline.com
125
Sindrome di Silver Russel
c
a
b
d
Figura 6 a, b, c, d: Modelli in gesso delle arcate dentarie: II classe dentale, affollamento superiore anteriore, morso profondo,
cross dell’elemento 12, inclinazione del piano occlusale, palato ogivale
L et al, 2009; Saal HM, 2002; Rizzo V et al, 2001;
Blissett J et al., 2000).
Dal punto di vista odontostomatologico, i pazienti con SSR hanno molteplici problemi che rendono necessari interventi odontoiatrici multispecialistici specifici (odontoiatra infantile, igienista
dentale, ortodontista, chirurgo maxillo-facciale).
Essendo i pazienti affetti da SSR ad elevato rischio
di patologia cariosa e parodontale è necessario
attuare, dal momento in cui viene formulata la
diagnosi, interventi di prevenzione primaria mi-
rati, con particolare riferimento a informazioni sui
rischi derivanti da una alimentazione cariogenica,
all’igiene orale domiciliare e professionale, all’utilizzo di fluoro topico e quando indicato sistemico,
alle visite periodiche trimestrali/semestrali e alla
sigillatura dei solchi, delle fessure e dei fori ciechi.
Considerando l’elevata prevalenza di patologie
ortopedico-ortodontiche, è importante una diagnosi precoce nell’obiettivo di stabilire il piano di
trattamento più efficace.
Per favorire uno sviluppo cranio-facciale armonico
www.buponline.com
126
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
dal punto di vista funzionale ed estetico è necessario risolvere la discrepanza cranio-scheletrica in età
evolutiva (6-12 anni). Nei quadri clinici caratterizzati da palato ogivale in un mascellare superiore
ipoplasico l’approccio precoce prevede l’utilizzo di
un apparecchio di tipo ortopedico che sfrutta l’immaturità suturale per stimolare la crescita ossea sul
piano trasversale (disgiuntore rapido del palato).
Nelle II classi scheletriche al fine di stimolare
l’avanzamento mandibolare sono indicati di attivatori funzionali; i tempi e i risultati dipendono
dalla collaborazione del paziente. Il timing ideale
di trattamento è il periodo immediatamente precedente il picco di crescita puberale; nei pazienti
in terapia con GH, in cui il picco di crescita viene
indotto farmacologicamente, è quindi necessaria
una stretta collaborazione tra ortodontista ed auxologo.
Nei casi di II classe non trattati e nei casi di mancata risposta alle terapie finalizzate alla stimolazione
della crescita mandibolare, può essere necessario
un intervento di chirurgia maxillo-facciale; in letteratura è descritto un caso di ipomandibolia trattato chirurgicamente mediante osteodistrazione
della mandibola prima della terapia ortodontica
(Kisnisci RS et al., 1999).
BIBLIOGRAFIA
Abrahm E, Altick H, Lubicky JP. Muscoloskeletal manifestations of SSR. J Pediatric Orthop.
2004;24:552-64
Abu-Amero S, Monk D, Frost J, Preece M, Stanier P, Moore GE. The genetic aetiology
of Silver-Russell syndrome. J Med Genet.
2008;45:193-9
Anoderson J, Viskchil D, O’Gorman M, Gonzales C. Gastrointestinal complications of Russell-Silver syndrome: a pilot study. Am J Med
Genet. 2002;113:15-9
Bartholdi D, Krajewska-Walasek M, Ounap K,
Gaspar H, Chrzanowska KH, Ilyana H, Kayserili H, Lurie IW, Schinzel A Baumer A. Epigenetic mutation imprinted IGF2-H19 domain
in Silver-Russel Syndrome (SRS): result from a
large cohort of patient with SRS and SRS-like
phenotypes. J Med Genet. 2009;46:192-7
Bedi R, Moody GH: A primary double molar
tooth in a child with Russell-Silver syndrome.
Br Dent J. 1991;171:284-6
Bergman A, Kjellberg H, Dahlgren J: Craniofacial
morphology and dental age in children with
Silver-Russel Syndrome. Orthod Craniofacial
Res. 2003;6:54-62
Blissett J, Harris G, Kirk J. Effect of growth hormone therapy on feeding problems and food
intake in children with growth disorders. Acta
Pædiatr. 2000;89:644-9
Bruckheimer E, Abrahamov A. Russel-Silver syndrome and Wilms tumor. J Pediatr.
1993;122:165-6
Cullen CL, Wesley RK. Russell-Silver syndrome:
microdontia and other pertinent oral findings.
SSDC J Dent Child. 1987;54:201-4
Chitayat D, Friedman JM, Anderson L, Dimmick
JE. Hepatocellular carcinoma in a child with
familial Russel-Silver syndrome. Am J Med
Genet. 1988;31:909-14
Christoforidis A, Maniadaki I, Stanhope R. Managing children with Russell-Silver syndrome:
more than just growth hormone treatment?. J
Pediatr Endocrinol Metab. 2005;18:651-2
Draznin MB, Stelling MW, Johanson Aj. Silver-Russel syndrome and craniopharyngioma.
J Pediatr. 1980;96:887-9
Eggermann T. Silver-Russel and Beckwith-Wiedemann syndrome:opposite (epi)mutations
in 11p15 result in opposite clinical pictures.
Horm Res. 2009;71:30-5
Eggermann T, Begemann M, Binder G, Spengler
S. Silver-Russell syndrome: genetic basis and
www.buponline.com
127
Sindrome di Silver Russel
molecular genetic testing. Orphanet J Rare
Dis. 2010;5:19
Kisnisci RS, Fowel SD, Epker BN. Distraction osteogenesis in Silver Russel syndrome to expand
the mandible. Am J Orthod Dentofacial Orthop. 1999;116:25-30
Kotilainen J, Hölttä P, Mikkonen T, Arte S, Sipilä
I, Pirinen S. Craniofacial and dental characteristics of Silver-Russell syndrome. Am J Med
Genet. 1995;56:229-36
Kulkarni ML, Venkataramana V, Sureshkumar C,
Shabeer HM: Russell-Silver syndrome: a study
of 3 cases. Ann Dent. 1995;54:56-60
Lahiri A, Lester R. Hand anomalies in Russell Silver syndrome. J Plast Reconstr Aesthet Surg
2009;62:462-5
Lai K, Skuse D, Stanhope R, Hindmarsh P. Cognitive abilities associated with the Silver-Russell
syndrome. Arch Dis Child. 1994;71:490-6
Mazzanti L, Tamburrino F, Bergamaschi R, Scarano E, Montanari F, Torella M, Ballarini
E, Cicognani A. Developmental syndromes:
growth hormone deficiency and treatment.
Endocr Dev. 2009;14:114-34
Netchine I, Rossignol S, Duforg MN, Azzi S,
Rousseau A, Perin L, Houang M, Steunou V,
Esteva B et al. 11p15 ICR1 imprinting center region 1 loss of methylation is a common
and specific cause of typical Russel-Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Clin Endocrinol
Metab. 2007;92:3148-54
Noeker M, Wollmann HA. Cognitive development
in Silver-Russell syndrome: a sibling-controlled
study. Dev Med Child Neurol. 2004;46:340-6
Perkins RM, Hoang-Xuan MT. The Russell-Silver syndrome: a case report and brief review of the literature. Pediatr Dermatol.
2002;19:546-9
Price SM, Stanhope R, Garrett C, Prece MA,
Trembath RC. The Spectrum of Silver-Russel sindrome: a clinical and molecular genetic
study and new diagnostic criteria. J Med Genet. 1999;36:837-42
Rizzo V, Traggiai C, Stanhope R. Growth Hormone Treatment Does Not Alter Lower Limb
Asymmetry in Children with Russell-Silver
Syndrome. Horm Res. 2001;56:114-6
Rubenstein LK, Vitzky PL. Dental management
of patients with SSR. J Pedod. 1988;12:215-9
Saal HM. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews™
[Internet]. Seattle (WA): University of Washington, Seattle; 1993-2002 Nov 02 [updated
2011 Jun 02].
Saal HM, Pagon RA, Bird TC, Dolan CR, Stephens K, editors: Russell-Silver Syndrome. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2002 Nov 02,
updated 2011 Jun
Wakeling EL, Amero SA, Alders M, Bliek J, Forsythe E, Kumar S, Lim DH, MacDonald F,
Mackay DJ, Maher ER, Moore GE, Poole
RL, Price SM, Tangeraas T, Turner CL, Van
Haelst MM, Willoughby C, Temple IK, Cobben JM. Epigenotype-phenotype correlations
in Silver-Russel syndrome. J Med Genet.
2010;47:760-8
www.buponline.com
www.buponline.com
12. Sindrome di Noonan
Sinonimo: Sindrome di Turner maschile
Codice ICD 10: Q87.1
L. Armuzzi, I. Cremonesi, T. Tagariello, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Sindrome di Noonan (SN), descritta per la
prima volta nel 1963 dalla cardiologa pediatra
Jacqueline Noonan, è una malattia genetica rara
caratterizzata da un quadro clinico malformativo
peculiare: bassa statura, dismorfia facciale caratteristica, cardiopatie congenite (Noonan JA et
al., 1963).
L’incidenza è stimata tra 1 su 1.000 e 1 su 2.500
nati vivi per anno in assenza di predilezione razziale, etnica, geografica e di genere (Allanson JE et
al., 2011).
Gli indici di morbilità e di mortalità sono simili
alla popolazione generale in assenza o in presenza
di forme lievi-moderate di cardiopatie congenite;
cardiopatie congenite gravi riducono la sopravvivenza media (Allanson JE, 1987).
GENETICA E DIAGNOSI
La SN è una patologia congenita per il 50% a base
sporadica e per il 50% a carattere ereditario autosomico dominante con prevalente trasmissione
materna.
La principale causa genetica è stata scoperta nel
2001 con l’individuazione della mutazione del
gene PTPN11 nel cromosoma 12 (locus 12q24),
presente nel 40-50% dei pazienti Noonan (Tartaglia M, 2001). Tra il 2006 e il 2007 sono stati
scoperti altri 4 geni malattia: KRAS, SOS1, RAF1
e MEK1, presenti rispettivamente nel 2,3%, 21%,
10% e 4,3% dei pazienti senza mutazioni nel gene
PTPN11 (Jorge AA et al., 2009). Dal 2007 ad oggi
sono stati scoperti altri otto geni coinvolti nella SN
(KRAS, SOS1, RAF1, MEK1, SHOC2, NRAS,
BRAF, CBL). Sulla base di queste recenti scoperte,
la diagnosi di SN può essere geneticamente confermata nel 75% dei pazienti affetti.
I geni malattia codificano per proteine coinvolte
nelle vie di trasduzione del segnale RAS-MAPK
implicate in numerosi processi cellulari: divisione
cellulare, differenziamento, espressione genica, organizzazione del citoscheletro, traffico vescicolare
e trasporto nucleo-citoplasmatico (Tartaglia M et
al., 2011).
Nei casi di accertamento di una mutazione responsabile di SN in un componente della famiglia
si può effettuare la diagnosi prenatale mediante villocentesi o amniocentesi (Van Der Burgt I,
2007).
www.buponline.com
130
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
La diagnosi di SN può essere sospettata in epoca
prenatale nel II e III trimestre all’ecografia per la
presenza di igroma cistico (accumulo patologico di fluido dietro la nuca fetale), in particolare
quando associato a cardiopatia congenita, edema
fetale e polidramnios (aumento del quantitativo
di liquido amniotico) (Edwards PC et al., 2005;
Tartaglia M et al., 2011). La presenza di anomalie
prenatali visibili all’ecografia può far sospettare la
diagnosi di SN ma non può far predire tutte le
caratteristiche fenotipiche postnatali (Baldassarre
G et al., 2011).
Nonostante i significativi progressi sulle basi genetiche della SN, la diagnosi è primariamente clinica e si basa sulle caratteristiche fenotipiche del
paziente. Nel neonato il quadro clinico è spesso
sfumato e in assenza di cardiopatia congenita e/o
di anamnesi familiare positiva la diagnosi è più tardiva (Ammond P, 2005). La diagnosi clinica può
essere confermata da indagini genetiche.
Un’accurata diagnosi differenziale risulta fondamentale dal momento che altre patologie, in
particolare la Sindrome di Turner, la Sindrome
Cardio-Facio-Cutanea, la Sindrome di Costello,
la Sindrome di Leopard, sono caratterizzate da
fenotipi sovrapponibili alla SN (facies, cardiopatie congenite, bassa statura) (Schubbert S et al.,
2007).
MANIFESTAZIONI CLINICHE SISTEMICHE
Le caratteristiche somatiche comuni nei pazienti affetti da SN sono molte nonostante l’estrema
eterogeneità fenotipica correlabile all’eterogeneità genetica e alla variabilità fenotipica del singolo
paziente nelle differenti età. Le caratteristiche somatiche della SN si modificano infatti con l’età:
i neonati presentano un’eccessiva quantità di cute
a livello nucale a causa dell’igroma cistico prenatale; da 1 a 3 anni la testa è relativamente larga,
gli occhi prominenti, il collo corto; a 3-4 anni il
corpo diventa più robusto e il torace prominente;
nella seconda infanzia i tratti somatici diventano
grossolani; durante l’adolescenza le caratteristiche
facciali si fanno più nette fino al raggiungimento
dell’età adulta, in cui possono attenuarsi (Noonan
JA, 2006).
Le manifestazioni cliniche associate alla SN sono
descritte nella Tabella 1 (Jorge AA et al., 2009).
Caratteristica dei pazienti affetti da SN è la bassa statura. Alla nascita peso e altezza sono nella
norma ad eccezione dei pazienti con edema sottocutaneo che presentano un peso elevato. Dopo la
nascita la crescita è inferiore alla norma; l’altezza
media definitiva è di 162,5 cm nei maschi e 152,7
cm nelle femmine, valori che appartengono al terzo percentile. La crescita prepuberale è ridotta e
in alcuni casi l’accelerazione di crescita puberale è
assente. La pubertà è ritardata sia nei maschi che
nelle femmine: l’inizio avviene circa 2 anni dopo
la media della popolazione generale. Nei maschi il
criptorchidismo (mancata discesa dei testicoli nel
sacco scrotale), spesso causa di infertilità, è di frequente riscontro (60-69%); in alcuni casi è presente marcato ipogonadismo.
Nella prima infanzia sono descritti difficoltà alla
suzione (da ipotonia dei muscoli del cavo orale)
ed episodi di reflusso gastroesofageo (RGE); con
la crescita possono presentarsi problemi digestivi, scarso appetito e frequenti episodi di vomito
(Van Der Burgt I, 2007).
Di riscontro molto frequente sono le cardiopatie
congenite (50-70%): stenosi della valvola polmonare (50%), cardiomiopatia ipertrofica ostruttiva
(20%), difetti del setto atriale (10%), difetti del
setto ventricolare (5%).
Di frequente riscontro sono alterazioni della coagulazione: il 55% dei pazienti (soprattutto in età
infantile) presenta una lieve-moderata tendenza
www.buponline.com
131
Sindrome di Noonan
Tabella 1: Manifestazioni cliniche della Sindrome di Noonan
MANIFESTAZIONI CLINICHE
CRESCITA
Bassa statura
APPARATO
CARDIOVASCOLARE
Cardiopatie congenite: stenosi della valvola polmonare, cardiomiopatia
ipertrofica ostruttiva, difetti del setto atriale, difetti del setto ventricolare,
insufficienza mitralica, stenosi aortica
TORACE
Anomalie toraciche: torace deformato, petto carenato superiormente e/o
scavato inferiormente
APPARATO
GENITO-URINARIO
Criptorchidismo
APPARATO SCHELETRICO
Cubito valgo
Anomalie nelle mani: clinodattilia, brachidattilia, polpastrelli smussi
APPARATO NEUROLOGICO
Ritardo nello sviluppo motorio
Ritardo mentale, generalmente di lieve entità
Ritardo nel linguaggio
Deficit nell’apprendimento
APPARATO EMATOLOGICO
Lieve/moderata tendenza al sanguinamento
Deficit dei fattori della coagulazione XI e XII
Malattia di Von Willebrand
Trombocitopenia
Anomala funzione piastrinica
Leucemia mielocitica
ALTRI
Pubertà ritardata
Epatosplenomegalia (non in relazione alla patologia cardiaca)
Difetti dell’udito (complicanza di otiti medie)
Linfedema generalizzato
Linfedema periferico
Anomalie oculari (errori di rifrazione, strabismo, ambliopia)
al sanguinamento e nel 3% sono descritte emorragie severe. Gli esami della coagulazione possono
evidenziare tempo di sanguinamento prolungato,
deficit dei fattori VIII, XI e XII, trombocitopenia,
difetti della funzione piastrinica, isolati o in combinazione.
Il 25-35% dei pazienti affetti da SN presenta ritardo mentale, solitamente di lieve entità e tale da
non compromettere un buon inserimento sociale.
Generalmente il ritardo si manifesta con compromissione dell’area del linguaggio, che può essere
aggravata da deficit uditivo, complicanza di otiti
ricorrenti o di tipo neurosensoriale, e con deficit
nell’apprendimento.
www.buponline.com
132
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
CARATTERISTICHE ORO-FACCIALI
Le caratteristiche facciali tipiche della SN si modificano con l’età: sono poco marcate alla nascita,
si accentuano nell’infanzia e possono attenuarsi in
età adulta.
Alla nascita la faccia è caratterizzata da fronte alta
e ampia, ipertelorismo, epicanto, rime palpebrali inclinate verso il basso, orecchie ruotate
posteriormente con elice spesso, collo corto con
pterigio, eccesso di pelle nucale, bassa attaccatura posteriore dei capelli.
Durante l’infanzia il viso diventa triangolare e
presenta un aspetto miopatico: gli occhi sono
prominenti con ptosi palpebrale (unilaterale o
bilaterale); le labbra sono spesse con filtro nasolabiale prominente (Figura 1 a, b, c).
b
a
c
Figura 1 a, b, c: Bambino di 7 anni affetto da SN. Facies caratteristica: fronte alta e ampia, ipertelorismo, epicanto,
orecchie ruotate posteriormente con elice spesso
www.buponline.com
133
Sindrome di Noonan
Nell’adolescente e nel giovane adulto il viso diventa sempre più triangolare, con lineamenti affilati, occhi meno prominenti con ptosi palpebrale, naso sottile, a punta, con radice schiacciata
e narici ipoplasiche, sopracciglia rade.
Con l’avanzare dell’età il paziente adulto presenta
caratteristiche facciali meno specifiche: l’attaccatura anteriore dei capelli è alta, il filtro nasolabiale è prominente con solcatura evidente, le rime
palpebrali sono spesse, la pelle è raggrinzita (Jorge AA et al., 2009).
Per quanto concerne le patologie odontoiatriche,
in letteratura è reperibile solo un esiguo numero di
lavori su pazienti affetti da SN.
Un’elevata prevalenza di lesioni cariose multiple e
destruenti della dentatura decidua e permanente
è descritta in uno studio su tre bambini Noonan,
alla cui anamnesi sono emerse assunzione frequente e prolungata di alimenti zuccherati e difficoltà
nelle manovre di igiene orale domiciliare a causa
di deficit psicomotori (Barberia Leache E, 2003)
(Figura 2 a, b).
Elevati indici di placca e di infiammazione gengivale ed elevato DMFt sono riportati in uno studio su due pazienti con SN di 13 e 14 anni (Ortega
Ade O et al., 2008).
La nostra esperienza clinica, condotta su undici
pazienti con SN di età compresa tra 6 e 22 anni,
ha evidenziato una elevata prevalenza di patologia
cariosa della dentatura sia decidua che permanente
e la presenza in 2 pazienti affetti da SN positivi alla
mutazione del gene PTPN11 di anomalie dentarie
di numero (in un caso di due agenesie e in uno di
un elemento soprannumerario).
Uno studio descrive due pazienti con SN in età
evolutiva affetti da parodontopatia severa (Torres-Carmona MA, 1991).
Un case report descrive un paziente con SN affetto da xantoma orale e cheratocisti odontogene
multiple (Condor JM, 1982).
In pazienti affetti da SN sono descritte lesioni mul-
a
b
Figura 2 a, b: Bambina di 6 anni affetta da SN. Carie
destruenti di elementi decidui
tiple a cellule giganti localizzate nella mandibola
e più raramente nel mascellare superiore. Cohen
e Gorlin hanno definito “Noonan-like/multiple
giant cell lesion sindrome” (NS/MGCLS) una
sindrome contrassegnata dalle caratteristiche della
SN e da lesioni multiple a cellule giganti nel cavo
orale (Cohen MM Jr et al., 1991). Essendo state
evidenziate nella NS/MGCLS mutazioni nei geni
PTPN11 e SOS1, le lesioni a cellule giganti fanno
parte dello spettro clinico della SN e non rappresentano un’entità separata (Bufalino A et al., 2010;
Hanna N et al., 2009; Lee JS et al., 2005).
Per quanto concerne le patologie ortopedicoortodontiche, nella Tabella 2 sono riportati i dati
relativi ai case report presenti in letteratura.
www.buponline.com
134
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Tabella 2: Patologie ortopedico-ortodontiche in pazienti affetti da SN
Crescita
iperdivergente
Ierardo G
et al.,
2010
(1 pz)
Emral
ME et
al.,
2009
(1 pz)
x
x
I classe scheletrica
Ortega
Ade O
et al.,
2008
(2 pz)
x
Asokan
S et al.,
2007
(1pz)
x
Buccheri
A et al.,
2006
(2 pz)
x
x
Okada M
2003
(1 pz)
x
x
II classe scheletrica
Sugar AW
et al.,
1994
(1 pz)
x
x
x
III classe scheletrica
x
x
x
x
Protrusione incisivi
superiori e retrusione
incisivi inferiori
Diminuzione angolo
interincisivo
x
Aumento angolo
interincisivo
x
x
x
Prognatismo
mandibolare
Ipoplasia mandibolare
x
Biretrusione
x
x
Biprotrusione
x
Crossbite posteriore
x
x
Affollamento
Morso aperto
x
x
Palato ogivale
Morso profondo
x
x
x
x
x
x
x
x
x
x
x
x
x
www.buponline.com
x
135
Sindrome di Noonan
Dai case report emerge che le patologie ortopedico-ortodontiche più frequenti nella SN sono malocclusione scheletrica di II classe da retrusione
mandibolare, palato ogivale, morso aperto anteriore, crescita iperdivergente (Figura 3).
Figura 3: Teleradiografie latero-laterale: morso aperto scheletrico con iperdivergenza delle basi, protrusione bimascellare, accentuazione della lordosi cervicale
LINEE GUIDA DI TERAPIA
I pazienti con SN sono affetti da numerose alterazioni e patologie; la diagnosi, il follow-up e le
terapie richiedono la presenza di un team multi
e interdisciplinare adeguatamente formato sulla
sindrome.
La bassa statura, il carattere fenotipico più frequente della SN, è trattata con ormone della crescita
(GH) che promuove la crescita a breve e a lungo
termine, determinando un aumento staturale anche
di 9-13 cm (Romano AA et al., 2010).
Per quanto concerne le cardiopatie congenite, a
seconda del tipo e della gravità si intervene con monitoraggio cardiologico, con terapie mediche e/o
chirurgiche e, in rari casi, con trapianto di cuore.
Il criptorchidismo deve essere diagnosticato precocemente per attuare tempestivamente la terapia
farmacologica o chirurgica (Romano AA et al.,
2010).
Durante la prima infanzia spesso sono presenti
problemi di alimentazione ma solo in rari casi è
necessario ricorrere al sondino nasogastrico (Van
Der Burgt I, 2007).
In caso di frequenti episodi di RGE è necessario
istruire i genitori sulla necessità di tenere in posizione eretta il bambino durante e dopo i pasti
e, nei casi più gravi, somministrare farmaci antireflusso (Gaviscon, Ranitidina). È inoltre necessario informare i genitori sul rischio di erosioni e di
patologia cariosa che il RGE comporta.
Per quanto concerne le patologie odontostomatologiche in un’ottica di interdisciplinarietà, è
compito del pediatra, dal momento in cui si effettua la diagnosi, inviare il paziente all’odontoiatra
infantile perché informi la famiglia sull’importanza
della salute orale e sulla necessità di interventi di
prevenzione.
Per quanto concerne la patologia cariosa, essendo i
pazienti affetti da SN ad elevato rischio di patologia
cariosa e, quando affetti da cardiopatia congenita, a
rischio di Endocardite Batterica (EB), è necessario
attuare, dal momento in cui viene formulata la diagnosi, interventi di prevenzione primaria mirata,
con particolare riferimento all’alimentazione non
cariogenica, all’igiene orale domiciliare, all’utilizzo
di fluoro topico e quando indicato sistemico, alle
visite periodiche trimestrali/semestrali, alla sigillatura di solchi, fessure e fori ciechi.
In presenza di patologie cardiovascolari a rischio di EB, prima di effettuare procedure odontoiatriche che provocano sanguinamento è necessario effettuare la profilassi antibiotica (vedi
capitolo 2).
In presenza di deficit della coagulazione, la necessità della preparazione farmacologica (fattori
di sostituzione emoderivati, antifibrinolitici, de-
www.buponline.com
136
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
smopressina) deve essere valutata in ogni singolo
paziente in funzione della gravità della coagulopatia e del rischio emorragico che le terapie odontoiatriche comportano. Sono considerate ad alto
rischio emorragico tutte le manovre chirurgiche,
l’anestesia tronculare, la detartrasi (soprattutto in
presenza di infiammazione gengivale), la levigatura delle radici e, in generale, tutte le manovre
che comportano sanguinamento. È opportuno
evitare un costante posizionamento dell’aspirasaliva in bocca, che può essere traumatico per i
tessuti del pavimento orale. Dopo l’esecuzione
di manovre chirurgiche i lembi devono essere
correttamente suturati; è raccomandato l’uso di
materiali che inducono la coagulazione (tamponi imbevuti di acido tranexamico, spugne o
garze di fibrina o di collagene, cemento chirurgico); è sempre indicata l’applicazione di impacchi
di ghiaccio, che limitano il sanguinamento e la
formazioni di ematomi. Nel caso si manifesti un
ematoma, oltre all’applicazione di ghiaccio, è indicata una immediata terapia farmacologica sostitutiva. In caso di terapie conservative è necessario
ricorrere ad alcuni accorgimenti, in particolare
utilizzo della diga di gomma per la protezione
dei tessuti molli e molta attenzione nella scelta
e nel posizionamento di uncini per la diga e di
matrici metalliche. Per quanto riguarda le terapie endodontiche è possibile un sanguinamento
pulpare difficilmente controllabile, soprattutto
in elementi con infiammazione pulpare. L’emostasi è di solito ottenibile tramite irrigazione con
ipoclorito di sodio, acqua ossigenata, idrossido di
calcio in soluzione; se non si ottiene il controllo
in sede intraoperatoria, è opportuno ricorrere ad
una medicazione intermedia con idrossido di calcio in pasta. Per evitare una sovrastrumentazione,
causa di emorragia, è necessario effettuare un’attenta valutazione della lunghezza canalare tramite
radiografie pre- ed intra- operatorie e localizzatore
d’apice.
Il paziente non deve essere dimesso dalla struttura odontoiatrica fino a quando non sia assicurata
un’emostasi ottimale.
Per quanto concerne il controllo del dolore postoperatorio, in tutti i pazienti con deficit coagulativo è controindicato l’uso di salicilati e di FANS.
Paracetamolo, aril-propionici, destropropossifene,
pentazocina rappresentano valide alternative.
Nei pazienti che per il deficit di coagulazione necessitano di preparazione farmacologia, se lo consente
il livello di collaborazione (parametro fondamentale per stabilire la durata delle sedute), è opportuno
impostare il piano di trattamento per quadranti,
per ridurre il numero delle preparazioni.
Nei casi in cui l’assenza di collaborazione renda necessario intervenire in anestesia generale, è necessaria una attenta valutazione anestesiologica in relazione al rischio di complicanze legate a patologie
cardiache e a difetti della coagulazione. L’intervento in AG deve essere considerato come il punto di
partenza per un programma di controlli periodici
ravvicinati, nel corso dei quali attuare igiene orale professionale e intercettare le lesioni allo stadio
iniziali, quando le terapie sono più semplici e di rapida esecuzione, quindi più accettate dal paziente.
Considerando l’elevata prevalenza di patologie
ortopedico-ortodontiche è importante una diagnosi precoce nell’obiettivo di stabilire il piano di
trattamento più efficace.
Per favorire uno sviluppo cranio-facciale armonico
dal punto di vista funzionale ed estetico è necessario risolvere la discrepanza cranio-scheletrica in età
evolutiva (6-12 anni).
Nei quadri clinici caratterizzati da palato ogivale
in un mascellare superiore ipoplasico l’approccio
precoce prevede l’utilizzo di un apparecchio di tipo
ortopedico che sfrutta l’immaturità suturale per
stimolare la crescita ossea sul piano trasversale (disgiuntore rapido del palato).
Anche l’iperdivergenza scheletrica, che può manifestarsi con morso aperto anteriore, si avvale di
www.buponline.com
137
Sindrome di Noonan
un approccio ortopedico mediante l’applicazione di
una trazione extra-orale alta su morsetto applicato ai
molari superiori o inferiori (Tallonette); trattandosi
di un dispositivo rimovibile i risultati dipendono dal
livello di collaborazione offerto dal bambino e dalla
sua famiglia. Nei casi di iperdivergenza nei quali è
indicata l’espansione ortopedica del mascellare superiore è consigliato l’utilizzo di un REP su slitte
(bite-block) per contrastare l’azione di apertura del
morso. In alcuni casi l’iperdivergenza richiede un
approccio chirurgico a fine crescita.
Nelle II classi scheletriche al fine di stimolare l’avanzamento mandibolare sono indicati attivatori
funzionali di diverso tipo; anche in questi casi i
tempi e i risultati dipendono dalla collaborazione
del paziente. Il timing ideale di trattamento è il
periodo immediatamente precedente il picco di
crescita puberale; nei pazienti in terapia con GH
in cui il picco di crescita viene indotto farmacologicamente è quindi necessaria una stretta collaborazione tra ortodontista ed auxologo.
Per necessità funzionali e/o estetiche in fase di
dentizione permanente può essere indicata l’applicazione di apparecchiature ortodontiche fisse
multibrackets; l’elevato rischio di carie che questi
dispositivi comportano deve essere affrontato con
interventi di prevenzione mirati (igiene orale domiciliare molto accurata, utilizzo di fluoro topico
domiciliare, applicazione ambulatoriale di gel o
vernici al fluoro, sigillatura dei solchi, delle fessure
e dei fori ciechi).
BIBLIOGRAFIA
Allanson JE. Noonan syndrome. J Med Genet.
1987;24:9-13
Allanson JE, Roberts AE. Noonan Syndrome. In:
Pagon RA, Bird TC, Dolan CR, Stephens K,
editors GeneReviews. Seattle (WA): University
of Washington, Seattle; 1993-2003
Ammond P. Discriminative power of localized
three-dimensional facial morphology. Am J
Hum Genet. 2005;77:999-1010
Asokan S, Muthu MS, Rathna PV. Noonan syndrome: a case report. J Indian Soc Pedod Prevent Dent. 2007;25:144-7
Baldassarre G, Mussa A, Dotta A, Banaudi E, Forzano S, Marinosci A, Rossi C, Tartaglia M,
Silengo M, Ferrero GB. Prenatal features
of Noonan syndrome: prevalence and prognostic value. Prenat Diagn. 2011;31:949-54
Barberia Leache E. Etiopathogenic analysis of the
caries on three patients with Noonan Syndrome. Med Oral. 2003;8:136-42
Buccheri A, Ceccano A. Caratteristiche dentoscheletriche della sindrome di Noonan. Mondo
Ortodontico. 2006;6:417-24
Bufalino A, Carrera M, Carlos R, Coletta RD. Giant Cell Lesions in Noonan Syndrome: Case
Report and Review of The Literature. Head
Neck Pathol. 2010;4:174–7
Cohen MM Jr, Gorlin RJ. Noonan-like/multiple
giant cell lesion syndrome. Am J Med Genet.
1991;40:159-66
Condor JM. Multiple odontogenic keratocysts in a
case of the Noonan syndrome. Br J Oral Surg.
1982;20:213-6
Edwards PC Fox J, Fantasia JE, Goldberg J,
Kelsch RD. Bilateral central giant cell granulomas of the mandible in an 8-year-old girl
with Noonan syndrome (Noonan-like/multiple giant cell lesion syndrome). Oral Surg
Oral Med Oral Pathol Oral Radiol Endod.
2005;99:334-40
Emral ME, Akcam MO. Noonan syndrome: a case
report. J Oral Sci. 2009;51:301-6
Hanna N, Parfait B, Talaat IM, Vidaud M, Elsedfy
HH. SOS1: a new player in the Noonan-like/
multiple giant cell lesion syndrome. Clin Genet. 2009;75:568-71
Ierardo G, Luzzi V, Panetta F, Sfasciotti GL, Poli-
www.buponline.com
138
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
meni A. Noonan sindrome: a case report. Eur J
Paediatr Dent. 2010;11:97-100
Jorge AA, Malaquias AC, Arnhold IJ, Mendonca
BB. Noonan syndrome and related disorders:
a review of clinical features and mutations in
genes of the RAS/MAPK pathway. Horm Res.
2009;71:185-93
Lee JS, Tartaglia M, Gelb BD, Fridrich K, Sachs S,
Stratakis CA, Muenke M, Robey PG, Collins
MT, Slavotinek A. Phenotypic and genotypic
characterisation of Noonan-like/multiple giant
cell lesion syndrome. J Med Genet. 2005;42:11
Noonan JA, Ehmke DA. Associated non cardic malformations in children with congenital
heart disease. J Pediatr. 1963;63:468
Noonan JA. Noonan syndrome and related disorders: alterations in growth and puberty. Rev
Endocr Metab Disord. 2006;7:251-5
Okada M. Oral findings in Noonan syndrome: report of a case. J Oral Sci. 2003;45:117-21
Ortega Ade O, Guaré Rde O, Kawaji NS, Ciamponi AL. Orofacial aspects in Noonan syndrome: 2 case report. J Dent Child (Chic).
2008;75:85-90
Romano AA, Allanson JE, Dahlgren J, Gelb BD,
Hall B, Pierpont ME, Roberts AE, Robinson
W, Takemoto CM, Noonan JA. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010;126:74659
Schubbert S, Shannon K, Bollag G. Hyperactive
Ras in developmental disorders and cancer. Nat
Rev Cancer. 2007;7:295-380
Sugar AW, Ezsias A, Bloom AL, Morcos WE.
Orthognathic surgery in a patient with Noonan’s syndrome. J Oral Maxillofac Surg.
1994;52:421-5
Tartaglia M. Mutations in PTPN11, encoding the
protein tyrosine phosphatase SHP-2, cause
Noonan syndrome. Nat Genet. 2001;29:465-8
Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract
Res Clin Endocrinol Metab. 2011;25:161-79
Torres-Carmona MA. Periodontal disease in
Noonan’s syndrome. Bol Med Hosp Infant
Mex. 1991;48:271-4
Van der Burgt I. Noonan syndrome. Orphanet J
Rare Dis. 2007;14;2-4
www.buponline.com
13. Sindrome di Marfan
Sinonimo: MFS
Codice ICD 10: Q87.4
M. Taddei, T. Tagariello, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Sindrome di Marfan (SM), descritta per la prima volta nel 1896 dal pediatra francese Antoine
Bernard-Jean Marfan, è una malattia genetica rara
caratterizzata da disordini sistemici dei tessuti connettivali (Marfan MA-B, 1896). Le manifestazioni
principali includono eccessiva crescita in lunghezza
delle ossa associata ad altre anomalie scheletriche,
ectopia del cristallino, aneurisma dell’aorta, dissezione dell’aorta, prolasso della valvola mitrale
(Keane MG et al., 2008; Hecht F et al., 1972).
L’incidenza è stimata di 2-3 su 10.000 nati vivi
per anno, in assenza di predilezione razziale, etnica, geografica e di genere (Pyeritz RE, 2007).
L’indice di mortalità è elevato; le patologie cardiovascolari sono la causa più frequente di morte (Judge
DP et al., 2005). Tuttavia negli ultimi anni la sopravvivenza di questi pazienti è notevolmente migliorata
e da una aspettativa media di vita di 45 anni si è passati ad una di 72 anni (Pyeritz RE et al., 2012).
EZIOLOGIA E GENETICA
La SM classica, di tipo I, è causata da mutazio-
ni nel gene FBN1 che codifica per una proteina
della matrice extracellulare, la fibrillina-1, componente di una classe di microfibrille extracellulari
necessarie per la formazione dei tessuti elastici ed
in grado di fornire le proprietà elastiche ai tessuti
stessi (Dietz HC et al., 1991). Le mutazioni del
gene FBN1 alterano la struttura della fibrillina-1 o
ne diminuiscono la sintesi, provocando la compromissione delle proprietà tensili delle microfibrille
(Frydman M, 2008). Sono state descritte più di
1.000 mutazioni di FBN1; non sono emerse correlazioni certe tra genotipo e fenotipo, tuttavia
mutazioni nella regione centrale del gene sembrano correlate a patologie cardiovascolari più severe.
La SM di tipo II, meno frequente, è causata da una
mutazione del gene TGFBR2, che codifica per un
recettore del TGF-beta (Dean JCS, 2007).
Circa il 75% dei pazienti Marfan eredita la patologia dai genitori affetti; il rimanente 25% presenta
una mutazione ex novo. Un individuo affetto ha il
50% di probabilità di trasmettere ai figli la mutazione responsabile della malattia.
Nonostante i significativi progressi sulle basi genetiche e molecolari della SM, la diagnosi è primariamente clinica (Ammash NM et al., 2008;
De Peape A et al., 1996). Il test genetico presen-
www.buponline.com
140
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
ta infatti vari limiti: la mutazione nel gene per la
fibrillina-1 può causare altre patologie simili alla
SM; nessuno degli attuali metodi identifica tutte le
mutazioni che causano la SM; membri di una famiglia con la stessa mutazione possono presentare
un’ampia varietà di manifestazioni cliniche (Ammash NM et al., 2008).
La diagnosi prenatale su villocentesi o amniocentesi è possibile nelle famiglie in cui è stata identificata la mutazione causale.
MANIFESTAZIONI CLINICHE
Molte sono le caratteristiche cliniche comuni ai
pazienti affetti da SM, nonostante l’estrema variabilità fenotipica anche nell’ambito della stessa
famiglia.
Sono coinvolti principalmente i sistemi muscolo
scheletrico, oculare e cardiovascolare.
Le manifestazioni scheletriche sono spesso i
primi segni della malattia e includono dolicostenomelia (eccessiva lunghezza degli arti), statura
alta, aracnodattilia, deformazioni scoliotiche,
acetabolo protruso, deformità toraciche (pectus
carenatum, pectus excavatum), ipermobilità articolare, dolicocefalia, ipoplasia malare e altre
alterazioni facciali.
Le manifestazioni oculari includono ectopia del
cristallino e miopia assiale, che può essere complicata da distacco della retina (Keane MG et al.,
2008; Dean JCS, 2007). L’ectopia del cristallino,
unico criterio maggiore dell’apparato oculare, sem-
bra essere assente nella SM di tipo II, anche se il
numero di casi geneticamente identificati è troppo basso per confermare il dato (Disabella E et al.,
2006).
Le manifestazioni cardiovascolari includono aneurisma dell’aorta, con aumentato rischio di dissezione aortica (che condiziona la prognosi), prolasso
della valvola mitrale, che può essere complicato da
aritmie, endocardite insufficienza cardiaca; manifestazioni più rare sono dilatazione dell’arteria polmonare e calcificazione della valvola mitrale.
Manifestazione a livello dell’apparato respiratorio è il pneumotorace spontaneo.
Manifestazioni a livello cutaneo sono smagliature non associate a perdita di peso o gravidanza ed
ernie recidivanti.
La manifestazione a livello del sistema nervoso è
l’ectasia della dura madre a livello lombosacrale.
La diagnosi di SM, non sempre semplice in particolare nei primi mesi di vita, per molti anni si è
basata sulla nosologia di Ghent, che comprende
criteri maggiori ad alta specificità diagnostica (segni e sintomi di raro riscontro nella popolazione
generale) e criteri minori (segni e sintomi relativamente comuni nella popolazione generale) riportati nella Tabella 1 (De Peape A et al., 1996). Nei
casi sporadici la diagnosi richiedeva la presenza di
almeno due criteri maggiori in apparati diversi ed il
coinvolgimento di un terzo apparato; nei casi familiari era richiesta la presenza di un singolo criterio
maggiore o la dimostrazione di una mutazione nel
gene FBN1 (Frydman M, 2008).
www.buponline.com
141
Sindrome di Marfan
Tabella 1: Criteri di Ghent maggiori e minori per la diagnosi di SM
APPARATI
CRITERI MAGGIORI
-
-
SCHELETRICO
Necessaria la presenza di
almeno quattro criteri
maggiori
-
-
-
-
-
Petto carenato o escavato che
necessita di chirurgia
Rapporto tra apertura
massima delle braccia e altezza
> 1,05
Segni del polso e del pollice
Scoliosi >20° o spondilolistesi
(patologia della colonna
vertebrale caratterizzata dallo
scivolamento di una vertebra
sull’altra)
Ridotta estensione dei gomiti
(<170°)
Piedi piatti
Protrusione dell’acetabolo
-
-
-
-
-
-
-
OCULARE
Necessaria la presenza di
almeno due dei criteri
minori
CRITERI MINORI
Ectopia del cristallino
-
-
-
CARDIOVASCOLARE
Sufficiente la presenza di
solo uno dei criteri minori
-
-
Aneurisma dell’aorta
ascendente e coinvolgimento
del seno di Valsalva
Dissezione dell’aorta
RESPIRATORIO
Sufficiente la presenza di
solo uno dei criteri minori
-
Nessuno
-
Nessuno
CUTANEO
Sufficiente la presenza di
solo uno dei criteri minori
www.buponline.com
-
-
-
Petto escavato
Ipermobilità articolare
Palato ogivale con affollamento
dentario
Alterazioni facciali (dolicocefalia,
ipoplasia malare, enoftalmo,
retrognazia, palpebre inclinate verso
il basso)
Cornea piatta
Aumentata lunghezza assiale del
globo oculare
Ipoplasia dell’iride o ipoplasia del
muscolo ciliare
Insufficienza aortica
Prolasso della valvola mitralica con o
senza rigurgito
Dilatazione dell’arteria polmonare
prima dei 40 anni
Calcificazione della valvola mitrale
prima dei 40 anni
Dilatazione o dissezione dell’aorta
addominale prima dei 50 anni
-
-
Pneumotorace spontaneo
Bolle polmonari apicali
-
Smagliature (non associate a perdita
di peso o gravidanza)
Ernie recidivanti
-
142
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
NERVOSO
FAMILIARITÀ/
GENETICA
Uno costituisce un criterio
maggiore
-
Ectasia della dura madre a
livello lombosacrale
-
Nessuno
-
Parente di primo grado con
criteri Marfan
Presenza di una mutazione
genetica nel gene FBN1
Presenza di un marker
genetico, vicino a FBN1,
trasmesso con la malattia nella
famiglia
-
Nessuno
-
-
Attualmente la diagnosi di SM si basa sulla nosologia di Ghent revisionata, semplificata rispetto alla precedente, che valuta i due aspetti
clinici cardinali della SM (aneurisma della radice
aortica o dissezione aortica e ectopia lentis), la
positività al test genetico ed un “parametro sistemico”, termine con cui si intende l’insieme
di tutte le altre manifestazioni cardiovascolari
ed oculari tipiche della sindrome e di quelle che
coinvolgono tutti gli altri apparati ed organi (Tabella 2) (Loeys BL et al., 2010).
Il “parametro sistemico” accerta il coinvolgimento sistemico quando sui 20 punti totali disponibili si ha un valore pari o superiore a 7.
Nei casi sporadici la diagnosi di SM si effettua
nei casi di:
1. diametro aortico a livello del seno di Valsalva caratterizzato da Z-score ≥2 (indice che
consente una corretta diagnosi di aneurisma
della radice aortica) o presenza di dissezione
aortica e presenza di ectopia lentis;
2. diametro aortico a livello del seno di Valsalva caratterizzato da Z-score ≥2 o presenza di
dissezione aortica e presenza di mutazione
genetica del gene FBN1;
3. diametro aortico a livello del seno di Valsal-
va caratterizzato da Z-score ≥2 o presenza di
dissezione aortica e “parametro sistemico”
positivo (≥ 7 punti)
4. presenza di ectopia lentis e presenza di mutazione genetica del gene FBN1.
Nei casi familiari la diagnosi di SM si effettua
nei casi di:
1. presenza di ectopia lentis
2. “parametro sistemico” positivo (≥ 7 punti)
3. diametro aortico a livello del seno di Valsalva caratterizzato da Z-score ≥2 in età > 20
anni e ≥3 in età < 20 anni.
Nonostante la nosologia di Ghent revisionata sia
semplificata, per una corretta diagnosi della SM
è sempre necessario un approccio multidisciplinare, che prevede la partecipazione di cardiologo, ortopedico, oculista, pediatra e genetista.
CARATTERISTICHE ORO-FACCIALI
Palato ogivale con affollamento dentario ed
alterazioni facciali (dolicocefalia, ipoplasia malare e retrognazia, enoftalmo, rima palpebrale rivolta verso il basso) sono inclusi tra i criteri minori di Ghent a livello dell’apparato scheletrico.
Le caratteristiche craniofacciali descritte in letteratura sono faccia lunga, retrusione mascellare
www.buponline.com
143
Sindrome di Marfan
Tabella 2: Nosologia di Ghent revisionata: manifestazioni cliniche e relativo punteggio del parametro sistemico
MANIFESTAZIONE
PUNTEGGIO
- Segni del polso e del pollice
- Segno del polso
- Segno del pollice
3
1
1
- Petto carenato o escavato che necessita di chirurgia
- Petto escavato o asimmetrico
2
1
- Deformità della porzione posteriore del piede
- Piedi piatti
2
1
- Pneumotorace
2
- Ectasia della dura madre
2
- Protrusione dell’acetabolo
2
- Ridotto rapporto tra la porzione corporea superiore ed
inferiore, aumentato rapporto tra apertura massima delle
braccia ed altezza, scoliosi non severa
1
- Scoliosi >20° o cifosi toracolombare
1
- Ridotta estensione dei gomiti (<170°)
1
- Almeno 3 alterazioni facciali su 5 (dolicocefalia, ipoplasia
malare, enoftalmo, retrognazia, palpebre inclinate verso il
basso)
1
- Smagliature non associate a perdita di peso o gravidanza
1
- Miopia: 3 diottrie
1
- Prolasso della valvola mitralica
1
e mandibolare, tendenza al morso aperto, II
classe scheletrica, palato ogivale, cross-bite
posteriore da deficit del mascellare superiore, denti stretti e lunghi (Khonsari RH et al.,
2010; De Coster PJ et al., 2004; De Coster PJ et
al., 2002; Cistulli PA et al., 2001; Westling L et
al., 1998; Gorlin RJ et al., 1990; Baden E et al.,
1965) (Figura 1 a, b, c, d, e, f ). La mandibola
retrusa e il palato ogivale sono concause dell’elevata prevalenza di apnee ostruttive notturne
(OSAS) (Cistulli PA et al., 2001).
In pazienti con SM sono state descritte palatoschisi e ugola bifida (Lynas MA, 1958; Wilson
R, 1957).
L’ipermobilità articolare, inclusa tra i criteri minori di Ghent a livello dell’apparato scheletrico,
www.buponline.com
144
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
b
a
Figura 1 a, b: Paziente di 17 anni affetta da SM. Facies caratteristica: faccia allungata, retrusione mandibolare e mascellare, assenza di sigillo labiale
può essere responsabile di disfunzioni dell’articolazione temporomandibolare quali sublussazione, dislocamento anteriore del disco articolare, osteoartrosi (Bauss O et al., 2004; Gorlin RJ
et al., 1990).
Per quanto concerne le anomalie dentarie, in letteratura sono state descritte anomalie di numero
(in difetto e in eccesso), di forma (deformazioni
radicolari), di struttura (ipoplasie dello smalto,
dentinogenesi imperfetta) (Khonsari RH et al.,
2010; Utreja A et al., 2009; De Coster PJ et al.,
2002; Sachdev MS et al., 1986; Baden E et al.,
1965) (Figura 2 a, b, c; Figura 3 a, b, c).
Un case report descrive un paziente con SM affetto da oligodonzia (agenesie dentali multiple) e
da aniridia (assenza congenita dell’iride) bilaterale
(Sachdev MS et al., 1986).
Per quanto concerne la struttura della polpa e dei
tessuti gengivali, studi al microscopio elettronico
evidenziano la presenza di fibre elastiche anomale
e dilatazione delle strutture vascolari (Temtamy
SA et al., 1989). In pazienti Marfan giovani e adulti è inoltre descritta la frequente presenza di calcificazioni a livello della polpa (Bauss O et al., 2008).
www.buponline.com
145
Sindrome di Marfan
c
d
e
f
Figura 1 c, d, e, f: Paziente di 17 anni affetta da SM. Modelli in gesso. Assenza dei primi molari permanenti estratti per
carie destruenti, morso aperto anteriore, palato ogivale
www.buponline.com
146
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
b
a
Figura 2 a, b: Paziente di 18 anni affetto da SM. Facies caratteristica: faccia allungata, retrusione mascellare e mandibolare, tendenza al morso aperto, II classe scheletrica, palato ogivale, denti stretti e lunghi
Per quanto concerne la patologia cariosa in pazienti con SM è descritta un’elevata prevalenza,
in particolare della dentatura decidua (De Coster
PJ et al., 2002).
Per quanto concerne la malattia parodontale sono
scarsi i dati reperibili in letteratura. In un case report, in cui si descrive un paziente affetto da SM
con un quadro severo di parodontopatia profonda,
si ipotizza che la suscettibilità alla malattia parodontale sia incrementata da alterazioni del tessuto
connettivo (Straub AM et al., 2002).
LINEE GUIDA DI TERAPIA
I pazienti con SM sono affetti da numerose alterazioni e patologie che rendono necessari interventi multispecialistici. La diagnosi, il follow-up e le
terapie richiedono la presenza di un team multi
e interdisciplinare adeguatamente formato sulla
sindrome, che include cardiologi, chirurghi, ortopedici, oculisti, psicologi, pediatri ed odontoiatri.
Le patologie cardiovascolari sono le più gravi,
su tutte la dissezione aortica, e sono responsabili
del 90% dei decessi; attualmente, grazie ai progressi nella diagnosi e nelle terapie farmacologi-
www.buponline.com
147
Sindrome di Marfan
a
b
c
Figura 3 a, b, c: Paziente di 14 anni affetta da SM: ipoplasie dello smalto
che (β-bloccanti, ACE-inibitori) e chirurgiche, le
aspettative di vita sono migliorate (Ammash NM
et al., 2008).
Le patologie dell’apparato scheletrico, in particolare la scoliosi progressiva e le deformità del torace, spesso richiedono interventi chirurgici (Judge
DP et al., 2005).
L’ectopia del cristallino è corretta con occhiali o
lenti a contatto; solo occasionalmente è necessario
l’intervento chirurgico (Ehud Raanani MD et al.,
2008).
Per adattare il paziente alla propria situazione e favorire il suo inserimento nella società è utile l’intervento di uno psicologo.
Per quanto concerne le patologie di pertinenza
odontoiatrica, essendo i pazienti affetti da SM ad
elevato rischio di patologia cariosa è necessario
attuare, dal momento in cui viene formulata la diagnosi, interventi di prevenzione primaria mirati,
con particolare riferimento all’alimentazione non
cariogenica, all’igiene orale domiciliare, all’utilizzo
di fluoro topico e, quando indicato sistemico, alle
visite periodiche trimestrali/semestrali, alla sigillatura dei solchi e delle fessure degli elementi posteriori.
In presenza di patologie cardiovascolari a rischio
di EB, prima di effettuare procedure odontoiatriche che provocano sanguinamento è necessario effettuare la profilassi antibiotica (vedi il capitolo
Cardiopatie congenite).
Prima di procedere con interventi a rischio di sanguinamento è necessaria un’accurata anamnesi sui
farmaci assunti, in particolare anticoagulanti.
In caso di terapie endodontiche, è necessario valutare su una Rx endorale preoperatoria l’eventuale
presenza di calcificazioni a livello del tessuto pulpare.
Considerando l’elevata prevalenza di patologie
ortopedico-ortodontiche, è consigliato stabilire
un protocollo diagnostico precoce nell’obiettivo
di promuovere uno sviluppo armonico cranio-fac-
www.buponline.com
148
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
ciale dal punto di vista sia funzionale che estetico.
La risoluzione tempestiva della discrepanza cranioscheletrica mediante il trattamento ortopedicoortodontico di espansione del mascellare superiore evita il consolidamento di una malocclusione
che comporterebbe terapie tardive e probabilmente
più invasive e complesse (estrazioni e/o chirurgia
ortognatica) (Figura 4 a, b, c). Un altro aspetto che
induce ad intervenire precocemente è la presenza di OSAS correlate ad anomalie cranio facciali
che, a causa dell’aumentata pressione intratoracica
notturna, possono contribuire alla progressiva e
rapida dilatazione dell’aorta (Verbraecken J et al.,
2003). Nella pianificazione della eventuale terapia
ortodontica va valutata la presenza di disfunzioni
dell’articolazione temporomandibolare, che necessitano di una valutazione gnatologica specialistica.
In caso di terapie ortodontiche in soggetti con SM
è stato segnalato un rischio aumentato di riassorbimento radicolare e di necrosi pulpare (Bilodeau
JE, 2010).
BIBLIOGRAFIA
Ammash NM, Sundt TM, Connolly HM. Marfan
syndrome-diagnosis and management. Curr
Probl Cardiol. 2008;33:7-39
Baden E, Teaneck NJ, Spirgi M. Oral manifestations of Marfan’s syndrome. Oral Surg Oral
Med Oral Pathol. 1965;19:757-70
Bauss O, Neter D, Rahman A. Prevalence of pulp
calcifications in patients with Marfan syndrome. Oral Surg Oral Med Oral Pathol Oral
Radiol Endod. 2008;106:56-61
Bauss O, Sadat-Khonsari R, Fenske C, Engelke W,
Schwestka-Polly R. Temporomandibular joint
dysfunction in Marfan syndrome. Oral Surg
Oral Med Oral Pathol Oral Radiol Endod.
2004;97:592-8
Bilodeau JE. Retreatment of a patient with Marfan
a
b
Figura 4 a, b: Paziente di 7 anni affetto da SM:
malocclusione dento-scheletrica di II classe, palato
ogivale, vestibolo-inclinazione degli incisivi centrali
superiori
Figura 4 c: Paziente di 7 anni affetto da SM
in trattamento ortopedico-ortodontico espansivo
mediante disgiuntore palatale rapido
www.buponline.com
149
Sindrome di Marfan
Syndrome and sever root resorption. Am J Orthod Dentofacial Orthop. 2010;137:123-34
Cistulli PA, Gotsopoulos H, Sullivan CE. Relationship between craniofacial abnormalities
and sleep-disordered breathing in Marfan’s
syndrome. Chest. 2001;120:1455-60
De Coster PJ, De Pauw G, Martens L, De Paepe
A. Craniofacial structure in Marfan syndrome:
a cephalometric study. Am J Med Genet A.
2004;15;131:240-8
De Coster PJ, Martens LC, De Paepe A. Oral manifestations of patients with Marfan syndrome:
a case-control study. Oral Surg Oral Med Oral
Pathol Oral Radiol Endod. 2002;93:564-72
De Peape A, Devereux RB, Dietz HC. Revised diagnostic criteria for the Marfan Syndrome. Am
J Med Genet. 1996;62:417-26
Dean JCS. Marfan syndrome: clinical diagnosis and Management. Eur J of Hum Genet.
2007;15:724-33
Dietz HC, Cutting GR, Pyeritz RE. Defects in the
fibrillin gene cause the Marfan syndrome; linkage evidence and identification of a missense
mutation. Nature. 1991;352:337-9
Disabella E, Grasso M, Marziliano N, Ansaldi S,
Lucchelli C, Porcu E, Tagliani M, Pilotto A,
Diegoli M, Lanzarini L, Malattia C, Pelliccia A, Ficcadenti A, Gabrielli O, Arbustini E.
Two novel and one known mutation of the
TGFBR2 gene in Marfan syndrome not associated with FBN1 gene defects. Eur J of Hum
Genet. 2006;14:34-8
Ehud Raanani MD, Probal Ghosh MD. The
multidisciplinary approach to the Marfan patient. IMAJ. 2008;10:171-4
Frydman M. The Marfan syndrome. Isr Med Assoc
J. 2008;10:175-8
Gorlin RJ, Cohen MM, Levin LS. Syndromes of
head and neck. Oxford (UK): Oxford Univ
Press. 1990;267-72
Hecht F, Beals RK. “New” syndrome of con-
genital contractural arachnodactyly originally described by Marfan in 1896. Pediatrics.
1972;49:574-9
Judge DP, Dietz HC. Marfan’s syndrome. Lancet.
2005;366:1965-76
Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation.
2008;117:2802-13
Khonsari RH, Corre P, Boukerma-Vernex Z, Schmidt J, Martin A, Maillet R, Riethmuller D,
Renaudin K, Frayssé C, Gayet-Delacroix M,
Khau Van Kien P, David A. Extreme oral manifestations in a Marfan-type syndrome. Int J
Oral Maxillofac Surg. 2010;39:622-5
Loeys BL, Dietz HC, Braverman AC, Callewaert
BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM,
Pyeritz RE, Sponseller PD, Wordsworth P, De
Paepe AM. The revised Ghent nosology for the
Marfan syndrome. J Med Genet. 2010;7:47685
Lynas MA. Marfan’s syndrome in Northern Ireland; an account of thirteen families. Ann
Hum Genet. 1958;22:289-309
Marfan MA-B. Un cas de deformation congenitale des quatre membres, plus prononcee aux
extremites, caractrerisee par l’allongement des
os avec un certain degre d’amincissement. Bull
Mem Soc Med Hop Paris. 1896;13:220-6
Pyeritz RE. Marfan syndrome and related disorders. Churchill Livingstone. 2007;3579-624
Pyeritz RE, Loeys B. The 8th international research
symposium on the Marfan syndrome and related conditions. Am J Med Genet. 2012;158:42-9
Sachdev MS, Sood NN, Kumar H, Ghose S. Bilateral aniridia with Marfan’s syndrome and dental anomalies: a new association. Jpn J Ophthalmol. 1986;30:360-6
Straub AM, Gravame R, Scully C, Tonetti MS. Severe periodontitis in Marfan’s syndrome: a case
report. J Periodontol. 2002;73:823-6
www.buponline.com
150
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Temtamy SA, Nassar AM, Ismail SR, Aboul-Ezz
EHA, Magid N. Comparative ultrastructure
studies of gingiva and pulp in the Marfan syndrome and homocystinuria. Am J Med Genet.
1989;32:242
Utreja A and Evans CA. Marfan Syndrome - An
Orthodontic Perspective. Angle Orthodontist.
2009;79:394-400
Verbraecken J, Paelinck BP, Willemen M, et al.
Aortic root diameter and nasal intermittent
positive airway pressure treatment in Marfan’s
syndrome. Clin Genet. 2003;63:131-4
Westling L, Mohlin B, Bresin A. Craniofacial manifestations in the Marfan syndrome: palatal dimensions and a comparative cephalometric analysis. J Craniofac Genet Dev Biol. 1998;18:211-8
Wilson R. Marfan’s syndrome: description of a family. Am J Med. 1957;23:434-44
www.buponline.com
14. Neurofibromatosi Tipo 1
Sinonimo: malattia di Von Recklinghausen
Codice ICD 10: Q85.0
L. Kondo, F. Skendo, M. Taddei, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Neurofibromatosi 1 (NF1) o malattia di Von
Recklingausen, dal nome del medico che la descrisse per la prima volta nel 1882, è una malattia
genetica rara caratterizzata da macchie caffelatte,
neurofibromi, lentiggini, noduli di Lish, displasia ossea e deficit cognitivo (Von Recklinghausen
FD, 1882; National Institutes of Health Consensus Development Conference, 1988).
L’incidenza è stimata di 1 su 4.000-5.000 nati vivi
per anno, in assenza di predilezione razziale, etnica,
geografica e di genere (Friedman JM et al., 1999).
Gli indici di morbilità e mortalità sono superiori
rispetto alla restante popolazione; le cause di morte
più frequenti sono le patologie cardiovascolari (Rasmussen SA et al., 2001).
GENETICA E DIAGNOSI
La NF1 è causata dalla mutazione del gene NF1
localizzato sul braccio lungo del cromosoma
17 nella regione 17q11.2 il cui prodotto proteico, la neurofibromina, è un soppressore tumorale
ad espressione ubiquitaria ampiamente presente
nel sistema nervoso (Viskochil D et al., 1990).
Nel 50% dei casi la malattia viene ereditata da un
genitore affetto secondo una modalità di tipo autosomico dominante; nel 50% la mutazione compare ex novo in figli di genitori sani (casi sporadici)
(Stephens K et al., 1992).
La diagnosi clinica di NF1 si formula in base alla
presenza di almeno 2 dei 7 criteri diagnostici stabiliti dalla National Institutes of Health Consensus
Development Conference nel 1988 (Tabella 1).
In assenza di anamnesi familiare positiva, nella
maggior parte dei casi la NF1 viene diagnosticata
intorno ai 3 anni in base a criteri clinici.
I test genetici, identificando la mutazione, permettono di confermare la diagnosi di NF1 nel
95% degli individui affetti. Nei casi familiari è
possibile la diagnosi prenatale su villocentesi o
amniocentesi mediante la ricostruzione dell’aplotipo (approccio semidiretto) o la ricerca diretta della
mutazione quando identificata in uno o più dei familiari affetti (Messiaen LM et al., 2000).
MANIFESTAZIONI CLINICHE SISTEMICHE
I pazienti affetti da NF1 presentano molte caratte-
www.buponline.com
152
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Tabella 1: Criteri diagnostici della Neurofibromatosi 1
1. sei o più macchie cutanee caffelatte di diametro >5 mm in epoca pre-puberale e >15 mm in epoca post-puberale
2. due o più neurofibromi cutanei e/o sottocutanei o un neurofibroma plessiforme
3. lentiggini alle ascelle e/o all’inguine
4. due o più noduli di Lisch
5. lesione scheletrica caratteristica (displasia dell’ala dello sfenoide, assottigliamento delle ossa lunghe con o
senza pseudoartrosi)
6. glioma del nervo ottico
7. un parente di primo grado affetto da NF1 (genitore, figlio, fratello)
ristiche comuni, nonostante l’estrema variabilità
fenotipica anche nell’ambito della stessa famiglia.
Le macchie cutanee caffelatte (Figura 1), sebbene possano essere presenti alla nascita, di norma si
sviluppano tra i primi mesi e i 2 anni di vita. Nella
maggior parte dei casi rappresentano la prima manifestazione della NF1. Il numero (a volte alcune
decine) e la dimensione (in media tra 1 e 3 cm)
delle macchie non sono correlati alla severità della patologia. La distribuzione è casuale, con localizzazione più frequente a livello del tronco e degli
arti e rara a livello del volto. Non hanno tendenza
alla trasformazione maligna (Korf BR, 2002).
I neurofibromi sono neoplasie composte da cellule di Schwann, fibroblasti, cellule perineurali e
mastociti. Il numero (solitamente limitato) e la localizzazione variano tra gli individui affetti, anche
all’interno della stessa famiglia. In base all’aspetto
e alla localizzazione sono classificati in 4 gruppi:
cutanei (focali o diffusi), sottocutanei, plessiformi (nodulari o diffusi), spinali (Williams VC et
al., 2009).
I neurofibromi cutanei compaiono tipicamente
nella tarda infanzia o nella prima adolescenza; raramente causano dolore e deficit neurologici; non si
trasformano in tumori maligni (Friedrich RE et al.,
Figura 1: Ragazza di 11 anni affetta da NF1. Macchie
cutanee caffelatte su viso e collo
2012; Williams VC et al., 2009; Ferner RE, 2007)
(Foto 2 a, b).
I neurofibromi sottocutanei sono spesso responsabili di dolore e di deficit neurologici (Ferner RE,
2007).
www.buponline.com
153
Neurofibromatosi
a
b
Figura 2 a, b: Bambina di 8 anni affetta da NF1. Tumefazione della guancia sede di neurofibroma
I neurofibromi plessiformi, visibili clinicamente
nel 30% dei pazienti con NF1, sono frequentemente congeniti e caratterizzati da ricca vascolarizzazione e fasci nervosi multipli; possono essere
responsabili di deficit neurologici (Friedrich RE et
al., 2012; Williams VC et al., 2009; Ferner RE et
al., 2007).
I neurofibromi spinali interessano le radici dei
nervi spinali crescendo in modo caratteristico attraverso un forame intervertebrale e producendo
masse intra- ed extraspinali (tumori “a clessidra”);
la componente intraspinale può comprimere il midollo causando deficit neurologici (Williams VC et
al., 2009).
Nel 7-12% dei casi, solitamente con partenza da
un neurofibroma plessiforme, a livello della guaina
di nervi periferici si sviluppa un processo tumorale maligno (Ferner RE, 2007).
Le lentiggini alle ascelle e/o all’inguine (segno di
Crowe), presenti in più del 60% dei casi, spesso
compaiono tra i 3 e i 5 anni; sono piccole (<3mm
di diametro) e possono essere localizzate anche su
palpebre, collo e seno (Korf BR, 2002).
I noduli di Lisch, amartomi melanocitici dell’iride, si sviluppano tra i 5 e i 10 anni e non provocano problemi di vista; sono patognomonici della
NF1 (Korf BR, 2002).
Le manifestazioni dell’apparato scheletrico comprendono ridotta densità ossea (nel 50%), bassa
statura (nel 30%), scoliosi idiopatica o distrofica
(nel 10%) e difetti ossei congeniti quali pseudoartrosi, displasia dell’ala dello sfenoide, macrocefalia. La scoliosi distrofica, più grave e ad esordio più
precoce della idiopatica, può essere responsabile di
gravi complicanze neurologiche e respiratorie. La
pseudoartrosi della tibia, evidente già alla nascita o
nei primi mesi di vita, è associata a fratture in seguito a traumi anche di scarsa entità, caratterizzate
da guarigione ritardata (Ferner RE, 2007).
Il glioma del nervo ottico è la manifestazione maligna intracranica più frequente nei pazienti affetti
www.buponline.com
154
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
da NF1; si sviluppa prevalentemente a livello del
chiasma ottico o in posizione postchiasmale; si
manifesta nel 15% dei bambini prima dei 6 anni
ed aumenta di volume durante la prima decade; in
circa 1/3 dei pazienti è responsabile di esoftalmo,
di deficit visivi e di pubertà precoce per invasione
dell’ipotalamo (Williams VC et al., 2009).
Le malattie cardiovascolari includono cardiopatie congenite (la più frequente è la stenosi della arteria polmonare) e vasculopatie, spesso localizzate
al sistema arterioso (stenosi, aneurismi, malformazioni artero-venose); la stenosi dell’arteria renale,
la più frequente, è causa di ipertensione (Williams
VC et al., 2009).
Il deficit cognitivo è una tra le manifestazioni più
frequenti associate alla NF1 ed è caratterizzato da
deficit di attenzione, iperattività, disordini dello
spettro dell’autismo, comportamenti anomali e
problematiche psicosociali. La maggior parte dei
pazienti presenta un quoziente intellettivo medio-basso.
Deficit di coordinazione motoria (sia fine che
grossolana), viso-spaziale e viso-motoria e disordini del linguaggio (verbali e non verbali) sono
presenti nel 30-65% dei bambini con NF1 (Williams VC et al., 2009; Ferner RE, 2007).
CARATTERISTICHE ORO-FACCIALI
Le lesioni caratteristiche della NF1 a livello del distretto cefalico possono essere localizzate nelle ossa
mascellari (neurofibromi solitari, ipoplasia, assenza di strutture ossee) e nei tessuti molli (fibromi
e neurofibromi della mucosa orale e della lingua)
(Sigillo R et al., 2002). Dall’analisi della letteratura
la prevalenza di queste lesioni risulta estremamente
variabile: 22% (Adkins JC et al., 1977), 72% (Shapiro SD et al., 1984), 92% (D’Ambrosio JA et al.,
1988). Tale variabilità è attribuibile all’eterogeneità
dei campioni esaminati ma soprattutto alle diverse
metodiche diagnostiche utilizzate e/o alla mancata
diagnosi per assenza di valutazione radiografica o
per difetti di imaging (Sigillo R et al., 2002; Kaplan I et al., 1994; Gorlin RJ et al., 1990).
I neurofibromi ossei si evidenziano radiograficamente come lesioni radiotrasparenti con margini o
scarsamente o ben definiti (Kaplan I et al., 1994).
Le caratteristiche radiografiche associate alla
presenza di neurofibromi nelle aree mascellari
includono: ipoplasia del processo coronoide e del
condilo, aumento dell’incisura coronoide, assottigliamento e concavità del ramo, diminuzione
dell’angolo mandibolare, incisura antigoniale accentuata, allargamento fusiforme e/o ramificazione
del canale mandibolare, forame mandibolare allargato e basso, denti non erotti con lesioni di tipo
cistico, aree di radiotraspareza periapicali a livello
di denti vitali, malformazioni e riassorbimenti radicolari (Friedrich RE et al., 2003; Sigillo R et al.,
2002; Holtzman L, 1998; Lee L et al., 1996) (Foto
2 c, d). Pur essendo ancora utilizzata la radiologia
tradizionale nella diagnosi e nella caratterizzazione
delle lesioni ossee extracraniche, per definire la localizzazione e le dimensioni delle lesioni a livello
della base cranica e delle ossa facciali sono utilizzate la Risonanza Magnetica e la Tomografia Assiale
Computerizzata, che presentano migliore qualità
diagnostica (Kaplan I et al., 1994).
In pazienti con neurofibromi ossei localizzati a livello delle ossa mascellari sono descritti 12 segni
clinici e radiografici patognomonici (Lee L et al.,
1996):
1. deformazione dell’orecchio esterno (dislocato
inferiormente) sul lato affetto;
2. minimo gonfiore della guancia o della mucosa
intraorale;
3. ridotta traslazione del condilo sul lato affetto;
4. deviazione della mandibola verso il lato affetto;
5. denti affollati a livello del mascellare superiore
sul lato affetto;
www.buponline.com
155
Neurofibromatosi
c
e
d
Figura 2 c, d: Ortopantomografia : formula di dentizione mista; estesa area radiotrasparente a livello dell’emiarcata inferiore
destra causata dalla presenza di un neurofibroma, responsabile
di deformazione a livello del condilo e dell’incisura coronoide,
inclusione degli elementi 16 e 46 e dislocazione degli elementi
17 e 47; elementi 26 e 36 di aspetto taurodontico
6. denti affollati a livello della mandibola sul lato
affetto;
7. ipoplasia del mascellare superiore e dello zigomo sul lato affetto;
8. deformazione dell’incisura coronoide (dislocata inferiormente);
9. pseudoallungamento del processo condilare/
coronoide;
10. ipoplasia deformante del ramo ascendente con
difetti perforanti;
11. angolo goniaco piatto o mancante;
12. deformazione o ipoplasia del corpo della mandibola.
Per quanto riguarda i neurofibromi con localizza-
f
Figura 2 e, f: Tessuto neurofibromatoso in sede 46
zione a livello dei tessuti molli, le sedi intraorali
più frequenti sono la lingua e la mucosa orale
(Foto 2 e, f ) (Neville BW et al., 1991); sono descritte localizzazioni anche a livello di labbra (Pollack RP, 1990), gengiva (Allen CM et al., 1997) e
pavimento della bocca; palato e guance sono raramente interessati (Bongiorno MR et al., 2006;
Tripi TR et al., 1998).
I neurofibromi a livello della lingua, quasi sempre
nodulari e unilaterali, sono causa di macroglossia
e di allargamento delle papille fungiformi. La lesione di solito è asintomatica e la diagnosi spesso
viene effettuata quando il paziente lamenta il disagio di una massa che aumenta; la sintomatologia
dolorosa è legata ad un trauma secondario (Sigillo
www.buponline.com
156
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
R et al., 2002). Quando la sede è la base della lingua la sintomatologia è caratterizzata da odinofagia, disfagia, cambiamento della voce, dolore alle
orecchie; clinicamente si evidenzia una massa a
livello del collo (Keutel C et al., 1997).
I neurofibromi orali si presentano come masse
dure, sottomucose, a crescita lenta, di dimensioni comprese tra pochi millimetri (piccoli noduli) e
alcuni centimetri (grandi masse peduncolate).
Nella NF1 sono descritte proliferazioni neoplastiche di cellule perineurali e di Schwann a livello
della gengiva responsabili di iperplasia gengivale,
causa di dislocazioni, affollamenti e inclusioni dentali e di elevati indici di placca (Doufexi A et al.,
2005; Clementi M et al., 1990).
I neurofibromi a livello parodontale possono
manifestarsi clinicamente sotto forma di ascesso
parodontale asintomatico (Cunha KS et al., 2004)
o essere diagnosticati radiograficamente come lesione radiotrasparente uniloculare circoscritta a livello radicolare (Powell CA et al., 2006). È descritto un caso di iperplasia gengivale diffusa unilaterale
con localizzazione sia mandibolare che mascellare
in cui la gengiva ipertrofica, fibrosa, non dolente,
non sede di infiammazione, era responsabile di
dislocazioni dentali con formazione di diastemi
e deviazione della linea mediana (Bekisz O et al.,
2000).
Sono riportati in letteratura due casi di neurofibromi localizzati a livello del tessuto pulpare
(Fani MM et al., 2005; Curtin JP et al., 1997).
In pazienti affetti da NF1 è descritta eruzione
anticipata dei denti decidui (Lammert M et al.,
2007).
Per quanto riguarda la prevalenza di patologia cariosa in pazienti affetti da NF1, in letteratura sono
presenti dati contrastanti (Bardellini E et al., 2011;
Visnapuu V et al., 2011; Tsang et al., 2010; Tucker
T et al., 2007). La nostra esperienza clinica evidenzia come gli indici di patologia cariosa non mostrino differenze statisticamente significative rispetto
alla popolazione con anamnesi medica negativa.
Per quanto riguarda le patologie ortopedico-ortodontiche, i dati presenti in letteratura relativi alla
prevalenza di malocclusioni nei pazienti affetti da
NF1 sono scarsi; nessuno rileva differenze significative rispetto alla restante popolazione eccetto
che per la relazione molare, essendo descritta nei
pazienti affetti da NF1 una prevalenza di malocclusioni dentali di III classe più elevata rispetto ad
un gruppo controllo di soggetti sani (Bardellini E
et al., 2011).
LINEE GUIDA DI TERAPIA
La diagnosi, il follow-up e le terapie necessitano
della presenza di un team multi e interspecialistico adeguatamente formato sulla sindrome per
monitorare il paziente e sottoporre a terapia le patologie dal momento in cui si manifestano (Huson
SM, 1999).
In età evolutiva è necessario un costante monitoraggio clinico, con controlli annuali, per individuare l’insorgenza di deficit psicomotori, di glioma
del nervo ottico, di neurofibromi e di scoliosi. I
bambini devono essere sottoposti tempestivamente a valutazione neuropsicologica per impostare il
piano educativo più idoneo da attuare in stretta
collaborazione tra pediatri, psicologi e insegnanti
(Williams VC et al., 2009). Studi clinici su animali da laboratorio positivi a NF1 hanno evidenziato un miglioramento dei deficit cognitivi dopo
trattamento con lovostatina (Li W et al., 2005);
sono tuttavia necessarie ulteriori ricerche per il suo
utilizzo nell’uomo. Esami complementari, come
esame radiografico dello scheletro (cranio, forame
ottico, colonna vertebrale) e visita otorinolaringoiatrica vengono eseguiti quando i segni clinici lo
richiedono (Becelli R et al., 2002). Un’eccezione
va fatta per la risonanza magnetica del nervo ottico, da eseguire sistematicamente per valutare la
www.buponline.com
157
Neurofibromatosi
presenza di un glioma aggressivo in pazienti in cui
l’esame oftalmologico sia di difficile esecuzione a
causa della giovane età e/o di deficit cognitivi. Appena la collaborazione del bambino lo permette, è
sufficiente eseguire un esame oftalmologico annuale valutando acutezza visiva e campo visivo (Pinson
S et al., 2001).
Gli adulti che non presentano patologie possono
essere esaminati ogni 2-3 anni.
Per il trattamento dei neurofibromi plessiformi
sono stati proposti farmaci antistaminici, farmaci
chemioterapici (interferone), farmaci antifibrotici (pirfenidone) (Babovic-Vuksanovic D et al.,
2006 Riccardi VM, 1993). Nessuno di questi ha
dimostrato una reale efficacia anche se gli antistaminici si sono dimostrati utili per il trattamento
di sintomi, come il prurito. E’consigliato applicare
emollienti sui neurofibromi ed evitare l’eccessivo calore per prevenirne l’irritazione (Ferner RE,
2007). Quando causano disagi al paziente, anche
dal punto di vista estetico, possono essere asportati
chirurgicamente (Ferner RE, 2007).
Per quanto concerne le patologie di pertinenza
odontostomatologica, in un’ottica di interdisciplinarietà, è compito del pediatra, dal momento
in cui si effettua la diagnosi, inviare il paziente
all’odontoiatra infantile perché informi la famiglia
sull’importanza della salute orale e sulla necessità
di interventi di prevenzione (igiene orale domiciliare, alimentazione non cariogenica, utilizzo di
fluoro topico e, quando indicato, sistemico, visite periodiche trimestrali/semestrali, sigillatura dei
solchi, delle fessure e dei fori ciechi dei molari permanenti).
In ambiente odontoiatrico, nei pazienti con NF è
importante utilizzare un approccio psicologico e
comportamentale mirato in relazione al possibile
deficit neurocognitivo. Di fondamentale importanza è individuare i canali comunicativi più efficaci per conquistare la fiducia e la collaborazione
del paziente.
In presenza di patologie cardiovascolari a rischio
di EB, prima di effettuare procedure odontoiatriche che provocano sanguinamento è necessario effettuare la profilassi antibiotica (vedi il capitolo
Cardiopatie congenite).
Essendo i pazienti affetti da NF1 ad elevato rischio
di sviluppare neurofibromi nel distretto cefalico, è
importante stabilire un programma di follow-up
dedicato.
Le visite odontoiatriche devono essere periodiche, a cadenza almeno semestrale. Nel corso
dell’esame obiettivo intra- ed extra-orale si valuta
la presenza di gonfiori della guancia e dei tessuti
molli intraorali, deviazioni della mandibola, noduli multipli pigmentati, lesioni orali polipoidi,
ipertrofia gengivale, anomalie di eruzione e dislocazioni dentarie.
Esami radiografici (in particolare l’ortopantomografia) devono essere eseguiti a partire dai 6 anni
di età, con una frequenza da concordare con il pediatra curante in base al livello di rischio.
La necessità di un programma di follow-up periodico è supportata dalle possibili e frequenti
complicanze locali e dal documentato rischio di
trasformazione maligna dei neurofibromi. In particolare, in caso di rapido incremento dimensionale
del neurofibroma e di comparsa di sintomatologia
dolorosa si deve sospettare una trasformazione maligna, che necessita di conferma bioptica.
Per quanto riguarda le patologie di pertinenza
ortopedico-ortodontica è necessario monitorare
i pazienti con NF1 e attuare, quando indicate, terapie di tipo intercettivo in dentizione decidua o
mista precoce per consentire un corretto sviluppo
delle basi ossee e terapie di tipo ortodontico al
termine della dentizione mista.
Per quanto riguarda le lesioni dentali di origine
traumatica, pur non esistendo in letteratura alcun
riferimento specifico, considerando i possibili deficit di coordinazione e di attenzione e l’iperattività,
è ipotizzabile una loro elevata prevalenza a livello
www.buponline.com
158
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
degli elementi sia decidui che permanenti; è quindi necessario fornire alla famiglia linee guida per
la loro gestione, in particolare sull’opportunità del
reimpianto immediato in caso di avulsione di denti
permanenti.
BIBLIOGRAFIA
Adkins JC, Ravitch MM. The operative management of von Recklinghausen’s neurofibromatosis in children, with special reference to lesions
of the head and neck. Surgery. 1977;82:342-8
Allen CM, Miloro M. Gingival lesion of recent onset in a patient with neurofibromatosis. Oral
Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:595-7
Babovic-Vuksanovic D, Ballman K, Michels V,
et al. Phase II trial of pirfenidone in adults
with neurofibromatosis type 1. Neurol.
2006;67:1860-2
Bardellini E, Amadori F, Flocchini P, Conti G, Piana G, Majorana A. Oral findings in 50 children
with neurofibromatosis type 1. A case control
study. Eur J Paediatr Dent. 2011;12:256-60
Becelli R, Renzi G, Cerulli G, Saltarel A, Perugini M. Von Recklinghausen neurofibromatosis
with palatal localization. Diagnostic and surgical problems in two clinical cases. Minerva
Stomatol. 2002; 51:391-7
Bekisz O, Darimont F, Rompen EH. Diffuse but
unilateral gingival enlargement associated with
von Recklinghausen neurofibromatosis: a case
report. J Clin Periodontol. 2000;27:361-5
Bongiorno MR, Pistone G, Aricò M. Manifestations of the tongue in Neurofibromatosis type
1. Oral Dis. 2006;12:125-9
Clementi M, Barbujani G, Turolla L, Tenconi R.
Neurofibromatosis-1: A maximum likelihood
estimation of mutation rate. Hum Genet.
1990;84:116-8
Cunha KS, Barboza EP, Dias EP, Oliveira FM.
Neurofibromatosis type I with periodontal
manifestation. A case report and literature review. Br Dent J. 2004;196:457-60
Curtin JP, McCarthy SW. Perineural fibrous thickening within the dental pulp in type 1 neurofibromatosis: a case report. Oral Surg Oral Med
Oral Pathol Oral Radiol Endod. 1997;84:4003
D’Ambrosio JA, Langlais RP, Young RS. Jaw and
skull changes in neurofibromatosis. Oral Surg
Oral Med Oral Pathol. 1988;66:391-6
Doufexi A, Mina M, Ioannidou E. Gingival overgrowth in children: epidemiology, pathogenesis, and complications. A literature review. J
Periodontol. 2005;76:3-10
Fani MM, Shahidi SH, Zamiri B, Daneshbod KH.
Neurofibroma of dental pulp. Case report. Indian J Radiol Imaging. 2005;15:47-8
Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of
individuals with neurofibromatosis 1. J Med
Genet. 2007;44:81-8
Ferner RE. Neurofibromatosis 1. Eur J of Hum
Genet. 2007;15:131-8
Friedman JM, Riccardi VM. Clinical and epidemiological features. In: Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. 3rd
ed., Baltimore, MD: Johns Hopkins University
Press. 1999:29-86
Friedrich RE, Giese M, Stelljes C, Froeder C,
Scheuer HA. Size of tooth crowns and position of teeth concerning the extension of facial plexiform neurofibroma in patients with
neurofibromatosis type 1. Anticancer Res.
2012;32:2207-14
Friedrich RE, Giese M, Schmelzle R, Mautner VF,
Scheuer HA. Jaw malformations plus displacement and numerical aberrations of teeth in
neurofibromatosis type 1: a descriptive analysis
of 48 patients based on panoramic radiographs
www.buponline.com
159
Neurofibromatosi
and oral findings. J Craniomaxillofac Surg.
2003;31:1-9
Gorlin RJ, Cohen MM Jr, Levin S. Syndromes
of the head and neck. 3rd ed., Oxford: Oxford
University Press. 1990;392-9
Holtzman L. Radiographic manifestation and
treatment considerations in a case of multiple
neurofibromatosis. J Endod. 1998;24:442-3
Huson SM. What level of care of neurofibromatoses? Lancet. 1999;353:1114-6
Kaplan I, Calderon S, Kaffe I. Radiological findings in jaws and skull of neurofibromatosis type 1 patients. Dentomaxillofac Radiol.
1994;23:216-20
Keutel C, Vees B, Krimmel M, Cornelius CP,
Schwenzer N. Oral, facial and cranial manifestations of von Recklinghausen neurofibromatosis (NF). Mund Kiefer Gesichtschir.
1997;1:268-71
Korf BR. Clinial features and pathobiology of neurofibromatosis 1. J Child Neurol. 2002;17:5737
Lammert M, Friedrich RE, Friedman JM, Mautner VF, Tucker T. Early primary tooth eruption
in neurofibromatosis 1 individuals. Eur J Oral
Sci. 2007;115:425-6
Lee L, Yan YH, Pharoah MJ. Radiographic features
of the mandible in neurofibromatosis: a report
of 10 cases and review of the literature. Oral
Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:361-7
Li W, Cui Y, Kushner SA, et al. The HMG-CoA
reductase inhibitor lovostatin reverses the
learning and attention deficits in a mouse
model of neurofibromatosis type 1. Curr Biol.
2005;15:1961-7
Messiaen LM, Callens T, Mortier G, Beysen D,
Vandenbroucke I, Van Roy N, Speleman F,
Paepe AD. Exhaustive mutation analysis of the
NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual
splicing defects. Hum Mutat. 2000;15:541-55
National Institutes of Health Consensus Development Conference: Neurofibromatosis Conference statement. Arch Neurol. 1988;45:575-8
Neville BW, Hann J, Narang R, Garen P. Oral neurofibrosarcoma associated with neurofibromatosis type I. Oral Surg Oral Med Oral Pathol.
1991;72:456-61
Pinson S, Créange A, Barbarot S, Stalder JF, Chaix
Y, Rodriguez D, Sanson M, Berheim A, d’Incam M, Doz F, Stoll C, Combemale P, Kalifa C, Zeller J, Teillac-Hamel D, Lyonnet S,
Zerah M, Lacour JP, Wolkenstein P. Réseau
NF- France. Guidelines for the management
of neurofibromatosis 1: Raccomandations de
prise en charge de la neurofibromatose 1. Ann
Dermatol Venereol. 2001;128:567-75
Pollack RP. Neurofibroma of the palatal mucosa. J
Periodontol. 1990;61:456-8
Powell CA, Stanley CM, Bannister SR, McDonnell HT, Moritz AJ, Deas DE. Palatal Neurofibroma associated with localized periodontitis. J
Periodontol. 2006;77:310-5
Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using
U.S. death certificates. Am J Hum Genet.
2001;68:1110-8
Riccardi VM. A controlled multiphase trial of ketotifen to minimize neurofibroma-associated pain
and itching. Arch Dermatol. 1993;129:577-81
Shapiro SD, Abramovitch K, Van Dis ML,
Skoczylas LJ, Langlais RP, Jorgenson RJ, et
al. Neurofibromatosis: Oral and radiographic manifestations. Oral Surg Oral Med Oral
Pathol. 1984;58:493-8
Sigillo R, Rivera H, Nikitakis NG, Sauk JJ: Neurofibromatosis type 1: a clinicopathological
study of the orofacial manifestations in 6 pediatric patients. Pediatr. Dent. 2002;24:575-80
Stephens K, Kayes L, Riccardi VM, Rising M,
Sybert VP, Pagon RA: Preferential mutation
www.buponline.com
160
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
of the neurofibromatosis type 1 gene in paternally derived chromosomes. Hum Genet.
1992;88:279-82
Tripi TR, Bonaccorso A. Recklinghausen neurofibromatosis. Report of a case. Minerva Stomatol. 1998;47:617-22
Tsang ES, Birch P, Friedman JM, Johnston D, Tucker T, Armstrong L. Prevalence of dental caries in children with neurofibromatosis 1. Clin
Oral Investig. 2010;14:479-80
Tucker T, Birch P, Savoy DM, Friedman JM. Increased dental caries in people with neurofibromatosis 1. Clin Genet. 2007;72:524-7
Viskochil D, Buchberg AM, Xu G, et al. Deletions
and a translocation inrterrupt a cloned gene
at the neurofibromatosis type I locus. Cell.
1990;62:187-92
Visnapuu V, Pienihäkkinen K, Peltonen S, Happonen RP, Peltonen J. Neurofibromatosis 1 and
dental caries. Clin Oral Investig. 2011;15:11921
Von Recklinghausen FD. Uber die multiplen fi
brome der haut und ihre Beziehung zu den
multiplen neuromen. Berlin: A Hirschwald.
1882
Williams VC, Lucas J, Babcock MA, Gutmann
DH, Korf B, Maria BL. Neurofibromatosis
type 1 revisited. Pediatrics. 2009;123:124-33
www.buponline.com
15. Sindrome di Alagille
Sinonimi: Sindrome da paucità dei dotti biliari,
Displasia arterioepatica, Delezione 20p11.2
Codice ICD 10: Q44.7
S. Bagattoni, F. Skendo, I. Cremonesi, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
GENETICA E DIAGNOSI
La Sindrome di Alagille (SAG), descritta per la
prima volta da Alagille, Habib eThomasin nel
1969, è una malattia genetica rara caratterizzata
da anomalie epatiche, vascolari, cardiache, oculari, scheletriche e facies caratteristica e, meno
frequentemente, da anomalie renali e neurologiche
(Kamath BM et al., 2010; Spinner NB et al., 2010;
Alagille D et al., 1987; Alagille D et al., 1969).
L’incidenza è stimata in 1 su 70.000 nati vivi per
anno, in assenza di predilezione razziale, etnica,
geografica e di genere (Kamath BM et al., 2010;
Garcia MA et al., 2005).
La prognosi, estremamente variabile in funzione
della gravità delle anomalie associate, è peggiore
nei bambini con ittero colestatico perinatale; la
presenza di insufficienza epatica e di patologie cardiovascolari aumenta il rischio di morte (Emerick
KM et al., 1999).
La SAG è una malattia a trasmissione autosomica
dominante, a penetranza ed espressività variabili anche all’interno di una stessa famiglia (Spinner
NB et al., 2010; Garcia MA et al., 2005).
Si distinguono due forme: tipo 1, la più frequente (89%), causata da mutazioni del gene JAG1
(20p12) e tipo 2, più rara, causata da mutazioni
del gene NOTCH2 (1p12).
Le mutazioni nel 30-50% sono ereditate e nel 5070% sono ex novo (Turnpenny PD et al., 2012).
La diagnosi è clinica e si formula in base alla presenza di almeno tre dei cinque criteri diagnostici
elencati nella Tabella 1 ed è confermata da test
genetici (Turnpenny PD et al., 2012; Kamath BM
et al., 2010; Spinner NB et al., 2010; Gridley T,
2003; Emerick KM et al., 1999).
www.buponline.com
162
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Tabella 1: Criteri clinici diagnostici di Sindrome di Alagille
CRITERI CLINICI
colestasi cronica con o senza ittero
ittero o iperbilirubinemia nel periodo neonatale
anomalie vascolari e cardiache
stenosi periferica dell’arteria polmonare, atresia
polmonare, difetto del setto atriale, difetto del setto
ventricolare, tetralogia di Fallot
anomalie scheletrice
vertebre a farfalla, occasionalmente emivertebre,
fusione di vertebre adiacenti, spina bifida
anomalie oculari
difetti del compartimento anteriore, embryotoxon
posteriore
facies caratteristica
forma del viso “a triangolo invertito”, fronte ampia
e prominente, occhi infossati con moderato ipertelorismo, ponte nasale piatto, naso diritto, sottile e a
bulbo, mento prominente e a punta, profilo piatto,
orecchie a impianto basso
La diagnosi prenatale può essere effettuata su villocentesi o amniocentesi (Jung C et al., 2007) ma
non è predittiva della severità delle manifestazioni
cliniche (Turnpenny PD et al., 2012). È descritto in letteratura un caso di diagnosi prenatale in
donna affetta da SAG sulla base di alterazioni fetali
all’ecografia (stenosi severa dell’arteria polmonare
e ritardo nella crescita severo e progressivo), confermata dopo la nascita (Albayram F et al., 2002).
MANIFESTAZIONI CLINICHE SISTEMICHE
Le manifestazioni cliniche tipiche della SAG sono
descritte nella Tabella 2 (Spinner NB et al., 2010;
Kamath BM et al., 2010; Garcia MA et al., 2005;
McElhinney DB et al., 2002; Hingorani M et al.,
1999; Emerick KM et al., 1999; Rosenfield NS et
al., 1980).
La sindrome può manifestarsi nel neonato con ittero prolungato causato dalla colestasi e/o con i
segni e i sintomi della patologia cardiaca.
La colestasi, presente nella quasi totalità dei pazien-
ti, è responsabile di ittero, epatosplenomegalia,
feci acoliche, urine ipercromiche, aumento degli
indici di colestasi e di danno epatico (bilirubina
diretta, transaminasi, γ-GT, fosfatasi alcalina), iperlipidemia (colesterolo totale e trigliceridi), deficit di
fattori della coagualazione. La metà dei casi evolve
in ipertensione portale (Spinner NB et al., 2010;
Kamath BM et al., 2010; Sze DY et al., 2008).
Nell’80% dei casi si associa ritardo di crescita causato da un bilancio energetico negativo secondario a ridotto introito calorico e a malassorbimento
lipidico; tra tutte le forme di colestasi i bambini
con SAG presentano il difetto di crescita più severo
(Rovner AJ et al., 2006; Emerick KM et al., 1999).
Il malassorbimento lipidico può causare deficit di
vitamine liposolubili (A, D, E, K). Il deficit di
vitamina A può causare rossore agli occhi e cecità
notturna; quello di vitamina D deficit di mineralizzazione e rischio di fratture a livello di ossa e radici
dentali; quello di vitamina E patologie del sistema
nervoso e dell’apparato muscolare; quello di vitamina K deficit della coagulazione.
Il prurito intenso, manifestazione della colestasi,
www.buponline.com
163
Sindrome di Alagille
Tabella 2: Manifestazioni cliniche nella Sindrome di Alagille
MANIFESTAZIONE SISTEMICA
PREVALENZA
CONSEGUENZE CLINICHE
Patologie epatiche
colestasi cronica
96%
atresia dei dotti biliari intraepatici
85%
iperbilirubinemia coniugata nel periodo neonatale
non specificata
prurito intrattabile
frequente
ipertensione portale
cirrosi
frequente
carcinoma epatocellulare
raro
epatosplenomegalia, ittero
15% necessita di trapianto
Anomalie vascolari e cardiache
atresia o stenosi delle arterie polmonari (67%)
tetralogia di Fallot (7-10%)
difetti interventricolari e interatriali
stenosi e coartazione aortica
90%
di gravità e sintomatologia molto
variabili
anomalie congenite della vascolarizzazione intracranica
non specificata
rischio di emorragie intracraniche
emorragie intracraniche
15%
nel 30-50% fatali
78-89%
moderata diminuzione dell’acuità
visiva, potenziale causa di glaucoma
Anomalie oculari
embryotoxon posteriore
anomalia di Axenfeld
anomalia di Rieger
retinite pigmentosa
anomalie papillari
anomalie del disco ottico
anomalie retiniche
anomalie corneali
anomalie dell’iride
ipopigmentazione del fondo oculare
57%
anomalie oculari non specifiche secondarie a deficit vitamine A e E
non specificata
www.buponline.com
164
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
MANIFESTAZIONE SISTEMICA
PREVALENZA
CONSEGUENZE CLINICHE
vertebre a farfalla
51%
asintomatiche
anomalie di:
colonna vertebrale
mani
costole
ossa pelviche
non specificata
deficit di mineralizzazione ossea (da ridotto
assorbimento di calcio e vitamina D)
non specificata
rischio di fratture patologiche
scarso assorbimento di lipidi, di acidi grassi essenziali e di vitamine liposolubili
non specificata
malnutrizione e alterazione di crescita
coagulopatia
(deficit vitamina K)
rachitismo
(deficit vitamina D)
retinopatia, miopatia e neuropatia
periferica (deficit vitamine A e E)
deficit di crescita intrauterino
50-90%
ritardo del picco di crescita
frequente
Anomalie scheletriche
Anomalie del sistema nervoso centrale
ritardo mentale (QI tra 60 e 80)
sindrome da iperattività,
disturbi dell’attenzione
30%
rari
distonie e tremori
non specificata
possono risolversi in seguito al
trapianto
Anomalie renali
anomalie di funzione aspecifiche
anomalie anatomiche
acidosi renale tubulare
40%
insufficienza renale
Manifestazioni cutanee
xantomi, teleangectasie
28-42%
www.buponline.com
165
Sindrome di Alagille
è frequente ed è solitamente il sintomo più debilitante; è causa di lesioni da grattamento (escoriazioni, lichenificazioni, mutilazioni cutanee), insonnia,
difficoltà di attenzione e riduzione del rendimento
scolastico (Turnpenny PD et al., 2012; Garcia MA
et al., 2005; Emerick KM et al., 2002).
Secondari a colestasi protratta e severa sono gli
xantomi, presenti nel 28-42% dei casi; la localizzazione più comune è la superficie estensoria delle
dita, seguita da solchi palmari, nuca, gomiti, ginocchia, zone glutea e perianale, fossa poplitea, inguine. Compaiono progressivamente a partire dai 4
anni, per regredire dopo i 10 anni. Meno frequenti
sono linfedema dell’estremità, eritema palmare,
secchezza cutanea, ipercheratosi follicolare e teleangectasie (Schwartz R et al., 2008; Garcia MA et
al., 2005).
Le anomalie vascolari e cardiache sono un altro
criterio diagnostico principale della sindrome di
Alagille; le più comuni sono la stenosi e l’atresia
delle arterie polmonari; altre sono la tetralogia di
Fallot, i difetti interventricolari e interatriali, la stenosi e la coartazione aortica (Ghidini A et al., 2007;
McElhinney DB et al., 2002). Meno frequenti sono
anomalie della vascolarizzazione intracranica, potenzialmente responsabili di emorragie intracraniche, che in più del 30% dei casi sono mortali.
Le anomalie oculari, responsabili di moderata riduzione dell’acuità visiva, comprendono embryotoxon
posteriore (ispessimento della linea di Schwalbe
nella giunzione tra l’iride e la cornea), anomalia di
Axenfeld (alterazione del segmento anteriore dell’occhio con dislocazione della linea di Schwalbe e formazione di sinechie anteriori periferiche), anomalia
di Rieger (anomalia dell’iride che si presenta in alcune porzioni adesa alla cornea con opacamento della
cornea stessa, e distorsione della pupilla), retinite
pigmentosa, anomalie papillari, anomalie del disco
ottico, della cornea, dell’iride (Turnpenny PD et al.,
2012; Narula P et al., 2006).
Tra le anomalie scheletriche quelle vertebrali sono
le più frequenti, interessando il 66% dei soggetti affetti da SAG, il 48% dei quali presenta un interessamento vertebrale multiplo. Le vertebre a farfalla,
le più frequenti, localizzate a livello T6-T9, sono secondarie a mancata fusione, completa o incompleta,
dell’arco vertebrale anteriore. Nella maggior parte
dei casi sono simmetriche, quindi clinicamente silenti, rappresentando un reperto non sempre semplice da identificare con indagini radiologiche. Le
rare forme sintomatiche sono quelle asimmetriche,
che possono presentarsi con scoliosi di grado variabile. Anomalie scheletriche meno frequenti sono
ipoplasia delle falangi distali delle dita (16%), ulna
corta (13%), anomalie di costole e ossa pelviche (58%) (Ryan RS et al., 2003; Sanderson E et al., 2002;
Delgado A et al., 1996).
Meno frequenti sono ritardo mentale, alterazioni
comportamentali, anomalie renali, ipotiroidismo
(Sze DY et al., 2008; Schwartz R et al., 2008).
Relativamente rare sono anomalie auricolari (alterazioni delle ossa temporali, displasia dei canali
semicircolari posteriori) (Koch B et al., 2006).
CARATTERISTICHE ORO-FACCIALI
Nella prima infanzia il volto del paziente con
SAG, definito “a triangolo invertito”, è caratterizzato da fronte ampia e prominente, occhi infossati con moderato ipertelorismo, ponte nasale piatto, naso diritto, sottile e a bulbo, mento
prominente e a punta, profilo piatto, orecchie
a impianto basso. Durante la crescita gli occhi
rimangono infossati ed aumenta il prognatismo,
risultando predominante la porzione inferiore del
volto. In rari casi sono state descritte macrocefalia
e craniosinostosi (Spinner NB et al., 2010; Kamath
BM et al., 2002; Kamath BM et al., 2010; McElhinney DB et al., 2002; Emerick KM et al., 1999).
Manifestazioni a livello dentale caratteristiche
della SAG sono correlate alla colestasi e alla con-
www.buponline.com
166
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
seguente iperbilirubinemia. Nei pazienti in cui nel
corso dell’odontogenesi la concentrazione sierica di
bilirubina è superiore a 30 mg/100 ml, l’accumulo
di bilirubina a livello dei germi dentali è causa
di discromie dentali di colore verde (Figura 1 a,
b). Sono interessati tutti gli elementi decidui e permanenti la cui dentinogenesi sia avvenuta prima
della risoluzione della colestasi (Amaral TH et al.,
2008; Guadagni MG et al., 2005; Guimares LP et
al., 2003; Al-Mutawa S et al., 2002). I denti si presentano con striature di pigmentazione (corrispondenti alle fasi di deposizione tissutale) di differenti
colorazioni di verde, che tendono a modificarsi nel
tempo per modificazioni della translucenza dello
smalto, che trasmette la colorazione verde della
dentina sottostante (Amaral TH et al., 2008; Guimares LP et al., 2003).
Un case report relativo ad un paziente con SAG descrive denti decidui taurodonti con estese decalcificazioni ed alterazioni della predentina e della dentina
interglobulare. Nello stesso studio l’analisi cefalometrica evidenzia ramo mandibolare ridotto in altezza
e angolo goniaco ampio (Kazuko I et al., 1998).
Nei pazienti affetti da SAG sottoposti a trapianto
epatico l’assunzione di farmaci immunosoppressori può comportare problematiche a livello orale. La Ciclosporina A ha come effetto collaterale
frequente l’ipertrofia gengivale; il Tacrolimus,
attualmente utilizzato, comporta minori complicanze gengivali in termini di prevalenza e di gravità (Guadagni MG et al., 2005; Sheehy EC et al.,
2000). Altre complicanze della terapia immunosoppressiva sono leucoplachie, micosi da Candida
Albicans, infezioni da Herpes virus (Al-Mutawa S
et al., 2002).
I pazienti affetti da SAG sono ad alto rischio di
patologia cariosa. I bambini hanno carenze nutrizionali che obbligano ad una frequente assunzione
di alimenti ad alto contenuto glucidico; i genitori, a causa dell’effetto alone provocato dalla grave
patologia sistemica, tendono a sottovalutare l’im-
portanza dell’igiene orale ed utilizzano soluzioni
zuccherate per favorire l’addormentamento, reso
spesso difficile dal prurito. Questi comportamenti
rappresentano fattori di rischio per la patologia cariosa e per quadri, anche gravi, di Early Childhood
Caries (ECC) (Amaral TH et al., 2008; Guadagni
MG et al., 2005).
Sono frequenti quadri di gengivite marginale, prevalentemente causati da insufficiente igiene orale
domiciliare, eventualmente aggravati, in pazienti
sottoposti a trapianto di fegato, dall’alterata risposta infiammatoria e dall’ipertrofia gengivale secondarie alla terapia immunosoppressiva (Sheehy EC
et al., 2000) (Figura 1 c, d).
La letteratura non evidenzia patologie ortopedicoortodontiche direttamente riconducibili alla SAG.
LINEE GUIDA DI TERAPIA
I pazienti con SAG sono affetti da numerose patologie che rendono necessari interventi multispecialistici. La diagnosi, le terapie e il follow-up
richiedono la presenza di un team multi e interdisciplinare adeguatamente formato sulla sindrome,
che include pediatri, cardiologi, chirurghi, ortopedici, oculisti ed odontoiatri.
La gestione di questi pazienti dipende dalla severità
delle patologie epatiche e cardiache.
Di primaria importanza è il controllo delle complicanze della colestasi. Per prevenire il progressivo
deterioramento della funzionalità epatica è necessario un corretto apporto nutrizionale, eventualmente per via parenterale (Ling SC, 2007). La dieta deve essere ipolipidica con supplementazione di
vitamine liposolubili, di acidi grassi essenziali e di
trigliceridi a catena media.
Per alleviare il prurito, che soprattutto nella prima
infanzia interferisce con il sonno, la crescita e la
nutrizione, sono utilizzati antistaminici, acido ursodeoxicolico, idrossizina; l’associazione colestira-
www.buponline.com
167
Sindrome di Alagille
a
c
d
b
Figura 1: Ragazzo di 18 anni affetto da SAG. a: All’età di 12 anni presentava discromie dentali di colore verde da accumulo
di bilirubina nel corso dell’odontogenesi. b, c, d: Lo stesso paziente all’età di 20 anni. Riabilitazioni protesiche estetiche
(faccette) posizionate da primo premolare a primo premolare, nell’arcata inferiore (f ) e superiore (g). Controllo a distanza
di 6 mesi. Gengivite marginale grave da insufficiente igiene orale domiciliare
mina-rifampicina si è dimostrata efficace nel ridurre il prurito intrattabile e la comparsa di xantomi
(Ling SC, 2007; Garcia MA et al., 2005; Martin
SR et al., 1996). Nelle forme che non rispondono
alle terapie farmacologiche, con funzionalità epatica ancora buona, si ricorre all’intervento chirurgico
di diversione biliare. L’ipercolesterolemia è comune
nei soggetti con SAG ma non se ne conoscono le
implicazioni cardiovascolari a lungo termine (Ling
SC, 2007). Il 15-31% dei soggetti affetti da SAG
necessita di trapianto epatico nella prima infanzia, rappresentando il 6% circa di tutti i trapianti di fegato in età pediatrica (Sze DY et al., 2008;
Schwartz R et al., 2008; Ling SC, 2007). Sono
indicazioni al trapianto epatico la cirrosi e il malassorbimento grave resistente alla nutrizione parenterale (Ling SC, 2007; Garcia MA et al., 2005;
Emerick KM et al., 1999). La mortalità post-operatoria per complicanze cardiovascolari riduce la
percentuale di pazienti sopravvissuti, sebbene siano
www.buponline.com
168
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
descritti casi di successo in soggetti trapiantati con
gravi patologie cardiache (Ling SC, 2007; Garcia
MA et al., 2005)
Per quanto riguarda le malformazioni cardiache,
frequentemente è necessaria una correzione cardiochirurgica; la prognosi dipende dalla severità
del coinvolgimento cardiaco ed epatico, con una
mortalità media del 17-30%.
In presenza di anomalie renali è necessario il monitoraggio continuo dell’apparato urinario mediante urinocolture ed ecografie (Ling SC, 2007;
Martin SR et al., 1996).
Dal punto di vista odontostomatologico, essendo i pazienti affetti da SA ad elevato rischio
di patologia cariosa e, quando affetti da cardiopatia congenita, a rischio di Endocardite Batterica (EB), è necessario attuare, dal momento in cui
viene formulata la diagnosi, interventi di prevenzione primaria mirati, con particolare riferimento
all’alimentazione non cariogenica, all’igiene orale
domiciliare, all’utilizzo di fluoro topico e quando
indicato sistemico, alle visite periodiche trimestrali/semestrali, alla sigillatura di solchi, fessure e fori
ciechi.
Nei pazienti con SAG a rischio di EB, gli interventi
odontoiatrici a rischio di sanguinamento debbono
essere eseguiti in un regime di profilassi antibiotica
(vedi il capitolo Cardiopatie congenite).
I bambini che necessitano di trapianto epatico, in
considerazione dei rischi che le infezioni a livello
del cavo orale comportano in corso di terapia con
immunosoppressori, devono essere sottoposti a
bonifica del cavo orale prima dell’intervento.
Qualora lo scarso livello di collaborazione del bambino renda necessario intervenire in anestesia generale, necessitano di attenta valutazione anestesiologica le funzionalità epatica e renale e la pervietà
delle vie d’accesso aeree (Yildiz TS et al., 2007).
In presenza di allergia al lattice, più frequente
nei soggetti sottoposti ad interventi chirurgici nei
primi anni di vita come i pazienti affetti da SAG,
Figura 2: Ragazzo di 10 anni affetto da SAG: edema de
labbro superiore da reazione allergica al lattice
è imperativo in ambiente odontoiatrico utilizzare
materiali latex free (guanti, fogli di diga, elastici
ortodontici, ecc…) (Guadagni MG et al., 2005)
(Figura 2).
Un eventuale deficit di Vitamina K può determinare un deficit di coagulazione, da valutare in caso
di estrazioni o altri interventi di chirurgia orale,
per prevenire complicanze emorragiche (Yildiz
TS et al., 2007).
Ogni trattamento farmacologico, inclusa la fluoroprofilassi sistemica, essendo le funzionalità epatica
e renale coinvolte nel metabolismo dei farmaci,
deve essere valutato con il medico curante.
In presenza di patologie ortopedico-ortodontiche,
la terapia è indicata quando è garantito il mantenimento di una corretta igiene orale domiciliare.
Le discromie dentali di colore verdastro, soprattutto quando interessano gli elementi permanenti
del settore frontale, comportano per il bambino/
adolescente importanti difficoltà psicologiche,
che rappresentano un’indicazione ad un intervento
precoce. Il primo approccio consiste in sedute di
sbiancamento dentale, i cui risultati spesso non
sono soddisfacenti. In fase di crescita possono essere utilizzate faccette provvisorie in composito con
una minima preparazione delle superfici dentali
www.buponline.com
169
Sindrome di Alagille
che può rendere difficoltoso mascherare la discromia, per cui in fase di cementazione è indicato l’utilizzo di un cemento opaco. In caso di trattamento
ortodontico, le faccette in composito hanno il vantaggio di essere facilmente utilizzate per il bonding
dei brackets. Al termine della crescita si esegue una
riabilitazione definitiva minimamente invasiva
con faccette estetiche in ceramica integrale che,
essendo più coprenti, garantiscono un’estetica migliore anche con spessori ridotti (Guadagni MG et
al., 2005; Guimares LP et al., 2003).
BIBLIOGRAFIA
Alagille D, Habib EC, Thomasin N. L’atrésie des
voies biliaires intra-hépatiques avec voies biliaires extra-hépatiques perméables chez l’enfant (à
propos de 25 observations). In: Royer P, editor.
Journées parisiennes de pédiatrie. Paris: Flammarion, 1969: 301-18
Alagille D, Estrada A, Hadchouel M, Gautier
2M, Odièvre M, Dommergues JP. Syndromic
paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of
80 cases. J Pediatr. 1987;110:195-200
Albayram F, Stone K, Nagey D, Schwarz KB,
Blakemore K. Alagille syndrome: prenatal diagnosis and pregnancy outcome. Fetal Diagn
Ther. 2002;17:182-4
Al-Mutawa S, Mathews B, Salako N. Oral findings
in Alagille syndrome. A case report. Med Princ
Pract. 2002;11:161-3
Amaral TH, Guerra Cde S, Bombonato-prado KF,
Garcia de Paula E Silva FW, de Queiroz AM.
Tooth pigmentation caused by bilirubin: a case
report and histological evaluation. Spec Care
Dent. 2008;28:254-7
Delgado A, Mokri B, Miller G. Butterfly vertebrae.
J Neuroimaging. 1996;6:56-8
Emerick KM, Rand EB, Goldmuntz E, Krantz
ID, Spinner NB, Piccoli DA. Features of
Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology.
1999;29:822-9
Emerick KM, Whitington PF. Partial external
biliary diversion for intractable pruritus and
xanthomas in Alagille syndrome. Hepatology.
2002;35:1501-6
Garcia MA, Ramonet M, Ciocca M, Cabrera H,
Lapunzina P, Alvares E, de Davila MT. Alagille syndrome: cutaneous manifestations in 38
children. Pediatr Dermatol. 2005;22:11-4
Ghidini A, Incerti M, Andreani M. Alagille syndrome: prenatal sonographic findings. J Clin
Ultrasound. 2007;35:156-8
Gridley T. Notch signaling and inherited disease
syndromes. Hum Mol Genet. 2003;12:9-13
Guadagni MG, Cocchi S, Tagariello T, Piana
G. Case report: Alagille sindrome. Minerva
Stomatol. 2005;54:593-600
Guimares LP, Silva TA. Green teeth associated with
cholestasis caused by sepsis: A case report and
review of the literature. Oral Surg Oral Med
Oral Pathol Oral Radiol Endod. 2003;95:44651
Hingorani M, Nischal KK, Davies A, Bentley C,
Vivian A, Baker AJ, Mieli-Vergani G, Bird
AC, Aclimandos WA. Ocular abnormalities in Alagille Syndrome. Ophthalmology.
1999;106:330-7
Jung C, Driancourt C, Baussan C, Zater M,
Hadchouel M, Meunier-Rotival M, Guiochon-Mantel A, Jacquemin E. Prenatal
molecular diagnosis of inherited cholestatic diseases. J Pediatr Gastroenterol Nutr.
2007;44:453-8
Kamath BM, Loomes KM, Oakey RJ, Emerick
KE, Conversano T, Spinner NB, Piccoli DA,
Krantz ID. Facial Features in Alagille Syndrome: specific or colestasis facies? Am J Med
Genet. 2002;112:163-70
www.buponline.com
170
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Kamath BM, Loomes KM, Piccoli DA. Medical
management of Alagille syndrome. J Pediatr
Gastroenterol Nutr. 2010;50:580-6
Kazuko I, Hiroshi Kuroda, Hiroshi M. Dental
findings and care of children with Alagille Syndrome: report of 4 cases. Japanese Journal of
Pediatric Dentistry. 1998;5:932-8
Koch B, Goold A, Egelhoof J, Benton C. Partial
absence of the posterior semicircular canal in
Alagille syndrome: CT findings. Pediatr Radiol. 2006;36:977-9
Ling SC. Congenital cholestatic syndromes: what
happens when children grow up? Can J Gastroenterol. 2007;21:743-51
Martin SR, Garel L, Alvarez F. Alagille’s syndrome
associated with cystic renal disease. Arch Dis
Child. 1996;74:232-5
McElhinney DB, Kratz ID, Bason L, Piccoli DA,
Emerick KM, Spinner NB, Goldmuntz E.
Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with
a JAG1 mutation and/or Alagille syndrome.
Circulation. 2002;106:2567-74
Narula P, Gifford J, Steggall MA, Lloyd C, Van
Mourik IDM, Mckiernan PJ, Willshaw HE,
Kelly D. Visual loss and idiopathic intracranial hypertension in children with Alagille
syndrome. J Pediatr Gastroenterol Nutr.
2006;43:348-52
Rosenfield NS, Kelley MJ, Jensen PS, Cotlier E,
Rosenfield AT, Riely CA. Arteriohepatic dysplasia: radiologic features of a new syndrome.
AJR Am J Roentgenol. 1980;135:1217-23
Rovner AJ, Stallings VA, Piccoli DA, Mulberg AE,
Zemel BS. Resting energy expenditure is not
increased in prepubertal children with Alagille
syndrome. J Pediatr. 2006;148:680-2
Ryan RS, Myckatyn SO, Reid GD, Munk P.
Alagille syndrome: case-report with bilateral
radio-ulnar synostosis and a literature review.
Skeletal Radiol. 2003;32:489-91
Sanderson E, Newman V, Haigh SF, Baker
A, Sidhu PS. Vertebral anomalies in children with Alagille syndrome: an analysis
of 50 consecutive patients. Pediatr Radiol.
2002;32:114-9
Schwartz R, Rehder K, Parsons DJ, Morrell DS.
Intense pruritus and failure to thrive in Alagille
syndrome. J Am Acad Dermatol. 2008;58:911
Sheehy EC, Roberts GJ, Beighton D, O’Brien G.
Oral health in children undergoing liver transplantation. Int J Paediatr Dent. 2000;10:10919
Spinner NB, Hutchinson AL, Krantz ID, Kamath
BM. Alagille Syndrome. University of Washington, Seattle; 1993-2000 May 19, updated
2010 Jul 20
Sze DY, Esquivel CO. Alagille syndrome. J Vasc
Interv Radiol. 2008;19:1278-81
Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J
Hum Genet. 2012;20:251-7
Yildiz TS, Yumuk NO, Baykal D, Solak M, Toker
K. Alagille syndrome and anestesia management. Paediatr Anaesth. 2007;17:87-97
www.buponline.com
16. Displasia ectodermica ipoidrotica/anidrotica
Sinonimo: Sindrome da Christ-Siemens-Touraine
Codice ICD 10: Q82.4
F. Battelli, M. Montanari, I. Cremonesi, N. Alkhamis, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
La Displasia Ectodermica ipoidrotica/anidrotica,
descritta per la prima volta da Thurmann nel 1848,
fa parte di un ampio gruppo di malattie genetiche rare, le Displasie Ectodermiche (DE), la cui
caratteristica comune è rappresentata da anomalie
di derivati ectodermici (peli, capelli, denti, unghie,
ghiandole). Attualmente sono state classificate come
DE circa 200 condizioni cliniche differenti, l’80%
delle quali è rappresentato dalla DE ipoidrotica/anidrotica legata al cromosoma X (Irvine AD, 2009).
L’incidenza è stimata di 1 su 100.000 nati vivi per
anno, in assenza di predilezione razziale, etnica, geografica (Wright JT et al., 2011).
La morbilità e la mortalità sono correlate alle manifestazioni cliniche; la maggior parte dei pazienti ha
un’aspettativa di vita normale; in casi rari infezioni
respiratorie acute ed episodi di iperpiressia sono responsabili di danno cerebrale o di morte nella prima
infanzia (Blüschke G et al., 2010; Itin PH et al., 2004).
GENETICA E DIAGNOSI
Le basi molecolari delle DE coinvolgono geni im-
plicati nella morfogenesi epiteliale, nella comunicazione cellulare e nel controllo del ciclo cellulare
(Lamartine J, 2003).
Il gene alla base della DE ipoidrotica/anidrotica,
localizzato sul cromosoma X in posizione Xq12q13.1, codifica una proteina di membrana (ectodisplasina A) espressa nei cheratinociti, nei follicoli
piliferi e nelle ghiandole sudoripare, coinvolta nella
regolazione dell’interazione cellula-cellula e cellula-matrice (Blüschke G et al., 2010; Visinoni AF et
al., 2009; Cambiaghi S et al., 2000). La trasmissione avviene tramite la madre portatrice, nella
quale possono essere presenti alcuni segni clinici
in forma lieve. La penetranza, molto variabile, è
completa solo nel maschio.
La diagnosi, prevalentemente clinica, di solito non
viene fatta alla nascita ma durante l’infanzia quando
compaiono i sintomi e le manifestazioni caratteristici della patologia. Le manifestazioni dentali possono rappresentare il reperto clinico più significativo:
la mancata eruzione dei denti decidui è infatti
un’evenienza relativamente rara che può indirizzare
verso questa diagnosi. La diagnosi clinica viene confermata dai test di funzionalità delle ghiandole sudoripare, dalla biopsia cutanea e dai test genetici.
Le femmine portatrici dovrebbero sottoporsi a
www.buponline.com
172
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
consulto genetico e le madri portatrici a villocentesi o amniocentesi per una diagnosi prenatale.
MANIFESTAZIONI CLINICHE SISTEMICHE
La DE ipoidrotica/anidrotica è caratterizzata dalla
triade sintomatologica ipo-anidrosi (ridotta o assen-
te sudorazione), ipotricosi (ridotta presenza di peli e
capelli), ipo-oligo-anodontia (ipodontia: assenza di
meno di 6 elementi dentari; oligodontia: assenza di
6 o più elementi; anodontia: assenza di tutti gli elementi) e da altre molteplici manifestazioni cliniche di
gravità variabile, elencate nella Tabella 1 (Wright JT
et al., 2011; Blüschke G et al., 2010; Bal C et al.,
2008; Itin PH et al., 2004; Priolo M et al., 2000).
Tabella 1: Manifestazioni cliniche nelle DE ipoidrotica/anidrotica
Manifestazioni cutanee
cute secca, desquamata, marmorea, sottile, ipopigmentata, predisposta alle infezioni
cute perioculare iperpigmentata con rughe evidenti
eczemi cutanei, dermatite atopica
ipercheratosi cutanea a livello di articolazioni, palmo delle mani e pianta dei piedi
cute ispessita
Manifestazioni a livello ghiandolare
ipoplasia/aplasia ghiandole sudoripare
ipoplasia/aplasia ghiandole sebacee
ipoplasia/aplasia ghiandole salivari
Manifestazioni a livello dei follicoli piliferi
capelli radi, sottili, secchi; talvolta alopecia
sopracciglia e ciglia rade, sottili, talvolta assenti
peli del corpo scarsi, talvolta assenti
nel maschio crescita di barba e baffi spesso normale
Manifestazioni ungueali
iperconvesse, con superficie irregolare
facilmente soggette a frattura
talvolta assenti
infezioni croniche del perionichio
Manifestazioni oculari
ipoplasia ghiandole lacrimali (occhi secchi, abrasioni)
cataratta
congiuntiviti
panuveite bilaterale
displasia corneale
opacità corneale e lenticolare (rare)
www.buponline.com
173
Displasie Ectodermiche
Altre manifestazioni
grave intolleranza al caldo, iperpiressie ricorrenti
disfagia
ipogonadismo
patologie otorinolaringoiatriche: epistassi, otiti (medie ed esterne), ipoacusia
ritardo mentale (raro)
polidattilia, sindattilia (rare)
nanismo
epilessia
anomalie a livello di derivati mesenchimali (derma, ipoderma, cellule endoteliali cervico-facciali)
La cute è poco pigmentata, di aspetto marmoreo, sottile, delicata, predisposta ad infezioni (Figure 1; 2).
Figura 1: Bambino di 4 anni affetto da DE ipoidrotica/
anidrotica. Facies caratteristica: cute ipopigmentata, sottile;
sopracciglia rade e di colore chiaro; zona perioculare sede di
pigmentazioni
Figura 2: Bambino di 6 anni affetto da DE ipoidrotica/
anidrotica. Facies caratteristica: tipicamente vecchieggiante, con bozze frontali accentuate, profilo piatto, naso piccolo e a sella, ali nasali e regione malare ipoplasiche, labbra
protruse con frequente eversione del labbro inferiore, mento
prominente
www.buponline.com
174
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
I capelli e i peli sono radi, sottili, di colore molto chiaro, le ciglia e le sopracciglia sono scarse o
assenti; tuttavia la crescita della barba e dei baffi
nell’uomo spesso è normale.
Sono molto frequenti manifestazioni a carico delle
unghie, iperconvesse, con superficie irregolare,
facilmente soggette a frattura, con frequenti infezioni croniche del tessuto paraungueale; in alcuni
pazienti le unghie sono assenti.
Le ghiandole sudoripare sono presenti in numero ridotto o completamente assenti; questo causa
gravi problemi di termoregolazione che possono
causare convulsioni febbrili, potenziali responsabili
di danni di tipo neurologico e in rari casi di morte.
Le ghiandole sebacee e le ghiandole salivari possono essere ipoplasiche, talora assenti. Le ghiandole lacrimali possono essere ridotte in numero,
con conseguente secchezza agli occhi e predisposizione a sviluppare abrasioni e cataratte. A livello
dell’apparato respiratorio le ghiandole mucipare possono essere ipoplasiche o assenti; l’atrofia
della mucosa laringo-faringea può essere causa di
disfonia; infezioni respiratorie ricorrenti possono
rendere difficoltosa la respirazione.
Manifestazioni allergiche (eczema e/o asma) sono
spesso presenti (nel 65% dei casi).
CARATTERISTICHE ORO-FACCIALI
I pazienti affetti da DE ipoidrotica/anidrotica presentano caratteristiche di interesse odontoiatrico
peculiari: facies caratteristica, xerostomia, anomalie dentali.
La facies è tipicamente vecchieggiante, con bozze
frontali accentuate, profilo piatto, naso piccolo
e a sella, ali nasali e regione malare ipoplasiche,
padiglioni auricolari piccoli e sporgenti, labbra protruse con frequente eversione del labbro
inferiore, mento prominente (Dellavia C et al.,
2008; Bondarets N et al., 2002) (Figure 1; 2). La
a
b
Figura 3 a: Bambino di 3 anni affetto da DE ipoidrotica/
anidrotica: capelli radi, sottili e di colore molto chiaro. b:
Bambino di 3 anni affetto da DE ipoidrotica/anidrotica:
ciglia e sopracciglia rade e di colore chiaro; rughe profonde
nella zona perioculare
zona perioculare può essere sede di rughe e di pigmentazioni (Figure 1; 2; 3b).
In alcuni pazienti sono descritte riduzione dei seni
frontali, ipertrofia dei seni mascellari e anomalie
morfologiche delle ossa mascellari (Rhuin B et al.,
2001).
Le ghiandole salivari possono essere diminuite in
numero o sede di alterazioni infiammatorie e di
ectasia dei dotti. La ridotta secrezione salivare
è responsabile di xerostomia, che determina predisposizione alla patologia cariosa, difficoltà nella
fonazione, nella masticazione, nella deglutizione e
www.buponline.com
175
Displasie Ectodermiche
Figura 4: Ortopantomografia di bambino di 5 anni affetto da DE ipoidrotica/anidrotica: oligodonzia; presenza dei
germi dei molari inferiori
Figura 5: Ortopantomografia di bambino di 7 anni affetto
da DE ipoidrotica/anidrotica: oligodonzia; anomalie dentali di forma
nella ritenzione di protesi rimovibili (Lexner MO
et al., 2007b).
Le anomalie dentarie di numero, forma e struttura coinvolgono la dentatura sia decidua che permanente; la loro prevalenza è circa dell’80%.
Le anomalie di numero sono estremamente variabili, da quadri di agenesie singole fino all’anodonzia (Figure 4; 5; 6; 7). Le agenesie sono più frequenti a livello mandibolare (Tarjan I et al., 2005;
Nordgarden H et al., 2001).
Figura 6: Bambino di 7 anni affetto da DE ipoidrotica/
anidrotica: agenesie multiple, elementi dentali conoidi,
atrofia dei mascellari nelle zone edentule
Figura 7: Ragazzo di 10 anni affetto da DE ipoidrotica/
anidrotica: oligodonzia; atrofia dei mascellari nelle zone
edentule
Le anomalie di forma, molto frequenti, possono
coinvolgere sia la corona che la radice del dente.
I denti permanenti sono di dimensioni più piccole rispetto alla norma, i posteriori soprattutto
nel diametro mesio-distale. Molto comune è la
forma conoide (Figure 6; 8; 11a), principalmente a livello di incisivi e canini e talvolta di
premolari (Lexner et al., 2007a). Frequente è il
taurodontismo, che spesso colpisce il secondo
molare deciduo (Lo Muzio L et al., 2005). Le
radici dei molari possono essere parzialmente
www.buponline.com
176
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Dellavia C et al., 2008; Tarjan I et al., 2005; Ruhin
B et al., 2001).
LINEE GUIDA DI TERAPIA
Figura 8: Ragazzo di 12 anni affetto da DE ipoidrotica/
anidrotica: oligodonzia; denti di forma conoide
fuse o di forma piramidale (Lexner et al., 2007a)
(Figura 5).
Per quanto concerne le anomalie di struttura, lo
smalto può presentarsi ipoplasico, ipocalcificato,
immaturo.
Quanto più è alto il numero di agenesie tanto più
sono accentuate le anomalie facciali (Tarjan I et al.,
2005); l’assenza dei denti comporta infatti atrofia dei processi alveolari e perdita di dimensione verticale del terzo medio ed inferiore del viso
(Dellavia C et al., 2008) (Figura 2). La dimensione antero-posteriore è ridotta, con retrograzia
più accentuata a livello del mascellare superiore e
tendenza alla terza classe scheletrica per rotazione antero-superiore della mandibola (Dellavia C et
al., 2010; Bondarets N et al., 2002).
Gli studi longitudinali sulla crescita del complesso
craniofacciale in soggetti affetti da DE ipoidrotica/
anidrotica sono pochi; i dati presenti in letteratura
evidenziano che la crescita delle singole strutture
nei primi anni di vita è inferiore rispetto a quella di soggetti di pari età e sesso ma che durante
l’adolescenza tende a normalizzarsi, soprattutto se
correttamente stimolata mediante dispositivi ortodontici e protesi rimovibili (Dellavia C et al., 2010;
Il paziente affetto da DE necessita di un intervento interdisciplinare che coinvolge un team medico ed odontoiatrico che collabora nell’ambito delle
specifiche competenze, con l’obiettivo comune di
promuovere la salute del paziente e garantirne una
buona qualità di vita.
Il trattamento delle problematiche correlate alla sudorazione assente o scarsa è di tipo palliativo.
Per evitare fenomeni di iperpiressia è di fondamentale importanza evitare l’esposizione al calore,
frequentare ambienti condizionati ed indossare un
abbigliamento adeguato, soprattutto nei mesi estivi. Sono consigliati sport in cui la sudorazione sia
ridotta, come il nuoto.
Per la secchezza e la fragilità di cute e capelli sono
utili creme emollienti dermatologiche e prodotti
specifici per i capelli.
In relazione all’elevata incidenza di infezioni respiratorie sono consigliate misure preventive quali
copertura vaccinale annuale, follow-up otorinolaringoiatrici, screening audiometrici, pulizia dei
condotti uditivo e nasale con irrigazioni.
Per la secchezza oculare sono consigliate lacrime
artificiali. Per la xerostomia è consigliato il consumo di abbondanti quantità di liquidi, l’utilizzo di
scialagoghi e di saliva artificiale (Wright JT et al.,
2011; Lexner MO et al., 2007b).
Dal punto di vista odontoiatrico, è necessaria una
visita odontoiatrica all’età di due anni, prima che
il bambino inizi a frequentare l’asilo, al fine di riabilitare l’apparato stomatognatico per migliorare le
attività masticatoria e fonatoria e l’aspetto estetico,
favorendo l’inserimento sociale.
Nella fase iniziale, l’assenza di collaborazione rende
necessaria la contenzione dolce del bambino da
www.buponline.com
177
Displasie Ectodermiche
parte di uno dei genitori. Particolare attenzione va
rivolta al rischio di fenomeni di ipertermia provocati nel bambino dall’agitazione e da crisi di pianto; è necessario mettere in atto una serie di misure
precauzionali quali ambiente climatizzato, impacchi di acqua fredda, sospensione del trattamento in
caso di intense reazioni di pianto.
A partire dai tre-quattro anni per ottenere la collaborazione e la fiducia del paziente possono essere
utilizzate tecniche quali il tell-show-do, la desensibilizzazione e il rinforzo positivo, indispensabile per portare il paziente all’accettazione di un
trattamento protesico-ortodontico che lo accompagnerà per molti anni.
Qualora siano presenti elementi dentali in arcata, considerata la predisposizione alla patologia
cariosa, sono necessari interventi di prevenzione primaria mirati, con particolare riferimento
all’alimentazione non cariogenica, all’igiene orale
domiciliare, all’utilizzo di fluoro topico e quando
indicato sistemico, alle visite periodiche. Per prevenire la patologia cariosa è raccomandata la sigillatura dei solchi, delle fessure e dei fori ciechi nei
molari permanenti, in particolare dei taurodonti
nei quali gli spessori dei tessuti duri sono inferiori
rispetto al normale e, di conseguenza, la patologia
cariosa ha una rapida progressione verso il tessuto
pulpare; in questi elementi inoltre la terapia endodontica presenta difficoltà tecniche, a causa della
particolare anatomia del sistema endodontico.
Nei bambini/adolescenti con ipodonzia, in attesa della riabilitazione protesica definitiva a crescita
conclusa, si utilizzano tecniche adesive per il rimodellamento estetico degli elementi conoidi (Figura 11 a, b) (Lo Muzio L et al., 2005), terapie ortodontiche per rendere armonico lo sviluppo delle
basi ossee (Suri S et al., 2004) e, quando indicate,
terapie protesiche con protesi provvisorie fisse o
rimovibili (Cetiner D et al., 2001).
Nei pazienti con oligo/anodonzia le anomalie dentali comportano notevoli disagi e rappresentano la
menomazione più grave sotto il profilo psicologico, funzionale ed estetico. In questi pazienti si
rende necessaria una riabilitazione protesica precoce,
intorno ai due anni, tramite l’applicazione di protesi rimovibili costruite in modo da accompagnare e
promuovere la crescita del massiccio facciale (Battelli F et al., 2007; Lo Muzio L et al., 2005; Tarjan I
et al., 2005). La riabilitazione protesica stabilisce un
corretto piano occlusale, migliora la relazione intermascellare sul piano sagittale e verticale e fornisce
supporto ai tessuti molli (Dellavia C et al., 2010;
Tarjan I et al., 2005; Suri S et al., 2004; Gardel P et
al., 1985) (Figura 9 a, b, c, d; 10 a, b).
Il protocollo operativo prevede:
• documentazione fotografica intra ed extraorale
• rilevamento impronte di studio in alginato
• costruzione portaimpronte individuali
• rilevamento impronte in Eugenolato di Zinco
per modello maestro
• realizzazione di basi in resina con valli in cera
per registrazione rapporti intermascellari
• montaggio modelli maestro in articolatore
• montaggio denti anteriori e prova estetica su
paziente
• montaggio denti diatorici secondo la tecnica
di Gerber
• realizzazione protesi definitive in laboratorio
• consegna protesi superiore
• istruzioni ai genitori sulla modalità di utilizzo
e sul mantenimento igienico della protesi
• dopo un mese, per favorire l’adattamento del
bambino alla nuova situazione orale, consegna
protesi inferiore
• dopo 3 mesi, inserimento di viti di espansione
(a 3 vie nella superiore, a 2 vie nell’inferiore),
la cui attivazione permette di accompagnare e
stimolare la crescita del massiccio facciale (Figura 10 a, b)
• ribasatura mensile delle protesi
• periodica valutazione clinica e radiografica
dell’accrescimento
www.buponline.com
178
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
a
c
d
Figura 9 a, b, c, d: Protesi superiore ed inferiore e foto extra
ed intraorali con protesi in sede in ragazzo di 12 anni affetto da DE ipoidrotica/anidrotica
b
•
periodiche sostituzioni delle protesi per renderle
coerenti alla crescita dei mascellari e alle fasi della
dentizione, considerando i tempi fisiologici di
permuta e di eruzione dei denti permanenti.
La progettazione e la costruzione di una protesi totale rimovibile comportano problemi connessi alla
scarsa collaborazione di un bambino in età prescolare, alla situazione anatomica particolarmente
complessa determinata dall’iposviluppo delle basi
ossee e dalle creste alveolari piatte e alla necessità di
non inibire l’accrescimento cranio-facciale.
Anche una semplice manovra come il rilevamento
di un’impronta in alginato in un paziente di questa
età può essere estremamente difficoltosa e causare
reazioni di rifiuto: può essere utile consegnare ai genitori un portaimpronta da portare a casa, per permettere al bambino di familiarizzare con l’oggetto.
Per quanto riguarda l’accettazione delle protesi in
www.buponline.com
179
Displasie Ectodermiche
a
a
b
Figura 10 a: Protesi superiore con viti di espansione a 3
vie per bambino affetto da DE ipoidrotica/anidrotica di 3
anni. b: Protesi inferiore con viti di espansione a 2 vie per
bambino affetto da DE ipoidrotica/anidrotica di 3 anni.
genere, dopo una prima fase difficile, i bambini
si adattano bene; gli inconvenienti più frequenti
sono la perdita di ritenzione e le modificazioni occlusali a causa dell’eruzione dentale e della crescita
delle basi ossee (Lo Muzio L et al., 2005).
Genitori e bambini devono essere adeguatamente
istruiti sulla gestione domiciliare della protesi, al fine
di prevenire infezioni micotiche, e sulla necessità di
mantenere sani gli elementi dentari presenti in arcata.
Per risolvere i disturbi fonetici che l’inserimento
delle protesi comporta è necessario l’intervento
logopedico (Montanari M et al., 2009).
Il posizionamento di impianti osteointegrati in pa-
b
Figura 11 a, b: Rimodellamento estetico con materiale composito di elementi conoidi decidui in bambino di 3 anni
affetto da DE ipoidrotica/anidrotica.
zienti in età evolutiva non è raccomandato come
pratica di routine a causa della crescita scheletrica
cui il bambino va incontro e dell’insufficienza di
osso alveolare, fattori che predispongono ad una
maggiore incidenza di fallimento (Kramer FJ et al.,
2007). Tuttavia secondo alcuni autori è possibile
posizionare con successo impianti in pazienti affetti da DE con ipo/anodonzia anche in età evolutiva previa attenta valutazione dell’osso disponibile
(Kramer FJ et al., 2007; Bergendal T et al., 1991).
La riabilitazione con protesi rimovibili deve essere
considerata provvisoria ed accompagnare il bambino durante gli anni dell’accrescimento somatico, in
www.buponline.com
180
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
attesa del termine dello sviluppo osseo, momento
in cui è possibile la riabilitazione implantoprotesica, previa preparazione chirurgica delle basi
ossee (Figura 12 a, b, c, d, e) (Kramer FJ et al.,
2007; Güler N et al., 2005; Suri S et al., 2004).
In caso di scarse ritenzione e stabilità della protesi
rimovibile inferiore può essere presa in considerazione in età evolutiva l’inserimento di impianti e
la realizzazione di una protesi rimovibile con barra
di espansione per assecondare la crescita della base
ossea (http://amsdottorato.unibo.it/5519/1/battelli_fillippo_tesi.pdf ).
c
d
a
e
b
Figura 12: Fasi della riabilitazione implanto-protesica in ragazzo di 10 anni affetto da DE ipoidrotica/anidrotica. a. scopertura degli impianti inseriti in zona interforaminale; b. inserimento degli abutment (83) sugli impianti; c. inserimento
della barra di espansione sugli impianti e suo fissaggio; d. realizzazione della contro-barra e della protesi capace di allargarsi
a livello della linea mediana; protesi fissata alla barra di espansione tramite ball-attachment; e. posizionamento della protesi
definitiva.
www.buponline.com
181
Displasie Ectodermiche
BIBLIOGRAFIA
Bal C, Bal BT, Tüfekçioğlu D. Treatment considerations for a patient with hypohidrotic ectodermal dysplasia: a case report. J Contemp Dent
Pract. 2008; 9(3):128-34
Battelli F, Cetrullo N, Cocchi S, Carboni C, Piana
G. La riabilitazione protesica di un caso di Displasia Ectodermica Anidrotica (HED). Quint
Int. 2007; 23,41-9
Bergendal T, Eckerdal O, Hallonsten AL, Koch
G, Kurol J, Kvint S. Osseointegrated implants
in the oral habilitation of a boy with ectodermal dysplasia: a case report. Int Dent J. 1991;
41(3):149-56
Blüschke G, Nüsken KD, Schneider H. Prevalence
and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy.
Early Human Development. 2010; 86:397–9
Bondarets N, Jones RM, McDonald F. Analysis of
facial growth in subjects with syndromic ectodermal dysplasia: a longitudinal analysis. Orthod Craniofac Res. 2002; 5(2):71-84
Cambiaghi S, Restano L, Pääkkönen K, Caputo R,
Kere J. Clinical findings in mosaic carriers of
hypohidrotic ectodermal dysplasia. Arch Dermatol. 2000; 136(2):217-24
Cetiner D, Engel U, Tüter G, Yalim M. Clinical
management of ectodermal dysplasia with long
term follow up: two case reports. J Clin Pediatr
Dent. 2001; 25(3):187-90
Dellavia C, Catti F, Sforza C, Tommasi DG,
Ferrario VF. Craniofacial growth in ectodermal dysplasia. An 8 year longitudinal evaluation of Italian subjects. Angle Orthod. 2010;
80(4):733-9
Dellavia C, Catti F, Sforza C, Grandi G, Ferrario
VF. Non-invasive longitudinal assessment of
facial growth in children and adolescents with
hypohidrotic ectodermal dysplasia. Eur J Oral
Sci. 2008; 116(4):305-11
Gardel P, Leyder P, Molhant G. Christ-Siemens-Touraine
syndrome.
Completed
management is unrelated to the number of
year, Rev Stomatol Chir Maxillofac. 1985;
86(2):114-6
Güler N, Cildir S, Iseri U, Sandalli N, Dilek O.
Hypohidrotic ectodermal dysplasia with bilateral impacted teeth at the coronoid process: a
case rehabilitated with mini dental implants.
Oral Surg Oral Med Oral Pathol Oral Radiol
Endod. 2005; 99(5): 34-8
Irvine AD: Towards a unified classification of the
ectodermal dysplasias. Opportunities outweigh challenges. Am J Med Genet A. 2009;
149A:1970–2
Itin PH, Fistarol SK. Ectodermal dysplasias. Am
J Med Genet C Semin Med Genet. 2004;
15;131C(1):45-51
Kramer FJ, Baethge C, Tschernitschek H. Implants
in children with ectodermal dysplasia: a case report and literature review. Clin Oral Implants
Res. 2007;1 8(1):140-6
Lamartine J. Towards a new classification of ectodermal dysplasia. Clin Exp Dermatol. 2003;
28(4):351-5
Lexner MO, Bardow A, Hertz JM, Nielsen LA,
Kreiborg S. Anomalies of tooth formation in
hypohidrotic ectodermal dysplasia. Int J Paediatr Dent. 2007a; 17(1):10-8
Lexner MO, Bardow A, Hertz JM, Almer L, Nauntofte B, Kreiborg S. Whole saliva in X-linked
hypohidrotic ectodermal dysplasia. Int J Paediatr Dent. 2007b; 17(3):155-62
Lo Muzio L, Bucci P, Carile F, Riccitiello F, Scotti
C, Coccia E, Rappelli G. Prosthetic rehabilitation of a child affected from anhydrotic ectodermal dysplasia: a case report. J Contemp
Dent Pract. 2005; 15;6(3):120-6
Montanari M, Battelli F, Sangiorgi S, Nucci C,
Malucelli P, Guidotti M, Piana G. Analisi fonetica in bambini con displasia ectodermica ri-
www.buponline.com
182
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
abilitati protesicamente. Dental Cadmos 2009;
77(10):43-50
Nordgarden H, Reintoft I, Nolting D, Fischer-Hansen B, Kjaer I. Craniofacial tissues including tooth buds in fetal hypohidrotic ectodermal dysplasia. Oral diseases 2001; 7:163-70.
Priolo M, Laganà C. Ectodermal dysplasias: a new
clinical- genetic classification. J Med Genet
2001; 38:579-85
Priolo M, Silengo M, Lerone M, Ravazzolo R. Ectodermal dysplasias: not only ‘skin’ deep. Clin
Genet. 2000; 58(6):415-30
Rhuin B, Martinot V, Lafforgue P, Catteau B,
Manuvrier-Hanu S, Ferri J. Pure Ectodermal
dysplasia: retrospective study of 16 cases and
literature review. Cleft Palate Cranofac J. 2001;
38(5):504-18
Suri S, Carmichael RP, Tompson BD. Simultaneous functional and fixed appliance therapy
for growth modification and dental alignment
prior to prosthetic habilitation in hypohidrotic
ectodermal dysplasia: a clinical report. J Prosthet Dent. 2004; 92(5):428-33
Tarjan I, Gabris K, Rozsa N. Early prosthetic
treatment of patients with ectodermal dysplasia: a clinical report. J Prosthet Dent. 2005;
93(5):419-24
Visinoni AF, Lisboa-Costa T, Pagnan NAB, Chautard-Freire-Maia EA. Ectodermal dysplasias:
Clinical and molecular review. Am J Med Genet A. 2009; 149A:1980–2002
Wright JT, Grange DK, Richter MK. Hypohidrotic
Ectodermal Dysplasia. In: Pagon RA, Bird TC,
Dolan CR, Stephens K, editors GeneReviews.
Seattle (WA): University of Washington, Seattle;
1993-2003, Apr28 [Updated 2011 Dec 29]
http://amsdottorato.unibo.it/5519/1/battelli_fillippo_tesi.pdf
www.buponline.com
17. Distrofie muscolari
Codice ICD 10: G71.0
I. Cremonesi, N. Alkhamis, T. Tagariello, G. D’Alessandro, G. Piana
DEFINIZIONE ED EPIDEMIOLOGIA
Le Distrofie muscolari (DM) sono un gruppo di
malattie genetiche rare degenerative che interessano l’apparato neuromuscolare a trasmissione
autosomica dominante o recessiva, relativamente frequenti nella popolazione. Sono caratterizzate da debolezza muscolare diffusa, abitualmente
simmetrica e prevalentemente prossimale a livello
dei muscoli vicini all’asse mediano del corpo. La
rapidità di evoluzione è estremamente variabile,
in relazione all’interessamento dell’apparato respiratorio e del muscolo cardiaco.
Le DM sono causate da mutazioni di geni che codificano per proteine coinvolte nel mantenimento
dell’integrità delle fibre muscolari.
Le Distrofie di Duchenne (DMD) e di Becker
(DMB) sono determinate da mutazioni di un
gene localizzato nel cromosoma X che contiene
le informazioni per la produzione della proteina
distrofina. Le mutazioni, di vario tipo (sostituzioni nucleotidiche, delezioni), sono responsabili di assenza della distrofina, totale nella DMD e
parziale nella DMB. Per questo motivo DMD e
DMB sono definite distrofinopatie (Kaufmann P
et al, 2005). La Distrofia muscolare di Emery-
Dreifuss (DMED) è determinata da mutazioni di
un gene localizzato nel cromosoma X che contiene le informazioni per la produzione dell’emerina,
proteina che costituisce la membrana delle cellule
muscolari. Come tutte le malattie a trasmissione
autosomica recessiva legate al cromosoma X si manifestano solo nei maschi e sono trasmesse da donne sane portatrici del gene difettoso. Dall’unione
fra una donna portatrice sana e un uomo sano ad
ogni gravidanza un figlio maschio su 2 può nascere
malato ed una figlia femmina su due può nascere
portatrice sana.
La Distrofia Fascio-Scapolo-Omerale (DFSO),
la Distrofia Oculo-Faringea (DOF) e le Distrofie Miotoniche sono trasmesse prevalentemente
con modalità autosomica dominante, colpendo
soggetti di genere sia maschile che femminile. La
Distrofia Fascio-Scapolo-Omerale (DFSO) è
causata da una delezione subtelomerica a livello
del cromosoma 4q35; la severità della malattia è
proporzionale alle dimensioni del tratto genomico
coinvolto dalla delezione. La Distrofia Miotonica di Tipo 1 (malattia di Steinert) è determinata
da mutazioni di un locus sul cromosoma 19q132 (ripetizione anomala della tripletta CTG). La
Distrofia Miotonica di Tipo 2 (miopatia mio-
www.buponline.com
184
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
tonica prossimale) è determinata dall’espansione
della ripetizione CCTG nell’introne 1 del gene
CNBP (3q21) (Kaufmann P et al., 2005; Hamamoto DT, 2006).
Nella Tabella 1 sono elencate le forme più frequenti di DM e le relative prevalenza, modalità di
trasmissione e proteina alterata (Balasubramaniam
R et al., 2008; Hamamoto DT, 2006). Le DM di
Duchenne, di Becker, di Emery-Dreifuss e FascioScapolo-Omerale si manifestano nell’infanzia,
l’oculo-faringea e le miotoniche tipicamente in età
adulta (Cruz Guzmán Odel R et al., 2012).
Tabella 1: Forme più frequenti di DM e relative prevalenza, modalità di trasmissione e proteina alterata
Tipo di DM
Incidenza
(nati /anno)
Modalità di trasmissione
Proteina alterata
DUCHENNE
1:3.300
Cromosoma X - recessiva
Distrofina
BECKER
1:18.000 1:31.000
Cromosoma X - recessiva
Distrofina
EMERY-DREIFUSS
1:300.000
Cromosoma X - recessiva
Emerina
FASCIO-SCAPOLOOMERALE
1:20.000
Autosomica dominante
sconosciuta
OCULO-FARINGEA
Non specificata
Autosomica dominante
Proteina coinvolta nella
poliadenilazione
MIOTONICA di TIPO 1
1:20.000
Autosomica dominante
Proteinchinasi
MIOTONICA di TIPO 2
1:100.000
Autosomica dominante
Proteinchinasi
GENETICA E DIAGNOSI
La diagnosi prenatale delle forme più comuni di
DM si effettua mediante villocentesi o amniocentesi. Si consiglia la diagnosi prenatale alle donne
portatrici, con precedenti figli affetti, con storia
familiare di DM.
Nella maggior parte dei casi la diagnosi delle DM
viene effettuata sulla base dei segni clinici e di esami strumentali (elettromiografia ed elettrocardiogramma) ed istologici (biopsia muscolare). Elevati
livelli ematici di creatinin-chinasi possono indirizzare alla diagnosi, perché indicativi di danno mu-
scolare (Hsu YD, 2004). La diagnosi è confermata
dal test genetico.
MANIFESTAZIONI CLINICHE SISTEMICHE
Nelle DM il tessuto muscolare, essendo sostituito da tessuto fibroso e adiposo, va incontro
ad atrofia e perde progressivamente la capacità
contrattile; la conseguenza clinica è la perdita
progressiva della forza muscolare e delle abilità motorie.
www.buponline.com
185
Distrofie Muscolari
In molte forme è coinvolto il sistema nervoso centrale; può essere interessata sia la sostanza bianca
dell’encefalo sia l’architettura del sistema nervoso
centrale; il coinvolgimento può essere subclinico
(evidenziabile solo attraverso esami strumentali) o
clinicamente evidente.
Nei casi in cui il deficit motorio sia già in atto nella
vita intrauterina, alla nascita sono presenti retrazioni articolari, configuranti un quadro di artrogriposi
multipla congenita.
Le manifestazioni cliniche caratteristiche delle DM
sono elencate nella Tabella 2.
Tabella 2: Manifestazioni cliniche caratteristiche delle DM
MANIFESTAZIONI CLINICHE
APPARATO MUSCOLARE
Debolezza/atrofia muscolare
Pseudoipetrofia muscolare
Difficoltà/impossibilità nei movimenti
APPARATO SCHELETRICO
Scoliosi/iperlordosi
APPARATO CARDIACO
Cardiomiopatia dilatativa
Fibrillazione atriale
Tachiaritmie ventricolari
APPARATO POLMONARE
Pneumopatia restrittiva
Insufficienza respiratoria
Polmoniti infettive/ab ingestis
APPARATO NEUROLOGICO
Ritardo mentale
Difficoltà nell’apprendimento
Ritardo nell’acquisizione del linguaggio
APPARATO FARINGEO
Disfagia
Alterazioni del timbro di voce
APPARATO OCULARE
Difficoltà nei movimenti oculari
Ptosi palpebrale
Cataratta
ALTRO
Atrofia testicolare
Diabete Mellito
L’età di insorgenza, le manifestazioni cliniche e il
decorso di ogni singolo tipo di DM sono descritte
nella Tabella 3 (Hermans MCE et al., 2010; Balasubramaniam R et al., 2008).
La Distrofia muscolare di Duchenne, la più
frequente tra le forme di DM, è caratterizzata
da insorgenza precoce dei sintomi, intorno ai
3 anni di età. Il bambino manifesta difficoltà
nella corsa, nel salire le scale e nell’alzarsi da
terra a causa dell’interessamento dei muscoli
del cingolo pelvico, in particolare dei muscoli
glutei. Segno precoce è la pseudoipertrofia mu-
www.buponline.com
186
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
scolare dei polpacci, voluminosi e di aumentata
consistenza alla palpazione. Anche la lingua e i
muscoli dell’avambraccio sono frequentemente
sede di pseudoipertrofia. La biopsia muscolare
evidenzia la sostituzione del tessuto muscolare
da parte di tessuto fibroadiposo. Sono di frequente riscontro ritardata acquisizione del
linguaggio, difficoltà nella deglutizione e
lentezza nei movimenti oculari. Nelle forme a
insorgenza più precoce, i problemi relativi alla
sfera linguistica e cognitiva possono risultare
prevalenti rispetto a quelli motori, ritardando
l’inquadramento diagnostico. Con il progredire
dell’età le difficoltà motorie diventano sempre
più evidenti e verso i 5-6 anni il quadro clinico
è caratterizzato da pseudoipertrofia del quadricipite femorale, iperlordosi lombare, scapole alate,
andatura anserina. La patologia si aggrava progressivamente; verso i 12 anni causa la perdita
della deambulazione autonoma e, successivamente, la perdita di funzione degli arti superiori.
Altri problemi clinici rilevanti sono la scoliosi,
che aggrava i problemi posturali e la situazione
respiratoria, e le retrazioni articolari, che accelerano la perdita della funzionalità motoria. In seguito sono coinvolti anche i muscoli respiratori
e il cuore; i soggetti sono destinati a sviluppare
una sindrome disventilatoria restrittiva e in un
arco di tempo variabile da soggetto a soggetto
si rende necessaria una ventilazione meccanica
prima notturna poi anche diurna. Il coinvolgimento dei muscoli respiratori si esprime con
tosse debole e inefficace, infezioni polmonari recidivanti e diminuzione del volume respiratorio.
La debolezza faringea può portare ad episodi di
aspirazione e di rigurgito di liquidi dal naso e a
un timbro di voce aereo o nasale. Più variabile
per età e gravità è il coinvolgimento del cuore,
che viene interessato da una cardiomiopatia dilatativa, la cui gravità non è correlata al grado
di debolezza dei muscoli scheletrici. Nei casi in
cui il coinvolgimento respiratorio e cardiaco è
precoce, l’aspettativa di vita è molto ridotta. Il
deterioramento intellettivo si verifica in tutti
i pazienti ma solo il 20-30% ha un quoziente
intellettivo (QI) inferiore a 70. I soggetti con
QI normale possono avere lievi difficoltà legate
all’apprendimento della lettura e della scrittura.
Solitamente i pazienti affetti da DMD non riescono a sopravvivere più di 16-18 anni, solo un
25% raggiunge un’età di 25 anni. Il decesso è
per lo più causato da complicanze cardiache, infezioni polmonari e/o insufficienza respiratoria
(Ropper AH, 2005).
La Distrofia muscolare di Becker rispetto alla
DMD ha un esordio clinico più tardivo, in genere tra gli 8 e i 10 anni, e una velocità di progressione più lenta. L’interessamento cardiaco
è il problema principale e in alcuni pazienti costituisce il sintomo d’esordio; dalla gravità della
cardiomiopatia dilatativa dipende l’aspettativa
di vita. La distribuzione e la progressione del
deficit muscolare ricalcano, in forma più lieve e
più lenta, quelle della DMD (Hermans MCE et
al., 2010). La perdita della deambulazione avviene in genere nella tarda adolescenza o in età
giovane-adulta. Il coinvolgimento respiratorio
è raro e, quando presente, non così grave come
nella DMD. Anche l’interessamento cognitivo è
più raro e meno severo. L’aspettativa di vita, più
elevata, permette una sopravvivenza anche fino
alla 5a-6a decade (Voisin V, 2004).
Nella Distrofia muscolare di Emery-Dreifuss
(DMED) il sintomo iniziale, la difficoltà nella
deambulazione, compare prima dei 5 anni. La
caratteristica patognomonica è la comparsa precoce di contratture a livello dei muscoli flessori
del gomito, estensori del collo e posteriori del
polpaccio. La malattia causa inoltre debolezza
muscolare a livello omero-peronale ed un’alta
incidenza di paralisi. Le patologie cardiache,
che si manifestano prima dei 30 anni, sono fi-
www.buponline.com
187
Distrofie Muscolari
brillazioni atriali importanti, che necessitano di
pace-maker, e cardiomiopatie dilatative (Voit T
et al., 1988).
La Distrofia Fascio-Scapolo-Omerale (DFSO)
si manifesta tra i 6 e i 20 anni con l’incapacità
di elevare le braccia oltre il capo e di alzare le scapole. È caratterizzata da debolezza muscolare a
livello del viso, delle spalle, del busto e degli arti
prossimali, con limitazione dei movimenti dei
muscoli facciali (in particolare delle labbra), dei
bicipiti, dei tricipiti e delle spalle e, progressivamente, degli arti inferiori e dei piedi (Mathews
KD, 2003).
Le Distrofie Miotoniche (DM), di cui sono descritti diversi sottotipi, sono malattie a carattere
autosomico dominante; rappresentano le forme
più diffuse di DM ad insorgenza in età adulta.
Sono caratterizzate da un decorso lentamente
progressivo e da un quadro clinico ampiamente variabile caratterizzato da perdita di massa
muscolare, miotono, cataratta, difetti nel sistema
di conduzione cardiaco, alterazioni endocrine e
deficit cognitivi. Nella stessa famiglia l’esordio
tende a presentarsi ad un’età sempre più giovane
di generazione in generazione (fenomeno di anticipazione) (Höweler CJ et al., 1989).
La Distrofia Miotonica di Tipo 1 (malattia di
Steinert) è caratterizzata da età di insorgenza e
severità sono molto variabili. Le manifestazioni
d’esordio sono atrofia dei piccoli muscoli della
mano e degli estensori dell’avambraccio e ptosi
delle palpebre. Rispetto a tutte le altre forme di
DM, interessa numerosi muscoli del distretto
cranio-facciale. Segni distintivi del viso, definito
“ad accetta”, sono l’ipotonia dei muscoli facciali,
l’atrofia dei temporali e le rughe frontali; l’atrofia dello sterno-cleido-mastoideo provoca un’esagerata curvatura del collo, definito “a cigno”.
Le patologie cardiache includono disturbi nella
conduzione, fibrillazione atriale, tachiaritmie
ventricolari. Possono essere presenti endocri-
nopatie (atrofia testicolare, diabete mellito). La
cataratta si sviluppa quasi in tutti i pazienti ma
la sua comparsa può essere tardiva. Sono descritti anche disturbi del sonno e calvizie. I pazienti
colpiti dalle forme più severe possono presentare
disturbi cognitivi. L’aspettativa di vita è limitata
per l’alta mortalità associata principalmente a
complicanze infettive polmonari e nel 20-30%
dei casi a complicanze cardiache (Mathieu J et
al., 1999).
La Distrofia Miotonica di Tipo 2 (miopatia
miotonica prossimale) esordisce sempre in età
adulta (40-50 anni). L’espressione è variabile ed
è caratterizzata da deficit motorio prossimale
(che interessa il cingolo pelvico e scapolare),
spesso associato a mialgia, suggestiva per la diagnosi. Possono essere presenti patologie cardiache (aritmie, anomalie della conduzione e, in
alcuni casi, cardiomiopatia) ma in forma lieve e
l’aspettativa di vita dei pazienti affetti da DM2
non è solitamente legata a complicanze cardiache (Finsterer J, 2002). Sono decritti inoltre
tremori (20-30%), coinvolgimento dei muscoli
facciali (12%), cataratta, endocrinopatie (atrofia
testicolare, diabete). L’insufficienza respiratoria
raramente è grave. La capacità di camminare è
spesso conservata fino ai 60 anni.
La Distrofia Oculo-Faringea (DOF) è caratterizzata da un’insorgenza tardiva dei sintomi,
solitamente dopo i 45 anni. La debolezza muscolare colpisce i muscoli dell’occhio e del faringe e
le manifestazioni cliniche principali sono ptosi palpebrale bilaterale e disfagia. I pazienti
lamentano debolezza progressiva dei muscoli
elevatori delle palpebre e per sopperire mantengono i muscoli frontali cronicamente contratti.
L’interessamento dei muscoli faringei causa cambiamenti nel tono della voce e difficoltà nella deglutizione. Negli stadi più avanzati sono interessati anche i muscoli delle spalle e pelvici (Ropper
AH et al., 2005).
www.buponline.com
188
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
Tabella 3: Distrofie muscolari: età di insorgenza, manifestazioni cliniche, decorso
Tipo di DM
Età d’esordio
Segni e Sintomi
Decorso clinico
3-5 anni
Difficoltà a camminare (andatura
ondeggiante), correre, salire le scale
Lordosi
Ritardo mentale
Alterazioni del ritmo cardiaco
Cardiomiopatia dilatativa
Ipertrofia miocardica
Relativamente rapido
Morte solitamente nella tarda
adolescenza per complicanze
cardiache (10-20%), infezioni
polmonari, insufficienza
respiratoria
5-15 anni
Difficoltà a camminare (andatura
ondeggiante), correre, salire le scale
Lordosi
Ritardo mentale
Lievi alterazioni del ritmo cardiaco
Cardiomiopatia dilatativa
Morte solitamente nella quinta
decade, nel 50% per complicanze
cardiache
5-30 anni
Contrattura dei muscoli flessori
del gomito, estensori del collo e del
polpaccio
Cardiomiopatia severa associata a
difetti di conduzione,
Cardiomiopatia dilatativa
Dilatazione atriale e/o ventricolare
Decorso generalmente
benigno ma condizionato dalla
cardiomiopatia
6-20 anni
Difficoltà ad alzare le braccia e
muovere le scapole
Debolezza dei muscoli facciali
Difficoltà a sorridere e a chiudere gli
occhi
Coinvolgimento cardiaco raro
Progressione lenta, con periodi di
stasi
Il 15% dei pazienti perde la
capacità di deambulazione
DISTROFIE
MIOTONICHE
20-50 anni
Atrofia muscolare e miotonia,
debolezza facciale, ptosi,
atrofia del muscolo massetere;
Faccia “ad accetta”
Collo a cigno
Voce nasale
Insufficienza ventricolare sinistra
Bradicardia e blocco atrioventricolare
Ritardo mentale di grado mediomoderato
DOF
20-50 anni
Ptosi
Disfagia
Cambiamenti del tono della voce
DMD
DMB
DMED
DFSO
www.buponline.com
Progressione lenta
Dopo 20 anni dall’esordio
pazienti perdono la capacità di
deambulazione
Morte causata da infezioni
polmonari e insufficienza cardiaca
(DM1: Morte da insufficienza
20-30% dei casi)
Progressione lenta
189
Distrofie Muscolari
CARATTERISTICHE ORO- FACCIALI
Nei pazienti con DM sono descritte anomalie
dentali: un significativo ritardo nell’eruzione
(in media di un anno), agenesie, macrodonzia
ed ipoplasie dello smalto localizzate prevalentemente a livello dei premolari (Mielnik-Blaszczak
M, 2007).
A causa delle difficoltà nelle manovre di igiene orale domiciliare per l’interessamento della
muscolatura degli arti superiori, nei pazienti
con DM sono di frequente riscontro elevate
quantità di placca e di tartaro, responsabili di
alta prevalenza di patologia cariosa e parodontale (Engavall M et al. 2007; Symons AL et al.,
2002).
L’alta prevalenza di carie è causata anche
dall’aumentato tempo di contatto degli zuccheri
con i denti causato dalla diminuita attività masticatoria. Nella masticazione i pazienti presentano infatti una funzionalità dimezzata a livello
del massetere e del muscolo temporale anteriore
(mentre l’attività del muscolo temporale posteriore non presenta differenze significative rispetto a individui sani) e, di conseguenza, necessitano di più tempo e di più cicli masticatori per
deglutire il cibo (Engavall M et al, 1997).
I soggetti affetti da DM sono ad alto rischio di
lesioni di origine traumatica all’apparato dente-tessuti di sostegno in relazione ai problemi
di deambulazione.
L’atrofia e la perdita progressiva della capacità
contrattile dei muscoli della testa e del collo nei
pazienti affetti da DM possono essere causa di
alterazioni cranio-facciali e di malocclusioni
dentarie (Staley RN et al., 1992).
Nei pazienti affetti da DFSO, la debolezza dei
muscoli periorali può causare difficoltà nella
masticazione e nella fonazione; in questi pazienti la forma del volto è tipicamente dolicofacciale (viso allungato e magro), con palato
Figura 1: Ragazza di 18 anni affetta da DM (di tipo non
identificato); tipologia dolicofacciale (viso allungato e magro).
ogivale e malocclusioni dentarie (Guler AU et
al., 2003) (Figure 1, 2 a, b, c, 3, 4).
Nelle Distrofie Miotoniche sono descritte malocclusioni causate dalla ridotta funzionalità
della muscolatura periorale e masticatoria; la
postura bassa della mandibola e della lingua,
causata dall’ipotono dei muscoli elevatori, fa sì
che i pazienti sviluppino una respirazione orale, associata a morso aperto anteriore e crossbite laterale (Kiliaridis S et al, 1998; Kiliaridis
et al., 1989; Gazit E et al, 1987). In letteratura
sono inoltre descritte a livello dell’articolazione
temporo-mandibolare superfici corticali erose,
appiattite ed irregolari, responsabili di ridotta apertura della bocca e di sublussazioni nei
movimenti di massima apertura (Wilson A et al.,
1989; Gold GN, 1966).
Nei pazienti con DMD sono descritte malocclusioni causate dalla compromissione dei muscoli
oro-facciali, dalla respirazione orale e dalla macroglossia, spesso presente. Il diminuito tono del
muscolo massetere e la posizione abbassata della
mandibola possono determinare l’estrusione dei
denti posteriori e il quadro di morso aperto anteriore. Le problematiche muscolari e respiratorie
www.buponline.com
190
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
sono responsabili di palato ogivale, con possibile morso crociato posteriore. (Kiliaridis S et al.
1998; Eckardt L et al., 1996).
LINEE GUIDA DI TERAPIA
I pazienti sono cronicamente in terapia con
cortisone per rallentare il decorso della malattia
(Chakkalakal JV et al, 2005). Per controllare le
complicanze sistemiche sono somministrati farmaci antiaritmici, calcio-antagonisti, antidepressivi e benzodiazepine.
Al fine di diminuire la debolezza e le contratture
muscolari, sono attuati interventi di riabilitazione
fisioterapici e posizionamento di dispositivi ortopedici.
Per quanto concerne le patologie di pertinenza
odontostomatologica, in un’ottica di interdisciplinarietà, è compito del pediatra, dal momento
in cui si effettua la diagnosi, inviare il paziente
all’odontoiatra infantile perché informi la famiglia
sull’importanza della salute orale e sulla necessità
di interventi di prevenzione (igiene orale domiciliare, alimentazione non cariogenica, utilizzo
di fluoro topico e, quando indicato, sistemico, visite periodiche trimestrali/semestrali, sigillatura
dei solchi, delle fessure e dei fori ciechi dei molari
permanenti).
L’obiettivo primario è condurre il bambino a provvedere alla cura della propria persona utilizzando
programmi di istruzione e coinvolgendo la famiglia, gli educatori e gli operatori sanitari coinvolti nella riabilitazione, in particolare logopedisti e
fisioterapisti.
Nei casi di deficit motorio importante, è indispensabile istruire le persone che accudiscono il paziente affinché mettano in atto tutte le manovre di igiene orale domiciliare.
Per le problematiche cardiache e respiratorie che
questi pazienti presentano, le terapie a livello
a
b
c
Figura 2 a, b, c: Palato ogivale, prognatismo mandibolare e
malocclusione di II classe scheletrica.
www.buponline.com
191
Distrofie Muscolari
Figura 3: Bambina di 7 anni affetta da Distrofia Miotonica
di tipo 1; malocclusione di III classe dento-scheletrica con
morso aperto anteriore e morso crociato monolaterale destro.
Figura 4: Ragazzo di 17 anni affetto da DMD; malocclusione di III classe scheletrica, morso aperto anteriore e morso
crociato bilaterale.
ambulatoriale in anestesia locale sono da preferire all’utilizzo dell’anestesia generale. In ambiente
odontoiatrico per un corretto approccio psicologico è indispensabile valutare la eventuale presenza
di ritardo mentale, di disturbi di linguaggio, di
deficit comunicativi e di apprendimento, il grado
di collaborazione e la gravità del deficit motorio.
Dal momento che pazienti sottoposti a frequenti ospedalizzazioni possono presentare una scarsa
accettazione nei riguardi del trattamento odontoiatrico, un approccio graduale e calibrato in base
all’età è indicato al fine di guadagnare la collaborazione necessaria all’attuazione delle terapie, nella
consapevolezza che risultati funzionali ed estetici
sono in grado di migliorare la qualità della vita del
paziente.
I pazienti affetti da DM necessitano di attenzioni
particolari, valutando le possibili complicanze cardiache e polmonari che possono essere scatenate
dall’intervento odontoiatrico.
Nei paziente affetti da cardiomiopatia è indicata la
profilassi antibiotica prima di attuare le manovre
odontoiatriche responsabili di batteriemia.
Nei casi di importante compromissione muscolare è necessario mantenere il paziente in posizione
seminclinata, al fine di ridurre al minimo il rischio
di fenomeni ab ingestis. Per lo stesso motivo è necessario l’uso della diga di gomma; è consigliabile
l’uso di apribocca in gomma per limitare lo sforzo
muscolare.
Nei pazienti affetti da DM non esistono controindicazioni al trattamento ortodontico. Controlli
frequenti del grado di igiene orale domiciliare e la
sensibilizzazione dei genitori agli stili di salute orale
permettono di attuare le stesse scelte terapeutiche
della popolazione sana. L’approccio più efficace è
l’intercettazione precoce e la terapia tempestiva
delle alterazioni di crescita cranio-facciale correlate
alla patologia muscolare nell’obiettivo di migliorare la respirazione; va comunque considerata la possibilità di recidive, in particolare del morso aperto
anteriore.
Dal momento che le lesioni traumatiche sono frequenti, genitori, educatori e operatori debbono
essere sensibilizzati nel prevenire gli eventi traumatici ed istruiti nella gestione delle lesioni, in
particolare sull’opportunità del reimpianto immediato in caso di avulsione di denti permanenti.
Quando a causa di assenza di collaborazione è indicato ricorrere all’anestesia generale è necessario
tenere in considerazione le complicanze intraoperatorie, tra queste la più rischiosa è l’ipertermia
www.buponline.com
192
Sindromi genetiche e cromosomiche e patologie del cavo orale in età evolutiva
maligna, che si manifesta con aumento della temperatura corporea, acidosi metabolica, rigidità muscolare generalizzata ed insufficienza respiratoria.
BIBLIOGRAFIA
Balasubramaniam R, Sollecito TP, Stoopler ET.
Oral health considerations in muscular dystrophies. Spec Care Dentist. 2008;28:243-53
Chakkalakal JV, Thompson J, Parks RJ, Jasmin BJ.
Molecular, cellular, and pharmacological therapies for Duchenne/Becker muscular dystrophies. FASEB J. 2005;19:880-91
Cruz Guzmán Odel R, Chávez García AL,
Rodríguez-Cruz M.: Muscular dystrophies at different ages: metabolic and endocrine alterations. Int J Endocrinol.
2012;2012:ID485376
Eckardt L, Harzer W. Facial structure and functional findings in patients with progressive
muscular dystrophy (Duchenne). Am J Orthod
Dentofacial Orthop. 1996;110:185-90
Engvall M, Birkhed D: Oral sugar clearance and
other caries-related factors in patients with
myotonic dystrophy. Acta Odontol Scand.
1997;55:111-5
Engvall M, Sjogreen L, Kjellberg H, Robertson A,
Sundell S, Kiliaridis S: Oral health in children
and adolescents with myotonic dystrophy. Eur
J Oral Sci. 2007;115:192-7
Finsterer J. Myotonic dystrophy type 2Eur J Neurol, 9 (2002), pp. 441–447
Gazit E, Bornstein N, Lieberman M, Serfaty V,
Gross M, Korczyn AD. The stomatognathic
system in myotonic dystrophy. Eur J Orthod.
1987;9:160-4
Gold GN. Temporomandibular joint dysfunction in myotonic dystrophy. Neurology.
1966;16:212-6
Guler AU, Ceylan G, Ozkoc O, Aydiin M, Cen-
giz N: Prosthetic treatment of a patient with
facioscapulohumeral muscular dystrophy: a
clinical report. J Prosthet Dent. 2003;90:321-4
Hamamoto DT: Muscular dystrophy. In Hupp J,
Williams T, Firriolo F, eds. Dental clinical advisor. St. Louis, MO: Mosby; 2006;144-6
Hermans MC, Pinto YM, Merkies IS, de DieSmulders CE, Crijns HJ, Faber CG. Hereditary muscular dystrophies and the heart.Neuromuscul Disord. 2010 Aug;20(8):479-92.
doi: 10.1016/j.nmd.2010.04.008
Höweler CJ, Busch HF, Geraedts JP, Niermeijer
MF, Staal A. Anticipation in myotonic dystrophy: fact or fiction? Brain, 112 (1989), pp.
779–797
Hsu YD: Muscular dystrophy: from pathogenesis
to strategy. Acta Neurol Taiwan. 2004;13:50-8
Kaufmann P, Weimer LH, Rowland LP: Progressive muscular dystrophies. In: Rowland LP, ed.
Merritt’s neurology, 11th ed. Philadelphia, PA:
Lippincott, Williams, & Wilkins; 2005;895911
Kiliaridis S, Mejersjo C, Thilander B. Muscle function and craniofacial morphology: aclinical
study in patients with myotonic dystrophy. Eur
J Orthod. 1989;11:131-8
Kiliaridis S, Katsaros C: The effects of myotonic
dystrophy and Duchenne muscular dystrophy
on the orofacial muscles and dentofacial morphology. Acta Odontol Scand. 1998;56:36974
Mathews KD: Muscular dystrophy overview: genetics and diagnosis. Neurol Clin. 2003;21:795-816
Mathieu J, Allard P, Potvin L, Prevost C, Begin P.
A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology, 52
(1999), pp. 1658–1662
Mielnik-Blaszczak M, Malgorzata B: Duchenne
muscular dystrophy–a dental healthcare program. Spec Care Dentist 2007;27:23-5
Ropper AH, Brown RH. The muscular dystro-
www.buponline.com
193
Distrofie Muscolari
phies: In: Ropper AH, Brown RH, eds. Adams
and Victor’s principles of neurology, 8th ed.
New York, NY: McGraw-Hill; 2005;1213-29
Staley RN, Bishara SE, Hanson JW, Nowak AJ:
Craniofacial development in myotonic dystrophy. Cleft Palate Craniofac J. 1992;29:456-62
Symons AL, Townsend GC, Hughes TE: Dental characteristics of patients with Duchenne
muscular dystrophy. ASDC J Dent Child.
2002;69:277-83,234
Voisin V, de la Porte S: Therapeutic strategies for
Duchenne and Becker dystrophies. Int Rev Cytol. 2004;240:1-30
Voit T, Krogmann O, Lenard HG, Neuen-Jacob
E, Wechsler W, Goebel HH et al. Emery–Dreifuss muscular dystrophy: disease spectrum
and differential diagnosis. Neuropediatrics, 19
(1988), pp. 62–71
Wilson A, Mackay L, Ord RA. Recurrent dislocation of the mandible in a patient with
myotonic dystrophy. J Oral Maxillofac Surg.
1989;47:1329-32
www.buponline.com
www.buponline.com