caricato da
david.camin
Energia e Spontaneità delle Trasformazioni: Termodinamica

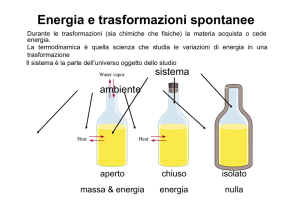
Energia e Spontaneità delle Trasformazioni Trasformazioni ed Energia Durante le trasformazioni (sia chimiche che fisiche) la materia acquista o cede energia. La termodinamica è quella scienza che studia le variazioni di energia in una trasformazione Sistema: è la parte dell’universo oggetto dello studio Intorno del sistema o ambiente: è la parte dell’universo esterna al sistema in cui le interazioni sono rilevabili Sistema aperto Sistema chiuso Sistema isolato massa & energia energia nulla Trasformazioni ed Energia L’interazione fra il sistema e l’ambiente può avvenire attraverso lo scambio di calore e lavoro -calore Q: energia trasferita tra il sistema e l’ambiente in conseguenza di una differenza di temperatura Q m c p (Tfin Tiniz ) cal o J ( 1cal = 4,186 J) m è la quantità di sostanza coinvolta ; Cp rappresenta la capacità termica specifica o calore specifico (cal (J) g-1 °C-1) della sostanza Tfin e Tiniz rappresentano rispettivamente la temperatura finale e iniziale della sostanza - Lavoro L: forma di trasferimento dell’energia che può essere di tipo meccanico, elettrico,…….. Trasformazioni ed Energia Il trasferimento di energia durante una trasformazione (calore e/o lavoro) richiedono da parte del sistema una variazione della sua energia, definita energia interna U U rappresenta l’energia totale del sistema ovvero la somma dell’energia cinetica (traslazionale, rotazionale, vibrazionale), e dell’energia potenziale (l’energia dei legami chimici , delle interazioni intermolecolari , delle forze di attrazione nucleo – elettroni e l’energia dei protoni e neutroni nel nucleo.) U E cin E pot U è una funzione di stato ovvero dipende solo dallo stato in cui si trova il sistema, ovvero temperatura, pressione, volume, tipo e quantità di sostanza, e non dal “percorso” fatto per raggiungerlo. 2H2(g) + O2(g) d=0,99820 g/ml CuSO4 * 5H2O H2O T= 298,15 K P= 1atm La densità rimane costante indipendentemente da come si sia arrivato ad un dato stato del sistema quindi è una funzione di stato Trasformazioni ed Energia L’ energia interna di un sistema non è facilmente stimabile, ma per una trasformazione fra uno stato 1 e uno stato 2 si può conoscere DU che sarà sempre costante Poiché U ha un valore unico in ogni stato anche DU dovrà avere un valore unico DUtotale = U2-U1+U1-U2 = 0 Trasformazioni ed Energia Principio di conservazione dell’energia: L’energia non può essere creata né distrutta ma solo trasformata. Se consideriamo la trasformazione da uno stato 1 (stato iniziale) ad uno stato 2 (stato finale) di un sistema chiuso. DU U2 U1 Q L I° principio della termodinamica Per convenzione: •Q assorbito dal sistema dall’ambiente ha segno positivo (+Q). La trasformazione si dice ENDOTERMICA •Q ceduto dal sistema all’ambiente ha segno negativo (-Q). La trasformazione si dice ESOTERMICA •L compiuto dal sistema vs l’ambiente ha segno positivo (+L) •L compiuto dall’ambiente sul sistema ha segno negativo (-L) Se consideriamo la trasformazione da uno stato 1 ad uno stato 2 di un sistema isolato. ΔU 0 ovvero l’energia totale è costante Trasformazioni ed Energia Sebbene U sia una funzione di stato Q e L non lo sono e il loro valore dipende dal percorso seguito quando un sistema subisce una variazione. Consideriamo una trasformazione isoterma di 0,1 moli di He alla T di 298,15 K da uno stato 1 (P1= 2,40 atm) ad uno stato 2 (P2 = 1,30 atm). L = F∙s = Pest∙A∙h= PDV = Pest (Vfin-Viniz) nRT Viniz 1,02 L Piniz nRT Vfin 1,88 L Pfin J L 1,30 (atm) (1,88 1,02 )( L) 101 110 J L atm Trasformazioni ed Energia Realizziamo la medesima trasformazione introducendo uno stato intermedio 2 1 Viniz 1,02 L Vint erm 1,36 L Vfin 1,88 L L L1 L 2 1,80 1,36 1,02 1,30 1,88 1,36 101 130 J Poiché DU= Q-L deve essere costante è evidente che l’aumento di L determina un aumento di Q Trasformazioni ed Energia Per una trasformazione che avviene a P costante la principale forma di lavoro può essere di contrazione (-L) o espansione (+L) L = PDV DU Q PDV Se la trasformazione avviene invece a volume costante (recipiente ermeticamente chiuso) PDV 0 DU Q v La variazione di energia interna corrisponde alla quantità di calore scambiata a volume costante Trasformazioni ed Energia Per una trasformazione a P costante si definisce entalpia H del sistema la somma dell’energia interna U e l’energia meccanica PV Come U anche H è una funzione di stato di cui non si conosce il valore assoluto H U PV Per una trasformazione da uno stato 1 (stato iniziale) ad uno stato 2 (stato finale) che avviene a P costante (generalità dei casi) DH H 2 H1 (U 2 PV2 ) (U1 PV1 ) DU PDV Poiché dal I° principio della temodinamica DU PDV Q p DH Q p La variazione di entalpia corrisponde alla quantità di calore scambiata a pressione costante Quando a P costante la trasformazione avviene senza variazione di volume DH DU PDV Qp Qv Trasformazioni ed Energia Trasformazioni che avvengono senza variazione di volume: H2(g) + I2(g)→2HI(g) N2(g) + O2(g)→2NO (g) Trasformazioni che avvengono con variazione di volume: •2H2(g) + O2(g)→2H2O (l) •CaCO3(s) →CaO(s) + CO2(g) •H2O(l) →H2O(g) •I2(s)→I2(g) Trasformazioni ed energia DU Q v DH Q p Q m c p (Tfin Tiniz ) Bomba calorimetrica Nella bomba calorimetrica si possono misurare solo Qv poichè le trasformazioni sono condotte in recipienti a V costante Qv +Qass(ceduto )H2O + Qass(ceduto)bomba=0 Esempio: 1,85 g di grafite vengono bruciati con eccesso di O2 in una bomba calorimetrica di acciaio avente una massa pari a 2,835 kg immersa in un recipiente perfettamente isolato contenete 3,20 kg di H2O. Un termometro immerso nell’acqua indica, dopo la reazione di combustione, un aumento di temperatura di 4,12 °C. Calcolare il DU e il DH della reazione di combustione in kcal. (cpacciaio= 0,114 cal g-1 °C-1; cpH2O(l) = 1,00 cal g-1 °C-1 ) C(s) + O2(g) CO2(g) La reazione è esotermica (-Q) come dimostra l’aumento di temperatura misurata nell’acqua per effetto del trasferimento del calore sviluppato nella reazione Qv +Qass(ceduto )H2O + Qass(ceduto)bomba=0 Qv = -QassorbitoH2O - Qassorbito bomba Q assorbitoH2O m ( H 2O ) c p ( H 2O ) (Tfin Tiniz ) Q assorbitobomba m ( bomba ) c p ( bomba ) (Tfin Tiniz ) kcal kcal 4,12(C) 2.835(kg) 0,114 4,12(C) 14,5kcal Q v 3,20(kg) 1,00 kg C kg C DU Q v 14,5kcal DH DU PDV C(s) + O2(g) CO2(g) Il lavoro PDV che il sistema può compiere è legato alla formazione e alla scomparsa delle specie gassose DH DU DnRT dove Dn = (ngas prodotto-ngas reagente) Poichè Dn = (nCO2-nO2) =0 DH DU 14,5kcal Esempio: Nella combustione del propano in una bomba calorimetrica DU =-2200kJ per mole di propano bruciato. Calcolare il DH della reazione di combustione alla temperatura di 25 °C e alla P = 1 atm. C3H8(l) + 5O2(g) 3CO2(g) + 4H2O(l) DH DU PDV DH DU DnRT Se la reazione venisse effettuata in un recipiente aperto il lavoro possibile è un lavoro di compressione poichè Dn = (nCO2-nO2) =3-5 =-2 J L atm J R 0,0821 101 8,31 mole K mole K L atm 8,31 kJ kJ kcal DH 2200 2(moli ) 298,15(K ) 2205 1000 mole K mole mole Spontaneità delle trasformazioni Il primo principio della termodinamica non pone limitazioni ai trasferimenti di energia purché sia rispettato il principio di conservazione. Pertanto non è possibile prevedere, con il principio di conservazione di energia, la direzione in cui una trasformazione chimica o fisica avviene spontaneamente ovvero senza alcun intervento esterno. Una trasformazione spontanea è anche irreversibile Le Transizioni di stato- Spontaneità Condensazione o liquefazione solidificazione brinamento DHfus >0 → DHsolidif <0 ← DHevaporazione >0 → DHcondensaz <0 ← Cosa causa l’espansione di un gas a T costante? Q0 DU D(PV ) Espansione isoterma Pa Va Pb Vb 3 Pa Va kT 2 L’espansione non è causata dal passaggio del sistema ad uno stato di minore energia Una descrizione più funzionale della causa determinate l’espansione è che per la stessa energia totale sono disponibili, nel maggior volume più livelli di energia traslazionale nei quali possono distribuirsi le molecole del gas. 3 Pb Vb kT 2 D(PV ) 0 DU 0 DH 0 Processi reversibili e irreversibili Processo reversibile quando le variabili di stato del sistema (volume, pressione) variano solo di una quantità infinitesima. Es: espansione reversibile di un gas Pi=Pest +dP Un processo reversibile può essere invertito in ogni momento tramite un cambiamento infinitesimo delle proprietà dell’ambiente. Processo irreversibile quando le variabili di stato del sistema variano di una quantità finita. Es: espansione irreversibile di un gas Pi=Pest +DP Un processo irreversibile non può essere invertito o arrestato e procede spontaneamente in una direzione. Processi reversibili e irreversibili Lavoro in un processo reversibile: Es: espansione reversibile di un gas Pi=Pest +dP dLrev = Pest ∙dV V2 L rev Pest dV Lrev V1 Poichè Pi ~ Pest V2 L rev L rev V2 V2 nRT dV Pi dV dV nRT V V1 V1 V1 V V2 nRT ln V1 Processi reversibili e irreversibili Lavoro in un processo irreversibile: Es: espansione irreversibile di un gas Pi=Pest +DP Lirrev V2 V2 V1 V1 L irrev Pest dV P2 dV P2 (V2 V1 ) Lrev > Lirrev Processi reversibili e irreversibili Calore in un processo reversibile e irreversibile: Processo reversibile Processo irreversibile Qirrev =DU+ Lirrev Qrev =DU+ Lrev Qrev - Qirrev = Lrev - Lirrev Poichè Lrev-Lirrev >0 Qrev-Qirrev >0 Qrev > Qirrev Entropia- Spontaneità La proprietà termodinamica collegata al modo in cui l’energia interna di un sistema è distribuita su livelli di energia microscopici è detta entropia Maggiore è il numero di configurazioni delle particelle microscopiche (atomi, ioni, molecole) tra i livelli di energia di un particolare sistema e maggiore è l’entropia del sistema Stato di disordine maggiore probabilità Stato di ordine minore probabilità La funzione di stato entropia S descrive quantitativamente la tendenza al disordine (maggiore probabilità di stato) di un sistema e un aumento di entropia accompagna i processi spontanei Equazione di Boltzmann per l’ Entropia L’entropia è correlata al numero di stati microscopici che lo descrivono secondo l’equazione: S = K ln W dove K è la costante di Boltzman (1,3807 10-23 J/K) W è il numero di stati microscopici del sistema. MAGGIORE E’ IL NUMERO DI STATI MICROSCOPICI MAGGIORE E’ LA PROBABILITA’ D’ ESISTENZA DEL SISTEMA STESSO L’entropia è una funzione di stato Previsioni qualitative delle variazioni di entropia E’ inoltre prevedibile una variazione anche per le trasformazioni chimiche; 2NH4NO3(s) 2SO2(g) + O2(g) CO(g) + H2O(g) 2N2(g) + 4 H2O(g) + O2 (g) 2SO3(g) CO2(g) + H2(g) DS=Sprodotti- S reagenti >0 DS=Sprodotti- S reagenti <0 DS= incerta Entropia di un sistema Se un sistema subisce una trasformazione isoterma, reversibile e infinitesima durante la quale scambia la quantità di calore dQrev con l’ambiente H2O(l) (0°C, 1 atm) H2O(s) (0°C, 1atm) il suo contenuto di entropia varia di una quantità infinitesima pari a : dQrev dS T Per una trasformazione tra lo stato 1 e lo stato 2 la variazione finita di entropia sarà: dQrev Qrev DS S 2 S1 dS T 1 1 T 2 2 cal K-1 Entropia di un sistema Se un sistema subisce una trasformazione irreversibile H2O(l) (0°C, 1 atm) H2O(s) (0°C, 1atm) Q irrev Q rev Q irrev Q rev Q irrev DS T T Disuguaglianza di Clausius In base a questi risultati in una trasformazione irreversibile si otterrebbe una quantità più piccola delle effettiva variazione di entropia del sistema Se il sistema si evolve attraverso una trasformazione irreversibile la variazione di entropia che esso subisce si calcola lungo il cammino di una qualunque trasformazione reversibile arbitrariamente scelta che consideri i medesimi stato iniziale e finale II° principio della termodinamica: Tutti i processi spontanei e irreversibili producono un aumento di entropia dell’universo DStotale= DS+DSamb>0 DS>0 DS<0 DS>0 DS<0 Stato iniziale Stato finale T 0< T T Per il sistema Sf < Si Per l’ambiente Sambf > Sambi Supponendo che Q scambiato non faccia variare T0 DS Q / T DS totale DSamb Q / To 1 Q Q 1 Q 0 T To T To Tanto più grande è T-T0 ossia tanto più ci si scosta dalle condizioni di reversibilità tanto maggiore è l’aumento di entropia; Se invece T-T0 tende a zero (trasformazione reversibile) la somma delle variazioni di entropia fra sistema e ambiente è zero Pertanto per valutare se un processo è spontaneo occorre valutare: - La variazione entropica del sistema - La variazione entropica dell’ambiente - La somma delle due Una semplificazione del problema si ottiene definendo una nuova funzione di stato “ENERGIA LIBERA” che ci permette di valutare: -Se un processo è spontaneo -Come un cambiamento di temperatura possa condizionare il carattere spontaneo di un processo Energia Libera di Gibbs Per un processo che avviene a T e P costante DStotale DS DSamb Q DS totale DS rev T TDStotale TDS Q rev TDStotale TDS DH TDStotale DH TDS Q rev DH Definita l’ Energia libera di Gibbs G come: G= H-TS G è una funzione di stato DG = DH-TDS DStotale >0 DG <0 DG= -TDStotale DStotale =0 DG =0 DStotale <0 DG >0 Trasformazione irreversibile o spontanea Trasformazione reversibile o all’equilibrio Trasformazione non spontanea Spontaneità ed effetto della Temperatura DG = DH-TDS DH>0 e DS>0 Solido DH< 0 e DS<0 spontanei ad alta temperatura Liquido Vapore o gas spontanei a bassa temperatura