- Giornale Italiano di Cardiologia")
CASO CLINICO
Linfoma di Burkitt cardiaco primitivo
in un giovane ragazzo africano
Enrico Donegani1, Jean Claude Ambassa1, Charles Mvondo1, Alessandro Giamberti1, Antonio Ramponi2,
Andrea Palicelli2, David Chelo3
1
St. Elizabeth Catholic General Hospital, Shisong Cardiac Centre, Camerun
S.C. di Anatomia ed Istologia Patologica, Azienda Ospedaliero-Universitaria “Maggiore della Carità”, Novara
3
Fondation Chantal Biya, Douala, Camerun
2
Burkitt lymphoma is a non-Hodgkin lymphoma that is endemic in the Equatorial Belt of Africa, usually affecting
children and adolescents with primary head-neck or abdominal involvement. Primary cardiac lymphomas are
rare entities (1.3% of all primary cardiac tumors) of difficult clinical identification. Delayed discovery contributes to significant mortality.
We report a case of a primitive Burkitt lymphoma in a 14-year-old Cameroonian immunocompetent child, presenting with signs and symptoms of severe right inflow impairment. Echocardiography revealed a right atrial mass involving the right atrial ventricular junction. Surgical excision and chemotherapy regimens, administered according to established protocols, were effective in inducing complete remission at 6 months.
Key words. Burkitt lymphoma; Cardiac neoplasm; Primary cardiac lymphoma.
G Ital Cardiol 2013;14(6):481-484
INTRODUZIONE
I linfomi primitivi cardiaci (LPC) sono rari, con un’incidenza
dell’1.3% di tutti i tumori cardiaci primitivi1. Il linfoma di Burkitt (LB) è una neoplasia dei linfociti B, endemico nei paesi della fascia equatoriale africana e in Papua Nuova Guinea, strettamente associato al virus di Epstein-Barr (EBV), e sporadico in
Europa e nelle Americhe. Esso prende il nome dal chirurgo Denis Burkitt che per primo lo descrisse nel 1958 mentre lavorava in Uganda2.
Più frequentemente, specie nei bambini, si osserva una presentazione clinica extralinfonodale, interessante il distretto testa-collo (le ossa del massiccio facciale sono il sito di esordio
della malattia in oltre il 50% delle forme endemiche, con disseminazione verso l’alto fino a coinvolgere l’orbita) o quello addominale (sede maggiormente coinvolta nelle forme sporadiche), con possibile localizzazione a livello di reni, ovaie, retroperitoneo e linfonodi addominali, mammella e, meno frequentemente, sistema nervoso centrale2. L’esordio linfonodale è invece più comune nelle forme sporadiche degli adulti e nelle forme associate ad immunodeficienza2.
Agli inizi degli anni 2000 risultavano descritti in letteratura
solo 50 casi di LPC (la maggior parte linfomi diffusi a grandi
cellule)3-5, riguardanti preferenzialmente maschi adulti, generalmente oltre i 40 anni di età5, solo pochi di essi insorti in pazienti immunocompetenti4. In base alle nostre ricerche bibliografiche sono stati descritti rari casi di LB a primitiva localizza-
© 2013 Il Pensiero Scientifico Editore
Ricevuto 01.10.2012; nuova stesura 15.11.2012; accettato 16.11.2012.
Gli autori dichiarano nessun conflitto di interessi.
Per la corrispondenza:
Dr. Enrico Donegani Strada Pecetto 257/4, 10131 Torino
e-mail: [email protected]
zione cardiaca1,3-10, alcuni dei quali in pazienti anziani1,3,7-9 o immunocompromessi8,10, solo pochi in bambini immunocompetenti4,5.
Presentiamo il caso di una forma endemica di LB primitivo
cardiaco (LBPC), riscontrato in un giovane ragazzo quattordicenne, camerunense, immunocompetente, ricoverato con gravi segni e sintomi di scompenso cardiaco destro.
CASO CLINICO
Un ragazzo africano di 14 anni veniva ricoverato presso il St. Elizabeth Catholic General Hospital, Shisong Cardiac Centre in
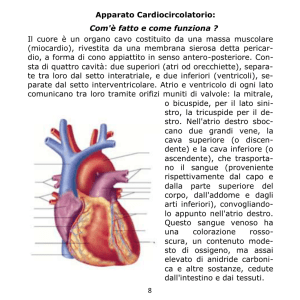
Camerun per grave dispnea e quadro clinico di scompenso cardiaco congestizio. La radiografia del torace rivelava ingrandimento dell’ombra cardiaca e versamento pleurico destro. L’ecocardiografia bidimensionale transtoracica mostrava una massa
che riempiva quasi completamente l’atrio destro con associata
occlusione intermittente dell’ostio della valvola tricuspide (Figura 1). I pochi ed essenziali esami ematochimici eseguiti risultavano non significativi. Il paziente risultava negativo ai test per
l’AIDS. Con il sospetto clinico preoperatorio di mixoma cardiaco in atrio destro, l’intervento chirurgico veniva eseguito con
criterio di emergenza. In bypass cardiopolmonare totale si procedeva alla rimozione della massa (macroscopicamente multilobulata, di colorito bianco-giallastro, friabile, di consistenza
gelatinosa, con piccole aree emorragiche) con la necessaria
asportazione del tetto e della parete libera dell’atrio destro
adiacente alla crista terminalis, sede d’impianto della neoformazione, strutture ricostruite poi con pericardio autologo. La
valvola tricuspide risultava indenne da lesioni. Il decorso postoperatorio del paziente fu regolare. Nell’impossibilità di poter
eseguire in loco l’esame istologico, il materiale venne fissato in
formalina ed inviato successivamente presso la Struttura ComG ITAL CARDIOL | VOL 14 | GIUGNO 2013
481
E DONEGANI ET AL
Figura 1. Immagine ecocardiografica bidimensionale apicale 4 camere che mostra una grossa massa occupante la cavità atriale destra con
impegno dell’ostio valvolare atrioventricolare.
plessa di Anatomia ed Istologia Patologica dell’Azienda Ospedaliero-Universitaria “Maggiore della Carità” di Novara.
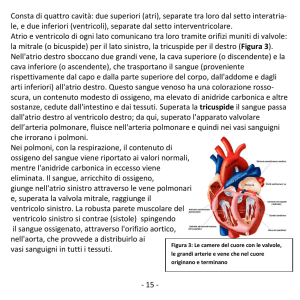
L’esame istologico mostrò una popolazione tumorale diffusamente monomorfa e di aspetto coeso, costituita da cellule di
medie dimensioni con citoplasma basofilo scarso, nuclei rotondi – di dimensione simile a quelli degli istiociti dispersi nell’infiltrato neoplastico con pattern “a cielo stellato” –, multipli piccoli nucleoli e cromatina addensata; numerosi i corpi apoptotici (alcuni fagocitati all’interno degli istiociti) e le figure mitotiche (Figura 2).
Le indagini immunoistochimiche rivelarono che le cellule tumorali erano elementi linfoidi B a fenotipo simile alle cellule
Figura 2. In alto a sinistra: sezione istologica della massa che mostra
una popolazione linfomatosa di medie dimensioni, mitoticamente attiva, con scarso citoplasma basofilo e multipli nucleoli, intervallati da
corpi apoptotici ed istiociti (ematossilina-eosina, 40x). In alto a destra:
microfocolaio di necrosi con numerosi corpi apoptotici (ematossilinaeosina, 40x). In basso a sinistra: la reazione immunoistochimica con anticorpo Ki-67 per cellule proliferanti evidenzia una positività nucleare in
oltre il 95% della popolazione neoplastica (40x). In basso a destra: si
evidenzia una marcata positività (granuli nucleari) degli elementi linfomatosi per EBV- EBER, mentre risultano negativi gli elementi istiocitari
ed endoteliali (ibridazione in situ, 40x).
482
G ITAL CARDIOL | VOL 14 | GIUGNO 2013
centrogerminative dei follicoli linfatici (CD20+, CD3-, bcl6+,
CD10+, bcl2-, MUM1-).
Sulla base dell’aspetto morfologico del clone neoplastico,
del suo immunofenotipo e dell’elevato indice di proliferazione
(>95%), stabilito grazie a metodica immunoistochimica con anticorpo Ki-67 (Figura 2), fu posta diagnosi di una diffusa infiltrazione tissutale da parte di linfoma B aggressivo, coerente
con LB.
L’utilizzo della metodica molecolare FISH (fluorescence in
situ hybridization) consentì di identificare la traslocazione reciproca t(8;14) (q24;q32), caratteristica della maggioranza dei
casi di LB. Inoltre fu possibile individuare con metodica di ibridazione in situ la presenza intratumorale di una diffusa positività ad EBV- EBER (Figura 2).
Ottenuta la diagnosi precisa, il paziente venne sottoposto a
trattamento chemioterapico secondo il protocollo terapeutico
CHOP (4 cicli comprendenti cortisonico, adriamicina, vincristina e ciclofosfamide) presso l’ospedale generale di Douala (Camerun). A 6 mesi di distanza dal termine della chemioterapia (9
mesi dall’intervento chirurgico), il paziente presenta completa
remissione della malattia, è in buone condizioni generali, senza segni clinici e strumentali di recidive.
DISCUSSIONE
Un interessamento cardiaco secondario da parte di linfomi maligni disseminati non è un evento infrequente (circa il 20% dei
casi autoptici)8, mentre l’identificazione di una localizzazione
unica e primitiva a livello del cuore e/o del pericardio risulta di
raro riscontro negli immunocompetenti (1.3% dei tumori cardiaci; 0.5% dei linfomi extranodali), più comune in pazienti immunodepressi (AIDS) o immunosoppressi (post-trapianto cardiaco o renale)1,5,8,10.
I LPC sono tipicamente di tipo non Hodgkin e quasi tutti
(l’80% dei casi studiati) sono neoplasie a cellule B aggressive4,8.
La prognosi è infausta per la loro rarità e per la presentazione
clinica aspecifica, che rende la diagnosi difficile; il riscontro è
frequentemente post-mortem8.
Un LPC può manifestarsi clinicamente con insufficienza cardiaca congestizia, dolore toracico, cardiomegalia, versamento
pleurico/pericardico, tamponamento cardiaco, pericardite, dispnea, aritmie, sindrome della vena cava superiore4,5,8,10. Possibili eventi sono anche infarto miocardico o morte improvvisa5.
Sedi di interessamento cardiaco da parte di LPC sono, in ordine decrescente di frequenza, l’atrio destro, il ventricolo destro, l’atrio sinistro, il setto interatriale e il setto interventricolare (talora simulando una cardiomiopatia ipertrofica)11, con un
coinvolgimento multicamerale in oltre il 75% dei casi8. Essi
dunque prediligono un’insorgenza nel cuore destro, mentre i
mixomi ed i sarcomi sono più frequenti nel cuore sinistro
(atrio)4,8. Differentemente, l’angiosarcoma si localizza come una
massa ben definita a sede preferenziale atriale destra (vicino alla vena cava inferiore sulla parete libera senza coinvolgere il setto) con frequente necrosi centrale (solitamente nei linfomi il riscontro di necrosi o emorragia è meno comune)8. Inoltre, in
confronto ai sarcomi, i linfomi raramente interessano il tessuto
valvolare cardiaco, sebbene talvolta possano intaccare la valvola tricuspide8.
Pattern di infiltrazione diffusa o nodulare da parte di LPC
possono esser identificati a livello di epicardio, miocardio e/o
endocardio, con occasionale riscontro di una massa polipoide
LINFOMA DI BURKITT PRIMITIVO IN GIOVANE AFRICANO
intracavitaria (come nel caso da noi descritto) con rischio di
ostruzione di flusso o di embolizzazione4,10.
Possibile inoltre l’infiltrazione del tessuto di conduzione cardiaco (ad es. nodo atrioventricolare) con manifestazione sintomatologica di disturbi della conduzione ed arresto cardiaco1,4,5,8,10.
Il pericardio può esser sede di localizzazione linfomatosa
oppure mostrare una pericardite reattiva con versamento pericardico o tamponamento cardiaco4,8.
Il LB è un linfoma aggressivo a frequente presentazione extranodale o leucemica acuta, esistente in tre varianti cliniche
(endemica, sporadica, immunodeficienza-associata)2. La variante endemica rappresenta la più comune forma di tumore
maligno pediatrico nell’Africa Equatoriale, con interessamento
mascellare-mandibolare e delle ossa facciali (anche orbitarie)
come sito di presentazione in circa il 50% dei casi, e possibile
coinvolgimento ileale distale, ciecale, omentale, gonadico, renale, tiroideo, mammario, delle ossa lunghe o delle ghiandole
salivari; rara la localizzazione midollare, assente la forma leucemica2. Nel 100% dei casi si osserva presenza di genoma di
EBV nella maggioranza delle cellule linfomatose2. La maggior
parte dei casi mostra una caratteristica attivazione dell’oncogene c-myc a seguito di una sua traslocazione dalla banda
8q24 al gene delle catene pesanti delle immunoglobuline (banda 14q32) o, con minor frequenza, a quello delle catene leggere λ (22q11) o κ (2p12)2.
Raramente in letteratura sono riportati casi di LBPC1,3-10.
Zyssman et al.6 individuarono multiple masse intracardiache
(atrio destro, ventricolo destro, atrio sinistro) da LBPC in un paziente di 29 anni. Carfagna et al.3 riferirono una massa atriale
destra inoperabile da LBPC in una donna di 78 anni con dispnea ed effusione pericardica e pleurica sinistra. Hoffmeier et
al.7 riscontrarono ecocardiograficamente, in un maschio di 85
anni con progressivi segni di deterioramento, un LBPC vegetante in sede atriale destra, prolassante fino a un terzo nel ventricolo destro (la valvola tricuspide non appariva chiaramente
distinguibile dalla massa alla tomografia computerizzata cardiaca). Chalabreysse et al.4 riportarono un caso di LBPC in un
maschio immunocompetente di 9 anni che presentava all’ecocardiografia transtoracica tre masse tumorali (submitralica con ampia base d’impianto sul setto interventricolare, atriale destra, infundibolare polmonare). Meshref et al.5 riscontrarono in un maschio di 10 anni la presenza di multiple localizzazioni intracardiache di LBPC (sede submitralica con incontinenza valvolare, setto interatriale, infundibolo polmonare, atri
destro e sinistro). De Filippo et al.8 descrissero un caso di LBPC
(definito linfoma Burkitt-like) in un maschio immunocompromesso di 70 anni con diabete mellito, cirrosi da virus dell’epatite C e cardiomiopatia dilatativa: le indagini radiologiche mostrarono una neoformazione atriale destra con invasione della vena cava superiore e diffusa infiltrazione del setto interatriale e dell’atrio sinistro8. Fatimi et al.9 riferirono di un paziente
anziano con LBPC non resecabile, a cui era stata posta inizialmente, in base alla sintomatologia clinica, diagnosi preoperatoria di trombo intracardiaco. Singh et al.10 descrissero un LBPC
in un bambino di 4 anni con AIDS, esordito con sintomatologia clinica di insufficienza cardiaca destra acuta a causa di
un’ostruzione di afflusso ventricolare destro provocata da una
massa linfomatosa friabile in atrio destro con embolizzazione
cronica ai vasi polmonari ed ipertensione polmonare fatale secondaria. Kamona et al.11 presentarono due casi di coinvolgimento cardiaco da LB, uno con associato interessamento dei
linfonodi addominali, l’altro senza precisarne la primitività cardiaca. Santini et al.1 descrissero un LBPC (definito LB atipico),
sporadico, in un maschio di 67 anni immunocompetente, esordito con un episodio sincopale da transitoria ipotensione: l’esame ecografico rivelò una massa endocavitaria biatriale con occlusione intermittente della vena cava superiore alla giunzione
atriale destra ed interferenze di flusso a livello delle valvole tricuspide e mitrale.
In conclusione, la diagnostica ecocardiografica è lo strumento di scelta per il riscontro della massa linfomatosa cardiaca; la tomografia computerizzata e la risonanza magnetica permettono di meglio definire l’infiltrazione del miocardio, l’interessamento pericardico e, soprattutto, accertare la presenza e
l’estensione di lesioni extracardiache8. Tuttavia, soltanto lo studio microscopico permette la precisa definizione istopatologica
della neoplasia, la sua prognosi e la successiva terapia. Il trattamento dei LPC non è ancora stato ben definito. La sola terapia radiante non sembra sortire alcun effetto sulla sopravvivenza12. L’asportazione chirurgica della massa tumorale da
LBPC, associata a opportuno trattamento chemioterapico, può
consentire la guarigione nel 90% dei casi nelle forme a basso
stadio e nel 60-80% nelle forme avanzate1.
RIASSUNTO
Il linfoma di Burkitt è un linfoma non Hodgkin endemico nei paesi della fascia equatoriale africana, interessante generalmente bambini ed adolescenti e caratterizzato da un primitivo insediamento a
livello del tratto testa-collo o addominale. Il reperto di un linfoma
a primitività cardiaca è di raro riscontro (incidenza dell’1.3% fra
tutti i tumori cardiaci) e di difficile ed infrequente diagnosi precoce, il che contribuisce alla sua alta mortalità.
Viene riportato il caso di un linfoma di Burkitt primitivo a localizzazione cardiaca riscontrato in un giovane ragazzo immunocompetente di 14 anni, camerunense, ricoverato con gravi segni e sintomi di scompenso cardiaco destro. All’ecocardiogramma si evidenziava una massa a livello dell’atrio destro e della giunzione
atrioventricolare. La rimozione chirurgica ed il successivo trattamento combinato chemioterapico secondo i protocolli standard
hanno determinato la buona remissione a 6 mesi del tumore.
Parole chiave. Linfoma cardiaco primitivo; Linfoma di Burkitt; Neoplasia cardiaca.
BIBLIOGRAFIA
1. Santini F, Innocente F, Gilioli E, et al. Primary bi-atrial Burkitt lymphoma with severe
inflow impairment in an immunocompetent
patient. Cardiovasc Pathol 2009;18:123-5.
2. Leoncini L, Raphaël M, Stein H, Harris
NL, Jaffe ES, Kluin PM. Burkitt lymphoma.
In: Swerdlow SH, Campo E, Harris NL, et al.,
eds. WHO classification of tumours of
haematopoietic and lymphoid tissues. Lyon:
WHO Press, 2008:262-4.
3. Carfagna P, Redondi A, Taglietti F, Battista M, d’Amati G, Brandimarte C. Difficulties in the diagnosis of primary cardiac lymphomas. Haematologica 2000;85:770-2.
4. Chalabreysse L, Berger F, Loire R, Devouassoux, Cordier JF, Thivolet-Bejui F. Primary cardiac lymphoma in immunocompetent patients: a report of three cases and review of the literature. Virchows Arch 2002;
441:456-61.
5. Meshref M, Sassolas F, Schell M, et al. PriG ITAL CARDIOL | VOL 14 | GIUGNO 2013
483
E DONEGANI ET AL
mary cardiac Burkitt lymphoma in a child.
Pediatr Blood Cancer 2004;42:380-3.
6. Zyssman I, Cantor A, Steyn M, Meyer T.
Multiple intracavitary cardiac masses; an uncommon presentation of African Burkitt’s
lymphoma. Int J Cardiol 1992;37:421-3.
7. Hoffmeier A, Semik M, Schmid CH, et
al. Primary Burkitt lymphoma of the heart diagnosis and therapy. Z Kardiol 2002;91:
347-51.
8. De Filippo M, Chernyschova N, Maffei
484
G ITAL CARDIOL | VOL 14 | GIUGNO 2013
E, et al. Primary cardiac Burkitt’s type lymphoma: transthoracic echocardiography,
multidetector computed tomography and
magnetic resonance findings. Acta Radiol
2006;47:167-71.
9. Fatimi S, Sheikh S, Shah Z, Shafiq M. Intra-cardiac Burkitt’s lymphoma mimicking
acute pulmonary embolism. J Coll Physicians Surg Pak 2006;16:536-7.
10. Singh AS, Dave DJ, Thanvi S, Atre DA,
Parikh P, Patel NH. Fatal secondary pul-
monary hypertension due to cardiac involvement in AIDS-associated Burkitt’s lymphoma. Indian J Med Sci 2006;60:380-4.
11. Kamona AA, El-Khatib MA, Swaidan
MY, et al. Pediatric Burkitt’s lymphoma: CT
findings. Abdom Imaging 2007;32:381-6.
12. Liang R, Yu CM, Au WY, Choy CK,
Kwong YL. Diagnosis in oncology. Case 2:
Secondary lymphoma of the heart manifesting as intracavitary masses. J Clin Oncol
2000;18:1998-9.
 - Giornale Italiano di Cardiologia")