F. Corelli e S. Pasquini
ANTIBATTERICI CHINOLONICI
ACIDO NALIDISSICO E CHINOLONI DI PRIMA GENERAZIONE
La storia degli antibatterici chinolonici ha avuto origine con una scoperta casuale all’inizio
degli anni sessanta. Nel corso di una sintesi alternativa della clorochina, i chimici della SterlingWinthrop isolarono come sottoprodotto il composto 1 (Figura 1), che risultò provvisto di una certa
attività antibatterica. Ripetute modificazioni chimiche di questo lead permisero l’ottenimento di una
varietà di analoghi strutturali che furono saggiati come antibatterici, arrivando così alla scoperta
dell’acido nalidissico (Figura 1). Questo composto, dotato di moderata attività contro i germi gramnegativi, ha rappresentato il capostipite della famiglia dei chinoloni e fu introdotto in terapia nel 1963
come chemioterapico delle vie urinarie.
L’esordio in terapia dell’acido nalidissico (specialità in vendita in Italia: Bexatina, Nalidixin,
Nalissina, Neg-Gram, Uralgin, Uri-Flor e Urogram) ha spronato un’intensa attività di ricerca nel campo
dei chinoloni e negli anni sessanta e settanta si ebbe lo sviluppo di una serie di analoghi che
costituiscono i cosiddetti chinoloni di prima generazione (Figura 2). Sebbene tutti questi composti
presentino un atomo di azoto alla posizione 1, la struttura naftiridonica dell’acido nalidissico fu
modificata ritornando al nucleo chinolonico (ad es. acido ossolinico) del lead originale 1. Si ebbe anche
l’inserimento di altri atomi di azoto in posizione 2 (cinossacina) e 6 (acidi piromidico e pipemidico),
mentre anelli addizionali vennero fusi alle posizioni 6 e 7 (acido ossolinico e cinossacina) o a ponte tra
le posizioni 1 e 8 (flumechina). L’introduzione in 7 di ammine cicliche ha prodotto gli acidi piromidico
e pipemidico. Il gruppo etilico in N1 fu mantenuto invece relativamente costante in quanto si riteneva
allora che nessun sostituente più grande potesse essere compatibile con una buona attività antibatterica.
Rispetto all’acido nalidissico, questi chinoloni di prima generazione presentano aumentata
attività contro i batteri gram-negativi, ma sono sostanzialmente privi di effetti verso i cocchi grampositivi, Pseudomonas aeruginosa e germi anaerobi. Tuttavia essi vengono ben assorbiti dopo
somministrazione orale e raggiungono elevate concentrazioni nel tratto urinario, il che li rende utili
come chemioterapici delle vie urinarie.
CHINOLONI DI SECONDA GENERAZIONE
Un progresso sensibile nel settore dei chinoloni si ebbe nel 1980, allorché i chimici della
Kyorin misero a punto la preparazione della norflossacina, derivante dalla combinazione di elementi
1
strutturali dell’acido pipemidico (7-piperazina) e della flumechina (6-fluoro) (Figura 3). La
norflossacina mostra una certa attività sui gram-positivi oltre ad una attività contro i gram-negativi
superiore rispetto agli agenti precedenti, ma in pratica è ancora indicata soltanto per le infezioni del
tratto urinario ed il trattamento di prostatiti ed infezioni trasmesse sessualmente (gonorrea), a causa dei
suoi bassi livelli ematici e della scarsa distribuzione tissutale. La norflossacina è stato il primo
fluorochinolone propriamente detto in quanto presenta non solo il fluoro in posizione 6, ma anche
l’altro elemento strutturale fondamentale che è la piperazina in 7 (si dovrebbe forse parlare di
fluoropiperazochinoloni). L’atomo di fluoro in 6 serve sia ad aumentare l’attività intrinseca nei
confronti della DNA girasi che a facilitare la penetrazione all’interno del batterio. La sua funzione è
cruciale, tanto che da allora in poi è stato mantenuto costante in tutti i chinoloni (fluorochinoloni).
Alcune delle modifiche apportate alla struttura della norflossacina (Figura 3) sembrano
piuttosto modeste, ma in realtà sono in grado di alterare sensibilmente le proprietà dei composti
risultanti, a dimostrazione del fatto che la struttura chimica influisce potentemente sulla attività
biologica in questa classe di farmaci. Ad esempio, l’aggiunta di un gruppo metilico all’azoto distale
della piperazina trasforma la norflossacina in peflossacina, che presenta una semi-vita più che doppia
rispetto al farmaco madre. La enossacina, analogo naftiridonico della norflossacina, possiede grosso
modo la stessa attività, ma migliore biodisponibilità. La contemporanea introduzione del metile nella
piperazina e di altri atomi di fluoro porta alla flerossacina ed alla lomeflossacina, caratterizzati da
lunghi tempi di semi-vita, tanto che entrambi questi composti vengono somministrati in dose unica
giornaliera per il trattamento di svariate infezioni sistemiche.
La sostituzione del gruppo etilico in N1 con un gruppo ciclopropilico, modificazione
strutturale apparentemente modesta, ha di fatto rappresentato un altro passo avanti fondamentale. La
ciproflossacina (Figura 3) possiede attività contro i germi gram-negativi e gram-positivi (chinolone ad
ampio spettro). Introdotta in terapia nel 1987, è stato il primo chinolone ad essere largamente prescritto
non solo per la cura delle solite infezioni delle vie urinarie, ma anche per trattare infezioni delle basse
vie respiratorie, della pelle e delle articolazioni. Il gruppo ciclopropile della ciproflossacina ha
soppiantato completamente l’etile come sostituente in N1 nello sviluppo dei fluorochinoloni successivi.
Nei chinoloni di seconda generazione l’anello piperazinico rimane relativamente immodificato, mentre
subirà modifiche anche notevoli nei composti di terza generazione.
I chinoloni di seconda generazione sono caratterizzati da attività buona o eccellente nei
riguardi dei germi gram-negativi (la più potente è la ciproflossacina), ma attività modesta contro lo
Staphylococcus aureus e solo marginale contro Streptococcus pneumoniae ed anaerobi. Queste
deficienze nello spettro di azione hanno dato impulso allo sviluppo dei chinoloni di terza generazione.
CHINOLONI DI TERZA GENERAZIONE
2
Questi nuovi composti (Figura 4) sono caratterizzati da aumentata novità e complessità
strutturale, che hanno determinato nuove ed utili proprietà biologiche, quali attività contro cocchi
gram-positivi (ed in particolare S. pneumoniae) e, in certi composti della serie, contro anaerobi e
patogeni atipici. In alcuni casi la maggior potenza di azione e l’ampiezza dello spettro di azione si
coniuga con proprietà farmacocinetiche più favorevoli che permettono di somministrare questi
composti una sola volta al dì.
In Figura 5 sono riportate schematicamente le variazioni strutturali principali dei chinoloni di
terza generazione e dei composti attualmente in sviluppo. In sintesi, la sostituzione in 8 (alogeno o
metossi) incrementa generalmente l’attività sui gram-positivi e negativi, probabilmente migliorando la
capacità di penetrazione della celllula batterica. Tuttavia, l’aumento di lipofilia che è stato attuato nei
chinoloni di terza generazione tende ad aumentare di solito più l’azione sui gram-positivi che quella sui
gram-negativi. Ciò può essere conseguenza degli effetti opposti che la idrofobicità ha sulla
penetrazione: l’aumento del logP tende ad aumentare l’accumulo nello S. aureus diminuendo, invece,
la penetrazione nei gram-negativi. La lipofilia può essere aumentata anche con l’introduzione di
sostituenti alchilici al C5 e/o nel nucleo piperazinico, come pure con la sostituzione della piperazina
con 3-amminopirrolidine.
E’ bene puntualizzare che, sebbene certe sostituzioni possano migliorare per se una particolare
proprietà biologica o chimica, le caratteristiche complessive di una molecola derivano tuttavia dalla
interazione di tutti i sostituenti uno con l’altro e con lo specifico nucleo utilizzato.
PROPRIETÀ CHIMICHE DEI FLUOROCHINOLONI
Il comportamento farmacocinetico e farmacodinamico dei chinoloni è strettamente correlato
alle caratteristiche chimiche di questi composti come la solubilità, il carattere acido-base, la capacità
chelante e la fotoreattività.
SOLUBILITÀ IN ACQUA
I fluorochinoloni sono composti poco solubili in acqua. Presentano punti di fusione elevati
(generalmente > 200 °C) a causa delle forme cristalline molto stabili che si formano (π-stacking tra gli
anelli aromatici). Per superare questo problema sono stati preparati alcuni profarmaci (Figura 6) aventi
gruppi funzionali che conferiscono una maggiore solubilità in acqua, ma che nell’organismo liberano
facilmente il principio attivo. La trovaflossacina è stata trasformata nell’alatroflossacina in cui il
gruppo amminico libero è stato fatto reagire con la coppia amminoacidica L-Ala-L-Ala dando origine
al derivato ammidico che possiede una maggiore solubilità in acqua. In vivo l’alatroflossacina viene
idrolizzata a dare la trovaflossacina, vero principio attivo.
3
ACIDITÀ E BASICITÀ
I fluorochinoloni hanno proprietà acide conferite dal gruppo carbossilico in posizione 3; il pKa
compreso tra 5,5-6,3 li rende meno acidi dell’acido acetico che possiede invece un pKa di 4,2. Inoltre,
grazie alla presenza di uno o più centri basici, questi composti hanno proprietà basiche con pKa di 7,69,3, che varia a seconda del sostituente presente sul sostituente eterociclico, e che li rende meno basici
della trietilammina (pKa = 10,7). Poiché i fluorochinoloni possiedono sia caratteristiche acide che
basiche sono considerati sostanze anfotere. In Figura 7 è riportata lo schema di
protonazione/deprotonazione della ciproflossacina, che si trova nella sua forma protonata in ambiente
acido e in quella deprotonata in ambiente basico, mentre a pH neutro si trova in equilibrio con la sua
forma zwitterionica dalla quale dipende la sua scarsa solubilità in acqua a pH fisiologico.
CHELAZIONE DI CATIONI METALLICI
I fluorochinoloni hanno la capacità di chelare cationi con costanti di affinità crescenti
nell’ordine Ca2+> Mg2+> Fe3+> Al3+. La chelazione di ioni di- o trivalenti avviene a livello della
funzione carbossilica e del vicino gruppo carbonilico in posizione 4: la coplanarità che esiste tra i due
gruppi rende possibile la formazione di chelati molto stabili. Questi chelati presentano un ridotto
assorbimento a livello intestinale, in conseguenza non tanto di una ridotta solubilità (molti chelati sono
più solubili dei fluorochinoloni liberi), quanto piuttosto della diminuita lipofilia che ne riduce la
biodisponibilità. Inoltre le capacità chelanti di fluorochinoloni possono dare origine ad interazioni
farmaco-farmaco (interazione con antiacidi) e farmaco-cibo. Nel caso della spaflossacina il problema
dell’assorbimento intestinale e dell’interazione con antiacidi è stato in parte superato in quanto il
gruppo amminico presente in posizione 5 interagisce con il carbonile in 4 rendendolo meno disponibile
alla chelazione (Figura 8).
FOTOREATTIVITÀ
I fluorochinoloni, in particolare quelli contenenti atomi di alogeno in posizione 8, sono
composti fotoreattivi specialmente in condizioni neutre o acide. Se opportunamente stimolati da
radiazioni elettromagnetiche, i fluorochinoloni danno origine a specie radicaliche di diversa natura a
seconda della struttura del chinolone e, infine, portano alla formazione di radicali dell’ossigeno molto
reattivi. I fluorochinoloni assorbono lunghezze d’onda di 350-425 nm che fanno parte della regione del
visibile dello spettro elettromagnetico, per questo motivo pazienti che assumono fluorochinoloni
possono manifestare reazioni di fotosensibilità a seguito dell’esposizione alla luce solare.
4
NOMENCLATURA DEI CHINOLONI
5
6
7
8
5
6
7
N
O
8a
3
3
N1
H
4
8a
3
N1
H
N
1,4-diidro-1,8-naftiridin-4-one
2
O
6
8a
1
N8
H
N
a
5,8-diidropirido[2,3-d]pirimidin-5-one
4
e
f
5
4a
N
2
1,4-diidrochinolin-4-one
2
O
4a
8
4
4
4a
7
b
3
d
N
2
c
N
5
6
1
pirimidina
piridina
SINTESI DEI FLUOROCHINOLONI
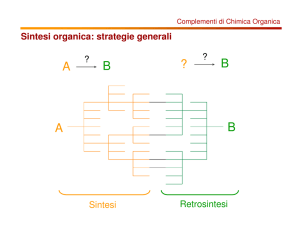
I fluorochinoloni vengono sintetizzati secondo due procedure generali in accordo con i
seguenti approcci retrosintetici:
Metodo A: disconnessione N1-C2 e C4-C4a
F
6
5
Metodo B: disconnessione C2-C3 e N1-C8
O
4a
4
COOH
F
6
5
O
4a
4
3
R1
7
8
8a
N1
R
2
COOH
3
R1
7
8
8a
N1
R
2
5
Metodo A: Sintesi di Gould-Jacobs. Vedi esempio in Figura 9.
La sintesi prevede una reazione di condensazione tra una anilina opportunamente sostituita e
l’etossimetilenmalonato dietilico (EMME) con la quale si ha la formazione del legame N1-C2. Poi, in
seguito a ciclizzazione in difeniletere ad elevata temperatura (ciclizzazione di Lappin) si ha la
connessione C4-C4a e la formazione del derivato 4-chinolonico. Il sostituente in posizione N1 si
introduce mediante reazione di alchilazione utilizzando l’opportuno alogenuro alchilico in presenza di
una base (SN2). In seguito, si ha l’idrolisi, in ambiente basico, dell’estere etilico presente in posizione 3
e per successiva acidificazione si ottiene l’acido chinolin-4-on-3-carbossilico. L’ultima reazione è una
sostituzione nucleofila aromatica in cui l’atomo di cloro viene spiazzato da un nucleofilo come la
piperazina per dare il prodotto finale. Il gruppo carbonilico in posizione 4 attiva le posizioni orto e para
alla sostituzione nucleofila aromatica, per questo motivo è soltanto l’atomo di cloro ad essere spiazzato
mentre l’atomo di fluoro in posizione 6 non reagisce.
Questo metodo di sintesi è utile per la preparazione di fluorochinoloni che presentano in N1 un gruppo
alchilico primario, che può essere introdotto tramite reazione di sostituzione nucleofila SN2. Pertanto
non può essere impiegato nella sintesi di moderni fluorochinoloni che presentano in N1 un gruppo
ciclopropilico od arilico.
Metodo B. Vedi esempio in Figura 10.
Questo metodo prevede l’utilizzo di opportuni acetofenoni. E’ di impiego generale in quanto permette
di ottenere composti a struttura chinolonica con sostituenti in posizione 1 di vario tipo, come gruppi
alchilici 1°, 2° o 3° o gruppi arilici.
La sintesi inizia con la reazione di condensazione di Claisen che nel primo passaggio si effettua tra un
acetofenone ed il dietilcarbonato, mentre nel secondo passaggio si ha la reazione con trietil
ortoformiato con la quale si ottiene la prima connessione C2-C3. Successivamente si ha l’introduzione
del gruppo amminico mediante reazione di addizione-eliminazione e poi, con la ciclizzazione in
presenza di una base forte come il NaH, la seconda connessione N1-C8a. Infine attraverso una
sostituzione nucleofila aromatica si introduce il sostituente in posizione 7 e, in seguito a idrolisi basica,
si ottiene il prodotto finale.
Una variante di questo metodo è riportato in Figura 11. In questo caso si parte da un cloruro acido
opportunamente sostituito che reagisce prima con il dimetilamminoacrilato di etile e con
ciclopropilamina poi per dare lo stesso intermedio che si ottiene anche con la sintesi precedente e che
viene sottoposto alla stessa sequenza di reazioni sopra riportate.
MECCANISMO D’AZIONE DEI FLUOROCHINOLONI
6
I fluorochinoloni interagiscono con il DNA del batterio bloccandone la replicazione. Il DNA
batterico è lungo circa 1300 µm, quindi affinché possa essere contenuto nella cellula batterica (2 µm x
1 µm) deve essere superavvolto (Figura 12). La DNA girasi è l’enzima preposto a tale scopo ed è il
bersaglio dei fluorochinoloni.
La DNA girasi è specifica dei batteri ed è il comune bersaglio dei fluorochinoloni in molti
gram-negativi. Un altro enzima, la topoisomerasi IV, presente anche nell’uomo, è invece il bersaglio in
molti gram-positivi. La DNA girasi appartiene alla famiglia delle topoisomerasi II, così chiamate
perché sono in grado di tagliare i due filamenti di DNA batterico e di cucirlo di nuovo una volta creato
il superavvolgimento. La DNA girasi è un tetrametro costituita da quattro subunità, due subunità A e
due subunità B che sono coinvolte nel trattamento del DNA batterico. In particolare, le due subunità A
si uniscono alle due subunità B nel momento in cui il DNA è pronto per essere processato (Figura 13).
Il primo frammento entra nella subunità B, viene tagliato e mantenuto in modo che i due frammenti
rimangano distanti l’uno dall’altro. Allo stesso tempo, un altro frammento di DNA viene passato
attraverso la rottura creata e in seguito i due frammenti vengo risaldati. In questo modo si crea un
superavvolgimento (Figura 14).
SELETTIVITÀ D’AZIONE DEI FLUOROCHINOLONI E TOSSICITÀ GENETICA
La selettività dei fluorochinoloni è conseguente alla inibizione selettiva della DNA girasi
batterica rispetto al corrispondente enzima topoisomerasi II dei mammiferi.
L’aumento della lipofilia dei fluorochinoloni comporta:
> attività su batteri Gram positivi (cambia il bersaglio di azione da DNA girasi a topoisomerasi IV?)
< attività su batteri Gram negativi (diminuisce la penetrazione)
< selettività tra DNA girasi e topoisomerasi II umana
In particolare la citotossicità è aumentata dalla presenza di
• C-7 pirrolidine (anziché piperazine)
• N-1 ciclopropile
• C-8 cloro, fluoro, metossi
Tuttavia, opportune modifiche strutturali possono ridurre od eliminare la citotossicità anche in
presenza di N-1 ciclopropile e/o sostituenti al C-8 (vedi per un esempio Figura 15).
METABOLISMO E FARMACOCINETICA DEI FLUOROCHINOLONI
I fluorochinoloni vengono somministrati per via orale; quelli altamente idrosolubili trovano
utilizzazione per via parenterale. I fluorochinoloni vengono eliminati a livello renale (filtrazione
glomerulare e secrezione tubulare attiva) ed intestinale. Le principali trasformazioni metaboliche si
7
hanno a carico del nucleo piperazinico e sono riportate in Figura 16. Pertanto, modifiche strutturali
dell’anello piperazinico (od amminopirrolidinico) che ostacolano la trasformazione metabolica possono
aumentare la semivita plasmatica dei fluorochinoloni: molti fluorochinoloni di terza generazione hanno
t1/2 tali da consentire una unica somministrazione giornaliera.
EFFETTI COLLATERALI DEI FLUOROCHINOLONI
Dati ottenuti dalle impressioni soggettivi dei pazienti (qualsiasi dose)
Fluorochinolone
ADRs
totale (%)
ADRs
tratto GI (%)
ADRs
SNC (%)
ADRs
Pelle (%)
Enossacina
6,2
1,2
1,2
0,6
Norflossacina
9,1
3,9
4,4
0,5
Peflossacina
8,0
5,6
0,9
2,2
Ciproflossacina
5,8
3,4
1,1
0,7
Oflossacina
4,3
2,6
0,9
0,5
Levoflossacina
3,3
1,8
0,5
0,2
Lomeflossacina
Non riportato
5,1
5,5
2,4
Avvertenze
specifiche
Interazioni con
xantine,
NAIDs
Non riportate
Fototossicità,
interazione
con teofilline
Interazioni con
teofilline
Problemi al
SNC ad alte
dosi
Non riportati
Alta incidenza
di fototossicità
e problemi al
SNC
Dati oggettivi ottenuti in seguito a questionari sottoposti ai pazienti
Fluorochinolone
ADRs
totale (%)
ADRs
tratto GI (%)
ADRs
SNC (%)
ADRs
pelle (%)
Sparflossacina
(200-400 mg)
22-32
6,4-10
2-3
1,8
Trovaflossacina
(100-200 mg)
27
4-7
Vertigini: 12
Mal di testa: 5
1
Avvertenze
specifiche
Fototossicità e
prolungamento
dell’intervallo
QT
Non reazioni
serie: vertigini
ad alte dosi
solo in alcuni
pazienti
8
PRINCIPALI APPLICAZIONI TERAPEUTICHE DEI FLUOROCHINOLONI
Infezioni delle vie urinarie:
• sono molto efficaci nel combattere le infezioni senza complicanze e rappresentano i farmaci di scelta
nei casi in cui la resistenza batterica compromette la terapia con beta-lattamici di routine
• efficaci contro infezioni croniche da P. aeruginosa
• efficaci nelle prostatiti batteriche generalmente resistenti a tetracicline, cefalosporine, trimetoprimasulfametossazolo
Malattie trasmesse sessualmente:
tutti i fluorochinoloni sono efficaci nel trattamento mono-dose di varie forme di gonorrea, ma non nei
casi di sifilide
Infezioni delle vie respiratorie:
• efficaci nelle infezioni pneumococciche resistenti ai beta-lattamici ed ai macrolidi
• nelle forme esacerbate di bronchite cronica, ciproflossacina, oflossacina e sparflossacina si sono
dimostrati equivalenti o superiori a tutta una serie di altri antibiotici
Infezioni gastrointestinali:
• i fluorochinoloni rappresentano un passo avanti molto importante per il trattamento di molte forme di
infezioni enteriche da moderate a gravi e sono i farmaci di elezione nei casi di febbre tifoide sia negli
adulti che nei bambini la ciproflossacina si è dimostrata attiva contro il colera al pari delle tetracicline.
9
O
COOH
Cl
N
Me
N
N
N
N
HN
acido pipemidico
ciproflossacina
F
COOH
N
N
N
F
COOH
N
HN
acido nalidissico
norflossacina
O
F
F
flumechina
chinoloni di prima generazione
CH3
O
F
COOH
N
N
N
O
levoflossacina
O
COOH
N
COOH
N
N
H
N
O
N
COOH
N
N
F
CH3
oflossacina
chinoloni di seconda generazione
N
F
Me
sparflossacina
O
O
O
COOH
N
N
HN
HN
H3C
F
COOH
F
COOH
N
1
NH2 O
O
O
H2N
H
trovaflossacina F
chinoloni di terza generazione
Figura 1. Evoluzione storica dei chinoloni
10
COOH
N
H3C
O
O
O
COOH
N
N
N
N
F
COOH
N
N
HN
acido pipemidico
acido nalidissico
O
O
COOH
N
N
N
N
acido piromidico
flumechina
COOH
O
O
O
N
acido ossolinico
COOH
O
O
N
N
cinossacina
Figura 2. Chinoloni di prima generazione
11
O
O
F
COOH
N
H3C
O
O
N
HN
O
COOH
N
F
N
enossacina
F
N
N
HN
peflossacina
N
COOH
N
N
COOH
F
N
norflossacina
F
H3C
COOH
N
N
HN
O
F
F
N
COOH
N
F
N
HN
CH3
F
ciproflossacina
lomeflossacina (NY-198)
flerossacina (AM-833)
O
O
F
F
COOH
COOH
N
N
H3C
N
N
S
CH3
ruflossacina
H3C
N
N
O
CH3
oflossacina
Figura 3. Chinoloni di seconda generazione
12
O
NH2 O
O
F
F
H3C
OH
N
O
H3C
N
HN
OH
N
HN
O
N
O
F
H3C
F
OH
N
N
HN
OCH3
CH3
Grepaflossacina
(OPC-17116)
Sparflossacina
(AT-4140)
Gatiflossacina
(AM-1155)
O
O
O
F
N
N
O
O
CH3
OH
N
N
N
F
S
H2N
CH3
H
F
Trovaflossacina
(CP-99,219)
O
O
OH
N
H
Pezuflossacina
(T-3761)
O
H2N
N
S
F
N
OH
H2N
Levoflossacina
(DR-3355)
F
O
F
OH
N
H3C
O
O
O
F
O
F
OH
OH
N
N
F
N
Cl
H2N
F
Tosuflossacina
(T-3262)
N
N
Cl
S
DU-6859a
H2N
R
O
F
Clinaflossacina
(Cl-960, AM-1091
Figura 4. Chinoloni di terza generazione
13
O
O
X
Y
O
OH
X
N
R
X
Y= H
Me
X
N
R
amine cicliche
Y= derivati piperazinici
R= Et
Y
O
OH
OH
X= CH, NH, CF
X= CH, NH
O
F
F
Y
Z
O
X
N
R
X= CH, NH, CF CCl COMe
Y= derivati piperazinici,
N
R=
F
F
Z= NH2 Me
prima generazione
seconda generazione
terza generazione
= caratteristiche strutturali non pi¯ utilizzate
= caratteristiche strutturali introdotte
Figura 5. Evoluzione strutturale dei chinoloni
14
O
O
F
H
N
O
H
F
H2N
F
OH
N
H
F
Trovaflossacina
solubilit in acqua < 0.01 mg/ml a pH neutro
O
L-Ala-L-Ala N
H
N
OH
N
F
H
F
Alatroflossacina
molto solubile in acqua
Figura 6. Solubilit in acqua della trovaflossacina e del profarmaco alatroflossacina
15
O
F
N
COOH
N
H2N
pH acido
O
F
N
O
COO
F
N
N
H2N
COOH
N
HN
pH basico
O
F
N
COO
N
HN
Figura 7. Protonazione/deprotonazione della ciproflossacina
16
R
N
N
R1
N
O
F
O
O
M
O
O
F
O
N
R1
H
N
H
O
F
H3C
N
NH2 O
OH
F
O
N
N
H3C
F
HN
N
R
CH3
NH2 O
F
O
N
HN
OH
N
F
CH3
H3C
H
O
O
N
N
F
HN
CH3
Sparflossacina
Figura 8. Chelazione di cationi metallici
17
COOEt
EtO
O
COOEt
F
Cl
NH2
(EMME)
F
(reazione di
Gould-Jacobs)
Cl
N
H
COOEt
COOEt
Δ, difeniletere
F
(ciclizzazione
di Lappin)
Cl
connessione N1-C2
EtI, base
(reazione SN2)
O
N
R
N
O
COOH
N
Et
N
H
connessione C4-C4a
R
N
F
COOEt
N
H
F
HCl
Cl
COOH
N
Et
1. OHŠ, H2O
F
2. H+
O
Cl
N
Et
COOEt
R= H: Norflossacina
R= Me: Peflossacina
Figura 9.
18
O
F
Me
Cl
Cl
O
CO(OEt)2
EtONa
O
F
(condensazione
di Claisen)
HC(OEt)3
EtONa
OEt
Cl
O
F
COOEt
(condensazione Cl
di Claisen)
Cl
Cl
OEt
connessione C2-C3
H2N
H
N
O
F
N
O
COOEt
N
N
H
F
HCl
Cl
O
COOEt
NaH
Δ
N
F
Cl
COOEt
Cl
NH
HN
connessione N1-C8a
OHŠ, H2O
O
F
N
COOH
N
HN
Ciproflossacina
Figura 10.
19
Me
N
Me
O
F
O
Cl
Cl
F
COOEt
Cl
Cl
NH2
O
Cl
O
OEt
F
Me
N
Me
Cl
COOEt
Cl
NH
connessione N1-C2
O
F
COOH
N
HN
O
O
Š
N
K2CO3
R
N
OH
F
H2O
N
COOEt
N
N
H
F
HCl
Cl
COOEt
N
HN
Ciproflossacina
connessione N1-C8a
Figura 11.
20
(a)
(b)
cellula
batterica
cromosoma
batterico
Figura 12. (a) Superavvolgimento del DNA; (b) rapporto in scala tra cellula batterica e DNA batterico
21
Figura 13. Meccanismo d'azione della DNA girasi
22
Figura 14. Funzione della DNA girasi
23
NH2 O
F
Me
COOH
N
HN
F
F
COOH
Me
N
F
NH2 O
NH2 O
N
HN
N
F
Me
COOH
N
HN
N
F
Me
Sparflossacina
inattiva verso la topoisomerasi II
Bis(demetil)-sparflossacina
potente inibitore della topoisomerasi II
Isomero trans della sparflossacina
inibitore della topoisomerasi II
non citotossica
citotossica
citotossica
Figura 15.
24
O
H3C N
N
OHC N
N-metil-oxo
H3C
N
N
H3COC N
N-formil
N
H3C N
N
N-acetil
N
HN
N
O
N-ossido
O
H3COC NH HN
HN
acetilmmino
N
HO3S N
oxo
H2N
HN
etilendiammino
N
N-solfonil
H2N
ammino
Figura 16. Trasformazioni metaboliche a carico del nucleo piperazinico
25