Fisica dello Stato Solido
Richiami di fisica classica
Corso di Laurea Magistrale in
Ingegneria Elettronica
a.a.10-11
http://www.de.unifi.it/FISICA/Bruzzi/fss.html
Sommario
Termodinamica
• Primo principio della Termodinamica
• Gas Ideale – Teoria Cinetica dei gas ideali
• Calori specifici molari
• Secondo principio della termodinamica
• Entropia
Onde elettromagnetiche
• Spettro delle Onde elettromagnetiche
• Diffrazione
• Interferenza
• Diffrazione con Raggi X su cristalli e legge di Bragg
Primo principio della termodinamica
Sistema termodinamico : insieme di uno o piu’ corpi di composizione nota che si
trovano in una regione dello spazio delimitata da superfici ideali o reali che li
distinguono fisicamente dagli altri corpi o sistemi con cui essi possono interagire e
che costituiscono l’ambiente circostante del sistema.
Universo = sistema + ambiente
Sistema aperto Se tra sistema e ambiente avviene scambio di energia e materia
Sistema chiuso Se tra sistema e ambiente avviene solo scambio di energia
Sistema isolato Se tra sistema e ambiente non avvengono scambi di energia e
materia
Lo stato del sistema termodinamico viene descritto mediante un insieme di
grandezze fisiche misurabili dette coordinate o variabili termodinamiche. Il numero
minimo di grandezze fisiche necessario a descrivere completamente uno stato
non è fissato, ma dipende dalle caratteristiche chimico.fisiche del sistema. Nel
caso che vedremo di gas ideale questo numero è 3, ad esempio p,V,T.
Considero un sistema termodinamico descrivibile con tre coordinate
termodinamiche macroscopiche (e.g. p,V,T). Il sistema è in uno stato di equilibrio
termodinamico se, ferme restando le condizioni esterne dovute all’ambiente, le
coordinate termodinamiche non variano. Perciò lo stato termodinamico è detto di
equilibrio quando le variabili termodinamiche che lo caratterizzano sono costanti
nel tempo.
In uno stato di equilibrio sussiste in generale una precisa relazione tra le coordinate
termodinamiche: f(pV,T) = 0, tale relazione viene chiamata equazione di stato.
Se viene meno lo stato di equilibrio avviene una trasformazione termodinamica
ed il sistema passa da uno stato iniziale A ad uno finale B, considereremo A e B
stati di equilibrio.
Supponiamo di avere eseguito una trasformazione A → B e di voler riportare il
sistema allo stato iniziale: se nel fare ciò anche l’ambiente è ritornato allo stato
iniziale allora la trasformazione si dice reversibile, altrimenti la trasformazione si
dice irreversibile.
In una trasformazione reversibile si conoscono tutti gli stati intermedi assunti dal
sistema nel passare dallo stato iniziale a quello finale, è quindi possibile
ripercorrere la trasformazione all’inversoIl sistema passa da una molteplicità di stati
di equilibrio mediante trasformazioni infinitesime con variazioni dp, dV, dT. Tutto ciò
non vale per la irreversibile.
Due sistemi in diversi stati termodinamici possono interagire tra loro. Nel caso in cui tra loro vi
sia una parete diatermica ( conduttore termico ) essi evolvono spontaneamente verso un nuovo
stato, detto di equilibrio termico. Se i due sistemi sono invece separati da una parete
adiabatica ( isolante termico ) essi restano nei loro stati termodinamici iniziali ( a meno che non
venga compiuto lavoro meccanico su di essi).
Per caratterizzare l’equilibrio termico fra i sistemi si introduce una nuova grandezza, detta
temperatura. Per definizione quindi, due sistemi che sono in equilibrio termico fra loro hanno la
stessa temperatura.
Principio zero della Termodinamica: Due sistemi separatamente in equilibrio termico con
un terzo sistema sono in equilibrio termico tra di loro.
Sulla base del principio zero è possibile procedere alla misura della temperatura utilizzando un
sistema campione: il termometro.
Consideriamo ora un sistema racchiuso da pareti adiabatiche, esso non subirà variazioni di
temperatura e le interazioni con l’ambiente esterno dovranno essere di natura
esclusivamente meccanica. In generale sappiamo che il lavoro effettuato dall’ambiente sul
sistema dipende sia dagli stati finale ed iniziale che dal tipo di trasformazione attuata. Nel caso
della trasformazione adiabatica invece, si verifica sperimentalmente che il lavoro dipende
solamente dagli stati iniziale e finale. E’ quindi possibile definire una grandezza, detta
energia interna Ui, tale che:
W = - ∆Ui = Uiiniziale - Uifinale
Una proprietà importante che ne discende è che l’energia interna è una
funzione di stato del sistema, definita a meno di una costante additiva, in
quanto la sua definizione operativa fornisce la grandezza come differenza
tramite il lavoro adiabatico.
Consideriamo invece il caso in cui siano consentiti scambi anche di tipo termico
(pareti diatermiche). Eseguendo trasformazioni diverse che portano dallo stato
iniziale allo stesso stato finale si osserva che il lavoro dipende sia dagli stati
iniziale e finale che dal tipo di trasformazione adottata. Inoltre, se la
trasformazione non è adiabatica, si osserva che:
W ≠ − ∆U i
Si introduce quindi una nuova grandezza fisica, detta calore, Q, tale che valga, per
qualsiasi trasformazione compiuta tra lo stato iniziale e quello finale la relazione
seguente:
Q − W = ∆U i
Primo Principio della Termodinamica
Usualmente, (ma non sempre ! vedi e.g. trasformazione a T = costante ) un corpo che
scambia calore varia anche la sua temperatura.
Consideriamo un corpo che
scambiando il calore Q vari la sua temperatura da Ti a Tf. Si definiscono le quantità:
δQ
C=
δT
Capacità termica
1 δQ
c=
m δT
Calore specifico
1 δQ
c=
n δT
Calore specifico molare
Osserviamo inoltre che il lavoro eseguito per passare da uno stato a Volume
V1 ad uno stato a Volume V2 può essere sempre valutato come:
sB
sB
V2
sA
sA
V1
W = ∫ F ⋅ ds =
∫ pAds = ∫ pdV
Gas ideale
Considero un sistema di N particelle contenute in un recipiente chiuso e fermo in
un sistema di riferimento inerziale. In generale le particelle risulteranno in
movimento e interagiranno tra loro con forze che supponiamo conservative.
Consideriamo in particolare che siano valide le seguenti assunzioni:
1) l’interazione tra le particelle ha raggio d’azione trascurabile, esse si muovono
perciò come particelle indipendenti l’una dall’altra, cioè (tra un urto con le pareti
e il successivo) di moto rettilineo uniforme. Tale movimento è del tutto casuale
visto che non esistono posizioni o direzioni privilegiate.
2) Gli urti delle molecole con le pareti del recipiente sono praticamente istantanei
e completamente elastici ( pareti lisce e di massa infinita ).
3) Il volume occupato dalle particelle è trascurabile rispetto a quello del
recipiente.
Tale modello è ragionevolmente applicabile ai gas rarefatti , cioè in condizioni di
bassa pressione e temperatura elevata rispetto al punto di liquefazione. In tal
caso parliamo di gas ideale.
Teoria cinetica dei gas ideali
Considero per semplicità un recipiente cubico di lato L ed un gas ideale
composto da particelle identiche di massa m.
Una particella urta contro una parete piano (y,z) con velocità v1, dopo l’urto ha
velocità v2, se la parete è liscia le componenti lungo y e z sono inalterate mentre
la vx si è invertita, dato che la parete ha massa infinita e l’urto è elastico v1 = v2.
La variazione di quantità di moto della particella i-esima nell’urto è:
y
v1
∆pi = -2mvxiux
La particella va avanti e indietro urtando le pareti, non urta
le altre particelle ( gas rarefatto ) e si muove tra urti successivi
di moto rettilineo uniforme . In un intervallo ∆t esegue
∆t
un numero di urti pari a :
N =
∆t
v2
x
2 L / v xi
ed in ciascun urto scambia l’impulso Ii = 2mvxi. Quindi l’impulso scambiato in ∆t da
tutte le particelle è:
I = ∑ I i =∑ 2mvxi N ∆t = ∑ 2mvxi
∆t
∆t
= ∑ mvxi2
2 L / vxi
L
L’impulso per unità di tempo è pari alla forza media esercitata dal gas sulla
parete, essa è pari a:
m
Fx =
v
∑
L
2
xi
A tale forza corrisponde una pressione sulla parete:
p=
Fx m
m
mN 1
= 3 ∑ v xi2 = ∑ v xi2 =
A L
V
V N
mN 2
2
v
=
∑ xi V vx
Ripetendo le stesse considerazioni per le altre pareti otteniamo risultati analoghi.
Poiché sperimentalmente risulta che la pressione è la stessa su tutte le pareti,
deve valere che:
2
2
2
vx = v y = vz
Essendo inoltre
v 2 = v x2 + v y2 + v z2
Otteniamo:
v x2 =
v2
3
mN 2 . Sia U = 1 mv 2 energia cinetica media delle particelle
K
v
Risulta perciò: p =
2
3V
Otteniamo:
2
pV = NU K
3
Equazione di stato dei gas ideali
Si verifica che in un sistema idrostatico di massa costante le tre coordinate
macroscopiche pressione, temperatura e volume non sono indipendenti, ma deve
esistere una relazione analitica che lega le tre coordinate: f(p,V,T) = 0: ad essa
viene dato il nome di equazione di stato di quel particolare sistema.
Definiamo:
mole = numero di atomi contenuti in 12g dell’isotopo del carbonio avente numero di massa 12
n= numero di moli = massa espressa in grammi / peso molecolare o atomico = m/mA
Numero di Avogadro NA = Numero di atomi o molecole contenute in una mole = 6.02x1023 mol-1
Per il gas ideale valgono le leggi sperimentali:
pV = cos t. Legge di Boyle valida per le trasformazioni a temperatura costante
VT = V0 βT Legge di Charles o I legge di Gay Lussac
valida per le trasformazioni a pressione costante
pT = p0 βT II legge di Gay Lussac
valida per le trasformazioni a volume costante
Con: β =
1
C −1
273.15
p0 e V0 pressione e volume del gas a 0 C, T in Kelvin.
Valgono inoltre le due leggi di Avogadro:
-Una mole di qualsiasi sostanza contiene NA = 6.02x1023 molecole/ atomi
-Volumi uguali di gas diversi nelle stesse condizioni di temperatura e pressione,
contengono lo stesso numero di molecole ( e quindi lo stesso numero di moli ).
Dalle leggi sopra viste otteniamo:
1
p
n,T costanti
Vα T
n,p costanti
Vα
Vα n
Da cui segue:
p,T costanti
pV = cos t.nT
Si trova cioè l’equazione di stato dei gas perfetti
pV = nRT
R = costante universale dei gas, dal valore sperimentale R = 8.31 J/molK
Scriviamo anche:
nR = nN A K B = NK B
con KB = costante di Boltzmann = 1.38x10-23 J/K
N = numero di particelle del gas
L’equazione di stato: pV = NK BT
unita alla relazione:
pV =
2
NU K
3
Ci porta ad una relazione tra temperatura del gas ideale ( grandezza macroscopica)
ed energia cinetica media delle particelle ( grandezza microscopica ):
UK =
3
K BT
2
Energia cinetica totale del gas ideale alla Temperatura T:
U Ktot = NU K =
3
nRT
2
Calori specifici dei gas ideali
Consideriamo alcune trasformazioni interessanti del gas ideale
Isocora ( volume costante ):
W = 0;
Q = ∆Ui
1 δQ
1 dU i
cV =
=
n δT V =cos t n dT
Poiché Ui è funzione di stato deduciamo che , per qualsiasi altra
trasformazione, possiamo sempre scrivere : dU = nc dT
i
che si traduce nell’espressione del primo principio:
Differenziamo la: pV = nRT
Si ha:
ottenendo:
δQ = ncV dT + nRdT − Vdp
V
δQ = ncV dT + pdV
pdV + Vdp = nRdT
Isobara ( pressione costante ):
W = p∆V;
Definiamo calore specifico a pressione costante
Allora vale :
E quindi:
1 δQ
cp =
n δT p =cos t
nc p dT = ncV dT + nRdT
c p = cV + R
Relazione di Mayer
Abbiamo mostrato prima come per una particella di cui non si consideri la
struttura interna ( molecola monoatomica ) l’energia cinetica media si può
esprimere come
UK =
3
K BT
2
In tal caso l’energia interna del gas si ottiene moltiplicando tale energia cinetica
per il numero di atomi che compongono il gas
3
3
U i = NK BT = nRT
2
2
Poiché una molecola monoatomica ha tre gradi di libertà è come se ciascuna
particella contribuisse in media all’energia interna con un’energia:
1
2
ε = K BT
Per ciascun grado di libertà. Questa affermazione può essere effettivamente
giustificata da un punto
di vista statistico attraverso il Teorema di
equipartizione dell’energia utilizzabile quando al sistema si può applicare la
termodinamica statistica classica:
In un sistema che si trovi in equilibrio termodinamico alla temperatura T
ogni termine quadratico indipendente della sua energia interna ha un
valore medio pari a 1
.
2
K BT
Da ciò, per un gas ideale monoatomico:
cV =
3
R
2
cp =
5
R
2
Per un gas biatomico, è necessario considerare i due atomi con ciascuno tre
gradi di libertà della posizione, però con una relazione tra essi che fissa costante
la loro distanza ( 5 gradi di libertà in tutto ). Entro questi limiti:
gas ideale biatomico:
cV =
5
R
2
cp =
7
R
2
Una evoluzione del modello prevede che la molecola biatomica possa vibrare, quindi
vanno aggiunti altri due contributi, l’energia cinetica vibrazionale e quella potenziale
elastica, da cui:
7
cV = R
2
cp =
9
R
2
Accade perciò che per temperature basse (e.g. per la molecola di H2 T minori di
40K) il calore specifico è quello della molecola monoatomica, per T intermedi (
sempre per H2 tra 250K e 500K ) il calore specifico è quello della molecola
biatomica a distanza interatomica fissa, per T superiori il calore specifico tiene
conto anche della componente vibrazionale.
Notiamo che questi comportamenti possono
essere descritti solo in termini quantistici, infatti
da un punto di vista classico l’energia dovuta alla
rotazione e quella dovuta alla vibrazione possono
assumere valori continui, anche piccoli a piacere,
quantisticamente
invece,
essendo
l’energia
quantizzata, i termini di rotazione e vibrazione
danno un contributo apprezzabile solo se la
temperatura supera un valore di soglia.
cv
7/2R
5/2R
3/2R
T
II Principio della Termodinamica
Enunciato di Kelvin-Planck
E’ impossibile realizzare una qualsiasi trasformazione il cui unico risultato
sia quello di convertire completamente in lavoro il calore prelevato da un
solo serbatoio.
La macchina termica piu’ semplice preleva Q1 da una
sorgente calda a T1 e cede calore Q2 alla sorgente fredda
a T2 producendo il lavoro W. La variazione di energia
interna è nulla perché ho ciclo. Convenzione sui segni:
T1
Q1
Q1 > 0
W
W<0
W>0
Q2
Q2 < 0
T2
Allora per il primo principio della termodinamica: W = Q1 + Q2
Rendimento del ciclo :
η=
W Q1 + Q2
Q
=
= 1+ 2 < 1
Q1
Q1
Q1
La macchina termica descritta nella slide precedente può essere
riprodotta utilizzando un ciclo di Carnot isoterma di espansione a T1 da A
a B, adiabatica BC, isoterma di compressione a T2 e adiabatica DA.
p
A
D
T2
Si dimostra che il ciclo di Carnot ha rendimento pari a : η C = 1 −
T1
B
T2
Q2 da cui otteniamo: Q1 Q2
= 1+
+
=0
Allora: 1 −
T1
Q1
T1 T2
C
V
Si può dimostrare inoltre che vale il Teorema di Carnot :
Il rendimento di una macchina termica generica non può essere maggiore di quello di
una macchina di Carnot:
η ≤ ηC
η = ηC
Se la macchina è reversibile, η < ηc se la macchina è irreversibile
ENTROPIA
In generale quindi la relazione diviene:
Q1 Q2
+
≤0
T1 T2
Dove l’uguaglianza vale per trasformazione ciclica reversibile. Il Teorema di
Clausius generalizza tale espressione al caso in cui il sistema durante il ciclo
scambia calore con piu’ sorgenti, in tal caso vale:
∫
δQ
T
≤0
integrale di Clausius
Dove l’uguaglianza vale per trasformazione ciclica reversibile:
δQ
∫ T rev = 0
Ma allora, nel caso di trasformazione ciclica reversibile, è possibile definire una
funzione di stato, detta entropia, S, tale che:
finale
∆S =
δQ
∫
T
rev
iniziale
Relazione tra integrale di Clausius e entropia
Considero una trasformazione ciclica formata dalla
trasformazione irreversibile da A a B piu’ una
reversibile da B ad A.
irreversibile
B
B
δQ
∫ T
A
= ∫ T
A
reversibile
A
δQ
La trasformazione reversibile può essere invertita:
+∫
irr
B
A
δQ
∫
B
Inoltre l’ integrale è pari alla
variazione di entropia:
B
∫
A
B
Otteniamo:
∫
A
δQ
T
< SB − S A
irr
δQ
T
T
δQ
T
<0
rev
B
= −∫
rev
A
δQ
T
= − ∆S
rev
= S B − S A = ∆S
rev
Come caso particolare di questa relazione
Consideriamo il sistema isolato termicamente
B
In generale allora vale la:
∆S = S B − S A ≥ ∫
A
δQ
T
Dove l’uguaglianza vale per la trasformazione reversibile, la disuguaglianza
per quella irreversibile.
Come caso particolare di questa relazione consideriamo il sistema
isolato termicamente. Poiché non si ha scambio termico allora δQ = 0
e quindi
∆S = S B − S A ≥ 0
Che è noto come principio dell’aumento di entropia:
L’entropia di un sistema isolato termicamente aumenta se esso
esegue una trasformazione irreversibile, resta costante se la
trasformazione è reversibile.
L’universo è sicuramente un sistema isolato, quindi vale sempre:
Poiché:
∆Su ≥ 0
∆Su = ∆S sistema + ∆S ambiente
se la trasformazione è ciclica ∆Ssist =0 quindi:
∆Su = ∆S Amb ≥ 0
Denominazione
Spettro
Elettromagnetico
ν [Hz]
[ ]
λ
Onde radio
< 3 109
> 10 cm
Microonde
3 109 – 3 1011
10 cm – 1 mm
Infrarossi
3 1011 – 428 1012
1 mm – 700 nm
Luce visibile
428 1012 – 749 1012
700 nm – 400 nm
Ultravioletti
749 1012 – 3 1016
400 nm – 10 nm
Raggi X
3 1016 – 3 1018
10 nm – 1 pm
Raggi gamma
> 3 1018
< 1 pm
2 . Diffrazione
Consideriamo una sorgente di onde elettromagnetiche S piane, i cui fronti d’onda
incontrano un ostacolo come l'apertura in uno schermo opaco (fenditura). La
fenditura abbia dimensioni lineari dello stesso ordine di grandezza della
lunghezza d’onda della radiazione elettromagnetica. Consideriamo il caso
particolare Diffrazione di Fraunhofer ) dove la sorgente S e lo schermo C dove si
visualizza il fenomeno della diffrazione siano a grande distanza dalla fenditura
che supponiamo rettilinea, di larghezza a e lunghezza L>>a.
S
k
a
Fronti d’onda piana
Schermo opaco con fenditura
Schermo C
Suddividiamo la fenditura in N strisce ciascuna di larghezza ∆y =a/N. Ciascuna
striscia funge da sorgente di onde secondarie ( principio di Huygens-Fresnel)
contribuendo con ampiezza ∆E al campo risultante Ep in un punto P dello
schermo, individuato dai raggi uscenti ad angolo θ rispetto alla normale al
piano della fenditura.
I contributi relativi a due strisce adiacenti hanno nel punto P la differenza
di fase, derivante dalla differenza di cammino ∆ysenθ
θ:
Metodo dei fasori
Possiamo rappresentare l’onda
armonica
come un vettore, detto FASORE,
di modulo E0/r, che ruota intorno
all’origine con velocità angolare
ω. La proiezione del fasore
sull’asse verticale dà, istante
per istante, il valore E1(t).
Con riferimento alla figura, gli N fasori che
rappresentano le ampiezze ∆E delle singole sorgenti
secondarie, in cui è suddivisa la fenditura,
costituiscono una poligonale di N lati. L’angolo
formato tra ciascun fasore e il successivo è dato da :
La differenza di fase tra l’onda emessa
dall’estremo B e l’estremo A è :
Per ∆y → 0 ed N → ∞ la poligonale diventa un arco di circonferenza
di raggio ρ con angolo al centro pari a α. Dalla figura l’ampiezza del
campo elettrico risultante è pari alla corda che sottende l’arco:
Esercizi sulla diffrazione
3.
1.
2.
1.
2.
3.
3. Interferenza di onde: esperimento di Young
In questo esperimento la luce uscente dalla sorgente S viene diffratta alle
fenditure S1 ed S2. La luce emessa da S1 ed S2 produce su uno schermo C, posto a
distanza L >> d ( d = separazione fenditure) una figura di interferenza consistente
in strisce chiare (massimi di intensità luminosa ) e scure (minimi) alternate, detta
figura di interferenza.
Siano E1, E2 onde prodotte dalle sorgenti S1 ed S2:
La differenza di fase tra le due onde è:
I massimi di interferenza si hanno quando la differenza di percorso dsenθ
θè
un multiplo intero della lunghezza d’onda λ. In questa condizione le due
onde risultano infatti in fase.
Esercizi sull’ interferenza
1.
3.
2.
1.
2.
3.
4. Diffrazione X dei Cristalli
Abbiamo visto come i solidi, in forma cristallina, si dispongano in strutture
tridimensionali ordinate. Un reticolo cristallino molto comune in natura è per esempio
il reticolo cubico a facce centrate (FCC).
a (Å)
C ( diamante )
Si
Ge
α-Sn
GaAs
a
3.57
5.43
5.66
6.49
5.65
Cu
Si 2 FCC
compenetrati di ¼
della diagonale di
corpo
NaCl 2 FCC
compenetrati di 1/2
lato del cubo
E’ possibile esplorare la struttura microscopica
dei cristalli utilizzando un fascio di raggi X,
radiazione elettromagnetica con lunghezza d’onda
di circa 1Ǻ, lo stesso ordine di grandezza della
costante reticolare a nei cristalli. La teoria della
diffrazione X è stata sviluppata da Sir William
Bragg nel 1913. Bragg mostrò che un piano di
atomi nel cristallo riflette la radiazione nello
stesso modo nel quale la luce viene riflessa da uno
specchio, percui l’angolo in uscita θr è uguale
all’angolo incidente θi.
k
cristallo
Fronte onda piana
Fascio
incidente
Fascio
riflesso
Θr = Θi
Θi
a
Piano di Bragg
Legge di Bragg
Se si considera la radiazione come riflessa da piani di Bragg paralleli e
successivi, è possibile che i fasci riflessi dai vari piani interferiscano
costruttivamente.
Perché si abbia interferenza
costruttiva, la differenza di
cammino tra le due onde
riflesse deve essere tale
che:
θ
A
θ
θ
d
AB + BC = nλ
λ
C
ossia deve valere la legge di
Bragg:
B
2d sen θ = nλ
λ
Poiché la distanza tra piani d corrisponde a qualche Å il fenomeno non si
osserva con luce visibile ( ~ 5000 Å). E’ necessario usare fotoni X.
Esercizi sulla Diffrazione nei cristalli
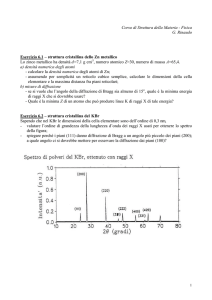
1. Lo ioduro di potassio ha stessa struttura cristallina di quella del NaCl, con d =
0.353 nm. Un fascio monocromatico di raggi X mostra un massimo di diffrazione
per primo ordine quando l’angolo di incidenza è 7.6°. Calcolare la lunghezza
d’onda dei raggi X.
λ = 0.934nm
2. Un fascio monocromatico di raggi X incide sulla superficie di un cristallo di
NaCl. Nel fascio riflesso il massimo del secondo ordine si trova ad un angolo di
20.5° tra il fascio incidente e la superficie. Determinare la lunghezza d’onda dei
raggi X.
λ = 0.984nm
3. Raggi monocromatici X di lunghezza d’onda λ = 0.166nm incidono su un
cristallo di KCl. Se la distanza tra i piani è di 0.314nm a quale angolo rispetto alla
superficie del cristallo bisogna dirigere il fascio per poter osservare un massimo
del secondo ordine ?
α = 32°
32°