Universitá del Salento
FACOLTÀ DI SCIENZE MATEMATICHE, FISICHE E NATURALI
Corso di Laurea Triennale in Fisica
Dall’entropia termodinamica
all’entropia di entanglement
Candidato:
Relatore:
Francesco Foggetti
Prof. Luca Girlanda
Anno Accademico 2012–2013
Indice
1 Introduzione
3
2 L’entropia termodinamica
2.1 L’entropia in senso macroscopico . . . . . . .
2.2 L’entropia in senso microscopico . . . . . . .
2.2.1 Legge dell’aumento dell’entropia . . .
2.3 Entropia di Gibbs ed Entropia di Boltzmann
2.4 Entropia e Disordine . . . . . . . . . . . . . .
2.4.1 Il paradosso di Gibbs . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
4
4
7
9
10
14
16
3 L’entropia quantistica
3.1 Discretizzazione dell’energia e ipotesi di Planck . . . .
3.2 Matrice densità . . . . . . . . . . . . . . . . . . . . .
3.3 Entropia di Von Neumann . . . . . . . . . . . . . . . .
3.4 Entanglement quantistico ed entropia di entanglement
3.5 Entropia e tempo . . . . . . . . . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
18
18
20
24
26
29
4 Entropia ed informazione
4.1 Entropia di Shannon e compressione di informazione
4.2 Proprietà dell’entropia . . . . . . . . . . . . . . . . .
4.2.1 Entropia Binaria . . . . . . . . . . . . . . . .
4.2.2 Entropia relativa . . . . . . . . . . . . . . . .
4.2.3 Mutua informazione . . . . . . . . . . . . . .
4.3 Teoremi di Shannon . . . . . . . . . . . . . . . . . .
4.4 Il diavoletto di Maxwell e il principio di Landauer. .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
33
33
34
34
34
35
35
37
2
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
Capitolo 1
Introduzione
Questa tesi si presenta in tre parti fondamentali, l’entropia nella termodinamica, nella meccanica quantistica e infine nella teoria dell’informazione:
nella prima parte riprendiamo la definizione classica di entropia[1] che incontriamo nei primi corsi di fisica richiamando alcuni risultati già noti. Entriamo
inoltre nel campo della meccanica statistica[2] analizzando con il suo formalismo le caratteristiche dei sistemi macroscopici. Infine in questa parte proviamo
ad esplicitare il legame tra entropia e disordine ed analizziamo il paradosso di
Gibbs riguardante l’entropia di miscelamento;
nella seconda parte, oltre ad introdurre alcuni concetti chiave della meccanica quantistica, affrontiamo il problema di definie l’entropia a partire dalla
conoscenza che possiamo avere dei sistemi quantistici in termini di funzione
d’onda o di matrice densità provando anche a chiarire il legame tra entropia
quantistica ed entanglement tra le particelle di un sistema: Definiamo inoltre
una possibile misura per quantificare il grado di entanglement di un sisetma
composto; infine analizziamo il legame tra l’ entropia ed il tempo citando il
paradosso di Loschmidt ed interpretando i risultati ottenuti;
la terza parte comprende i concetti fondamentali della teoria dell’informazione e si pone il problema dell’immagazinare l’informazione in sistemi fisici;
anche da questo punto di vista l’entropia “d’informazione” risulta essere un indicatore fondamentale della nostra possibilità di immagazzinare informazione
in un sistema fisico. La tesi si conclude ricollegando i risultati ricavati nell’ambito della termodinamica e della teoria dell’informazione all’interno della
risoluzione del paradosso del “diavoletto di Maxwell”.
3
Capitolo 2
L’entropia termodinamica
2.1
L’entropia in senso macroscopico
Supponiamo di avere un certo sistema soggetto ad una trasformazione ciclica. Supponiamo che tale trasformazione avvenga scambiando calore con una
serie di n termostati le cui temperature sono T1 , T2 , ... Tn .
Siano Q1 , Q2 , ...Qn i calori ceduti o assorbiti dai rispettivi termostati con segno
positivo se il calore è assorbito dal sistema e negativo se è ceduto dal sistema.
Introduciamo un altro termostato a temperatura T0 ed n macchine termiche
reversibili tali da avere n cicli di Carnot C1 , C2 , ...Cn ; nel ciclo Ci viene assorbito il calore −Qi alla temperatura Ti e ceduto il calore Qi0 alla teneratura T0 .
Risulta quindi nell’i -esimo ciclo
Qi0
T0
=− .
Qi
Ti
(2.1)
Il sistema complessivo (macchine termiche incluse quindi) scambierà calore solo
con il termostato a temperatura T0 . Il calore assorbito complessivamente sarà
quindi
n
n
X
X
Qi
.
(2.2)
Q0 =
−Qi0 = T0
T
i=1
i=1 i
Inoltre poiché il sistema e le macchine reversibili considerate tornano tutti nel
loro stato iniziale l’unico risultato del ciclo è la trasformazione del calore scambiato in lavoro, tale calore non può che essere negativo, altrimenti contradirebbe
il secondo principio della termodinamica nella formulazione di Kelvin, risulta
quindi
n
X
Qi
≤ 0.
(2.3)
T
i=1 i
L’uguaglianza vale se il ciclo cui è sottoposto il sistema è reversibile, se cosı̀
fosse infatti potremmo applicare il ragionamento precedente procedendo dallo
stato finale a quello iniziale e otterremmo
n
X
i=1
−
Qi
≤ 0,
Ti
4
(2.4)
2.1. L’ENTROPIA IN SENSO MACROSCOPICO
cioé
n
X
Qi
i=1
Ti
≥ 0;
5
(2.5)
poiché deve valere sia la (2.5) che la (2.3) deve essere valida necessariamente
l’uguaglianza.
n
X
Qi
= 0.
(2.6)
T
i=1 i
L’equazione si generalizza al caso in cui avvengano scambi di calore infinitamente piccoli δQ tra il sistema e la sorgente di calore a temperatura T
I
δQ
≤ 0,
(2.7)
T
valida in ogni caso, in particolare nel caso di trasformazione reversibile
I
δQ
= 0.
T
(2.8)
Il risultato che abbiamo ottenuto è generalmente noto come “Teorema di Clausius”.
Sottoponiamo ora il nostro sistema ad una trasformazione ciclica che porta
il sistema dal suo stato A nello stato B, per poi riportarlo di nuovo in A
−
attraverso due differenti cammini reversibili →
γ1 (rosso) e ←
γ−2 (nero):
Figura 2.1: Generica trasformazione ciclica nel piano di Clapeyron
sappiamo che
I
δQ
= 0,
T
(2.9)
−
l’integrale può essere scomposto nell’integrale sui due cammini →
γ1 e ←
γ−2
Z
Z
δQ
δQ
+
=0
(2.10)
−
→
←
−
γ1 T
γ2 T
6
CAPITOLO 2. L’ENTROPIA TERMODINAMICA
essendo reversibili possono essere percorsi al contrario:
Z
Z
δQ
δQ
=−
,
←
−
−
→
T
γ2
γ2 T
da cui
Z
−
→
γ
1
δQ
=
T
Z
−
→
γ
2
δQ
.
T
(2.11)
(2.12)
L’integrale tra gli stati A e B non dipende dal cammino percorso; tale quantità,
funzione solo degli stati tra cui è calcolato l’integrale, definisce la varizione
d’entropia del nostro sistema tra gli statiA e B. Dalla nostra definizione segue
che l’entropia di un certo stato è definita a meno di una costante additiva e che
è la differenza di entropia tra due stati ad essere univocamente determinata,
inoltre date le proprietà di linearità dell’integrale l’entropia risulta essere una
grandezza estensiva, cioè è una grandezza additiva del sistema, si può calcolare
sommando i contributi a tale grandezza dati da ogni parte del sistema stesso.
Mostriamo ora un’altra importante proprietà dell’entropia, supponiamo di
sottoporre il sistema che studiamo ad una trasformazione ciclica irreversibile,
il sistema procede dal suo stato A a B in maniera irreversibile, poi però si porta
da B ad A in maniera reversibile come mostrato in fig.2.2.
Figura 2.2: trasformazione irreversibile
Vale l’equazione
I
δQ
≤0
T
(2.13)
che si scompone nei termini
Z
irr
δQ
+
T
Z
A
B
δQ
≤ 0,
T
ma per l’integrale calcolato sul cammino reversibile vale la relazione
Z A
δQ
= S(A) − S(B)
T
B
(2.14)
(2.15)
2.2. L’ENTROPIA IN SENSO MICROSCOPICO
7
quindi
Z
S(B) − S(A) ≥
irr
δQ
.
T
(2.16)
In particolare per un sistema isolato δQ = 0 per cui risulta S(B) ≥ S(A), cioè
l’entropia di un sistema non può mai diminuire.
2.2
L’entropia in senso microscopico
Consideriamo un sistema in equilibrio formato da un gran numero di particelle, il loro moto è casuale e non ci sono direzioni privilegiate in cui possono
muoversi. Supponiamo di dividere il sistema in un gran numero di più piccoli
sottosistemi e consideriamone uno qualunque tra di essi, tale sottosistema sarà
il nuovo sottosistema oggetto di studio. Sia ω la funzione di distribuzione del
sistema in esame nello spazio delle fasi, a partire dal teorema di Liouville
∂ω
= {H, ω}
∂t
(2.17)
si dimostra che ω può essere espressa come combinazione linerare delle costanti
del moto del sistema, in particolare essa può essere espressa come funzione della
sola energia E del sistema[2]; nota ω(E) possiamo quindi ottenere la probabilità W (E) che l’energia del sistema appartenga all’intervallo [E, E + dE];
per ottenere tale probabilità ci basta moltiplicare la ω(E) per il numero di stati
del sistema la cui energia appartiene all’intervallo considerato.
Detto dΓ(E) tale numero, risulta:
W (E)dE = dΓ(E)ω(E),
(2.18)
essendo Γ(E) la funzione che ad un valore di E associa il numero di stati del
sistema considerato; risulta dΓ(E) = dΓ(E)
dE dE, quindi:
dΓ(E)
ω(E),
dE
R
vale inoltre la condizione di normalizzazione W (E)dE = 1.
W (E) =
(2.19)
È possibile dimostrare che per sistemi come quello considerato, composti da
un gran numero di particelle (limite termodinamico), la funzione W (E) è fortemente piccata attorno al suo massimo E = Ē, detto perciò ∆E l’intorno di
Ē in cui W (E) è non nulla e per cui vale
W (E)∆E = 1
(2.20)
ω(E)∆Γ = 1
(2.21)
dΓ
∆Γ
=
∆E
dE
(2.22)
otteniamo:
dove
8
CAPITOLO 2. L’ENTROPIA TERMODINAMICA
Osserviamo che ∆Γ corrisponde al numero di stati microscopici associati all’intervallo d’energia ∆E il cui ordine di grandezza è quello delle fluttuazioni
dell’energia che avvengono nel sistema. Per poter contare questi stati è necessario discretizzare lo spazio delle fasi introducendo la cella elementare nello
spazio delle fasi:
∆p∆q
(2.23)
∆Γ =
(2πh̄)ν
dove ν è il numero di gradi di libertà del sistema. Questa operazione oltre
che ad essere un artificio matematico è ben motivata fisicamente dall’ esistenza
del quanto elementare di azione h. Possiamo infine chiamare la grandezza ∆Γ
peso statistico del sistema; la relazione che lega entropia termodinamica a ∆Γ
risulta, come sarà discusso nel prossimo capitolo:
S = k log ∆Γ = k log
∆p∆q
.
(2πh̄)ν
(2.24)
Un’osservazione che è necessario fare è che l’argomento del logaritmo è una
quantità adimensionale, se non fossimo partiti dal peso statistico ∆Γ ma avessimo definito l’entropia a partire dal volume nello spazio delle fasi ∆p∆q l’argomento del logaritmo avrebbe avuto la dimensione di un’azione, quindi al
cambiare di sistema di unità di misura nell’espressione dell’entropia dovrebbe
comparire una costante additiva diversa da zero, infatti l’entropia termodinamica classica (macroscopica) è definita a meno di una costante. Il valore di
questa costante nella formulazione microscopia è determinato nell’ambito della
meccanica quantistica.
Ricordiamo ora che avevamo considerato solo un sottositema di un più grande
sistema isolato, il nostro obiettivo è quindi calcolarne l’entropia; consideriamo
ora un n-esimo sottosistema del sistema di partenza, dallo studio delle caratteristiche di tali sistemi, detta ω(En ) la funzione di distribuzione del sottosistema,
risulta[2]:
log ω(En ) = α + βEn
(2.25)
con α e β coefficienti reali; essendo tale espressione lineare
< log ω(En ) >= log ω(E n ) = α + βE n ,
(2.26)
il simbolo < > indica il valore medio della grandezza; ricordando che ω(E)∆Γ =
1 l’entropia del sistema risulta:
Sn = − log ω(E n )
(2.27)
analogamente l’entropia totale:
S = − log ω(E)
(2.28)
per la definizione di valor medio risulta:
S=−
X
n
ωn log ωn
(2.29)
9
2.2. L’ENTROPIA IN SENSO MICROSCOPICO
Considerando ora l’intero sistema siano ∆Γ1 , ∆Γ2 , ...∆Γn i pesi statistici di
tutti i sottostistemi in cui è diviso, il peso statistico totale sarà
∆Γ =
Y
∆Γn ,
(2.30)
n
passando ai logaritmi risulta che l’entropia totale del sistema è una grandezza
additiva
X
S=
Sn .
(2.31)
n
2.2.1
Legge dell’aumento dell’entropia
Nell’analisi precedente abbiamo considerato un sistema in stato di equilibrio; supponiamo di avere un sistema non in stato di equilibrio, esso evolverà
nel tempo fino a raggiungere tale condizione, il sistema passerà da stati di minore probabilità a stati di maggiore probabilità. Il primo passo nello studiare
il comportamento di un sistema che si porta nello stato di equilibrio è richiamare la distribuzione microcanonica mediante la quale possiamo descrivere le
proprità statistiche di un sistema isolato
dω = costante δ(E − E0 )
Y
dΓn ,
(2.32)
n
dove dΓn è il differenziale della funzione Γ(En ) che fornisce il numero di stati
del sottosistema con energia compresa nell’intervallo [En , En + dEn ]. Posso
riscrivere l’espressione:
dω = costante δ(E − E0 )
Y dΓn
dEn
dEn
n
(2.33)
∆Γn
dΓn
= ∆E
e ricordando la definizione di entropia in
nell’approssimazione che dE
n
n
S
termini
Q 1 di peso statistico risulta e = ∆Γ, inoltre la dipendenza del termine
n ∆En da E può essere trascurata rispetto alla dipendenza esponenziale del
termine eS (E), per cui può essere inglobato nella costante, quindi:
dω = costante δ(E − E0 )eS
Y
dEn
(2.34)
n
La probabilità di un certo stato è perciò legata all’entropia del sistema, intoltre, poiché il sistema tende a variare da stati di minore probabilità a stati di
maggiore probabilità ed essendo la dipendenza esponenziale in S possiamo dire
che i processi che avvengono in un sistema che raggiunge l’equilibrio fanno sı̀
che esso passi in modo continuo da stati ad entropia minore a stati ad entropia
maggiore finché un equilibrio non viene raggiunto e l’entropia raggiunge il suo
valore massimo.
Abbiamo cosı̀ espresso la legge dell’aumento dell’entropia, essa afferma che
l’evoluzione più probabile di un sistema avviene in stati successivi in cui l’entropia cresce sempre o, al limite, resta invariata.
Bisogna osservare che la questa legge, parlando di conseguenza più probabile, considera che la probabilità che S aumenti è incredibilmente più elevata
10
CAPITOLO 2. L’ENTROPIA TERMODINAMICA
rispetto alla probabilità che diminuisca. Altro elemento da puntualizzare è l’applicazione della suddetta legge al mondo circostante e all’universo in generale;
secondo tale legge l’universo, e ogni sua parte macroscopica, dovrebbe trovarsi
in uno stato di equilibrio dopo un intervallo di tempo sufficientemente lungo. Senza introdurre argomentazioni troppo formali possiamo affermare che,
citando la relatività generale, considerando le enormi dimensioni dell’universo non possiamo trascurare gli effetti gravitazionali che influenzano la metrica
spazio temporale dell’universo stesso, essendo l’universo in espansione tale metrica varia nel tempo, quindi i campi gravitazionali possono considerarsi come
condizioni esterne non stazionarie per l’universo. La legge dell’aumento dell’entropia non si traduce quindi nel raggiungimento immediato dell’equilibrio
del sistema universo.[3]
2.3
Entropia di Gibbs ed Entropia di Boltzmann
La definizione di entropia che fin’ora abbiamo utilizzato è dovuta a Gibbs.
Storicamente[4] si è data un’altra definizione per la stessa grandeza, dovuta a
Boltzmann, utilizzata per la formulazione del suo teorema H.
Considerando un gas monoatomico di N particelle la densità di probabilità
WN nello spazio delle fasi risulta:
WN = WN (x1 , p1 , ...xn , pn ; t);
la funzione HG introdotta da Gibbs è definita come
Z
HG = WN log WN dτ
(2.35)
(2.36)
dove dτ raccoglie tutte le coordinate della spazio delle fasi, mentre la corrispondente funzione HB di Boltzmann è definita mediante la probabilità di singola
particella,
wi = wi (xi , pi ; t),
(2.37)
come
Z
HB = N
w1 log w1 dτ1 .
(2.38)
dτ1 si riferisce solo le coordinate della particella 1 che è equivalente a tutte le
altre N − 1 particelle.
In particolare risulta:
Z
Z
3
3
3
3
w1 (x1 , p1 ; t) = WN d x2 d p2 ...d xn d pn = WN dτ−1
(2.39)
con dτ−1 = d3 x2 d3 p2 ...d3 xn d3 pn .
Sia w1 che WN sono normalizzate ad 1, poiché WN è simmetrica rispetto a
permutazioni delle particelle, ricordando la forma di w1 dalla (2.39) possiamo
riscrivere la HB come
Z
HB = N WN log w1 (x1 , p1 )dτ
Z
=
WN log[w1 (x1 , p1 )w2 (x2 , p2 )...wn (xn , pn )]dτ,
(2.40)
2.3. ENTROPIA DI GIBBS ED ENTROPIA DI BOLTZMANN
11
ed essendo le varie distribuzioni di singola particella equivalenti risulta
Z
w1 (1)...w1 (N )
HB − HG = WN log
dτ.
(2.41)
WN (1, ..., N )
con (xi , pi ) = i.
Ora, essendo w e W distribuzioni positive, per le proprietà dei logaritmi risulta
che log x ≤ x − 1 per cui risulta
Z
w1 (1)...w1 (N )
− 1 dτ
(2.42)
HB − HG ≤ WN
WN
segue quindi che
HB ≤ HG ,
(2.43)
dove l’uguaglianza vale se WN = w1 , ...wN , cioè se WN è fattorizabile. La
differenza HB −HG dipende dalla funzione di distribuzione del sistema. Siamo
interessati al caso dell’equilibrio termico, in questo caso la distribuzione è quella
canonica
WN ∝ e−βH ,
(2.44)
dove β = 1/kT e H è l’hamiltoniana del sistema
H=
N
X
p2i
+ V (x1 , ...xN ),
2m
i=1
(2.45)
con V funzione simmetrica rispetto alle coordinate delle particelle. Detta Z
una costante di normalizzazione tale che
Z
Z = Z(β, Ω) = Ω e−βV d3 x2 ...d3 xN
(2.46)
con Ω volume ottenuto integrando sulle coordinate del centro di massa, essa è
legata alla energia libera di Helmotz F dalla relazione
1
= eβF .
Z
(2.47)
Risulta quindi
WN =
β
2πm
3N
2
Z −1 e−β
p2
i
i=1 2m −βV
PN
,
(2.48)
otteniamo la distribuzione per singola particella
w1 (x, p) =
β
2πm
3/2
p2
Ω−1 e−β 2m
(2.49)
sostituendo quest’equazione nella precedente otteniamo
w1 (1)...w1 (N )
= ZΩ−N eβV ,
WN (1...N )
(2.50)
HB − HG = log Z − N log Ω + β < V >
(2.51)
da cui
12
CAPITOLO 2. L’ENTROPIA TERMODINAMICA
dove < V > indica la media di V calcolata sulla distribuzione canonica, infatti
Z
WN βV dτ = β < V > .
(2.52)
Ricaviamo ora due relazioni ben note dell’ensemble canonico; il valor medio di
V
Z
< V >= WN V dτ
(2.53)
utilizzando la forma esplicita di WN risulta
< V >=
β
2πm
32 N
1
Z
Z
e−β
p2
i
i=1 2m −βV
PN
V dτ.
(2.54)
Integrando la parte dipendente solo dagli impulsi e ricordando che V non
dipende dalle coordinate del centro di massa ottengo
Z
Ω
< V >=
e−βV d3 x2 ...d3 xN ;
(2.55)
Z
ora considero la quantità log Z e ne calcolo la derivata in β,
Z
∂ log Z
1 ∂Z
Ω
=
=−
e−βV d3 x2 ...d3 xN ;
∂β
Z ∂β
Z
per cui:
< V >= −
(2.56)
∂ log Z
.
∂β
(2.57)
∂ log Z
.
∂Ω
(2.58)
Dall’equazione (2.47) si deduce inoltre
β < P >=
Adesso, sfruttando le equazioni appena ricavate, studiamo la differenza HB −
HG per cambi di stati infinitesimi
d(HB − HG ) = β < P > dΩ− < V > dβ −
N
dΩ + βd < V > + < V > dβ;
Ω
(2.59)
inoltre
N
= βP0 ,
(2.60)
Ω
con P0 pressione del gas ideale con volume e temperature pari a quella del
nostro sistema; risulta quindi
d(HB − HG ) = βd < V > +β(< P > −P0 )dΩ.
(2.61)
Possiamo introdurre le entropie S associate alle rispettive funzioni H
S = −kH,
(2.62)
con k costante di Boltzmann e integrando la (2.61) su un cammino reversibile.
Z 2
d < V > +(< P > −P0 )dΩ
(SG − SB )2 − (SG − SB )1 =
(2.63)
T
1
2.3. ENTROPIA DI GIBBS ED ENTROPIA DI BOLTZMANN
13
Dal momento che la forma di w1 è nota è possibile ricavare l’entropia di
Boltzmann
3
3
(2.64)
SB = N k log(2πmkT ) + N K log Ω + N k.
2
2
Ricavo da questa espressione che:
∂SB
3 N kdT
d<K>
dT =
=
(2.65)
∂T Ω
2 T
T
Nk
P0 dΩ
∂SB
=
dΩ =
(2.66)
∂Ω T
Ω
T
dove < K >= 3/2N kT è l’energia cinetica del sistema; integrando sullo stesso
cammino reversibile otteniamo:
Z 2
d < K > +P0 dΩ
(2.67)
(SB )2 − (SB )1 =
T
1
e per differenza con l’equazione (2.63) otteniamo l’entropia di Gibbs,
Z
(SG )2 − (SG )1 =
1
2
d < K + V > + < P > dΩ
.
T
(2.68)
essendo Ω il volume e K + V l’energia totale del sistema, invocando il primo
principio della termodinamica risulta
Z 2
δQ
.
(2.69)
(SG )2 − (SG )1 =
T
1
Quindi dalla nostra analisi emerge che l’entropia di Boltzmann è l’entropia di
un gas con le stesse caratteristiche del nostro sistema ma che non tiene conto delle forze intermolecolari delle particelle, mentre l’entropia di Gibbs che
ne tiene conto è l’entropia sperimentalmente osservata che si riconduce all’entropia come definita macroscopicamente da Clausius; infine possiamo affermare che la differenza tra i due tipi di entropie non è trascurabile se gli effetti
delle forze intermolecolari del sistema hanno effetti osservabili sulle proprietà
termodinamiche del sistema.
14
2.4
CAPITOLO 2. L’ENTROPIA TERMODINAMICA
Entropia e Disordine
Descriviamo adesso con un esempio come il concetto di entropia si ricolleghi
al concetto di disordine e quale legame ci sia tra i due. Consideriamo un
sistema molto semplice, formato da poche particelle distinguibili, ad esempio
4. Il sistema è posto inizialmente in un contenitore costituito da due cavità
separate da un setto removibile e completamente concentrato in una delle due
cavità (a).
Figura 2.3:
Alla rimozione del setto le particelle spontaneamente diffonderanno verso la
cavità (b) fino a raggiungere l’equilibrio, in questo caso rappresentato da due
particelle per cavità; vedremo inoltre che a questa configurazione del sistema è
anche associata l’entropia maggiore.
Figura 2.4:
Osserviamo come tale configurazione sia la più probabile semplicemente
elencando tutte le possibili configurazioni come mostriamo in tabella 2.1.
15
2.4. ENTROPIA E DISORDINE
a)
4
3
2
1
0
b)
0
1
2
3
4
n.combinazioni
1
4
6
4
1
probabilità
1/16
4/16
6/16
4/16
1/16
Tabella 2.1: configurazioni e probabilità
La configurazione in cui le molecole sono equi-distribuite è la più probabile
ed è lo stato d’equilibrio ad entropia maggiore cui il sistema tende; infatti,
come visto in precedenza, l’entropia è proporzionale al logaritmo del numero
dei microstati, e dove le particelle sono equidistribuite il numero dei microstati
del sistema considerato è massimo.
Più in generale fissato il numero totale N di particelle, supposto molto grande, sia m il numero di particelle nella cavità (a); Il numero di possibili combinazioni di m particelle prese tra N totali risulta essere dato dal coefficiente
binomiale
N!
N
=
.
(2.70)
m
m!(N − m)!
Vogliamo ora ricavare il valore di m per cui tale numero di combinazioni, e
con esso la probabilità che il sistema si trovi in quello stato, è massimo. È più
semplice studiare il logaritmo di questa quantità ed esprimere il comportamento
dei fattoriali mediante la formula di Stirling:
1
1
log n! ' n +
(2.71)
log n − n + log 2π,
2
2
tale approssimazione è tanto più accurata quanto più è grande il numero n
considerato. Nel nostro caso, trattandosi di un sistema macroscopico, N è un
numero dell’ordine del numero di Avogadro ed m, trattandosi del numero di
particelle dello stato più probabile, sarà anche esso un numero molto grande.
Ricordiamo che, essendo il logaritmo una funzione monotona, il massimo della
quantità originale, e quello del suo logaritmo coincidono.
Risulta quindi:
N
log
m
'
1
1
N+
log N − N − m +
log m + m
2
2
1
− N −m+
log(N − m) + (N − m)
2
(2.72)
dove abbiamo trascurato il contributo del termine log 2π;
Imponendo che la derivata in m sia nulla otteniamo
− log m + log(N − m) −
1
1
+
= 0,
2m 2(N − m)
(2.73)
e nell’ipotesi di N ed m molto grandi posso trascurare i termini senza i logaritmi, per cui ottengo
N −m
log
=0
(2.74)
m
16
CAPITOLO 2. L’ENTROPIA TERMODINAMICA
che è soddisfatta per m = N/2.
Abbiamo mostrato perciò che lo stato più probabile è quello in cui le particelle sono distribuite ugualmente tra le due cavità del recipiente. Il concetto di
disordine si ricollega a questo, inizialmente le particelle erano state tutte ordinate nella stessa parte del recipiente, tuttavia rimuovendo il setto separatore il
sistema ha avuto la possibilità di evolvere spontaneamente, si è portato quindi ad uno stato ad entropia maggiore, in cui tutte le molecole erano disposte
casualmente tra le due cavità, in uno stato visibilmente più disordinato senza
alcuna imposizione specifica sul dove tali particelle dovessero essere.
Va fatto notare che, preso un certo sistema in equilibrio, è possibile, spendendo energia, riportarlo in uno stato di ordine a minore entropia, tuttavia, il
bilancio totale dell’entropia sistema-ambiente risulta comunque in positivo.
2.4.1
Il paradosso di Gibbs
Un altro argomento da discutere in relazione al legame tra entropia e disordine è il paradosso di Gibbs. Tale paradosso si riferisce al mescolamento di
due gas alla stessa temperatura e pressione inizialmente separati da un setto
removibile.
Consideriamo come sistema isolato un contenitore di volume V separato da un
setto removibile in due sottovolumi V1 e V2 contenenti ciascuno un differente
gas con N1 e N2 particelle ma entrambi a temperatura T e pressione p. Rimuovendo il setto osserviamo l’espansione di entrambi i gas nel volume disponibile
e il reciproco mescolamento dei due fino a raggiungere uno stato di equilibrio.
Possiamo calcolare la variazione di entropia associata a questo processo a partire dalla variazione di entropia cui sono soggetti i due gas e sfruttando poi
l’additività dell’entropia. Sapendo che il mescolamento avviene a temperatura
costante, possiamo calcolare la variazione di entropia lungo una trasformazione
reversibile isoterma che collega gli stessi stati finale e iniziale, risulta
dS =
δQ
.
T
(2.75)
per gas ideali ∆E = 0 lungo trasformazioni isoterme, quindi per il primo
principio della termodinamica risulta
δL = δQ,
(2.76)
kN dV
pdV
=
T
V
(2.77)
essendo δL = pdV risulta
dS =
quindi, integrando tra gli stati iniziale e finale otteniamo:
- per il gas 1 ∆S1 = kN1 log
V
V1 ;
- per il gas 2 ∆S2 = kN2 log
V
V2 ;
2.4. ENTROPIA E DISORDINE
17
nel caso semplice di N1 = N2 = N e V1 = V2 = V risulta:
∆S = 2kN log 2.
(2.78)
È è importante osservare il significato di questo risultato, un’entropia di mescolamento diversa da zero indica che per riportare il sistema nello stato iniziale,
cioè per separare i due gas e riordinare il sistema, devo necessariamente compiere lavoro.
Altro elemento da puntualizzare è che nell’espressione finale dell’entropia il tipo
di gas in questione non fa diferenza ai fini del risultato. Considerando gas identici però esso sarebbe diverso, se infatti considerassi il sistema appena studiato
con l’unica differenza che in entrambi i vani del contenitore ci sia lo stesso gas
non ossererverei alcuna variazione di entropia tra stato finale ed iniziale.
Eppure ci aspettavamo un valore ben determinato di tale entropia, qual è il
problema?
Macroscopicamente questo risultato può essere inteso considerando che il sistema è sempre nello stesso stato, sia prima che dopo la rimozione del setto. I
valori delle quantità macroscopiche infatti non variano alla sua rimozione, inoltre trattandosi dello stesso gas se reinserissi il setto riotterrei lo stato iniziale
senza aver dovuto compiere lavoro, non è possibile infatti macroscopicamente
identificare le particelle che inizialmente si trovavano nell’uno o nell’altro vano.
Chiariamo ora perché si parli di paradosso considerando questo problema;
è assodato che al mescolamento di gas differenti è associata una variazione di
entropia non nulla, indipendente dalla natura dei gas, purché non identici. Se a
questo punto potessimo identificare genericamente le differenze tra i due gas e
fossimo in grado di modificarne uno in modo che tale differenza vari con continuità osserveremmo che la variazione d’entropia, finché i gas sono distinguibili
resta costante, ma che ad un certo punto cala bruscamente a zero non appena
essi non sono più distinguibili.
Matematicamente il paradosso risiede nel fatto che, detto λ il parametro che
quantifica la distinguibilità (o differenza) dei gas, al tendere con continuità di
λ a zero corrisponda una variazione discontinua della funzione entropia. Per
superare questa difficoltà concettuale occorre ricordare che l’entropia è legata
alla nostra conoscenza del sistema, supponendo di poter rendere sempre più
piccole le differenze tra i due gas e di poterle misurare per verificare se i gas
sono diventati indistinguibili o no ad un certo punto arriverei ad avere differenze cosı̀ piccole da non essere rilevate dai miei strumenti di misura, la mia
conoscenza del sistema in questo caso crolla bruscamente, per quanto ora posso
conoscere del sistema esso è composto di particelle indistinguibili, quindi la sua
entropia di mescolamento è nulla.
La soluzione del paradosso appare più chiara se considerata dal punto di
vista della meccanica quantistica, a stati macroscopicamente identici corrispondono stati microscopicamente identici; infatti quella che avevamo supposto essere una limitazione pratica nel distinguere i sistemi si rivela essere una
caratteristica intrinseca dei sistemi quantistici di particelle identiche.
Capitolo 3
L’entropia quantistica
3.1
Discretizzazione dell’energia e ipotesi di Planck
Uno degli elementi peculiari della meccanica quantistica è il concetto di
quanto per gli scambi di energia: diversamente che nel caso classico due sistemi che interagiscono possono scambiare una quantità di energia solo pari ad
un multiplo di una quantità fondamentale, appunto il quanto di energia. Si
dice cosı̀ che l’energia è scambiata in maniera discreta, contrariamente al caso
classico in cui è scambiata in maniera continua, cioè potendo assumere uno
qualunque dei possibili valori all’interno di un certo intervallo.
Il ruolo della discretizzazione può essere analizzato nel contesto della meccanica
statistica classica. Consideriamo infatti un sistema di N [5] particelle a volume
V fissato e immerso in un bagno termico a temepratura T . Ogni particella può
assumere i valori di energia compresi tra [0, ∞]. Vogliamo esprimere l’energia
e l’entropia del sistema in funzione della sua temperatura T nell’ambito ndell’ensemble canonico.
Risolveremo il problema sotto le due ipotesi di energia scambiata con continuità e di energia scambiata in maniera discreta.
Nel primo caso l’energia totale del sistema, detta k la costante di Boltzmann,
risulta essere:
R∞
E0
N 0 E 0 e− kT dE 0
E = R ∞ E0
(3.1)
e− kT dE 0
0
da cui
E = N kT.
(3.2)
È possibile a partire dall’energia ricavare l’entropia del sistema, essendo
∂S
dE = T dS − pdV risulta ∂T
= T1 ∂E
∂T quindi
S = N K log
e T0 compare come costante d’integrazione.
18
T
T0
,
(3.3)
3.1. DISCRETIZZAZIONE DELL’ENERGIA E IPOTESI DI PLANCK
19
Studiamo ora il caso in cui l’energia è scambiata per contributi discreti proporzionali ad una certa costante C. Quindi l’energia di ogni particella potrà
essere:
E = nC
con
n = 0, 1, 2...
(3.4)
La funzione di ripartizione del sistema risulta
Z=
∞
X
nC
e− kT ,
(3.5)
n=0
C
somma di una serie geometrica di ragione e− kT ; nota la funzione di ripartizione
si ricava l’energia libera:
F = −kN T log Z = −kN T log
1
C
1 − e− kT
,
(3.6)
da cui, ricordando le relazioni E = F − T S ed S = − ∂F
∂T , risulta:
E=
CN
e
C
kT
−1
C
ed S = −kN log(1 − e− kT ) −
1
NC
.
C
T e kT − 1
(3.7)
Osserviamo il comportamento delle quantità calcolate quando la temperatura
tende allo zero:
nel primo caso, mandando a zero la temperatura T , l’entropia diverge, nel
secondo caso invece l’entropia si annulla, proprio come atteso dalla terza legge
della termodinamica. Sempre nel secondo caso, supponendo di mandare a zero
la costante C per studiare il caso limite al continuo, osserviamo che l’energia
tende al valore kN T coerentemente al primo caso, mentre l’entropia diverge,
tuttavia è sempre possibile calcolare la differenza di entropia tra stati differenti.
Abbiamo mostrato con questo esempio che la discretizzazione dei microstati
accessibili al sistema gioca un ruolo determinante per definire un’entropia che
possegga le giuste proprietà termodinamiche. Nella meccanica quantistica questa operazione trova anche un fondamento fisico già a partire dalla spiegazione
dello spettro d’emissione del corpo nero ad opera di Planck. Prima del suo
contributo infatti, ipotizzando che l’energia potesse essere scambiata con continuità tra pareti del corpo nero e ambiente circostante, i tentativi di spiegare
tale spettro avevano portato alla formulazione della legge di Rayleigh-Jeans:
uν = kT
8π 2
ν dν
c3
(3.8)
con uν densità di energia per unità di fequenza di emissione ν. È facile vedere
che tale equazione non descrive correttamente il fenomeno, infatti provando
ad integrarla essa diverge per il contributo delle alte frequenze, portando alla
cosiddettacatastrofe ultravioletta. Planck risolse il problema introducendo il
quanto fondamentale d’azione h che porta il suo nome, e che determinò a
partire dalla sua famosa relazione di corpo nero
uν =
8π 2 hν
ν hν
.
c3 e kT
−1
(3.9)
20
3.2
CAPITOLO 3. L’ENTROPIA QUANTISTICA
Matrice densità
Oltre a fornire la base fisica per una corretta descrizione statistica dell’entropia termodinamica, e a introdurre la nozione di indistinguibilità (v. paradosso
di Gibbs) dei microstati, la meccanica quantistica permette anche di definire
un’entropia per sistemi microscopici.
A tale riguardo va considerato che in generale un sistema quantistico, visto
come parte di un sistema più esteso, non è sempre descrivibile in termine di
una singola funzione d’onda. È necessario in questo caso utilizzare quella che è
definita matrice densità. Questo si può verificare quando la procedura di preparazione del sistema in esame non determina univocamente lo stato. Risulta
in questo caso conveniente considerare una collezione di sistemi ciascuno dei
quali possa trovarsi in uno tra gli stati |ψii (i = 0, 1, 2...k), non necessariamente
ortogonali, con probabilità pi . È importante puntualizzare che tali probabilità
esprimono una indeterminazione di tipo classico; in particolare non si può dire
che il sistema si trovi in una combinazione degli stati |ψi i. Detta f una generica osservabile, cui è associato l’operatore fˆ e di cui vogliamo effettuare una
misura sul sistema, possiamo calcolare il valore aspettato di fˆ mediante una
media pesata sulle probabilità dei vari stati, risulta[6]
f¯ =
k
X
pi hψi | fˆ |ψi i .
(3.10)
i=0
Detti ora |a0 i e |a00 i elementi di una base nello spazio di Hilbert degli stati |ψi i
introduco nell’espressione del valore atteso di fˆ le relazioni di completezza dei
suddetti elementi
k XX
X
a0
i=0
pi hψi |a0 i ha0 | fˆ |a00 i ha00 |ψi i ,
(3.11)
a00
quindi risulta
k
XX X
(
pi ha00 |ψi i hψi |a0 i) ha0 | fˆ |a00 i .
a0
a00
(3.12)
i=0
Posso infine definire loperatore densità:
ρ=
k
X
pi |ψi i hψi | ,
(3.13)
ha00 | ρ |a0 i ha0 | fˆ |a00 i = Tr(ρfˆ)
(3.14)
i=0
per cui risulta
f¯ =
X
a0 ,a00
cioè posso ricavare i valori medi delle osservabili di un sistema mediante l’operatore densità associato ad esso.
Lo stesso tipo di descrizione può essere ottenuta, prescindendo da miscele
statistiche, considerando un sottosistema di un sistema quantistico in un determinato stato. Supponiamo di avere un sistema isolato cui è associato uno
spazio di Hilbert H. Al sottosistema in esame è associato lo spazio di Hilbert
21
3.2. MATRICE DENSITÀ
X sottospazio di H. Detto infine Q il sottospazio di H associato al rimanente
sistema risulta
H =Q⊗X
(3.15)
Riferendoci per semplicità ai sistemi considerati con gli spazi di Hilbert ad
essi associati si parla in questo caso di H come di un sistema composto dei
sistemi X e Q, matematicamente la composizione di più sistemi si esprime
mediante il prodotto tensoriale indicato dal simbolo ⊗. Se il sistema H si
troverà in un certo stato |ψi, in generale però non esistono due stati |ϕi e
|χi rispettivamente dei sistemi Q ed X tali che |ψi ne sia il prodotto, cioè in
generale non è fattorizzabile,
|ψi =
6 |ϕi |χi .
(3.16)
Allo stesso modo, dette |qi e |xi basi dei sistemi Q e X si possono associare
delle funzioni d’onda agli stati considerati tali che:
Z
|ψi = ψ(q, x) |qi |xi dqdx
(3.17)
Z
|ϕi =
ϕ(q) |qi dq
(3.18)
χ(x) |xi dx
(3.19)
Z
|χi =
e anche per le funzioni d’onda in generale risulta:
ψ(q, x) 6= ϕ(q)χ(x).
(3.20)
Sia ora f una certa grandezza osservabile cui è associato l’operatore fˆ che opera
solo sul sottosistema X
fˆ = fˆX ⊗ 1Q .
(3.21)
Il valore atteso di fˆ sullo stato considerato risulta
Z
¯
ˆ
f = hψ|f |ψi = dq 0 dx0 dq dx ψ ∗ (q, x)ψ(q, x) hq 0 , x0 |fˆ|q, xi ;
(3.22)
inoltre la fˆ agisce solo su X, per cui
Z
¯
f = dq 0 dx0 dq dx ψ ∗ (q, x)ψ(q, x) hx0 |fˆ|xi hq 0 |qi ,
(3.23)
ed essendo hq 0 |qi una delta di Dirac
Z
f¯ = dq dx0 dx ψ ∗ (q, x)ψ(q, x) hx0 |fˆ|xi .
(3.24)
Quindi posso definire l’ operatore densità
Z
0
ρ(x , x) = dq ψ ∗ (q, x0 )ψ(q, x),
(3.25)
in termini del quale si ha
f¯ =
Z
dx0 dx ρ(x0 , x) hx0 |fˆ|xi .
(3.26)
22
CAPITOLO 3. L’ENTROPIA QUANTISTICA
Nel caso discreto, dette |xi i e |qj i (i = 1, 2, ...n) (j = 1, 2, ...m) due basi
ortonormali di X e Q, uno stato del sistema totale sarà esprimibile come
|ψi =
X
λij |xi i |qj i .
(3.27)
i,j
Il valor medio della solita osservabile sarà
f¯ =
hxi0 , qj 0 |fˆ|xi , qj i λ∗i0 j 0 λij ,
X
(3.28)
i,j,i0 ,j 0
e dal momento fˆ agisce solo su X,
f¯ =
hqj 0 |qj i hxi0 |fˆ|xi i λ∗i0 j 0 λij .
X
(3.29)
i,j,i0 ,j 0
Posto ρi,i0 =
P
j
λ∗i0 j λij risulta
f¯ =
X
hxi0 |fˆ|xi i ρi,i0 = Tr(fˆρ).
(3.30)
ρi,i0 = hxi |ρ|xi0 i
(3.31)
i,i0
con
da cui
X
ρ=
λ∗nj λmj |xm i hxn | .
(3.32)
j,n,m
P
Osserviamo che, posto n λjn |xn i = aj |αj i, con |αj i normalizzato, ρ risulta
essere espresso in una forma equivalente a quella precedentemente ricavata:
ρ=
X
|aj |2 |αj i hαj | ,
(3.33)
j
infatti, dalla condizione di normalizzazione per |αj i, poiché anche |ψi è normalizzato risulta
X
X
|aj |2 =
|λnj |2 ≤
|λnj |2 = 1,
(3.34)
n
jn
quindi le due definizioni sono equivalenti; una miscela statistica e un sottostistema di un sistema dato sono descritti dallo stesso oggetto matematico.
Alcune proprietà dell’ operatore densità sono:
- l’operatore densità è un operatore hermitiano e positivo, infatti per ogni
|Ψi si ha hΨ|ρ|Ψi ≥ 0, come si vede dall’equazione (3.33);
- la traccia dell’operatore densità è uguale ad uno; infatti dalla (3.32)
possiamo calcolare la traccia di ρ
T r(ρ) =
X
k
hxk |
X
j,n,m
λ∗nj λmj |xn i hxm |xk i =
X
j,k
|λkj |2 = 1;
(3.35)
23
3.2. MATRICE DENSITÀ
alternativamente dalla (3.13), detta {|ϕi i} una base ortonormale nello spazio di Hilbert considerato, dato che gli |ψi i sono normalizzati, la
traccia risulta essere
X
j
hϕj |
k
X
pi |ψi i hψi |ϕj i =
i
k
X
pi
i
X
| hϕj |ψi i |2 = 1
(3.36)
j
L’operatore densità è usato quindi per descrivere stati di cui non abbiamo
completa conoscenza detti miscele statistiche o stati misti. Il caso specifico di
stati puri, di cui si ha completa conoscenza, è ottenuto quando le pi nell’equazione (3.13) sono tutte nulle tranne una. L’opertore densità corrispondente ad
uno stato puro si riduce quindi ad un proiettore, per cui T r(ρ2 ) = 1.
Analogamente nel caso in cui lo stato del sistema globale |ψi sia fattorizzabile
|ψi = |χi |ϕi ,
(3.37)
con |χi e |ϕi vettori normalizzati di X e Q,
|χi =
X
ai |xi i ,
|ϕi =
X
i
bj |qj i ,
(3.38)
j
risulta nell’equazione (3.32) λij = ai bj e quindi
ρ=
k
X
a∗n b∗j am bj |xn i hxm | = |χi hχ| ;
(3.39)
j,n,m
- possiamo quindi affermare che
T r(ρ2 ) ≤ T r(ρ) = 1,
(3.40)
dove l’uguaglianza è soddisfatta nel caso di stati puri;
- dall’equazione di Schröedinger di evoluzione temporale degli stati si ottiene
∂ρ
= [H, ρ]
(3.41)
∂t
La desrizione di un sottostitema nel caso in cui il sistema composto non
si trovi in uno stato puro si ottiene mediante una immediata generalizzazione
del procedimento illustrato nella sezione precedente[7]: detti A e B due sistemi
formanti un sistema composto AB descritto da una matrice densità ρAB , si
definisce l’operatore densità ridotto relativo al sistema A
ρA = TrB (ρAB )
(3.42)
e analogamente per il sistema B
ρB = TrA (ρAB ),
dove l’operazione di traccia parziale agisce ad esempio come segue:
X
TrB (ρ) =
hbi |ρ|bi i
i
essendo |bi i una base ortonormale dello spazio B.
(3.43)
(3.44)
24
CAPITOLO 3. L’ENTROPIA QUANTISTICA
3.3
Entropia di Von Neumann
Possiamo ora parlare di entropia nel senso quantistico del termine, detto A
un certo sistema fisico descritto da un operatore densità ρ, l’l’entropia di Von
Neumann S(ρ)ad esso associata è definita da
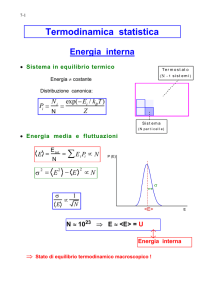
S(ρ) = −Tr(ρ log ρ),
(3.45)
dove per log ρ s’intende l’operatore che ha per autovalori il logaritmo degli
autovalori di ρ.
Nella base in cui ρ è diagonale siano pi , al variare di i, gli autovalori di ρ,
allora:
X
S=−
pi log pi .
(3.46)
i
Osserviamo che:
- L’entropia S(ρ) è positiva;
- se il sistema è in uno stato puro esiste un unico valore di i, ī per cui pī = 1
e tutti gli altri autovalori sono nulli, quindi l’entropia di Von Neumann è
nulla in uno stato puro;
- il valore massimo dell’entropia di Von Neumann si ottiene dalle seguenti
equazioni,
(
P
∂
∂pi [S − λ(P j pj − 1)] = 0
(3.47)
∂
j pj − 1)] = 0
∂λ [S − λ(
doveP
λ è un moltiplicatore di Lagrange inteso ad assicurare la condizione
che i pi = 1, da cui
log pi +P
1+λ=0
(3.48)
j pj = 1
e quindi ∀i pi = e−(λ+1) = N1 . Tutti gli stati sono quindi equiprobabili e
si ha
X 1
1
SMax = −
log
= log N.
(3.49)
N
N
Quindi un sistema privo di vincoli ha entropia massima quando la matrice
densità che lo descrive è proporzionale all’identità, si parla del sistema come di
una“miscela completamente incoerente”.
Possiamo ricavare anche l’entropia di un sistema all’equilibrio termodinamico ma sottoposto a vincoli, ad esempio un preciso valore dell’energia. Posto
hHi = U ed essendo il sistema in equilibrio risulta
∂ρ
= 0,
∂t
(3.50)
e dall’equazione (3.41) si ha [H, ρ] = 0: esiste perciò una base comune di
autovettori per ρ e H; essendo anche hHi = T r(ρH) e detti pi ed Ei gli
autovalori di ρ ed H risulta:
X
U=
pi Ei
i
25
3.3. ENTROPIA DI VON NEUMANN
che, assieme a
Lagrange
P
i
pi = 1 è un vincolo per l’equazione ai moltiplicatori di
X
X
X
∂
= [−
pj log pj − λ(
pj − 1) − β(
pj Ej − U )] = 0,
∂pi
j
j
j
(3.51)
si ricava che log pi = −βEi − λ − 1, da cui
pi = ceβEi
(3.52)
con c = e−λ−1 , inoltre dalla condizione di normalizzazione
e−βEi
pi = P −βEj .
je
(3.53)
La matrice densità risulta allora
P
i
ρ=
e−βEi |ψi i hψi |
P −βE
j
je
(3.54)
per cui, nota ρ possiamo ricavare l’entropia del sistema, essendo:
−βE1
0
···
0
−e
0
e−βE2 · · ·
1
0
(3.55)
ρ=
..
..
..
..
Z
.
.
.
.
0
0
· · · e−βEn
Pn
con n numero totale di stati e Z = i e−βEi , la funzione di ripartizione del
sistema. Calcoliamo la quantità ρ log ρ:
−βE1
e
1
ρ log ρ = −
Z
(βE1 + log Z)
0
.
.
.
0
0
e−βE2 (βE2 + log Z)
.
.
.
0
···
···
..
.
···
0
0
.
.
.
−βEn
e
(βE2 + log Z)
(3.56)
per cui essendo S = −T r(ρ log ρ) risulta
" n
#
1 X −βEi
S=
e
(βEi + log Z) ;
Z
i
(3.57)
separando la somma e ricordando la definizione di Z risulta
S = log Z +
ma la somma
Pn
i
n
β X
Ei e−βEi ,
Z i
(3.58)
Ei e−βEi rappresenta il valore medio U dell’energia quindi
S = log Z +
U
.
kT
(3.59)
Dunque è possibile legare l’entropia di Von Neumann a quella termodinamica
mediante la costante di Boltzmann:
ST d = kS(ρ)
(3.60)
26
CAPITOLO 3. L’ENTROPIA QUANTISTICA
per cui l’entropia termodinamica di un sistema all’equilibrio termico risulta:
ST d = k log Z +
U
T
(3.61)
equazione già nota dalla meccanica statistica se si osserva che il termine log Z
rappresenta l’energia libera del sistema.
3.4
Entanglement quantistico ed entropia di entanglement
Dato un sistema composto da due sottosistemi A e B, questo sarà descritto
da stati non necessariamente fattorizzabili in prodotto tensore di stati del sistema A e del sistema B. Si parla in questo caso di stati “entangled” (intricati,
aggrovigliati). Esempi tipici di stati entangled per sistemi composti da due
sstemi a due livelli sono
|00i ± |11i
√
2
|10i ± |01i
√
2
(3.62)
noti come stati di Bell ed epressi nella cosiddetta base computazionale |0i |1i
in ciascuno dei due sistemi. È facile vedere che le matrici densità associate
ai sottosistemi A e B sono proporzionali all’identità e danno quindi luogo a
valori massimi dell’entropia di Von Neumann. Si parla in questo caso di stati
massimamente entangled, in effetti l’entropia di Von Neumann può essere usata come misura del grado di entanglement in un dato stato. Ciò è dovuto al
fatto che le entropie di Von Neumann dei due sottosistemi coincidono e misurano quindi non quantità relative ai singoli due sistemi ma comuni ad entrambi.
Supponiamo infatti che il sistema composto AB cui è associato lo spazio di
Hilbert H = Ha ⊗ HB si trovi in uno stato puro cioè, detto ρ l’operatore
densità ad esso associato,
ρ = |ψi hψ| .
(3.63)
Siamo interessati a studiare l’entropia dei due sottosistemi A e B che compongono il sistema complessivo. Possiamo esprimere lo stato |ψi mediante basi
ortonormali degli spazi HA ed HB , rispettivamente |αn i e |βk i:
|ψi =
N X
M
X
n
cnk |αn i |βk i
(3.64)
k
dove N ed M sono le dimensioni degli spazi di Hilbert associati ai sottosistemi
A e B. Posso porre:
X
|Bn i =
cnk |βk i ,
(3.65)
k
da cui
|ψi =
X
n
|αn i |Bn i ,
(3.66)
3.4. ENTANGLEMENT QUANTISTICO ED ENTROPIA DI ENTANGLEMENT27
dove gli |Bj i non sono necessariamente ortonormali;
supponendo che |αn i sia base di autovettori per ρA , dove ρA è la matrice densità
ridotta associata al sottosistema A, risulta
ρA =
X
pn |αn i hαn |
(3.67)
n
ed anche:
ρA = T rB (ρ) =
X
cnk c∗n0 k |αn i hαn0 | ;
(3.68)
nn0 k
dovendo le due espressioni coincidere segue che, avendo imposto che αn
fosse base per ρA , i termini con n 6= n0 non danno contributo, quindi
X
cnk c∗n0 k = δnn0 pn .
(3.69)
k
Da notare che nell’equazione (3.67), possiamo considerare la somma estesa ai
solo valori di n per cui pn 6= 0. Sia N̄ il numero di tali valori. Nel caso in cui
N > M la relazione (3.69) implica che N̄ ≤ N . Risulta quindi dalla (3.65),
ricordando che i |βk i sono ortonormali,
hBn |Bm i = pn δnm .
(3.70)
Definendo dei ket ortonormali
|Bn i
|bn i = √
pn
(3.71)
per i soli n per cui pn 6= 0 e completando il set per ottenere una base ortonormale, si ha, avendo ridefinito per comodità di notazione |ai i = |αi i,
|ψi =
N
X
√
pn |an i |bn i ,
(3.72)
n
nota come decomposizione di Smidth dello stato |ψi. Possiamo calcolare la
matrice densità ridotta per B
ρB =
M
X
pj |bn i hbn |
(3.73)
n
dove, come prima possiamo considerare la somma estesa ai soli valori di n per
cui pn 6= 0. Dal confronto con l’equazione (3.67), i due operatori densità hanno
gli stessi autovalori quindi essendo
SA = −T r(ρA log ρA )
(3.74)
SB = −T r(ρB log ρB )
(3.75)
risulta che le entropie di sottosistemi di un sistema in uno stato puro sono
uguali
SA = SB .
(3.76)
28
CAPITOLO 3. L’ENTROPIA QUANTISTICA
Al valore comune delle entropie viene assegnato il nome di entropia di entanglement. Se lo stato del sistema AB oltre ad essere puro è fattorizzabile,
cioè:
|ψi = |ϕi |χi
(3.77)
con |ϕi e |χi rispettivamente stati di A e B, gli operatori densità ridotti
risultano
ρA = |ϕi hϕ| ,
ρB = |χi hχ| ,
(3.78)
per cui SA ed SB sono entrambe nulle.
In generale la (3.76) non vale se il sistema non si trova in uno stato puro. Ad
esempio se A e B sono entrambi sistemi a due livelli sia ρ la matrice densità
del sistema composto AB data da
ρ=
1
1
|00i h00| + |00i h01| + |01i h00| + |01i h01| .
2
2
(3.79)
Le matrici densità ridotte risultano essere
ρA = |0i h0|
e
ρB =
1
1
|0i h0| + |0i h1| + |1i h0| + |1i h1| ;
2
2
(3.80)
(3.81)
per cui
1
(3.82)
2
cioè per stati non puri le entropie d’entanglement dei sottositemi non sono
uguali. Diverse misure di entanglement possono essere adottate nel caso di stati
non puri[8]. Una di queste fa riferimento all’entropia relativa S(ρ1 ||ρ2 ) tra due
miscele statistiche che sarà definita nel capitolo 4. A tale scopo si individua
nell’insieme T delle matrici densità che descrivono il sistema composto AB,
il sottoinsieme D delle matrici associate a stati non entangled. Una generica
matrice densità di D sarà espressa nella forma ρA ⊗ρB . Il grado di entaglement
tra i due sistemi A e B nello stato descritto dalla matrice densità ρ è dato da
S(ρA ) = 0,
S(ρB ) = log
E(ρ) = min S(ρ||ρD )
ρD ∈D
(3.83)
29
3.5. ENTROPIA E TEMPO
3.5
Entropia e tempo
Un altro importante, e controverso, aspetto riguardante l’entropia è la definizione di una “freccia del tempo”. Nelle equazioni della meccanica classica
cosı̀ come in quelle della meccanica quantistica (salvo per il collasso delle funzioni d’onda) non compare alcuna direzione privilegiata del tempo, cioè non è
specificato che un fenomeno fisico debba accadere nel senso delle t crescenti o
decrescenti e, dato un certo sistema, è possibile conoscerne l’evoluzione temporale nel futuro o la situazione in cui si trovava nel passato ed invertendo le
condizioni inziali è possibile un evoluzione del sistema “all’indietro”.
La seconda legge della termodinamica rompe questa simmetria affermando
che l’entropia dell’universo non può far altro che crescere, individuando quindi
una direzione obbligata che i fenomeni fisici devono seguire, identificando il
passato come gli stati ad entropia minore che si sono evoluti in stati ad entropia
maggiore, il futuro.
Partiamo dal caso classico[9]. Considerando l’entropia di Gibbs di un certo sistema essa è ricavata a partire dalla funzione HG di Gibbs. Detta ρ la
distribuzione di probabilità del sistema risulta
Z
HG = ρ log ρdτ,
(3.84)
dove in dτ sono presenti tutte le variabili del sistema; la relazione tra la
funzione HG e l’entropia di Gibbs è determinata dalla costante di Boltzmann
SG = −kHG .
(3.85)
La HG varia al variare della ρ, in particolare detta Se l’entropia ricavata
sperimentalmente risulta
SG ≤ Se .
(3.86)
L’uguaglianza vale quando la SG è calcolata sulla distribuzione canonica, che
può essere ricavata minimizzando HG con la condizione che il valor medio
dell’energia si U :
Z
hHi =
ρHdτ = U.
(3.87)
D’altra parte la HG e quindi la SG restano costanti nel tempo. Ciò che appare
strano è l’osservare come la HG e quindi anche la SG restino costanti nel tempo,
infatti
Z
dρ
dHG
=
log ρdτ = 0,
(3.88)
dt
dt
per il teorema di Liouville ρ è stazionaria, quindi HG è costante.
Bisogna quindi specificare il rapporto tra l’entropia di Gibbs e quella ricavata sperimentalmente. Al tempo t = 0 consideriamo un sistema in equilibrio
termodinamico, la sua distribuzione sarà quella canonica, quindi vale l’uguaglianza SG = Se . Se si forza il sistema ad un cambio di stato adiabatico la
sua SG non varierà, tuttavia, al tempo successivo t0 il sistema raggiungerà di
nuovo l’equilibrio e la sua funzione di distribuzione sarà in questo caso ρ(t0 ),
ancora canonica, ma differente rispetto alla precedente in dipendenza dal cambio di stato che è avvenuto. Detta quindi Se0 l’entropia di questo nuovo stato
d’equilibrio varrà la diseguaglianza stretta
SG < Se0 ;
(3.89)
30
CAPITOLO 3. L’ENTROPIA QUANTISTICA
inoltre, poiché SG è rimasta costante, risulta
Se < Se0 ,
(3.90)
che in pratica è la seconda legge della termodinamica. È importante osservare
come l’entropia di Gibbs resti costante nel tempo mentre quella sperimentale,
cioè quella che misuriamo, varia.
Nel caso quantistico invece avevamo calcolato l’entropia di un sistema preso in
esame a partire dalla sua matrice densità ρ, ci si ricollegava all’entropia termodinamica mediante la costante di Boltzmann. Per stati puri risulta S = 0; essendo ρ un operatore unitario nell’applicare l’equazione di Srödinger osserviamo
che:
ρ(t) = e−iHt ρ(0)eiHt
(3.91)
Gli autovalori di ρ non variano al variare del tempo, quindi la sua traccia, e
l’entropia del sistema, restano costanti, ciò accade anche per processi irreversibili perché l’evoluzione è unitaria.
Il carattere probabilistico della legge dell’aumento dell’entropia può essere illlustrato attraverso un semplice esempio. Immaginiamo di avere un contenitore
diviso in due parti e contente un numero molto basso di particelle, nel nostro
caso 50, tutte confinate in un’unica parte. Supponendo che sia possibile per
una a caso tra le particelle di passare da un lato all’altro del contenitore ad intervalli regolari e che ogni volta che ciò accade misuriamo il numero di particelle
nella metà contenitore inizialmente piena, otterremmo la situazione riprodotta
in fig.3.1, dove rappresentiamo il numero di particelle nella metà contenitore
in funzione del numero di misurazioni effettuate.
Figura 3.1: Andamento temporale del numero di particelle presente in una delle
due metà del recipiente.
E’ facile rendersi conto di come il sistema evolva verso una situazione di
equilibrio in cui il numero di particelle è bilanciato tra le due metà del con-
3.5. ENTROPIA E TEMPO
31
tenitore. Ci si rende subito conto di quale sia lo stato iniziale del sistema e
possiamo associare una direzione del tempo parallela alla direzione in cui il
sistema si porta in equilibrio, cioè se invertissimo il grafico “a specchio” si capirebbe che la situazione in cui tutte le particelle si distribuiscono in un’unica
metà necessita di un intervento esterno e non accade mai spontaneamente.
Se tuttavia considerassimo il sistema ormai all’equilibrio dimenticandoci
dello stato iniziale e cancellando dal grafico il contributo delle prime centinaia
di scambi non osserveremmo alcun comportamento particolare delle particelle,
ma solo le generiche fluttuazioni che avvengono all’equilibrio, invertendo specularmente il grafico non noteremmo la differenza, cioè in assenza di condizioni
iniziali il sistema è simmetrico rispetto al tempo, come mostrato in fig.3.2.
Figura 3.2: Andamento temporale del numero di particelle in una metà del
recipiente eliminando il contributo delle prime centinaia di scambi.
Ciò significa che il concetto di “freccia del tempo” è legato a quelle che noi
chiamiamo “condizioni iniziali” che coincidono spesso a stati poco probabili del
sistema. Imponendo queste condizioni rompiamo la simmetria temporale che il
sistema ha invece in stato di equilibrio. In linea di principio possiamo eliminare
l’azione di uno sperimentatore esterno, è possibile infatti supporre che il sistema in questione sia frutto non di una preparazione da parte dell’osservatore
ma di una “fluttuazione statistica estremamente rara”, anche in quest’ottica il
sistema resta però simmetrico rispetto al tempo in quanto rispetto alla sua storia precedente tale fluttuazione sarebbe speculare o equiprobabile all’evoluzione
che noi osserviamo. È possibile ampliare il discorso arrivando a ipotizzare che
l’intero universo possa essere frutto di una tale rarissima fluttuazione giustificando il fatto che la sua entropia totale è più bassa di quanto atteso o che tutto
sommato si tratta di un universo ancora molto ordinato nella sua struttura
fatta di aggregati di galassie e nebulose, ma questa resta una speculazione.
32
CAPITOLO 3. L’ENTROPIA QUANTISTICA
L’evoluzione verso l’equilibrio è descritta dalla master equation che determina l’evoluzione temporale della matrice densità ridotta per sistemi non isolati, vista come matrice ridotta di un sistema composto isolato, quest’ultimo
soggetto ad un l’evoluzione unitaria[10]. Il paradosso di Loschimdt, cioè l’emergere di una freccia del tempo da una dinamica simmetrica rispetto inversione
temporale, rimane se si considera un sistema isolato, al limite l’intero universo.
In ultima analisi la simmetria riguarda la nostra conoscenza di un sistema
fisico più che la sua dinamica: è il processo di acquisizione di informazioni sul sistema infatti ad essere asimmetrico. Questo ci porta ad esplorare il
collegamento tra entropia ed informazione.
Capitolo 4
Entropia ed informazione
Provando a dare una definizione semplice di informazione possiamo dire
che essa è ciò che è scambiato attraverso la comunicazione, è codificata in
un linguaggio (le parole, la scrittura) e viene trasmessa attraverso di esso.
Proveremo ad analizzare l’informazione e le problematiche ad essa legate dal
punto di vista fisico arrivando a definire un’entropia di informazione.
4.1
Entropia di Shannon e compressione di informazione
Nel definire e quantificare l’informazione sorge il problema di possibili ambiguità, ad esempio volendo quantificare tramite la sua lunghezza l’informazione
contenuta in un messaggio scritto quest’ultima dipende da molteplici fattori
quali l’alfabeto o la lingua in cui è espresso. Il contributo fondamentale dovuto a Shannon[11] alla teoria dell’informazione consiste nell’aver definito una
grandezza che risolve questa ambiguità.
Un semplice esempio può chiarire il concetto: supponiamo che l’informazione da trasmettere sia uno tra i valori 1, 2, 3, 4; possiamo sempre con 2
bit trasmettere univocamente i quattro valori considerati. Un messaggio di
lunghezza n sarà in generale costituito da una successione di n occorrenze di
uno di questi quattro valori. Poiché possiamo sempre con 2 bit trasmettere
univocamente i quattro valori considerati, saranno sufficienti 2n bit per trasmettere il messaggio; per bit intendiamo la minima quantità di informazione
che possiamo trasmettere in logica binaria. Tuttavia supponiamo che le occorrenze dei quattro valori non siano ugualmente probabili. Un esempio di questo
fenomeno si trova nel linguaggio in cui certe lettere di un alfabeto compaiono
più frequentemente di altre. Sia ad esempio
p(1) =
1
,
2
p(2) =
1
,
4
p(3) = p(4) =
1
.
8
In questo caso, al fine di risparmiare risorse, possiamo assegnare meno bit ai
valori più frequenti e più bit a quelli più rari; una buona configurazione risulta
essere
1→0
2 → 10
33
34
CAPITOLO 4. ENTROPIA ED INFORMAZIONE
3 → 110
4 → 111
Questa scelta garantisce l’individuazione univoca in una stringa delle singole
occorrenze. La media dei bit impiegati, pesata con la loro probabilità, risulta essere 7/4, quindi il messaggio inviato necessiterà si soli 74 n bit per essere
trasmesso. Risulta in effetti che questa è la scelta ottimale per minimizzare il
numero di bit utilizzati, come conseguenza del teorema di Shannon.
Si definisce infatti la cosiddetta entropia di Shannon associata ad una certa
variabile stocastica x come
X
H(x) = −
pi log2 pi
(4.1)
i
dove pi rappresenta la probabilità di occorrenza dell’i-esimo valoredi x. Nel
caso precedente il valore di H risulta essere proprio 7/4. L’entropia di Shannon
quantifica quindi il minimo numero di risorse necessarie alla trasmissione di
informazione. Formalmente l’entropia di Shannon è equivalente all’entropia di
Von Neumann a meno di una costante moltiplicativa pari al log 2; come già
visto in quel contesto, essa è massima nel caso in cui i pi siano tutti uguali.
Se nell’esempio precedente si fosse imposta la condizione pi = 1/4 per ogni
valore di i, sarebbe risultato H = 2 cioè l’informazione trasmessa da ogni cifra
è massima.
4.2
Proprietà dell’entropia
Analizzeremo le varie forme in cui l’entropia di Shannon può presentarsi e
le proprietà che ne derivano;
4.2.1
Entropia Binaria
Nel caso in cui la variabile assuma solo due valori si parla dientropia binaria,
cioè, detta x una variabile con due valori possibili, x1 e x2 con probabilità
rispettivamente di p e (1 − p) risulta :
H(x) = −p log2 p − (1 − p) log2 (1 − p)
(4.2)
È facile vedere che H è massima quando p = 1/2, cioè quando i valori sono
completamente casuali l’incertezza è massima; inoltre si dimostra facilmente,
derivando in p, che la funzione H è concava.
4.2.2
Entropia relativa
L’entropia di Shannon è un ottimo strumento anche per verificare la “vicinanza” di due differenti distribuzioni di probabilità relative allo stesso set di
indici di una certa variabile X; dette p(x) e q(x) due distribuzioni di probabilità
definiamo entropia relativa di p(x) rispetto q(x) la quantità:
H(p(x)||q(x)) =
X
x
p(x) log2
X
p(x)
= −H(X) −
p(x) log2 q(x)
q(x)
x
(4.3)
35
4.3. TEOREMI DI SHANNON
Si dimostra che l’entropia relativa è non negativa. Consideriamo infatti la
diseguaglianza
log2 x log 2 = log x ≤ x − 1
∀x > 0,
(4.4)
l’entropia relativa è
H(p(x)||q(x)) = −
X
p(x) log2
x
essendo − log2 x ≥
1−x
log2
q(x)
;
p(x)
(4.5)
risulta
1 X
q(x)
1 X
H(p(x)||q(x)) ≥
p(x) 1 −
=
[p(x) − q(x)]
log 2 x
p(x)
log 2 x
(4.6)
ed essendo entrambe le distribuzioni normalizzate:
H(p(x)||q(x)) ≥ 0
(4.7)
Come si è visto nella sezione 3.4 l’entropia relativa è utilizzata per quantificare
l’entanglement tra due sottostistemi nel caso in cui il sistema composto si trovi
in uno stato non puro.
4.2.3
Mutua informazione
Dette X e Y due variabili, il contenuto di informazione che trasmettono
può essere collegato, si definisce a tale scopo l’entropia congiunta:
H(X, Y ) = −
X
p(x, y) log2 p(x, y)
(4.8)
x,y
Se oltre alla variabile Y è nota l’entropia ad essa associata H(Y ) allora
abbiamo dell’informazione in più e possiamo definire l’entropia condizionata:
H(X|Y ) = H(X, Y ) − H(Y )
(4.9)
La quantità di informazione può essere intesa anche come il grado di sorpresa
rispetto alla conoscenza dell’esito dell’occorrenza di un certo valore della variabile stocastica. L’entropia condizionata quindi quantifica il grado di sorpresa
rispetto all’esito di X una volta noto l’esito di Y .
Infine possiamo calcolare l’informazione comune che due variabili X e Y
trasmettono; La somma H(X) + H(Y ) quantifica sia l’informazione comune
che quella individuale che trasmettono, considerando due volte però quella
comune. Quest’ultima si può ottenere quindi come mutua informazione
H(X : Y ) = H(X) + H(Y ) − H(X, Y ) = H(X) − H(X|Y ).
4.3
(4.10)
Teoremi di Shannon
Sia x1 , x2 , ...xn una sequenza di numeri casualmente estratti da un alfabeto
binario, e p la probabilità che un generico elemento della sequenza abbia valore
1 e (1 − p) la probabilità che esso abbia valore 0. Si dicono stringhe tipiche
36
CAPITOLO 4. ENTROPIA ED INFORMAZIONE
tutte quelle stringhe di n elementi che possiedono np elementi con valore 1 ed
n(1 − p) elementi con valore 0. Il numero massimo di stringhe tipiche è dato in
termini dell’entropia di Shannon da 2nH(x) . Consideriamo infatti una sequenza
tipica: essendo i vari elementi della sequenza indipendenti, la probabilità di
occorrenza della stringa è fattorizzabile, quindi
p(x1 , x2 , ...xn ) = p(x1 )p(x2 )...p(xn ) = pnp (1 − p)n(1−p) .
(4.11)
Passando ai logaritmi, e ricordando la definizione di H si ottiene
− log2 p(x1 , x2 ...xn ) = − log2 p − n(1 − p) log2 (1 − p) = nH(x),
(4.12)
dove per x si intende la variabile stocastica che assume i valori 0, 1. A questo punto risulta che la probabilità di ottenere una sequenza tipica tra le 2n
sequenze possibili è:
p(x1 , x2 , ...xn ) = 2−nH(x)
(4.13)
il che significa che al massimo possiamo ottenere 2nH(x) sequenze tipiche. Inoltre una sequenza tipica necessiterà qundi di un numero di bit pari ad nH(x),
questo è il contenuto del teorema di Shannon per la codifica attraverso un canale privo di rumore, in relatà qualunque canale di comunicazione è affetto da
disturbi, interviene in questo caso il teorema di Shannon per la codifica attraverso canale disturbato.
Introduciamo preliminarmente un modello di canale d’informazione affetto da
rumore, il canale simmetrico binario rappresentato in fig. 4.1:
Figura 4.1: Canale simmetrico binario
un canale di questo tipo inverte il valore trasmesso con probabilità p e lo
trasmette invariato con probabilità 1 − p; esso agisce allo stesso modo su tutte
le cifre, è per questo un canale senza memoria. Per trasmettere affidabilmente
informazione attraverso un canale disturbato la strategia da utilizzare è di
impiegare più bit del necessario. Per cui, volendo trasmettere una stringa di k
bit, se ne utilizzeranno n, con n > k, si definisce allora il tasso della codifica
k
.
(4.14)
n
Osserviamo che tra la stringa in entrata e quella in uscita da un canale disturbato c’è in genere una differenza di np bit, con un’approssimazione tanto più
R=
4.4. IL DIAVOLETTO DI MAXWELL E IL PRINCIPIO DI LANDAUER.37
accurata quanto più n è grande; questa differenza di bit è detta “distanza di
Humming” tra le stringhe, e tutte le stringhe che distano da quella iniziale
di np bit si possono considerare appartenere ad una certa sfera di Humming
centrata nella nostra stringa di partenza. Affinché la codifica utilizzata per
trasmettere le stringhe sia affidabile esse devono distare più di np bit l’una
dall’altra, cioè le varie sfere di Humming centrate in ogni stringa inviata non
devono intersecarsi. Affinché tale condizione sia verificata basta imporre che
2k 2nH(p) = 2n[R+H(p)] ≤ 2n
(4.15)
per cui otteniamo una condizione sul tasso di codifica R:
R ≤ 1 − H(p).
(4.16)
Possiamo definire la capacità di un canale disturbato come il massimo valore
di R
C(p) = 1 − H(p).
(4.17)
Il teorema di Shannon per codifica attraverso canale disturbato è la generalizzazione del risultato riferito al canale simmetrico binario mirata a definire la
capacità di un generico canale disturbato senza memoria. Questo è caratterizzato da una certa distribuzione di probabilità condizionata p(y|x) dove x è la
generica occorrenza di una variabile stocastica X, distribuita secondo una p(x)
ed Y la variabile stocastica in uscita dal canale. La capacità del canale è data
da
C = max H(X : Y )
(4.18)
p(x)
dove il massimo è effettuato su tutte le possibili variabili in ingresso.
4.4
Il diavoletto di Maxwell e il principio di
Landauer.
Partendo dall’entropia termodinamica abbiamo mostrato come il concetto
di entropia sia legato alla conoscenza che abbiamo di un sistema fisico, quindi all’informazione relativa ad esso. È interessante osservare che il punto di
vista della teoria dell’informazione ha una precisa rilevanza anche sulla termodinamica. Per mostrare questo concetto descriveremo quello che è noto come
il paradosso del diavoletto di Maxwell, che risolveremo proprio in termini di
informazione immagazzinata in un certo sistema[12]. Il ponte tra teoria dell’informazione e termodinamica è dato dal principio di Landauer.
Il paradosso
Il paradosso nel trattato “Teoria del calore” di Maxwell:
Immaginiamo un essere le cui capacità siano cosı̀ acute che possa seguire qualunque molecola nel suo corso, tale essere, le cui capacità sono essenzialmente
finite, come le nostre, sarebbe capace di fare ciò che per noi è impossibile.
Poiché abbiamo visto che le molecole in un contenitore pieno d’aria a temperatura uniforme si muovono muovono a velocità niente affatto uniforme, tuttavia
38
CAPITOLO 4. ENTROPIA ED INFORMAZIONE
Figura 4.2: Schema semplificato del paradosso fi Maxwell[12]
.
la velocità media di qualunque gruppo di molecole sufficientemente grande, arbitrariamente selezionato, è quasi esattamente uniforme. Ora supponiamo che
tale contenitore sia diviso in due parti, A e B, da una parete che presenta una
piccola apertura, e che tale essere, che può vedere le molecole singolarmente,
apra e chiuda questa apertura, cosı̀ da permettere solo alle molecole più veloci
di passare da A a B, e solo a quelle più lente da B ad A. Allora, senza compiere lavoro, egli aumenterà la temperatura di B e diminuirà la temperatura du
A contrariamente alla seconda legge della termodinamica
Una versione semplificata di questo paradosso consiste nel supporre di avere una certa macchina termica che opera con un gas costituito da un’unica
particella, come in fig. 4.2. Inizialmente (a) il gas è libero di muoversi in un
volume V . Nel paradosso si suppone che un essere microscopico e senziente
(più realisticamente un computer di dimensioni molto piccole o comunque un
particolare strumento) inserisca una partizione (b) nel volume V , poi misurando in quale delle due sezioni del volume si trovi la particella memorizzi dove
essa si trovi, quindi la partizione viene sostituita con un sistema di pistoni e
pesi (c), infine il gas è messo in contatto con un termostato e si espande isotermicamente spostando il pistone e compiendo lavoro convertendo unicamente il
calore fornito dal termostato in lavoro (d), poi tornando al volume e allo stato
iniziale contradirrebbe apparentemente la seconda legge della termodinamica.
Risoluzione : Il principio di Landauer
La soluzione al paradosso sta nel notare che in realtà nel sistema formato
dal diavoletto e dalla macchina stato iniziale e finale non coincidono; infatti
il diavoletto ha ancora immagazzinata l’informazione riguardo alla posizione
della particella, per tornare allo stato iniziale quest’informazione deve essere
necessariamente cancellata.
Secondo il principio di Landauer la cancellazione di informazione necessita di un
costo in energia, la stessa quantità che il diavoletto guadagnerebbe dall’espansione del gas considerato; per cui, infine, non c’è nessun guadagno d’energia e
nessuna diminuzione di entropia nel sistema.
4.4. IL DIAVOLETTO DI MAXWELL E IL PRINCIPIO DI LANDAUER.39
Il principio di Landauer può essere illustrato immaginando un sistema di conservazione di informazione binario formato da un gas di un’unica particella e
da un volume separato da un setto.
1. particella nello stato 0 o nello stato 1
2. per cancellare l’informazione rimuoviamo il setto separatore e
comprimiamo isotermicamente il gas
3. indipendentemente dallo stato iniziale adesso la particella si trova nello
stato 0, l’informazione è stata cancellata.
Questa compressione necessita di un lavoro minimo pari a kT log 2 che viene
trasferito all’ambiente sotto forma di calore. Corrispondentemente l’entropia
del gas diminuisce di di k log 2.
Quindi il paradosso è risolto considerando anche l’informazione contenuta
nel sistema, c’è infatti una grande differenza nell’avere un generico gas in un
40
CAPITOLO 4. ENTROPIA ED INFORMAZIONE
contenitore e nell’avere un gas di cui si conosce la posizione e il moto di ogni
particella, nel secondo caso infatti è possibile sfruttare tale informazione per
estrarre energia dal gas.
Bibliografia
[1] E. Fermi, Thermodynamics, Dover Publication, Inc. New York 1956.
[2] L. D. Landau, E. M. Lifshitz, Fisica Teorica Volume 5 Meccanica statistica,
Editori Riuniti Edizioni Mir 1986.
[3] [2] Cap. I § 8.
[4] E. T. Janes, Gibbs vs Boltzmann Entropies, Am. J. Phys. 33 (1965) 391.
[5] L. Benguigui, The different paths to entropy, Eur. J. Phys. 34 (2013)303.
[6] J. J. Sakurai, Meccanica Quantistica Moderna, Zanichelli 1990.
[7] M. A.Nielsen, I. L. Chuang, Quantum Computation and Quantum Information, Cambridge University Press 2000. ry Physics, 42 (2001)
25.
[8] V. Vedral, M. B. Plenio, M. A. Rippin, P. L. Knight, Quantifying
Entanglement, Phys. Rev. Lett. 78 (1997) 2275.
[9] [4]
[10] S. Kryszewski, J. Czechowska-Kryszk, Master Equation-tutorial approach
arXiv:0801.1757
[11] [7]
[12] M. B. Plenio V. Vitelli, The physics of forgetting: Landauer’s erasure
principle and information theory, Contempora
41