LA METODOLOGIA DELLA
RICERCA CLINICA
Dr Marzia Lazzerini
IRCCS Burlo Garofolo, Trieste
[email protected]
Dr Marco Ercolani
Ibn Hindu (10th-11th century CE )
Miftah al-tibb wa-minhaj al-tullab
[The key to the science of medicine and the students' guide].
Conoscenza medica acquisita “ per caso”
Camminando nei campi, un gruppo di ragazzi
raccolse delle foglie di alloro, uno di loro ne
assoggiò una. Egli fu in seguito morso da un
serpente, e non accusando alcun sintomo, si
capì che furono i semi di alloro a salvarlo.
Questa fu la prova della loro efficacia come
antidoto, e da allora furono inclusi negli
antidoti per i veleni .. (p 50)
1
Ibn Hindu (10th-11th century CE )
Miftah al-tibb wa-minhaj al-tullab
[The key to the science of medicine and the students' guide].
Conoscenza medica acquisita
“attraverso esperimenti con un obiettivo”
"…provando diverse medicine, una per
una in corpi di diversa natura, volta dopo
volta, e registrando per ogni medicazione
l’effetto che ricorre dopo il suo uso...
Così dopo ripetuti usi, si provò che
“sakamuniya” libera dalla bile gialla, e
“afitimun” libera dalla bile nera” (p 51)
Ben Cao Tu Jing (1061).(Atlas of Materia Medica).
Song Dynasty (960-1279).
(960
(960-1279).
E’ detto che per valutare gli effetti del genuino ginseng Shangdang, due
persone vennero fatte correre, una dopo aver assunto il ginseng,
l’altra senza.
Dopo aver corso approsimativamente 3-5 li [1500-2500 metri], quello
senza ginseng sviluppò una severa mancanza di respiro, mentre
quello che aveva preso ginseng respirava bene e con tranquillà ...
2
Nettleton T (1722).
Physician at Halifax.
Letter To Dr. Jurin R.S Secr, on
Inoculation of the Small Pox
(varicella)
Philosophical Transactions.
32:209-212.
1753, Lind
Trial controllato a quattro bracci...
bracci...
sui marinai con lo scorbuto
Presi quattro pazienti con lo
scorbuto a bordo della
Salisbury.
I casi erano simili, giacevano
assieme a terra, ed avevano
una dieta comune
Quindi diedi ad ogni coppia:
•
Un quarto di sidro al giorno
•
25 gocce di elisir vitriolo
•
Due cucchiai di aceto
•
Arancia e limone
Il più immediato e visibile effetto
fu percepito dall’uso
dell’arancia e limone
3
TB randomised
controlled trial
STREPTOMYCIN
• Strepto
TREATMENT OF
PULMONARY
TUBERCULOSIS
(BMJ 1948:ii:769-782)
A MEDICAL RESEARCH COUNCIL
INVESTIGATION LONDON
OCTOBER 30 1948
19411941- clinical trial “sistematico”
sistematico”
19481948- 1° clinical trial randomizzato
4
1938 - USA : introduzione dei primi requisiti formali per la
sperimentazione animale
1948 - UK: prima sperimentazione clinica controllata ( STREPTO)
1950: primo trial con placebo (ANTISTAMINICI)
19601960-63 UK e USA : caso talidomide >> nascita del sistema
farmacovigilanza, commissione sulla sicurezza dei medicinali
1969 – USA (FDA): la sperimentazione clinica
diviene obbligatoria per ottenere
l'autorizzazione all’
all’immissione in commercio .
1970 - USA: introduzione “Good Clinical Practice”
Practice”
19801980-90 - Europa: introduzione delle GCP (Italia: 1992)
1992)
Dal 1980:
Avanzamento nella metodologia ricerca clinica,
clinica, EBM
METANALISI
MEGATRIALS
Da 2001: Linee guida di reporting (CONSORT, STARD, STROBE)
Dal 2004: Obblighi di registrazione trials
WHO new standards for registration of all human medical research
19 MAY 2006 | BRUSSELS/GENEVA
5
Tipi di studi clinici
STUDI OSSERVAZIONALI
STUDI SPERIMENTALI
DESCRITTIVI
•
Non controllati
•
Controllati
-non randomizzati
•
Case report
•
case series
- randomizzati
ANALITICI
•
Studi caso-controllo
•
Studi di coorte
- Senza corte parallela (retrospettici o
prospettici)
- Con corte parallela (prospettici)
•
Studi trasversali
Sia gli studi sperimentali che osservazionali possono essere controllati
controllati o meno.
OSSERVAZIONE vs ESPERIMENTO
Osservazione
Senza intervenire sul
fenomeno,
fenomeno,
studio e misuro cio’
cio’
che osservo rispetto al
fenomeno di interesse
Sperimentazione
Intervengo sul fenomeno,
fenomeno,
costruendo un
meccanismo tale da poter
rispondere a domande
riguardo al fenomeno di
interesse
6
Gerarchia delle evidenze
Scelta del disegno
sperimentale
• Efficacia nel rispondere all’obiettivo
dello studio
• Prevenzione dei possibili bias
• Fattibilità, Efficienza
7
Studio clinico/Clinical Trial:
definizione
• Qualsiasi forma di esperimento pianificato
• che coinvolge soggetti umani (volontari sani o
pazienti)
• e ha come obiettivo quello di fornire informazioni
sull’efficacia e/o tollerabilità di un trattamento
• da somministrare ai pazienti in una data condizione.
Principi dell
’inferenza
dell’inferenza
statistica
Popolazione
generale
Popolazione
Campione
Dato un campione rappresentativo della popolazione generale,
attraverso i metodi dell’inferenza statistica,
le informazioni ottenute sul campione
possono essere estese con una certa approsimazione alla popolazione generale.
8
Fasi per realizzare una ricerca
FASE
PRE
START
POST
FASE
PRE
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
Individuazione dell’
dell’oggetto della ricerca
Esame della conoscenza disponibile
Formulazione di domande specifiche
Pianificazione della ricerca (metodi,
metodi, tempi e costi)
costi)
Stesura del protocollo
Approvazione CE
Accordi tra le parti
Registrazione
FoundFound-raising
Preparazione del materiale per conduzione (CRF)
1.
2.
3.
Esecuzione (scientifica+
scientifica+ amministrativa)
amministrativa)
Monitoraggio
Analisi preliminare,
preliminare, interim (interim reports)
1.
2.
Analisi conclusiva
Reporting
Metodologia della
ricerca clinica
E’ fondamentale pianificare studi
metodologicamente corretti
– garanzia di risultati attendibili
– garanzia di eticita’
9
FASE
PRE
Standard scientifici
• Non e’
e’ etico condurre ricerca mal pianificata o mal
eseguita.
eseguita.
• Caratteristiche che identificano studi mal pianificati /
eseguiti:
– Debolezza del confronto (placebo/dose)
– Debolezza degli outcomes (soft, combinati etc)
– Fattori di rischio per bias ( es.
es. mancati/inadeguato
randomizzazione,
randomizzazione, allocation concealment, blinding)
– Scarsa (o eccessiva) numerosita’
numerosita’
– Grosse perdite al follow up
– Mancata pubblicazione dei risultati
FASE
PRE
Elementi fondamentali
di una ricerca clinica
OBIETTIVI
•
Devono essere clinicamente rilevanti
•
Devono essere dichiarati nel protocollo,
protocollo, in modo chiaro e non ambiguo.
ambiguo.
•
Deve essere dichiarata la gerarchia (primari e secondari) in modo chiaro.
•
Non possono cambiare durante lo studio
•
In termini generali, e’
e’ sempre meglio limitare il numero di obiettivi
CONFRONTO
. Trattamento di provata efficacia (in dose e tempi efficaci)
efficaci)
VALIDITA’
VALIDITA’ INTERNA (rigore
(rigore metodologico per ridurre bias)
VALIDITA’
VALIDITA’ ESTERNA (generalizzabilita
(generalizzabilita’’)
10
FASE
PRE
Pianificazione della ricerca
APPROCCIO CLINICO
• Popolazione, Criteri
inclu/exc
• Trattamento
• Trattamento di controllo
• Tempi di osservazione
• Rischi
APPROCCIO STATISTICO
• Dimensione del campione
• Disegno sperimentale
• Efficienza
• Prevenzione del bias
• Regole di analisi ad
interim
Scelta del miglior
disegno sperimentale per raggiungere
gli obiettivi con il minor numero di
pazienti, prevenendo il bias
FASE
PRE
Protocollo
Razionale.
Obiettivi:
E’
necessario per garantire
omogeneita’
omogeneita’ e riproducibilita’
riproducibilita’
Metodi:
•popolazione da selezionare
•Tipo d’intervento (allegati es Scheda tecnica)
•Tempi e modi del follow up
•Misure di outcome (primario e secondario)
•Numerosità campione
•Metodi di analisi statistica
•Sottogruppi pre-definiti
Responsabile scientifico, sponsor, riferimenti amministrativi, agreement
tra le parti, fonti di finanziamenti,
11
FASE
PRE
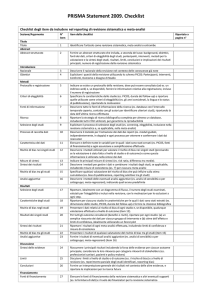
Bias - definizione
• Il “bias”
bias” indica una forma di distorsione introdotta
sui risultati.
risultati.
• l “bias”
bias” possono essere prevenuti attraverso un
adeguato disegno sperimentale e la sua corretta
esecuzione.
esecuzione.
FASE
PRE
LIMITI DI UNO STUDIO NON
CONTROLLATO
Non so se l’effetto osservato e’ realmente dovuto
• al trattamento,
o piuttosto
• al caso
• ad un miglioramento spontaneo della patologia
12
FASE
PRE
Importanza della scelta del
trattamento di controllo
TERAPIA SPERIMENTALE più efficace del PLACEBO
Ma ugualmente efficace rispetto al CONTROLLO ATTIVO
Studi controllati con placebo
– sono sempre studi di superiorita’
– Stimano “l’effetto placebo”, in questo senso
evitano sovrastime dell’effetto della terapia
sperimentale
– evidenziano l’attivita’ terapeutica specifica
del farmaco
– forniscono una piu’ diretta valutazione della
tollerabilita’
13
Quando osservo differenze
nell’effetto tra gruppi
(farmaco A > farmaco B)
Può dipendere da :
' differenze al basale tra i gruppi
(i pazienti del gruppo B erano piu’ gravi )
=SELECTION BIAS
' differenze nella gestione dei pz = PERFORMANCE BIAS
' Differenze nella valutazione dei pz = DETECTION BIAS
' Perdite di pz (effetti collaterali, compliance)= ATTRITION BIAS
* effettive differenze di efficacia tra i trattamenti
PREVENZIONE DEI BIAS
1- Randomizzazione
FASE
PRE
Randomizzazione = processo tramite il quale i pazienti vengono
assegnati casualmente a ricevere uno dei trattamenti previsti
OBIETTIVO: prevenzione del SELECTION bias
bilanciamento dei gruppi
relativamente a fattori prognostici noti ed ignoti
•
•
•
gravita’
gravita’ della patologia
fattori di rischio
terapie e patologie concomitanti
Metodi di randomizzazione:
Computer, tavole dei numeri casuali, Lancio della moneta o dadi.
NB: SONO SCONSIGLIABILI Assegnazione sistematica (giorni pari
e dispari, alternanza...)
14
FASE
PRE
PREVENZIONE DEI BIAS
2- Allocation concealment
OBIETTIVO: Nascondimento dell’
dell’allocazione
al gruppo di trattamento
Metodi di allocation concealment:
Confezioni contenenti trattamento, pre-numerate, di identico aspetto,
preparate da una farmacia indipendente
Randomizzzaione centrale in sede lontana ed indipendente rispetto
sede di arruolamento
Buste con numeri seriali, opache, chiuse
Od altre descrizioni che abbiano elementi convincenti per il
nascondimento del gruppo di allocazione.
INADEGUATE
Lista di allocazione aperta, buste non chiuse o non opache, od altri
approcci che potrebbero svelare il gruppo di trattamento al
momento dell’arruolamento
FASE
PRE
PREVENZIONE DEI BIAS
3- Blinding (cecità)
Nessuna cecita’
cecita’ (in aperto)
aperto)
– sia il paziente che lo sperimentatore conoscono il trattamento
assegnato
>>> conseguenza: possibile bias dell’osservatore nella
valutazione e possibile suggestione da parte del paziente
(DETECTION BIAS)
• Singolo cieco
– il paziente (o il medico) non conoscono il trattamento
• Doppio cieco
– Nè il pz nè il medico
•E’ necessario specificare sempre “chi” è in cieco.
•Preservare sia il il cieco del “ care giver” che “dell’ assessor”
•Preservare il cieco dello statistico che analizza i dati
•Per alcuni interventi (es ortopedici) è impossibile preservare il cieco
•Per alcuni outcome oggettivi ( es mortalità) può non essere rilevante
15
FASE
PRE
4- Dimensione della casistica
STUDI
SOVRADIMENSIONATI
• rappresentano uno spreco
di risorse
• posso ottenere
significativita’
significativita’ statistiche
anche in presenza di
differenze clinicamente
irrilevanti
• espongono al rischio della
sperimentazione un numero
eccessivo di pazienti
FASE
PRE
STUDI
SOTTODIMENSIONATI
• rappresentano uno spreco di
risorse
• posso NON ottenere
significativita’
significativita’ statistiche
anche in presenza di
differenze clinicamente
rilevanti
• espongono al rischio della
sperimentazione un numero
inutile di pazienti
La stima delle dimensioni del campione
Le domande da porsi:
¾ Qual è lo scopo principale dello studio?
¾ Qual è la misura principale di esito?
¾ Quale risultato si prevede di ottenere con il nuovo
trattamento?
¾ Qual è la differenza minima che si ritiene clinicamente
rilevante?
¾ Con quale grado di certezza?
16
FASE
PRE
5 – Analisi dei dati
per Intention to Treat
• ITT = Intention To Treat
Comprende TUTTI i pazienti
randomizzati in uno studio clinico
inclusi
– i pazienti che non completano lo studio
• usciti per effetti collaterali
– i pazienti violatori di protocollo
• con scarsa compliance
• inclusi erroneamente,
erroneamente, trattati erroneamente
FASE
PRE
Caratteristiche dell’ITT
• Oggettiva
• Fedele alla randomizzazione
• Pragmatica
(simula cio’ che potra’ accadere nella pratica clinica)
• Generalmente conservativa
(tende a sottostimare piuttosto che a sovrastimare gli effetti)
17
Conclusioni:
Pianificazione della ricerca-1
• Un adeguato disegno sperimentale permette di
controllare il bias.
• Un’
Un’adeguata analisi statistica permette di stabilire la
verosimiglianza di spiegazioni basate su fluttuazioni
casuali.
casuali.
• Solo avendo controllato tali aspetti, potrò,
potrò, al termine
dell’
dell’analisi dei risultati ,concludere per l’l’esistenza di
effettive differenze.
differenze.
Conclusioni:
Pianificazione della ricerca -2
La scrittura e presentazione di un protocollo di ricerca è una fase
fondamentale, da cui dipende il buon esito dell’intera
sperimentazione
Tuttavia
• Questa non è che la prima fase per l’avvio di una sperimentazione
• Al momento della stesura del protocollo è fondamentale pensare in
termini concreti alla fattibilità e sostenibilità dell’intero progetto sino
alla sua conclusione (fattibilità clinica, cooperativa, capacità di
arruolamento di ogni centro, sostenibilità economica, etc).
18
Esecuzione
ASPETTI CLINICI
• Reclutamento pazienti
• Trattamento dei pazienti
• Aspetti etici
• Verifica dei dati
ASPETTI STATISTICI
• Raccolta dei dati
• Gestione dei dati
• Monitoraggio dei dati
Correttezza ed
affidabilita’ dei dati
ottenuti
Flowchart
CONSORT
Esecuzione
Verifica criteri inclusione
Raccolta dati
demografici e dati basali
Consenso informato
Procedure
randomizzazione
Dispensazione del
trattamento
Follow up seriati
(modalità
e timing previsti)
Analisi
19
ESECUZIONE: Attività di
monitoraggio continuo di un trial
1. Flusso di arruolamento (aggiornamento su diagramma di flusso)
2. Censimento perdite al follow up/sospensioni/rifiuti.
3. Controllo in tempo reale delle CRF per:
9 Completezza > segnalazione ai centri (Queries) ed aggiornamento
9 Consistenza interna dati raccolti
9 Consistenza rispetto altre CRF dello stesso pz
9 Adesione a protocollo ( in particolare decisioni cliniche a Timing
richiesti)
9 Timing dei controlli ( rispetto data di compilazione)
9 Durata follow up
4.Veridicità CRF rispetto dati di origine: Site visits
5.Controllo invio campioni centralizzati
6. Verifica farmaco predisposto/consumato/restituito/distrutto
7. Archiviazione dati
8. Analisi ad interim.
9. Monitoraggio sottoprogetti
ESECUZIONE
- Procedure Good Clinical Practice (GCP)
(GCP)-• Arruolamento
– consenso informato
• Responsabilita’ nella conduzione
– Sponsor
– Investigatore
– Monitor
• Ispezioni regolatorie per assicurare
– adeguatezza dei sistemi
– accuratezza delle documentazioni
20
FASE
POST
Conclusione dello studio
•
• Analisi dei dati
• Confronto con risultati attesi
• Discussione rilevanza clinica
Discussione possibili fonti di bias, limiti dello studio
• Discussione della validità esterna
• Confronto con altre evidenze esistenti
• Presentazione dei risultati
La significatività statistica è un concetto ben
diverso dalla significatività clinica
21