Alcune classi particolari di malattie
genetiche
• Malattie Mitocondriali
• Imprinting (sindrome di Prader-Willi e
sindrome di Angelman)
• Malattie da triplette (X-fragile, SCA, Corea
di Huntington). Concetto di premutazione
ed anticipazione
Genetica mendeliana classica:
Le mutazioni sono stabili e vengono trasmesse
invariate da una generazione all’altra.
ECCEZIONE:
Mutazioni instabili dovute ad espansione di
DNA ripetuto Triplette trinucleotidiche in tandem sono abbastanza
comuni nel nostro genoma
Ci sono 10 possibili triplette:
AAC/GTT ACT/AGT
AAG/CTT AGG/CCT
AAT/ATT ACT/GAT
ACC/GGT CAG/CTG
ACG/ CGT CCG/CGG
Alcune triplette CAG/CTG e CCG/GGC mostrano un
comportamento anomalo: al di sopra di una certa soglia
diventano estremamente instabili in mitosi e meiosi MUTAZIONI DA DNA INSTABILE
……..CGGCGGCGG……..!
..…CGGCGGCGGCGGCGGCGGCGGCGG…..!
……..CAGCAGCAG……..!
..…CAGCAGCAGCAGCAGCAGCAGCAG…..!
AMPLIFICAZIONE DI TRIPLETTE DI DNA CHE AVVIENE
DI GENERAZIONE IN GENERAZIONE NUOVO MECCANISMO DI MUTAZIONE E DI TRASMISSIONE GENETICA
MECCANISMO!
Ci dovremmo aspettare sia espansioni
che riduzioni di triplette."
"
"
C’e’ una tendenza all’espansione!"
Genetica delle malattie da triplette:
• Concetto di premutazione
• Instabilita’ somatica e germinale
• Anticipazione
Concetto di Premutazione
Ripetute al limite superiore del normale, che di per se’ non
causano la malattia, ma possono espandersi e diventare
patogenetiche nella generazione successiva 22 29
82 22 83
>200
22 90
>200
29 80
~500
Instabilita’ genetica
normal: 5 - 37
premutation: 38 - 50 (minimal transition threshold: 35)
very late onset: 42 - 180
adult onset: few - several hundreds
congenital disease: greater than approx. 1000
Generic Pedigree of a Dominantly Inherited
Due to CAG Repeat Instability
SCA1
INSTABILITA’ GERMINALE
S. CHONG et al. 1995
Instabilita’ germinale
Origine parentale della tripletta può
essere importante:"
" "
• La trasmissione paterna ha un rischio aumentato di
espansione nel caso delle malattie da poliglutamine (CAG
codifica per glutamina): i casi piu’ gravi sono ereditati dal
padre.
• Le grandi espansioni di triplette alla base della distrofia
miotonica e della sindrome dell’X fragile non vengono in
genere trasmesse dal padre affetto: c’è probabilmente una
selezione a livello spermatico.
Es. nella distrofia miotonica, madri lievemente affette
possono avere figli con una gravissima forma di miopatia
congenita"
Anticipazione
= peggioramento della malattia da una generazione alla successiva Esempio nella distrofia miotonica:
I
I 1 = caratatta, a 50 anni insorge il il difetto muscolare
II
II 2 = debolezza muscolare insorta nell’
adolescenza, cataratta, cardiomiopatia
III
III 1 = neonato affetto da miotonia congenita ( insufficienza respiratoria ipotonia ecc.)
Zoghbi & Orr 1995
Relationship between the age of onset and the number of CAG
repeats on the expanded alleles of individuals affected with SCA1. PATHOGENIC EFFECT OF GLUTAMINE REPEATS
Relationship between the age of onset and size of the CAG repeat in SCA1-3
An inverse correlation is observed between the size of expansion and age of onset
The effect of expansion is different for each mutation
Qual e’ il meccanismo con cui
l’espansione da triplette causa la
malattia?"
1. Perdita di funzione
2. Acquisizione di una nuova funzione"
Posizione della tripletta nel gene
La tripletta espansa si trova nella regione codificante. Malattie di tipo I
HD
SCA1
DRPLA
MJD
121
Affected
Normal
DRF
3’ UTR
5’ UTR
Normal
Affected
SCA2
SBMA
SCA6
Alleli normali tra 10-30, alleli espansi tra 40-200
Il tratto poliglutaminico conferisce una nuova, patologica funzione
alla proteina
Type I Trinucleotide Disease
REDDY & HOUSMAN 1997
STORIA
• George Huntington
– medico americano; nel 1872 a 22 anni, descrisse il “ballo di
San Vito”
• “if by chance these children go through life
without it, the thread is broken and the children
and grand-children of the original shakers may
rest assured that they are free from the disease” George Huntington
Corea di Huntington"
"
Malattia neurologica devastante dovuta alla
degenerazione dei neuroni del corpo striato
Autosomica dominante
Eta’ di insorgenza intorno ai 40 anni
Sintomi iniziali irritabilita’, depressione; progredisce
con gravi disturbi del movimento (corea) e demenza"
"
COREA DI HUNTINGTON
• Autosomica dominante
• Anticipazione
• Origine paterna delle forme piu’ gravi
Numero CAG
<28
Normale, non a rischio
29-34
Normale, ma la
generazione successiva
e’ a rischio
35-39
Alcuni sviluppano la
malattia, la generazione
successiva e’ a rischio
>40
Sviluppano la malattia
Q"
Huntingtin"
"
"
• Espressa in tutti i neuroni"
• Funzione normale:
"regolazione della trascrizione"
"
"
"
"protezione dalla apoptosi"
"
Espansione poliglutamine: acquisizione di una nuova
funzione tossica per la cellula, tendenza a formare
aggregati nucleari"
FORMAZIONE DI AGGREGATI INTRANUCLEARI"
Caratteristiche comuni delle malattie
da poliglutamine:
• Progressivo fenotipo neurologico dovuto a perdita di neuroni
• Sono tutte caratterizzate da un’acquisizione di funzione della proteina dovuta al tratto poliglutaminico • Espressione ubiquitaria, ma fenotipo selettivo
• Il contesto proteico media la selettiva vulnerabilita’ neuronale
La tripletta si trova in regioni non
codificanti. Malattie di tipo II
FRAXA
MD
FA
Full mutation
Pre/protomutation
Normal
5’ UTR
Exon
Intron
Exon
Intron
Exon
3’ UTR
1997 Current Opinion in Cell Biology
Alleli normali tra 5-50 ripetute, alleli espansi hanno centinaia,
o migliaia di copie.
La ripetuta inibisce l’espressione del gene, causando una
perdita di funzione della proteina
Type II Trinucleotide Disease
REDDY & HOUSMAN 1997
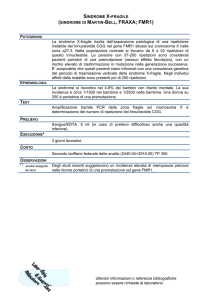
SINDROME DELL’X
FRAGILE
E’ una delle forme piu’ comuni di ritardo
mentale ereditario, e rappresenta il 15-20% dei
ritardi mentali legati all’X.
Incidenza: 1/4000 maschi; 1/8000 femmine
Si trasmette come tratto legato all’X recessivo
con penetranza ridotta (80% nei maschi, 30%
nelle femmine)
Fragile X Cromosome
Quando i linfociti in coltura vengono privati di folato si evidenziano i siti fragili
QUADRO CLINICO
• Ritardo mentale"
• Facies anormale con mandibola
prominente e grosse orecchie"
• Macroorchidismo"
• Anomalie comportamentali (iperattivita’
autismo) nei maschi"
Fragile X Syndrome
Fragile X Syndrome
20-50
(CGGCGGCGGCGG)
5’
ATG
3’
Gene FMR1
normale
3’
Gene FMR1
premutazione
STOP
50-200
(CGGCGGCGGCGGCGGCGGCGG)
5’
ATG
STOP
200-1000
(CGGCGGCGGCGGCGGCGGCGG)
5’
3’
ATG
STOP
Gene FMR1
pazienti
FMR1
• FMR1 codifica per una proteina con un
possibile ruolo nel trasportare mRNA dal
nucleo alla macchina traduzionale
• Piu’ del 99% delle mutazioni in FMR1 sono
dovute all’espansione di una sequenza
ripetuta CGG nel 5’ non codificante del
gene
Quando il numero di triplette supera la soglia di 230 l’estremita’ 5’ del gene FMR1
diventa ipermetilata e si blocca la trascrizione
Cause di variabilita’ fenotipica
• Dimensione della ripetuta CGG (premutazione/
mutazione)
• Metilazione
• Mosaicismo somatico (instabilita’ della ripetuta):
un maschio con mosaicismo somatico avra’
generalmente un ritardo mentale piu’ lieve rispetto
ad un individuo con la stessa espansione in tutte le
cellule.
X FRAGILE: LA genetica rivisitata
Legata all’X
Necessaria una mutazione in due step per l’espressione
della malattia
Non ci sono nuove mutazioni: tutti i maschi affetti hanno
madri portatrici
Alto rischio di trasformazione dalla premutazione alla
mutazione nella linea germinale femminile
Possibile trasmissione da parte di maschi normali:
attraverso le figlie portatrici sani ai loro nipoti
La tendenza all’espansione della CGG si verifica
solo quando la premutazione e’ trasmessa dalla
madre
Quando la premutazione e’ trasmessa dal padre rimane stabile quindi le
figlie femmine ricevono la premutazione senza che ci sia espansione ed
i figli maschi ricevono l’Y e non sono a rischio di ereditare la
premutazione
REGOLA
LE MALATTIE MONOGENICHE SI TRASMETTONO
SECONDO PATTERN BEN DEFINITI (AD, AR, XL)
ASSENZA DI RISCHIO DI RICORRENZA NELLE
MUTAZIONI DE NOVO
PER UN LOCUS AUTOSOMICO IL FIGLIO EREDITA
UN ALLELE DA CIASCUNO DEI DUE GENITORI
ENTRAMBI GLI ALLELI DI UN LOCUS AUTOSOMICO SONO UGUALMENTE ESPRESSI
LE MUTAZIONI SONO STABILI
ECCEZIONE
⇒
⇒
⇒
⇒
⇒
EREDITA’ MITOCONDRIALE
Es. Neuropatia ottica di Leber
MOSAICISMO GERMINALE
Es. Osteogenesis imperfecta
DISOMIA UNIPARENTALE
Es. PWS e AS
IMPRINTING
Es. Prader Willi /Angelman
MUTAZIONI IN STABILI
X Fragile
LA TERAPIA GENICA
1-Definizione e principi
2-Strumenti (Vettori)
3-Strategie
4-Trials Clinici
5-Successi e fallimenti
Terapia Genica
Trattamento di Malattie attraverso il Trasferimento e
l’Appropiata Espressione di una Informazione Genica
Funzionale in una Cellula Bersaglio
Terapia Genica
trattamento di malattie attraverso il trasferimento di
materiale genetico [DNA RNA]
Il Dogma:
DNA
DNA
nucleus
cytoplasm
RNA
mRNA
PROTEIN
AAAA
Strategie Terapeutiche
germinale non accettata su base Etico Scientifica
Strategie Terapeutiche
Terapia Genica In Vivo
Terapia Genica Ex Vivo
Strategie Terapeutiche
Trasferimento genico in vivo
Fibrosi Cistica
- Tessuto Specifico
- Gene Specifico
Emofilia
- Tessuto Aspecifico
- Gene Specifico
Tumore Cerebrale
- Cellula Specifico
- Gene Esogeno
1-Definizione e principi
2-Strumenti (Vettori)
3-Strategie
4-Trials Clinici
5-Successi e fallimenti
IDEALMENTE:
• Sistema di Trasferimento Genico Efficiente
e Specifico
• Espressione Stabile, Costante/Modulabile
• Mancanza di Tossicita’
• Non immunogeno
• Facilita’ di Produzione
Trasferimento Genico
Metodi per Introdurre Acidi Nucleici in Cellule
Vettore = Agente che Permette l’ Ingresso del Gene
Terapeutico
Vettori Virali
Interazione con un Recettore della Cellula Bersaglio
Vettori Non Virali
Interazione Fisica con la Membrana Cellulare
Vettori Non Virali
Infezione (Virus)
Trasduzione (Vettore)
I Virus Rappresentano Vettori Naturali Altamente Evoluti
per il Trasferimento di Informazione Genica Estranea
Ciclo Vitale del Virus
Infezione / Replicazione
Conversione di un virus in un vettore:
eliminazione dal genoma dei geni virali…..
…che vengono poi provvisti in trans nel processo
di produzione del vettore
Trasferimento Genico
Vettorologia: Sviluppo di Nuovi Agenti per il
Trasferimento Genico
Retrovirus
Adenovirus
Adeno-Associated virus
1-Definizione e principi
2-Strumenti (Vettori)
3-Strategie
4-Trials Clinici
5-Successi e fallimenti
Malattie Genetiche
Recessive
Dominanti
Malattie Multifattoriali
Cancro
Malattie Cardiovascolari
Diabete
Malattie Infettive
Espressione del Gene Terapeutico
Espressione Stabile
MALATTIE GENETICHE
Espressione Regolata
DIABETE
Espressione Transiente
TUMORI
Le Mutazioni Possono Causare:
MUTANT DNA
MUTANT mRNA
AAAA
MUTANT
PROTEIN
Perdita di Funzione
Acquisizione di Funzione
Strategia per Perdite di Funzione
Virus Ricombinante: Vettore
DNA
nucleus
mRNA
PROTEIN
cytoplasm
AAAA
Strategia per l’ Acquisizione di Funzione
RNAi
AAAA
control.RNAi
VEGF.RNAi
Decreased CNV w/ RNAi vs.
VEGF
Reich et al, 2003 Mol Vis
1-Definizione e principi
2-Strumenti (Vettori)
3-Strategie
4-Trials Clinici
5-Successi e fallimenti
La Terapia Genica Rimane Confinata Alla Medicina Sperimentale
Tuttavia …
1-Definizione e principi
2-Strumenti (Vettori)
3-Strategie
4-Trials Clinici
5-Successi e fallimenti
Ragazzo di 18 anni affetto iniettato con 6x10e11 particelle/kg
Segni di sofferenza mentale 18 ore dopo la somministrazione,
Coagulazione intravascolare disseminata e morte 96 ore dopo
polmone
fegato
milza
midollo
Il pz, OTC19, mostra innalzamento patologico di alcune
citochine pro-infiammatorie. Questo studio ha stabilito la
dose letale di Adenovirus ed ha raffreddato l’
entusiasmo nei confronti della terapia genica.
Terapia genica ex vivo in 10 pazienti con deficit di ADA e
Immunodeficienza Severa Combinata: a 8 anni di distanza
dal primo paziente trattato nessun serio evento avverso e tutti
I pazienti fanno una vita normale senza dover assumere ADA
Primo trial clinico di terapia genica per una malattia erditaria
della retina. Risultati a breve termine mostrano sicurezza ed
efficacia alla dose bassa del trattamento
In conclusione….
• Come in ogni altra terapia sperimentale
esistono difficolta’ tecniche e rischi…
ma si intravedono anche i primi
successi clinici!