Il Clonaggio
del DNA
Clonazione
In Biologia, clonare un organismo significa ottenere da un individuo
una popolazione di organismi geneticamente identici.
Clonaggio
In Biologia Molecolare clonare un tratto di DNA significa ottenere
un insieme di molecole di DNA identiche a quelle di partenza.
Tecnologia del DNA
Ricombinante
TECNOLOGIA DEL DNA RICOMBINANTE
Ha reso possibile il CLONAGGIO dei GENI permettendo di
ISOLARE
AMPLIFICARE
SEQUENZIARE
frammenti di DNA
Perché manipolare i geni?
1) Per facilitare lo studio dell’espressione genica e della regolazione
fisiologica;
2) per identificare il prodotto di un gene e/o per ottenerne la
sovraespressione;
3) per studiare la relazione fra struttura e funzione delle proteine;
4) per identificare componenti cellulari che interagiscono con particolari
sequenze di acidi nucleici o con particolari domini proteici
Applicazioni del Clonaggio del
DNA
Ricerca di base: studio dei maccanismi di replicazione genica e
dell’espressione nei procarioti e negli eucarioti.
Ricerca applicata: per ottenere microrganismi in grado di
produrre composti come l’insulina umana, l’interferone, ormoni
della crescita…
Terapia genica
PROCEDURA di CLONAGGIO
•ISOLAMENTO del gene
•INSERZIONE del gene in un VETTORE PLASMIDICO
•INTRODUZIONE del vettore plasmidico IN CELLULE
VIVENTI per propagarlo.
Cosa serve per un clonaggio ?
•Gene (DNA di interesse)
•Enzimi di restrizione
•DNA ligasi
•Vettore
•Cellula ospite
DNA Cloning
DNA cloning is a technique for
reproducing DNA fragments. It can be
achieved by two different approaches: (1)
cell based, and (2) using polymerase
chain reaction (PCR). In the cell-based
approach, a vector is required to carry the
DNA fragment of interest into the host
cell. The following figure shows the
typical procedure by using plasmids as
the cloning vector.
The essential steps in DNA cloning using plasmids as
vectors.
(a) DNA recombination. The DNA fragment to be cloned is
inserted into a vector. The recombinant vector must also
contain an antibiotic-resistance gene (not shown).
(b) Transformation. The recombinant DNA enters into the
host cell and proliferates. It is called "transformation"
because the function of the host cell may be altered. Normal
E. coli cells are difficult to take up plasmid DNA from the
medium. If they are treated with CaCl2, the transformation
efficiency can be significantly enhanced. Even so, only one
cell in about 10,000 cells may take up a plasmid DNA
molecule.
(c) Selective amplification. A specific antibiotic is added to
kill E. coli without any protection. The transformed E. coli is
protected by the antibiotic-resistance gene whose product can
inactivate the specific antibiotic. In this figure, the numbers
of vectors in each E. coli cell are not the same, because they
may also reproduce independently.
(d) Isolation of desired DNA clones.
Enzimi di
Restrizione
Sono endonucleasi di tipo II (classe delle idrolasi) in grado di tagliare
i legami fosfodiesterei del DNA per dare frammenti specifici.
L’attività endonucleasica e la funzione di metilazione del DNA sono
separate. Non hanno bisogno di ATP.
Tipo I: taglio casuale
Endonucleasi di tipo I e III: sono
enzimi bifunzionali e necessitano di
ATP come coenzima.
Tipo III: taglio in siti
specifici (24-26 coppie
di basi a valle del sito di
riconoscimento).
Sono enzimi di origine batterica necessari per degradare il DNA
virale dei batteriofagi
Riconoscono sequenze di 4,6, o 8 coppie nucleotidiche
Commonly used restriction enzymes.
Note: The "Recognition site" of a restriction enzyme is also called the restriction site. In this
column, the first line is from 5' to 3' and the second line is from 3' to 5'. The arrow indicates the
cleavage site. If the cleavage site is not at the center, the restriction enzyme will generate sticky ends
which can base-pair with other DNA fragments cleaved by the same restriction enzyme. If the
cleavage site is at the center, the restriction enzyme will generate blunt ends.
Gli enzimi di restrizione possono
generare estremità piatte (blunt
ends) oppure estremità coesive
(sticky ends).
Isoschizomeri: enzimi di
restrizione che riconoscono
la stessa sequenza.
Isocaudameri: riconoscono
siti diversi, ma lasciano
estremità compatibili (es.
BamHI e Sau3A).
5’-GGATCC-3’
5’-GATC-3’
3’-CCTAGG-5’
3’-CTAG-5’
Analisi del DNA tagliato dagli enzimi di restrizione:
Elettroforesi su gel di agarosio
Separa le molecole di DNA in base alla
lunghezza facendole migrare in un campo
elettrico.
Per visualizzare il DNA si usa Etidio Bromuro,
una molecola fluorescente alla luce
ultravioletta.
L’agarosio è un polisaccaride purificato dall’agar-agar, una
sostanza gelatinosa isolata dalle alghe. È un polimero lineare e
neutro formato da unità di D-galattosio e di 3,6-anidro-Lgalattosio legate alternativamente con legami glicosidici.
L’agarosio è un polisaccaride solubile in acqua alla temperatura
di ebollizione, mentre diventa solido man mano che si raffredda
formando un gel grazie alla formazione di una matrice
tridimensionale costituitasi attraverso dei legami ad idrogeno tra
le catene lineari.
Bromuro di Etidio
Le Mappe di Restrizione
E’ una rappresentazione grafica
delle localizzazioni dei siti di
restrizione all’interno di una
sequenza di DNA.
Plasmidi
Fagi
Cosmidi
YAC (Yeast Artificial Chromosome)
BAC (Bacterial Artificial Chromosome)
MAC (Mammalian Artificial Chromosome)
HAC (Human Artificial Chromosome)
Cloning Vectors
Vector" is an agent that can carry a DNA fragment into a host cell. If it is used for
reproducing the DNA fragment, it is called a "cloning vector". If it is used for expressing
certain gene in the DNA fragment, it is called an "expression vector".
Commonly used vectors include plasmid, Lambda phage, cosmid and yeast artificial
chromosome (YAC).
Plasmid
Plasmids are circular,
double-stranded DNA
molecules that exist in
bacteria and in the nuclei of
some eukaryotic
cells. They can replicate
independently of the host
cell. The size of plasmids
ranges from a few kb to
near 100 kb
A typical plasmid vector. It contains a polylinker which can recognize several different restriction
enzymes, an ampicillin-resistance gene (ampr) for selective amplification, and a replication origin
(ORI) for proliferation in the host cell.
Vettori
plasmidici
Molecola circolare di
DNA a doppio
filamento,
normalmente
presente nella cellula
batterica, in grado di
replicarsi
autonomamente
dal cromosoma.
PLASMIDI
Piccole molecole di DNA batterico extracromosomico, a
doppia elica circolare di dimensione variabili da 2 kb a 200 kb
(esistono anche plasmidi a doppia elica lineare).
In natura conferiscono vantaggi selettivi ai ceppi che li
contengono.
Ingegnerizzati per l’uso di laboratorio.
Per essere un buon vettore di clonaggio, un plasmide deve
avere:
l) Dimensioni contenute; 2) un sito di origine della replicazione;
3) un gene per la resistenza ad un antibiotico; 4) siti unici di
restrizione (polilinker)
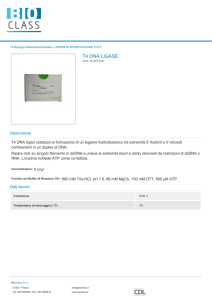
DNA LIGASI
Forma legami covalenti fra il gruppo fosfato 5’ di una estremità
ed il gruppo ossidrilico 3’ della catena adiacente.
L’enzima comunemente utilizzato è la T4 DNA Ligasi, perché
stabile e poco costosa.
CARATTERISTICHE DELLA REAZIONE:
Presenza di ATP;
Temperatura di 10°C o temperatura ambiente, per poche ore.
La bassa temperatura è consigliata in quanto, diminuendo l’energia cinetica
delle molecole, riduce la possibilità che le estremità appaiate si separino prima di
venir stabilizzate dalla ligazione.
Reazione
ligasica di
frammenti di
restrizione
Replicazione di un
plasmide
INSERIMENTO DI UN TRATTO DI DNA ESOGENO
IN UN VETTORE DI CLONAGGIO
Clonaggio in un
vettore
plasmidico
e trasformazione
I Plasmidi si dividono in:
• Plasmidi ad elevato numero di copie, la cui replicazione avviene più
frequentemente rispetto a quella del DNA cromosomico (alta resa);
• Plasmidi a basso numero di copie, la cui replicazione avviene con la
stessa frequenza del DNA cromosomico.
Possono essere classificati anche in base alla Specificità d’ospite:
Alcuni plasmidi sono in grado di replicare in un numero limitato di specie
batteriche e si dicono a specificità d’ospite limitata.
(per es. PBR322 e PUC18 si replicano solo in E.Coli)
Altri plasmidi sono in grado di replicarsi in una vasta gamma di specie
batteriche e si dicono a largo spettro d’ospite.
(possiedono geni per il riconoscimento dell’origine di replicazione e
dipendono meno dall’apparato replicativo dell’ospite batterico).
Requisiti dei Plasmidi per il Clonaggio
1) Piccole dimensioni per aumentare l’efficienza della trasformazione;
2) Devono avere un origine della replicazione (ORI);
3) Devono avere un buon numero di siti di restrizione unici (polylinker);
4) Devono avere dei marcatori
genetici specifici che consentano
la selezione delle cellule batteriche
In cui la trasformazione sia avvenuta
correttamente
Selezione per inattivazione
inserzionale
Bam HI
Il gene che conferisce resistenza
all’ampicillina codifica per l’enzima
β-lattamasi.
Il gene che conferisce resistenza
alla tetraciclina codifica per un
trasportatore di membrana che
espelle l’antibiotico dalla cellula.
Selezione per inattivazione
inserzionale
Il gene LacZ codifica per
l’enzima β-Galattosidasi, il
quale è in grado di demolire
il substrato cromogeno X-Gal
con la produzione di una
colorazione blu
Esempio di selezione bianco-blu
in pUC
INTERVALLO (Lag phase
phase))
4 - 5 ore
FASE LOGARITMICA
I batteri crescono esponenzialmente.
10 - 11 ore
5.0
Densità cellulare (OD600)
I batteri vengono diluiti nella coltura
iniziale; la divisione procede lentamente
in quanto le cellule si stanno adattando
al terreno fresco.
4.5
4.0
3.5
3.0
2.5
2.0
1.5
1.0
0.5
0
0
5
10
15
Tempo (h)
FASE STAZIONARIA
La densità cellulare rimane costante. La coltura può anche entrare in una
fase di declino in cui le cellule si lisano ed il DNA si degrada parzialmente.
20
Come evitare che il plasmide si richiuda su se
stesso
1) Digerire il frammento ed il plasmide con due enzimi diversi, in modo che le
estremità 5’ e 3’ del vettore non siano compatibili;
2) Alternativamente si può trattare il plasmide, prima della ligazione, con
fosfatasi alcalina che rimuove i gruppi fosfato al 5’
I batteri in grado di assumere DNA dall’ambiente
vengono definiti competenti.
Competenza naturale
Trattamento con
soluzioni fredde di
ioni bivalenti
(Ca2+).
Competenza artificiale
TRASFORMAZIONE delle CELLULE
BATTTERICHE:
Inserimento del plasmide nella cellula ospite
Cellule competenti
Solo le cellule che hanno incorporato il plasmide
formeranno delle colonie (selezione su terreno con
antibiotico).
METODI FISICI
METODI CHIMICI
• Shock termico
•Trasformazione con Cloruro
di Calcio
•Elettroporazione
•Metodo di Hanahan
•Microiniezione
METODI BIOLOGICI
•Lipofezione
• Le cellule batteriche si
raccolgono quando sono ancora
in fase di crescita logaritmica;
• Si centrifuga la coltura
batterica e si risospende in una
soluzione di CaCl2; le cellule
vanno tenute in ghiaccio per
almeno un’ora;
• Alle cellule competenti si
aggiunge il DNA della miscela di
ligazione e si lasciano le cellule
in ghiaccio (il DNA entra).
Heat shock (1 min a 42°C): favorisce
l’ingresso del plasmide;
Si lasciano le cellule a 37°C il tempo
necessario a far esprimere il gene
per la resistenza all’antibiotico;
Si dispensano aliquote di cellule su
piastre di terreno LB solido
contenenti l’antibiotico appropriato;
Dopo una notte in incubazione a
37°C si ottengono colonie da
vagliare per la presenza di DNA
ricombinante.
Elettroporazione:
Elettroporatore
ECM 830
1.
Le cellule batteriche vengono mescolate
con il DNA ricombinante plasmidico;
2.
Il mix ottenuto viene posto in cuvette
munite di elettrodi connessi ad un
alimentatore;
3.
Si induce un breve impulso elettrico che
apre transitoriamente dei pori nella
membrana cellulare attraverso i quali
entra il plasmide.
Lipofezione:
Il DNA può essere
incorporato entro
vescicole lipidiche
artificiali dette liposomi.
Sono stati sviluppati un certo numero di metodi allo scopo di
incapsulare il DNA in doppi strati lipidici sintetici che
assomigliano a membrane cellulari.
Questi liposomi sono essenzialmente delle sferette di membrane
sintetiche riempite di DNA, le quali si fondono spontaneamente
con le membrane cellulari, scaricando il loro contenuto nel
citoplasma.
Produrre dei liposomi ex novo è complicato, ma sono disponibili
in commercio dei reagenti che semplificano notevolmente la
procedura.
PURIFICAZIONE del DNA PLASMIDICO
1. RISOSPENSIONE
E. coli
3. NEUTRALIZZAZIONE
KAc
NaOH / SDS
RNasi
Centrifugazione
2. LISI
Supernatante
contenente il
DNA plasmidico
DNA cromosomico
DNA plasmidico
Precipitato
contenente DNA
genomico, proteine,
detriti cellulari
Eventualmente il DNA ricombinante può essere
ulteriormente purificato mediante cromatografia
a scambio ionico. Si utilizzano resine che hanno
sulla superficie gruppi dietil-amminoetilici (DEAE)
carichi positivamente. Il DNA con i gruppi fosfato
si legherà alla resina, e potrà quindi separato da
impurità cellulari che non si legheranno o si
legheranno debolmente. Infine il DNA potrà essere
elutio aumentando la concentrazione salina.
Vettori di Clonaggio: i FAGI
• I batteriofagi sono virus in grado di infettare i batteri
• Si utilizzano quando il frammento di DNA da clonare è troppo
grande per utilizzre vettori plasmidici (librerie di DNA).
Plasmidi fino a 10 Kb
Fagi fino a 20 -22 Kb
Batteriofago
(48.5 Kb)
ASS ORBIM
E
NTO
PA
RE
TE
BA
TT
ER
ICA
R
CICLO LITICO
A
CICLO
LISOGENICO
A
REPLICAZIONE
TARDIVA
(modello circolare)
REPLICAZIONE
PRECOCE
(bidirezionale)
COS
R
DNA
E. coli
RICOMBINAZ.
bio
GAL
ATT
CROMOSOMA
E. coli
ATT R COS
CON CATAMERO
GAL
TAGLI AD OPERA
PROTEINA A
A J ATT
RICOMBINAZ.
REPLICAZIONE CON
DNA CROMOSOMICO
PARTICELLA
FAGICA
MATURA
INCAPSIDAZIONE
LISI DELLA
PARETE BATTERICA
Cos: cohesive sequence
bio
CII = attivatore trascrizionale; CI = inibitore trascrizionale; CIII = proteina di legame,
protegge = inibitore trascrizionale.
CICLO LISOGENICO
Ad alto stato di infezione la cellula contiene alte concentrazioni di cIII:
Hfl è inibita
cII stimola la trascrizione di cI e della proteina Integrasi
cI è un repressore dei geni del ciclo litico e blocca la
sintesi di N, O, P e Q legandosi agli operatori OL e OR,
bloccando il ciclo litico
CICLO LITICO
A basse concentrazioni di cIII:
Hfl non inibita
riduce l’attività o la quantità di cII
blocco della sintesi di cI
la sintesi di N, O, P, Q non è più bloccata
Lambda phage
l phages are viruses that can infect
bacteria. The major advantage of the
phage vector is its high transformation
efficiency, about 1000 times more
efficient than the plasmid vector.
Schematic drawing of the DNA
cloning using phages as
vectors. The DNA to be cloned is first
inserted into the DNA, replacing a
nonessential region. Then, by an in
vitro assembly system the virion
carrying the recombinant DNA can be
formed. The genome is 49 kb in
length which can carry up to 25 kb
foreign DNA.
Il DNA isolato dalle particelle virali è a doppio filamento con
due estremità a singolo filamento di 12 nucleotidi complementari
fra loro, chiamati siti cos. Questi siti permettono al DNA di
ciclizzare dopo l’infezione della cellula ospite.
5’ GGGCGGCGACCTCGC-----------------ACG 3’
GCG-----------------TCGCCCGCCGCTGG
La mappa genetica del fago comprende circa 40 geni che
possono essere suddivisi in tre gruppi funzionali:
la parte sinistra, comprendente i geni da A a J, codifica per
proteine strutturali della testa e della coda.
la parte centrale, contiene geni responsabili per la lisogenia, cioé
il processo che porta all'integrazione del DNA virale ed altri
processi ricombinativi. Gran parte di questa regione non é
essenziale per la crescita litica e può essere eliminata per la
costruzione di vettori.
la parte destra contiene geni coinvolti nella replicazione del
DNA e nel ciclo litico (S, R), geni regolatori (cI, cII, cIII, cro, N,
Q).
Il fago Lambda λ
Inserimento
DNA
Proteine strutturali
della testa e della coda
Geni per la lisogenia
Cohesive ends
4.85 Kb
Limite massimo per l’impaccamento: 51 Kb
Limite minimo per fagi vitali: 37 Kb
Ciclo litico e
replicazione DNA
• Per l’assemblaggio
delle particelle fagiche
ricombinanti, il
packaging si fa in vitro.
The extreme ends of the λ DNA are known as COS
sites, each is single stranded, 12 nucleotides
long. Because their sequences are complementary
to each other, one end of λ DNA may base-pair
with the other end of a different λ DNA, forming
concatemers. The two ends of a λ DNA may also
bind together, forming a circular DNA. In the host
cell, the λ DNA circularizes because ligase may
seal the join of the COS sites.
In the assembly process of λ virions, two proteins
Nu1 and A can recognize the COS site, directing
the insertion of the λ DNA between them into an
empty head. The filled head is then attached to the
tail, forming a complete λ virion. The whole
process normally takes place in the host
cell. However, to prepare the λ virion carrying
recombinant λ DNA, the following in vitro
assembly system is commonly used.
Proteins Nu1 and A are encoded by the genes in the
λ genome. If the two genes are mutated, λ DNA
cannot be packaged into the pre-assembled
head. Because tails attach only to filled heads, the
cell will accumulate separate empty heads and tails,
which can then be extracted. When the extract is
mixed with recombinant λ DNA and proteins Nu1
and A, the complete λ virion carrying recombinant
λ DNA will be assembled.
The assembly process of the virion.
0
10
20
30
40
50 Kb
cI
gt10
braccio sinistro 32,7 Kb
braccio destro 10,6 Kb
EcoRI
lacZ
Charon16A
braccio destro 21,9 Kb
braccio sinistro 19.9 Kb
EcoRI
Stuffer
EMBL4
braccio sinistro 19.9 Kb
SalI, BamHI, EcoRI
braccio destro 8,8 Kb
SalI, BamHI, EcoRI
SalI
Stuffer
Charon40
braccio destro 9,6Kb
braccio sinistro 19.2Kb
polilinker
polilinker
COSMIDI
Sono PLASMIDI,
ma contengono, oltre alle
caratteristiche essenziali di un
plasmide, anche un
SITO COS
La presenza di questo elemento è determinante per consentire
l’inserimento della molecola di DNA ricombinante nella testa del virus
che, a sua volta, la inietta in una cellula batterica per infezione.
Cosmidi
• Un Cosmide è un plasmide di
circa 5 Kb contenente un sito cos;
• Permettono di inserire DNA fino a
45 Kb;
• Posseggono un origine di
replicazione e un gene per la
farmacoresistenza;
•Sono ad alto numero dicopie come
i plasmidi.
Cosmid
The cosmid vector is a
combination of the
plasmid vector and the
COS site which allows the
target DNA to be inserted
into the head. It has the
following advantages:
High transformation
efficiency.
The cosmid vector can
carry up to 45 kb whereas
plasmid and phage
vectors are limited to 25
kb.
Cloning by using cosmid vectors. (a) In addition to ampr, ORI, and polylinker as in the
plasmid vector, the cosmid vector also contains a COS site. (b) After cosmid vectors are
cleaved with restriction enzyme, they are ligated with DNA fragments. The subsequent
assembly and transformation steps are the same as cloning with phages.
Bacterial Artificial Chromosome - BAC
Plasmidi di dimensioni enormi (derivati dal plasmide F
di E. Coli), consentono l’inserimento di frammenti di
DNA umano fino a 300 Kb, (sono a basso numero di
copie).
Il DNA viene inserito nei batteri mediante processo di
elettroporazione: uno shock elettrico provoca la
formazione transitoria di pori sulla membrana
batterica attraverso i quali passa il DNA.
Fosmidi: cosmidi modificati con l’inserzione dell’origine di
replicazione del fattore F. Come i cosmidi possono clonare
frammenti di 40 Kb, ma sono molto più stabili. I fosmidi sono a
basso numero di copie.
Batteriofago P1: possono inserire frammenti di 70- 100 Kb.
Basso numero di copie per cellula.
Vettori PAC: cromosomi artificiali basati su P1.
Vettori di lievito: YAC Yeast Artificial Chromosome
Possono accettare inserti grandi fino a 2Mb!
I componenti essenziali di un vettore YAC sono:
• un centromero di lievito (CEN);
• una sequenza di replicazione autonoma (ARS);
• due set di sequenze telomeriche di lievito (TEL);
• marcatori di resistenza per stabilire una selezione positiva in E.Coli;
• un sito ORI;
• siti di restrizione unici;
• marcatori di selezione auxotrofica (ura3, trp1, leu2, his3).
Una delle principali differenze tra i vettori plasmidici e quelli di lievito risiede nei
marcatori di selezione. A differenza dei batteri, infatti, i lieviti sono molto meno resistenti
agli antibiotici e la selezione viene di solito effettuata sfruttando la complementazione di
mutazioni auxotrofiche nel ceppo di lievito ospite
YAC
The yeast artificial chromosome (YAC)
vector is capable of carrying a large
DNA fragment (up to 2 Mb), but its
transformation efficiency is very low.
Essential components of YAC vectors
Centromers (CEN), telomeres (TEL) and
autonomous replicating sequence (ARS) for
proliferation in the host cell.
ampr for selective amplification and markers
such as TRP1 and URA3 for identifying cells
containing the YAC vector.
Recognition sites of restriction enzymes
(e.g., EcoRI and BamHI)
Procedure
The target DNA is partially digested by
EcoRI and the YAC vector is cleaved by
EcoRI and BamHI.
Ligate the cleaved vector segments with a
digested DNA fragment to form an artificial
chromosome.
Transform yeast cells to make a large number
of copies.
Cloning by the yeast
artificial chromosome
(YAC) vector.
MAC (mammalian artificial chromosome)
MAC
Caratteristiche ed applicazioni dei diversi
vettori di clonaggio
Vettore
Base
Plasmide Plasmidi multicopia naturali
Dimensione
limite
dell’inserto
Applicazioni principali
< 10 kb
Subclonaggio, clonaggio di
cDNA e saggi di espressione
Fago
Batteriofago
5 – 20 kb
Clonaggio di DNA genomico
e di cDNA, librerie di
espressione
Cosmide
Plasmide contenete un sito cos del
batteriofago
35 – 45 kb
Costruzione di librerie
genomiche
BAC
Plasmide del fattore F di E. coli
75 – 300 kb
Analisi di grandi genomi
YAC
Centromero, telomeri e sequenza
che si replica autonomamente di S.
cervisiae
100 – 1000
kb
Analisi di grandi genomi, topi
transgenici YAC
MAC
Centromero, telomeri ed origine di
replicazione di mammiferi
Da 100 a > 1 Biotecnologie animali e
Mb
terapia genica umana
Per il sequenziamento del DNA;
Per la mutagenesi sito specifica in vitro.
Il DNA ricombinante a singolo filamento può essere
ottenuto mediante sistemi di clonaggio che si basano su
batteriofagi a singolo filamento come i fagi M13, fd ed f1.
Si possono usare anche plasmidi che contengono
sequenze fagiche in grado di produrre DNA a singolo
filamento.
Vettori M13: genoma di 6,4 Kb.
Vettori fagemidi: piccolo frammento di fago filamentoso a
singolo filamento inserito in un plasmide
Le strategie di base per il clonaggio
consistono in tre tappe:
1) Scelta della fonte di DNA da usarsi per il
clonaggio
DNA
cromosomico
cDNA
2) Frammentazione del DNA con enzimi di
restrizione
Libreria Genica o Genoteca
Genomica
a cDNA
Quando si clona l’intero genoma di un organismo si dice che si
costruisce una libreria (library) genomica
Produzione di frammenti di restrizione
sovrapponibili attraverso digestione parziale
con SauIII
Costruzione di una
libreria genomica
attraverso l’uso di
vettori fagici
Costruzione di una
libreria di cDNA:
1) Estrazione RNA totale;
2) Isolamento mRNA.
Costruzione di una libreria di cDNA
Sintesi del cDNA (metodo 1)
Preparazione di una
libreria di cDNA
utilizzando il
batteriofago λ
Exon 1
DNA
(gene)Promotor
Exon 2
...
Exon n
hn-mRNA
mRNA
AAAAAAAAA
protein coding region
poly A tail
nucleus
cytoplasm
protein
AAAAAAAAA
translation
3) Screening della libreria:
• uso di sonde oligonucleotidiche;
• uso di anticorpi specifici;
• saggi di funzionalità della proteina stessa.
Tre tipi principali di sonde:
Sonde oligonucleotidiche, che sono sintetizzate
chimicamente e marcate ad un’estremità.
2) Sonde di DNA, che sono DNA clonati e possono
essere marcarte ad un’estremità o marcate
internamente durante la replicazione in vitro.
3) Sonde di RNA (ribosonde), che sono marcate
internamente durante la trascrizione in vitro da
stampi di DNA clonato.
1)
Oligonucleotide: ogni molecola formata da un piccolo
numero di nucleotidi legati tra loro da legami fosfodiestere
5’-3’.
Il numero di nucleotidi in questi piccoli acidi nucleici a
singolo filamento è variabile, ma spesso nell’ambito da 6 a
24 nucleotidi (da esamero a 24mer).
Sonde oligonucleotidiche: oligonucleotidi sintetici o
naturali usati negli studi di ibridizzazione per identificare e
studiare specifici frammenti di acidi nucleici.
Questo frammento di DNA od RNA è di solito marcato con
un tracciante (di solito radioattivo), così da permettere al
biologo molecolare di seguire l’ibridizzazione della sonda
con un DNA od un RNA sconosciuto.
Sonde omologhe: è esattamente complementare alla
sequenza dell’acido nucleico che interessa
identificare.
Sonde eterologhe: è simile, ma non esattamente
uguale al filamento complementare della sequenza
dell’acido nucleico che interessa studiare.
Metodo di
marcatura
Tipo di marcatura
Enzima
Esempio di
applicazione
Primer casuali
Uniforme
DNA polimersi di
Klenow
Sonde di
ibridazione
Trascrizione in vitro Uniforme
RNA polimerasi del Sonde di
fago SP6, T7 o T3
ibridazione,
tracciamento della
localizzazione
dell’RNA
Riempimento di
Klenow
Marcatura
all’estremità 3’
DNA polimerasi di
Klenow
Oligonucleotidi
Marcatura
dell’estremità 5’ o
dell’estremità 3’
Polinucleotide
Sonde di
chinasi T4
ibridizzazione,
Trasferasi terminale EMSA*
*Saggio di spostamento della mobilità elettroforetica
Footprinting con
DNAasi
Radioisotopo
Simbolo
Molecola
marcata
Emivita
Applicazione
Trizio
3H
Basi azotate,
nucleotidi
12,28 anni
Marcatura di RNA
e DNA in vivo.
Sonde per
ibridizzazione in
situ.
Carbonio 14
14C
cloramfenicolo
5730 anni
Saggio CAT.
Fosforo 32
32P
Nucleotidi
trifosfato
14,29 giorni
Sonde di
ibridizzazione.
Zolfo 35
35S
Amminoacidi
87,4 giorni
Marcatura delle
proteine (in vivo
ed in vitro).
Iodio 125
125I
Proteine
60,14 giorni
Marcatura di
anticorpi.
Screening di una
libreria:
Colony hybridization
uso di sonde
oligonucleotidiche
Una sonda oligonucleotidica deve
essere lunga abbastanza così che la sua
sequenza complementare si ritrova solo nel
clone d’interesse e non in altri cloni.
Questa condizione è soddisfatta da
sonde contenenti circa 20 nucleotidi.
Questo perché una sequenza specifica di
20 nucleotidi si ritrova una volta ogni 420
( 1012) nucleotidi. Poiché tutti i genomi
sono molto più piccoli ( 3 x 10 nucleotidi
per l’uomo) una sequenza specifica di 20
nucleotidi in un genoma si ritrova una
sola volta.
Per prepare una sonda specifica si può utilizzare il metodo seguente:
1.
2.
3.
4.
Purificazione della proteina d’interesse e determinazione della sequenza
amminoacidica mediante degradazione di Edman o spettrometria di
massa.
Sulla basa del codice genetico, si può predire la sequenza
oligonucleotidiche che codifica una determinata sequenza peptidica.
Tuttavia, dobbiamo ricordarci che il codice genetico è degenerato, cioè
molti amminoacidi sono codificati da più di un codone.
Poichè non si conoscono i codoni specifici usati per codificare la nostra
proteina, si devono sintetizzare oligonucleotidi che contengono tutte le
possibili combinazioni di codoni, in modo da assicurarci che almeno una
di esse si appai al gene in maniera perfetta.
Si determina la sequenza di un frammento peptico di 6 -7 amminoacidi
che può essere codificato dal più piccolo numero di possibili sequenze di
DNA.
Quindi: una sonda degenerata 20-mer è costituita da
una mistura di tutti gli oligonucleotidi 20-mer possibili,
che possono codificare una porzione selezionata di una
sequenza peptidica.
Sintesi di sonde
oligonucleotidiche
basata sulla sequenza
aminoacidica e
marcatura con fosforo
radioattivo – Sonde
Degenerate
Le EST sono sequenze parziali di cDNA lunghe circa 200 -400 bp
(anche dette etichette “tags”, perchè rappresentano soltanto una breve
porzione del DNA genomico).
Questo metodo usa i dati delle sequenze parziali di cDNA conservati
in banche dati e resi disponibili alla comunità scientifica mediante
internet.
Il metodo permette di identificare un singolo oligonucleotide e non
una miscela degenerata.
Un programma informatico usa il codice genetico per tradurre un EST
in una sequenza parziale di amminoacidi. Se si trova una
corrispondenza con la proteina in esame , l’EST fornisce la sequenza
del DNA di quella porzione del cDNA.
Si può così sintetizzare una sonda ed usarla per identificare in una
libreria l’intero clone di cDNA o di DNA genomico.
Può quindi essere sintetizzata chimicamente e radiomarcata una
singola sonda specifica lunga fino a 100 basi che è
perfettamente complementare ad una porzione del EST.
Alternativamente si può utilizzare la PCR per sintetizzare una
sonda complementare all’intera lunghezza del EST.
Attualmente, il “database” delle EST umane è così grande che
possono essere identificati EST che codificano sequenze
parziali di amminocidi della maggior parte delle proteine
umane isolate.
Production of Recombinant Proteins
Many proteins which
may be used for medical
treatment or for research
are normally expressed
at very low
concentrations. Through
recombinant DNA
technology, a large
quantity of proteins can
be produced. This
involves the cloning of
the gene encoding the
desired protein into an
"expression vector"
Production of recombinant proteins. (a) The expression vector
which must contain a
contains the lac promoter and its neighboring lacZ gene
promoter so that the
protein can be expressed. encoding b-galactosidase. Lactose or its analog IPTG will
stimulate the expression of b-galactosidase. (b) If lacZ is
replaced by the gene encoding the protein of interest, lactose
or IPTG will stimulate the expression of desired proteins.
Vettori di espressione
inducibili in batteri
Affinchè la proteina venga espressa è necessario
rispettare alcuni requisiti:
1) Il gene di interesse viene clonato a valle di un promotore;
2) Il frammento di DNA deve essere in frame;
3) E’ necessario un sito di legame al ribosoma a monte del codone di
inizio del gene.
I fattori che influenzano il livello di espressione di un
gene clonato sono:
1) Numero di copie del plasmide
effetti tossici;
la sovra-espressione può avere
2) Solubilizzazione delle proteine espresse;
Limitazioni per gli eucarioti:
1) I batteri non sono in grado di rimuovere gli introni;
2) Se la proteina funzionale è glicosilata la cellula ospite deve essere
una cellula eucariotica.
Etichette di
purificazione e
rivelazione
Phage display
L’efficienza di integrazione è molto bassa, per cui le
rare cellule che portano l’inserto devono essere
selezionate, rispetto alle altre.
Due diversi approcci:
Complementazione funzionale;
Marcatori a selezione dominante (resistenza ad
antibiotici tossici sia per le cellule batteriche che per
gli eucarioti neomicina e G418).
Librerie di espressione:
ricerca della proteina di
interesse attraverso anticorpi
specifici.
Plasmidi e fagi si utilizzano
come vettori di espressione.
Si possono anche costruire
proteine di fusione.
Gel Electrophoresis
Gel electrophoresis is a
technique for separating
charged molecules with
different sizes. Two kinds of
gels are commonly used:
agarose and polyacrylamide.
Agarose gels can be applied to a
wider range of sizes than
polyacrylamide gels. By using
standard agarose
electrophoresis, nuclei acids up
to 50 kb (100 to 50,000 bp) can
be separated. If Pulsed Field
Gel Electrophoresis is used, the
upper limit can be extended to
10 Mb. Polyacrylamide gels
may separate nucleic acids that
differ in length by only 1
nucleotide if their length is less
than 500 bp.
In a gel (either agarose or polyacrylamide), the negatively charged DNA fragments move toward the positive electrode
at a rate inversely proportional to their length. After the electric field is applied for a certain period, DNA fragments with
different lengths will be separated, which can be visualized by autoradiography or by treatment with a fluorescent dye
(e.g., ethidium bromide). The relationship between the size of a DNA fragment and the distance it migrates in the gel is
logarithmic. Therefore, from the band positions, the lengths of DNA fragments can be determined.
Separation of DNA fragments of different lengths by gel
electrophoresis. A gel is prepared by pouring a liquid containing
either melted agarose or unpolymerized acrylamide between two
glass plates a few millimeters apart. As the agarose solidifies or
the acrylamide polymerizes into polyacrylamide, a gel matrix
(orange ovals) forms consisting of long, tangled chains of
polymers. The dimensions of the interconnecting channels, or
pores, depend on the concentration of the agarose or acrylamide
used to form the gel. Because the pores are larger in agarose gels
than in polyacrylamide gels, the former are used to separate large
DNA fragments (≈500 bp to ≈20 kb) and the latter to separate
small DNA fragments (1 nucleotide to ≈2 kb). The mixture of
DNA fragments to be separated is layered in a well at the top of
the gel and an electric current is passed through the gel. DNA
fragments move toward the positive pole at a rate inversely
proportional to the log of their length, forming bands that can be
visualized by autoradiography (if the fragments are radiolabeled)
or by addition of a fluorescent dye such as ethidium. Agarose
gels can be run in a horizontal orientation; in this case, the melted
agarose is allowed to harden on a single horizontal glass or
plastic plate. This is not easily done with polyacrylamide gels
because oxygen in the atmosphere inhibits polymerization of
acrylamide. Gels are generally depicted with the origin at the top
and migration downward.
Elettroforesi su gel di agarosio: i gels di agarosio hanno un ambito di
separazione molto ampio, ma un potere di risoluzione piuttosto basso.
Elettroforesi su gel in campo pulsante (PFGE): per frazionare grandi
molecole di DNA come per es. il vettore YAC, l’elettroforesi su gel di
agarosio è eseguita con un campo elettrico pulsante. Il campo elettrico
periodico provoca il riorientamento delle molecole di DNA. Applicando
questo metodo si possono separare molecole di DNA fino a 200 – 400 Mb.
Elettroforesi su gel di poliacrilammide (PAGE): i gels di poliacrilammide
hanno un ambito di separazione piuttosto piccolo, ma un potere di
risoluzione piuttosto alto. La poliacrillamide si usa per separare frammenti
più corti di 500 bp, ma in appropriate condizioni, si risolvono facilmente
frammenti di DNA che differiscono in lunghezza di un solo nucleotide.
Questa alta risoluzione è necessaria per alcune applicazioni, come il
sequenziamento del DNA, il footprinting con DNAasiI, ed i saggi di
spostamento della mobilità elettroforetica.
Membrane-hybridization
assay for detecting nucleic
acids. This assay can be used
to detect both DNA and RNA,
and the radiolabeled
complementary probe can be
either DNA or RNA.
Following gel electrophoresis, probes are often used to detect
specific molecules from the mixture. However, probes cannot be
applied directly to the gel. The problem can be solved by three types
of blotting methods: Southern blotting, Northern blotting and
Western blotting.
Southern blotting
Southern blotting is a technique for detecting specific DNA
fragments in a complex mixture. The technique was invented in
mid-1970s by Edward Southern. It has been applied to detect
Restriction Fragment Length Polymorphism (RFLP) and Variable
Number of Tandem Repeat Polymorphism (VNTR). The latter is
the basis of DNA fingerprinting.
Southern blotting. (a) The DNA to be analyzed is
digested with restriction enzymes and then
separated by agarose gel electrophoresis. (b) The
DNA fragments in the gel are denatured with
alkaline solution and transferred onto a
nitrocellulose filter or nylon membrane by blotting,
preserving the distribution of the DNA fragments
in the gel. (c) The nitrocellulose filter is incubated
with a specific probe. The location of the DNA
fragment that hybridizes with the probe can be
displayed by autoradiography.
The Southern blot technique for detecting the presence of
specific DNA sequences following gel electrophoresis of a
complex mixture of restriction fragments. The diagram depicts
three restriction fragments in the gel, but the procedure can be
applied to a mixture of millions of DNA fragments. A similar
procedure, called Northern blotting, is used to detect specific
RNA sequences. [See E. M. Southern, 1975, J. Mol. Biol.
98:508.]
Southern Blot
Fluorescence in situ hybridization (FISH), the assay of choice
for localization of specific nucleic acids sequences in native
context, is a 20-year-old technology that has developed
continuously. Over its maturation, various methodologies and
modifications have been introduced to optimize the detection of
DNA and RNA. The pervasiveness of this technique is largely
because of its wide variety of applications and the relative ease
of implementation and performance of in situ studies. Although
the basic principles of FISH have remained unchanged, high
sensitivity detection, simultaneous assay of multiple species,
and automated data collection and analysis have advanced the
field significantly.
State of the art in FISH. (A) Many transcription sites (10) detected using barcoded probes can
determine the gene expression pattern of each cell (Levsky et al., 2002) (B-D).
Levsky J M , Singer R H J Cell Sci 2003;116:2833-2838
©2003 by The Company of Biologists Ltd



![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)
![ESTRAZIONE DNA DI BANANA [modalità compatibilità]](http://s1.studylibit.com/store/data/004790261_1-44f24ac2746d75210371d06017fe0828-300x300.png)



![(Microsoft PowerPoint - PCR.ppt [modalit\340 compatibilit\340])](http://s1.studylibit.com/store/data/001402582_1-53c8daabdc15032b8943ee23f0a14a13-300x300.png)