INTRODUZIONE ALLA
BIOLOGIA DEI TUMORI
Il DNA dei cromosomi si compone di due filamenti avvolti l'uno attorno l'altro in una doppia elica. I filamenti sono formati da subunità: i NUCLEOTIDI. Ciascun nucleotide comprende un gruppo fosfato, uno zucchero e una base azotata
che puo’ essere di quattro tipi diversi (adenina, citosina, guanina, timina).
Esistono perciò quattro tipi di nucleotidi diversi a seconda della base azotata che contengono. E' l'ordine con cui si susseguono le basi all'interno della molecola di DNA che determina le caratteristiche della specie I cromosomi sono corpuscoli a forma di bastoncino contenuti nel
nucleo della cellula e visibili solo durante le fasi della divisione cellulare.
Sono costituiti da una lunga molecola di DNA e sono presenti in numero
costante per ogni specie (mappa cromosomica). Nel genere umano sono 46. Ciascun cromosoma contiene le informazioni ereditarie che vengono trasmesse alla prole. LA MITOSI
La mitosi é il processo riproduttivo della cellula.
Durante quest'evento essa va incontro a significative modificazioni che porteranno in seguito alla formazione di due cellule figlie identiche tra loro (fatta eccezioni per quelle rare mutazioni che compaiono).
La mitosi serve essenzialmente per la riproduzione cellulare e riguarda le cellule somatiche e quindi del soma.
Durante questo fondamentale processo esiste un problema di fondamentale importanza: tenere invariato il numero di cromosomi presente nelle cellule figlie.
Per questo si verifica, durante la riproduzione cellulare, un raddoppiamento del patrimonio genetico a (4n, tetraploide) che verrà poi riportato a (2n, diploide). Successivamente a tutto ciò le due cellule figlie saranno identiche alla
progenitrice e dotate di patrimonio genetico ancora (2n).
Questo tipo di riproduzione (adottata anche da organismi unicellulari come i batteri) presenta una scarsissima variabilità genetica, essendo data
essenzialmente dalle rare mutazioni genetiche.
DANNO GENETICO
PROTEINA MUTATA
PROTEINA DIMINUITA
ASSENZA DI
PROTEINA
DIMINUITA ATTIVITA'
INATTIVA
ALTERAZIONE FUNZIONE CELLULARE
PATOLOGIA DELLA CELLULA, DELL' ORGANO, DEL SISTEMA
Le al terazioni dell' equilibrio omeostatico che caratterizzano una
malattia
a) sono dovute a una o più cause;
b) sono riconducibili
agli effetti diretti dell'agente eziologico,
ai meccanismi di adattamento e di compenso,
ai fenomeni reattivi che si sviluppano secondariamente;
c)
si verificano in uno o più ben definiti distretti (molecole,
cellule, organi, apparati) e di conseguenza danno origine ad
uno specifico quadro di m odificazioni e sintomi a l ivello
molecolare, biochimico, anatomo-patologico e clinico;
d) hanno un andamento evolutivo;
e) possono dar luogo a c omplicazioni dovute all'instaurarsi di
alterazioni di equilibrio in altri distretti;
f ) possono terminare con la guarigione, con la cronicizzazione o
con la morte.
PATOLOGIA CELLULARE
Ö ALTERAZIONI DEGENERATIVE : la funzionalita’ cellulare viene alterata senza
pero’ arrivare alla necrosi, sono percio’ REVERSIBILI. Se la causa patogena persiste
esse culminano nella NECROSI:
- Rigonfiamento torbido, degenerazione vacuolare e degenerazione a
gocce ialine: legate a IPOSSIA cellulare (tipiche di cellule renali,
epatociti e miocardiociti
- Steatosi o degenerazione grassa: accumulo di lipidi
- Tesaurismosi o m. da accumulo
Ö MODIFICAZIONI DELLO STATO STAZIONARIO CELLULARE
(PROLIFERAZIONE./PROTEOSINTESI):
IPER/IPOTROFIA
MOLECOLARI
ATROFIA
IPER-IPOPLASIA
CELLULARI
APLASIA
Ö ALTERAZIONE DEI PROCESSI DI DIFFERENZIAZIONE
METAPLASIA= CAMBIAMENTO DIFFERENZIAZIONE, nell’ambito di uno stesso
foglietto embrionale
DISPLASIA: Comprende tutte le anomalie citologiche ed istologiche riguardanti la
differenziazione, la proliferazione e l’organogenesi. E’ un termine operativo.
ANAPLASIA: Immaturita’ associata ad anomalie della differenziazione tali da non riconoscere
piu’ morfologicamente il tessuto di origine. (tipica del cancro).
NEOPLASIA: "NUOVA CRESCITA DI CELLULE". Blocco differenziativo associato
a crescita incontrollata. Puo’ essere BENIGNA o MALIGNA
Ö ALTERAZIONE DELLA INFORMAZIONE GENETICA (EREDITATA o
ACQUISITA)
COSA E’ UNA NEOPLASIA
• La neoplasia risulta dalla crescita di una MASSA cellulare (tumore)
• Le noplasie si suddividono in due categorie principali: BENIGNE E
MALIGNE
• Anche se per tumore si intende letteralmente ogni tipo di massa,
generalmente ci si riferisce ad una neoplasia
• Il tumore puo essere molto piccolo e dare sintomi evidenti o diventare
enorme con pochi sintomi
• Il processo di trasformazione neoplastica si puo’ considerare
irreversibile
• Il tessuto neoplastico TENDE ad assomigliare al tessuto di origine (ma
esistono numerose eccezioni a questa regola), talore secerne le stesse
molecole con gravi conseguenze (es: insulina)
• La crescita neoplastica in genere non risponde ai normali processi
omeostatici (AUTONOMIA)
DIMENSIONI e SINTOMI
IL PROBLEMA CANCRO
DOCUMENTATO IN UN PAPIRO EGIZIO DEL 1600 A.C.
PROBLEMA DEL XX SECOLO: 87% DEI CANCRI INSORGE DOPO IL 57° ANNO
1 individuo su 3 (1 SU 2 MASCHI E 1 SU 2,6 FEMMINE) CONTRAGGONO UN CANCRO E 1 SU 5 NE MUORE
Il cancro e’ dovuto ad alterazioni genetiche acquisite somaticamente, talora associate a mutazioni
predisponenti
TUMORI BENIGNI E MALIGNI
COLON: ADENOMA
COLON: ADENOCARCINOMA
CONDROMA
CONDROSARCOMA
T. ADIPOSO: LIPOMA
T. ADIPOSO: LIPOSARCOMA
CARCINOMA A CELLULE SQUAMOSE
Localizzazione cutanea di
linfoma di Hodgkin
CARCINOMA
METASTATICO
CARATTERISTICHE DELLA CELLULA TUMORALE
● PERDITA DELLA INIBIZIONE DA CONTATTO
● MINORE ADESIVITA' ed AGGREGABILITA'
● PROLIFERAZIONE E SOPRAVVIVENZA IN ASSENZA DI ADESIONE
● NON NECESSITA DI FATTORI DI CRESCITA
● NON VA INCONTRO A SENESCENZA
● DIVERSE ANOMALIE GENETICHE, STRUTTURALI E METABOLICHE
● ATTRAVERSA LE MEMBRANE BASALI
● INDUCE NEOANGIOGENESI
● RESISTENTE ALLA REAZIONE IMMUNITARIA TRAMITE:
ANTIGENI TUMORALI SCARSAMENTE IMMUNOGENI, VARIABILI
DEPRESSIONE PROGRESSIVA DELLA RISPOSTA IMMUNITARIA
SOSTANZE ANTICHEMIOTATTICHE
INDUZIONE DI "SUICIDIO" DEI LINFOCITI CITOTOSSICI
SEGREGAZIONE (connettivi, piastrine, etc)
CLASSIFICAZIONE DEI TUMORI
TIPO TISSUTALE
COMPORTAMENTO
BENIGNO
MALIGNO
Squamoso stratificato
Ghiandolare
Transizionale
papilloma
adenoma
papilloma
cr. squamoso
adenocarcinoma
cr. a cellule transizionali
Tessuto fibroso
Tessuto adiposo
Muscolo liscio
Muscolo striato
Sinovia
Cartilagine
Osso
Vasi sanguigni
Cellule germinali
Neuroectoderma
fibroma
lipoma
leiomioma
rabdomioma
sinovioma
condroma
osteoma
emangioma
teratoma benigno
nevo
fibrosarcoma
liposarcoma
leiomiosarcoma
rabdomiosarcoma
sarcoma sinoviale
condrosarcoma
osteosarcoma
angiosarcoma
teratoma maligno
melanoma
EPITELIALE
NON EPITELIALE
DISTINZIONE TRA TUMORI BENIGNI E MALIGNI
CARATTERISTICHE
BENIGNE
MALIGNE
Velocità di crescita
bassa percentuale di mitosi
mitosi normali
nucleoli normali
simile al normale
mantenimento delle normali funzioni
incapsulati
no invasione
no metastasi
alta percentuale di mitosi
mitosi anormali
nucleoli ingranditi
spesso scarsa
funzioni perse o alterate
non capsulati
invasione locale
metastasi
Differenziazione
Diffusione
TUMORI ORIGINATI DA CELLULE MESENCHIMALI, NEUROENDOCRINE O
GERMINALI
CELLULA DI ORIGINE
TUMORE
Neuroblasto
Neuroblastoma, Retinoblastoma, Ganglioneuroma
Cellule della cresta neurale
Tumori neuroendocrini
Carcinoma a piccole cellule del polmone
Melanoma
Carcinoidi
Tumori endocrini del tratto G.I.
Precursori gliali
Glioblastoma/Astrocitoma/Oligodendroglioma
Cellule germinali
Teratomi/Seminomi/Coriocarcinomi
Cellule mesenchimali
SARCOMI:
Rabdomiosarcoma
Leiomiosarcoma
Osteosarcoma
Condrosarcoma
Liposarcoma
Linfangiosarcoma
Sarcoma di Kaposi
Emangiosarcoma
Cellule mesoteliali
Mesotelioma
Cellule di Schwann
Schwannoma
Fibroblasti delle meningi
Meningioma
LE BASI MOLECOLARI
DELLA TRASFORMAZIONE
NEOPLASTICA
Ovvero: come fa una cellula a perdere
il controllo?
PATOGENESI DELLE NEOPLASIE
ALTERAZIONE DI GENI CHE VIENE "FISSATA" NEL DNA E TRASMESSA ALLE
CELLULE FIGLIE
SONO NECESSARIE ALTERAZIONI DI CIRCA SEI DIVERSI GENI PER
PERMETTERE LO SVILUPPO DI UN CLONE CELLULARE TRASFORMATO
1) GENI CHE REGOLANO LA PROLIFERAZIONE IN SENSO POSITIVO:
PROTOONCOGENI
ONCOGENI
MECCANISMO: MUTAZIONE, FUSIONE GENICA O AMPLIFICAZIONE
2) GENI CHE REGOLANO LA PROLIFERAZIONE IN SENSO NEGATIVO:
GENI ONCOSOPPRESSORI
MECCANISMO: INATTIVAZIONE DELLE COPIE PATERNA E MATERNA (ES:
mutazione+delezione)
PROCESSO A TAPPE. SPERIMENTALMENTE SONO STATE OSSERVATE LE
SEGUENTI FASI:
1) INIZIAZIONE
2) PROMOZIONE
3) PROGRESSIONE (ACCUMULO DI DANNI GENETICI E SELEZIONE
CLONALE)
4) METASTATIZZAZIONE
Il cancro si sviluppa
a causa della combinazione di due
meccanismi:
• mutazioni che aumentano la proliferazione cellulare:
popolazione espansa di cellule in cui può verificarsi la
successiva mutazione
• mutazioni che diminuiscono la stabilità del genoma: aumento
del tasso di mutazione complessivo
Poiché il tumore dipende da questi stessi due meccanismi, si
sviluppa per stadi:
IPERPLASIA (crescita benigna) ⇒ TUMORE MALIGNO
L’instabilità cromosomica dei tumori maligni è ben visibile nei
cariotipi aberranti
DA: GENES, CHROMOSOMES & CANCER 1999, 24:213-221
EQUILIBRIO
GENI ONCO
SOPPRESSORI
(il freno)
PROTO-
ONCOGENI
(l’acceleratore)
MECCANISMI DI ATTIVAZIONE DEI
PROTOONCOGENI
•
•
•
Amplificazione:
– c-myc (adenocr, sarcomi)
– c-erbB2 (adenocr.)
– N-myc (adenocr., tumori neuro-end.)
– c-erbB-1 (cr. a cellule squamose)
Mutazione:
– N-ras
(AML)
– K-ras
(Adenocr.)
Traslocazione cromosomica
– c-myc (B-cell linfoma)
– c-abl (CML)
ONCOGENI
•
•
Classe I: Fattori di crescita (sis) Classe II: Recettori per fattori di crescita (erbB, fms, trk) • Classe III: Fattori di transduzione
intracellulari
(src, abl, raf, gsp, ras) • Classe IV: Fattori di trascrizione nucleari (jun, fos, myc) Classe I
Classe II
Classe III
AAAA
Classe IV
INSERZIONE
DELEZIONE
TRASLOCAZIONE
DUPLICAZIONE
I CROMOSOMI UMANI
UNA CELLULA NORMALE
UNA CELLULA NEOPLASTICA
Blood, 15 November 2000, Vol. 96, No. 10, pp. 3343-3356 The molecular biology of chronic myeloid leukemia Michael W. N. Deininger, John M. Goldman, and Junia V. Melo Blood, 15 November 2000, Vol. 96, No. 10, pp. 3343-3356 The molecular biology of chronic myeloid leukemia Michael W. N. Deininger, John M. Goldman, and Junia V. Melo EQUILIBRIO
GENI ONCO
SOPPRESSORI
(il freno)
PROTO-
ONCOGENI
(l’acceleratore)
Modello di cancerogenesi da soppressori tumorali
germinale
somatica
germinale
somatica
CANCRO
CARCINOMA
EREDITARIO
CARCINOMA
SPORADICO
CANCRO
GENI ONCOSOPPRESSORI
• RB: 13q14, fosfoproteina nucleare 105 kDa
• p53: 17q, checkpoint G1-S. 40-50% di tutti
i tumori studiati presentano mutazioni
puntiformi.
• BRCA-1: Coinvolto nella riparazione del
DNA?
• DCC: 18q21.3, molecola transmembrana di
190 kDa (recettore di adesione?)
• Altri (WT-1, APC, NF-1, inibitori delle
cdk: p15, p16, p21)
DNA
Oncogenesi
VIRALE
ALTERAZIONI STRUTTURALI
DEL GENOMA
DNA virus
FENOTIPO “MUTATORE”
Cancerogenesi CHIMICA/FISICA
RNA virus
Delez
.
Trasloc.
Invers.
MUTAZIONI PUNTIFORMI
Integrazione Attiv
proto-onc
oncogeni Per inserzione
(acuti)
(lenti)
Papill.
(E6,7)
SV40 (T)
Geni
ONCOSOPPRESSORI
ATTIV.
PROTOONC.
-ATTIVAZIONE
protoonc.
-Prot. CHIMERICHE
INSTABILITA’
GENETICA
INATT.
.
ONCOSOPPRESSORI
STORIA NATURALE DEI
TUMORI
LA CANCEROGENESI CHIMICA
Le fasi della trasformazione neoplastica
INIZIAZIONE: Processo rapido, additivo. Causata da agenti chimici, fisici o biologici in grado di danneggiare il DNA. Tali agenti NON inducono
proliferazione, anzi semmai la inibiscono. Le cellule iniziate sono
apparentemente indistinguibili dalle cellule normali
PROMOZIONE: Processo lento, effetto reversibile e non additivo. Sono agenti che stimolano la proliferazione cellulare (esteri del forbolo,
fenobarbital, ormoni, ferite, agenti irritanti quali soluzione fisiologica
instillata nei bronchi iniziati)
Tuattavia la maggioranza degli agenti cancerogeni sono sia iniziatori che promotori, Sono percio’ cancerogeni COMPLETI
PROGRESSIONE: meccanismo legato alla INSTABILITA’ GENETICA della cellula neoplastica che favorisce la l’insorgenza di danni genomici che danno luogo alla comparsa di nuovi cloni dotati di aggressivita’ clinica
crescente (METASTASI)
Cellula normale della mucosa
del colon
Delezione di APC sul crom. 5
Piccoli polipi
Mutazione di RAS sul crom. 12
Delezione di geni
oncosoppressori sul crom. 18
Grandi polipi con nidi di
cellule maligne
Delezione o mutazione di p53
sul crom. 17
Cr del colon
MARCATORI TUMORALI
Le LESIONI PRE-NEOPLASTICHE
Lesioni a potenziale evoluzione verso la neoplasia
Es: LEUCOPLACHIA: metaplasia cornea (cavo orale e genitali): 4-9% di potenzialita’ di cancerizzazione
IPERPLASIA DUTTALE O LOBULARE della mammella
NEVI DISPLASTICI:
èLENTIGO: melanociti solo nello strato basale
èNEVO GIUNZIONALE: melanociti nell’epidermide e giunzione dermo-epidermica
èNEVO COMPOSTO: melanociti in sede epidermica e dermica
èNEVO DISPLASTICO: presenza di anomalie citologiche: ad elevato rischio di trasformazione in melanoma
POLIPI: POLIPOSI INTESTINALE
1) ADENOMATOSA: proliferazione elementi staminali delle cripte (ALTO RISCHIO)
2) IPERPLASTICA: Proliferazione epiteliociti differenziati degli strati superiori dei villi
MASTOPATIA FIBROCISTICA: dilatazione cistiche della mammella (dotti)
10% delle DONNE: cisti secretive (APOCRINE): tipo I cisti piatte (trasudative): tipo II
Le cellule del tipo I secernono deidroepiandrosterone solfato e EGF. Rischio CR. INVASIVO.
Per DISPLASIA si intende una proliferazione disordinata ma non
neoplastica. E’ caratterizzata da pleomorfismo, ipercromatismo
cellulare e perdita della normale organizzazione strutturale di un
tessuto. I cambiamenti displastici si trovano solitamente negli
epiteli, soprattutto nella cervice uterina. Quando i cambiamenti
displastici sono marcati e coinvolgono l’intero spessore
dell’epitelio, la lesione e’ considerata una neoplasia preinvasiva e
viene definita come carcinoma in situ. Questa lesione e’ un
precursore, in molti casi, di un carcinoma invasivo. Una displasia non grave e’ tuttavia comune nella cervice uterina e
non porta sempre al carcinoma, essendo spesso reversibile quando
la causa scatenante (es: infiammazione cronica) viene rimossa.
Cervice Uterina
Prostata
SIL: Squamous Intraepithelial Lesion
CIN: Cervical Intraepithelial Lesion
MAMMELLA
microcalcificazione
Normale
Mastopatia fibrocistica
Le cellule neoplastiche acquisiscono una capacità proliferativa indefinita ed evitano la spinta verso la
differenziazione terminale e/o la quiescenza tipica delle cellule normali . Per acquisire queste
caratteristiche le cellule neoplastiche devono rendersi indipendenti dagli stimoli esterni. Le
alterazioni alla base della trasformazione neoplastica e della sua progressione verso una sempre
maggiore malignità passano attraverso l’alterazione dei seguenti meccanismi:
1)
Controllo della proliferazione
2)
Segnali di sopravvivenza
3)
Senescenza
4)
Angiogenesi
5)
Invasione e metastasi
6)
Geni riparatori del DNA CONTROLLO DELLA PROLIFERAZIONE
MECCANISMI:
1)
Produzione di fattori di crescita endogeni:cellule di glioblastoma e sarcoma producono FC (PDGF e TGF-α, quelle di melanoma FGF-2
2)
Attivazione costitutiva delle vie di trasduzione del segnale mitogenico tramite iperespressione o mutazione.
Es: ipersespr. EGF-r o HER-2/neu, mutazione o delezione attivatorie di EGF-r , mutazione k-ras (25% dei tumori), induzione di c-myc, alterazioni della via che controlla pRB da mutazioni/delez. di pRB stesso, da oncoproteine virali (SV40 T, HPV - E7, Adenov. E1A), da alterazione di geni coinvolti nella stessa
via di trasduzione (p16, ciclina D1 e cdk-4)
SEGNALI DI SOPRAVVIVENZA
MECCANISMI:
1)
Attiv oncogeni (ras, myc) induce anche segnali apoptotici mediati da p53 (inattivata in circa 50% dei tumori).
Es:é c-myc
é p19/
ARF
Legame e inattivazione mdm-2 (che media degradaz. p53)
é p53
Blocco ciclo cell. e induzione
apoptosi
2)
Aumentata espressione di fattori di crescita (IGFs, PDGFs, FGFs), geni anti-apoptotici (bcl-2) SENESCENZA
Le cellule normali, dopo un numero limitato di divisioni (60-70) vanno incontro crisi e,
conseguentemente, a arresto del ciclo cellulare ed entrata in uno stato di senescenza. Il numero di
cicli viene “contato” mediante l’accorciamento progressivo dei telomeri. Tali strutture sono
controllate dall’enzima TELOMERASI, inattivo nelle cellule somatiche normali e presente in circa
l’80% delle cellule neoplastiche. In effetti l’incidenza di tumori nei topi p16-/- diminuisce quando
sono incrociati con topi KO per mTR (mTR-/-)
Tuttavia tale enzima ha una funzione più complessa, come dimostrato dalla aumentata incidenza di
tumori in topi KO per p53. In questi casi la perdita di attività telomerasica correla con alto livello di
danno cromosomico e instabilità genomica.
ANGIOGENESI TUMORALE
Dipende dal bilanciamento fine tra fattori pro-angiogenetici (Es: VEGF-A) ed anti-angiogenetici (es:
trombospondina-1 indotta da p53, che induce apoptosi delle cellule endoteliali) prodotti dalle cellule
tumorali.
p53 è coinvolto nel processo angiogenetico anche tramite induzione di degradazione di HIF-1a, un
fattore di trascrizione che regola molti altri fattori inducibili da ipossia (tipica delle aree tumorali). In
seguito a perdita di funzione di p53, tale fattore non viene più degradato, favorando quindi
l’irrorazione della massa tumorale che può quindi meglio svilupparsi.
FENOTIPO MUTATORE
Il cambiamento mei meccanismi di regolazione di base quali la riparazione del DNA o la sua metilazione accelerano la progressione tumorale
Pazienti con HNPCC mostrano instabilita’ dei microsatelliti (MIN). I microsatelliti sono
sequenze di DNA altamente ripetitive che sono soggette ad accumulare errori durante la
replicazione del DNA
L’inattivazione di geni deputati al “mismatch repair”, MLH1 e MSH2 causa un aumento
degli errori spontanei da 1x10-6 a 1x10-3 - 1x10-2 . Tali sequenze sono state identificate All’interno di geni importanti nella trasformazione cellulare quali: APC, TGF-b RII,
TCF-4, bax e gli stessi geni deputati al riparo dei “mismatch”.
Instabilita’ dei cromosomi (CIN): si manifesta con aneuploidia. E’ dovuta a mutazioni
di geni coinvolti nella separazione dei cromosomi durante la mitosi (BRCA1 e 2, ATM)
INVASIONE E METASTASI
Stadi finali della trasformazione neoplastica. In tali fasi sono profondamente alterati i meccanismi di
controllo delle interazioni cellula-cellula e cellula-substrato.
E-caderina: proteina coinvolta nella formazione delle giunzioni aderenti delle cellula epiteliali. E’
inattivata funzionalmente in quasi tutti tumori maligni epiteliali (carcinomi) mediante: delezione,
mutazione, silenziamento del promotore, proteolisi. Inoltre la mutazione germinale predispone a cr
gastrico scarsamente differenziato. Forse agisce mediante rilascio di β-catenina che trasloca nel
nucleo ed attiva LEF-1/TCF
Metalloproteasi agiscono a vari livelli, essendo coinvolte nelle fasi di iniziazione, nel controllo della
proliferazione, nell’angiogenesi (MMP-2, uPA/uPAR)
Il tumore nei pazienti: Classificazione in stadi col sistema TNM
Proposto tra il 1943 e 1952 da Pierre Denoix
T: Identifica le dimensioni del tumore
N: Identifica lo stato dei linfonodi
M: identifica la presenza o meno di metastasi
EFFETTI SISTEMICI DIRETTI DELLE NEOPLASIE
EFFETTO
SINDROME CLINICA
COMPRESSIONE VASI
EDEMA
INVASIONE E EROSINE VASCOLARE
EMORRAGIA
INVASIONE LINFATICA
LINFEDEMA
INVASIONE FASCI NERVOSI
DOLORE, DISESTESIA, SONNOLENZA
METASTASI CEREBRALI
DEBOLEZZA, SONNOLENZA, CEFALEA,
ANORMALITA' di COORDINAZIONE,
ALTERAZIONI VISTA
COMPRESSIONE MIDOLLO SPINALE
DOLORE, PARALISI, INCONTINENZA
INVASIONE E DISTRUZIONE OSSEA
DOLORE, FRATTURE
OSTRUZIONE E PERFORAZIONE TUBO
NAUSEA, VOMITO, DOLORE
GASTROENTERICO
OSTRUZIONE VIE AEREE
DISPNEA, POLMONITE, PERDITA DI
VOLUME RESPIRATORO
OSTRUZIONE URETERALE
BLOCCO RENALE, INFEZIONI VIE
URINARIE
INVASIONE E METASTASI EPATICHE
INSUFFICIENZA EPATICA
METASTASI POLMONARI E PLEURICHE
DISPNEA, DOLORE TORACICO
INFILTRAZIONE MIDOLLO OSSEO
PANCITOPENIA, INFEZIONI, EMORRAGIE
Tappe salienti in oncologia
•
•
•
•
•
•
•
•
•
•
•
•
1902-14: Associazione tra alterazioni cromosomiche e neoplasie (T.
Boveri)
1910: Scoperta virus tumorali in modelli animali (P. Rous)
1915: Cancerogenesi chimica (K.Yamagiwa)
1953: Struttura del DNA di Watson-Crick
1960: Prima aberrazione cromosomica associata a neoplasia (“Cromosoma
Philadelphia” nella Leuemia Mieloide Cronica)
1962: Tecnica di “fusione cellulare”
1969: Dimostrazione esistenza di “tumor suppressor genes”
1971: “Two hits” (Knudson) e scoperta protooncogeni
1985: Sviluppo della PCR
1986: clonaggio del primo oncosoppressore (RB)
1990: Inizia il “Progetto Genoma”
2001: Viene resa pubblica le prima versione della sequenza genomica
umana
LE METASTASI
METASTASI
DEFINIZIONE:
primario e che si sviluppa nell’organismo ospite in un distretto
tissutale distante da quello sede della neoplasia primaria PATOGENESI:
FENOTIPO:
FASI:
Focolaio neoplastico secondario che trae origine da un focolaio
Una serie di anomalie acquisite solo da alcune cellule (cloni metastatici). Si tratta quindi di una conseguenza della PROGRESSIONE tumorale.
Minore adesivita’ ed aggregabilita’, maggiore motilita’, presenza di attivita’
proteasica associata alla membrana, espressione di recettori per proteine della
matrice extracellulare (laminina) o endoteli (recettori per chemochine).
Distacco
Disseminazione
Migrazione
Arresto
Impianto
FATTORI DELL’ OSPITE CHE CONDIZIONANO
LO SVILUPPO DELLE METASTASI
• circolazione dell’organo in cui si sviluppa il tumore
• movimento degli organi (rarissime nei muscoli)
• capacita’ difensive del sistema immunitario
• capacita’ di produrre risposta flogistica contro il tumore
• caratteristiche delle cellule endoteliali, che possono avere piu’ o meno
affinita’per le cellule tumorali
• capacita’ di produrre fattori angiogenetici, che favoriscono la
vascolarizzazione del tumore
• eta’, sesso, alimentazione, squilibri ormonali, stato di salute
TAPPE DELLA DIFFUSIONE METASTATICA
1. PRESENZA nella popolazione cellulare del tumore primitivo di
cellule con potenziale matastatico.
2. DISTACCO della cellule dal tumore primitivo, dovuto alla
diminuzione della adesività omotipica.
3. SUPERAMENTO della membrana basale ed invasione dei
tessuti adiacenti.
4. PENETRAZIONE delle cellule neoplastiche nel circolo
sanguigno o linfatico attraverso la parete di un capillare
5. FORMAZIONE nel torrente ematico di un embolo neoplastico
costituito da una o più cellule tumorali rico perte da un
rivestimento di fibrina.
METASTASI PERITONEALI
Localizzazione cutanea di
linfoma di Hodgkin
CARCINOMA
METASTATICO
IL CANCRO E L’AMBIENTE
CANCEROGENESI CHIMICA, FISICA e BIOLOGICA
Per “radiazione” si intende l’energia che
viene trasmessa sotto forma di onde o
particelle (λ=h/mv). Include le
radiazioni visibili, ultraviolette, le
microonde, radio onde, luce laser e
radiazione infrarossa. EMIVITA
Rappresenta il tempo impiegato da metà di un determinato Campione radioattivo a decadere.
L’emivita di un radioisotopo è unica per quell’isotopo e non viene modificata qualsiasi siano le condizioni chimico-fisiche
dell’ambiente esterno. EFFETTI SULLA MATERIA Energia del fotone Effetto
Fenomeni fisici
< 1 eV
Termico
Oscillazioni e dislocazioni atomiche
1-10 eV
Eccitante
Eccitazione e- di valenza con innesco
di reazioni chimiche (fotoattivazione)
> 10 eV
Ionizzante
Eccitazione di e- degli orbitali più interni
con conseguenti ionizzazioni atomiche e
molecolari
EBR
Tipo di radiazione
EBR
Gamma
1
Beta ed e- > 0.03 MeV
1
Beta ed e- < 0.03 MeV
1.7
Neutroni termici
3
Neutroni veloci
10
Protoni
10
Radiazioni alfa
10
Ioni pesanti
20
RADIAZIONI AMBIENTALI
• Terrestri (suolo e rocce)
• Cosmiche
• Interne
125 mrem/anno (media)
Radon da solo (polmoni):
circa 300 mrem/anno (dati U.S.A.)
• Esposizione medica: circa 100 mrem/anno
EFFETTI delle RADIAZIONI
IONIZZANTI
Fase fisica(10-16-10-13 sec): Cessione di energia, espulsione di e-
dalla molecola bersaglio
rottura di legami chimici con formazione di radicali liberi
Fase chimica (10-7-5 sec): : Interazione dei radicali liberi con macromolecole nucleofile Danno biologico: Diretto (sulle macromolecole) e indiretto
(da radiolisi dell’acqua)
EFFETTI BIOLOGICI
• EFFETTI SOMATICI
• A breve termine (eritema, radiodermite, alopecia,
alterazioni ematochimiche, sindrome acuta da
radiazioni con dosi > 100 rad in poche ore)
• A lungo termine: Aumento rischi di cr, cataratta,
effetti sull’embrione, diminuzione della spettanza di
vita.
• EFFETTI GENETICI: • Aumento della presenza di alterazioni genetiche
potenzialmente dannose nella popolazione
TIPO DI DANNO
• PROTEINE: denaturazione/inattivazione
• richiedono dosi 10x rispetto a DNA
• DNA: •
•
•
•
Modificazione o perdita di basi azotate (rottura legami glucosidici)
Rottura di legami H tra coppie di basi appaiate
Rottura dello scheletro pentoso-fosfato
Rottura di filamenti nucleotidici e formazione di legami crociati intra o
intermolecolari
RADIAZIONI IONIZZANTI
H2O
RADIOLISI
l
OH
CELLULE PROLIFERANTI
CELLULE NON PROLIFERANTI
DANNO
DANNO AL DNA
SUBLETALE
NON RIPARABILE
MUTAZIONI
APOPTOSI
GENETICHE
PEROSSIDAZIONE LIPIDI DI
MEMBRANA
PERDITA DI INTEGRITA' DELLA
MEMBRANA
Riparazione inefficace
CANCRO
MORTE CELLULARE
RADIAZIONI ECCITANTI
Tra 1000 e 200 nm (vicino infrarosso, visibile e UV).
Luce solare: 55% IR; 40% vis, 4,7% UV-A e 0,3% UVB
Picco di assorbimento da parte degli acidi nucleici e delle proteine:
260 e 280 nm rispettivamente. (UV-B: 315-280 nm/U-VC: 280-200 nm).
Rendono le molecole eccitate più facilmente ossidabili e riducibili.
EFFETTI GENOTOSSICI
Eccitazione delle basi pirimidiniche (T e C) che possono reagire con l’acqua e formare idrati o con basi contigue formando dimeri
pirimidinici. Questi ultimi sono stabili e possono causare, se
non rimossi, mutazioni all’atto della replicazione del DNA.
(es: transizione C
T dovuta ad appaiamento dei dimeri di C con A invece che con G)
Il sistema NER
CANCEROGENESI CHIMICA:
Alcune tappe salienti
• 1775: Sir Percival Pott associa cr. Scroto a fuliggine negli
spazzacamini inglesi
• 1915: Yamagiwa e Ichikawa pennellano catrame grezzo sulla cute di
conigli e causano neoplasie
• 1925: Kennaway dimostra che i componenti attivi del catrame
grezzo sono idrocarburi policiclici
• 1930: Sintesi chimica del primo cancerogeno (dibenzantracene)
• 1935: Cancerogenesi indiretta (coloranti azoici causano
cancerogenesi epatica)
Definizione di cancerogeno (J & E Miller)
Dicesi cancerogeno un agente che, somministrato a un animale previamente non trattato, induce, per azione genotossica diretta, un incremento Statisticamente significativo dell’incidenza di una data neoplasia rispetto agli animali di controllo (non esposti all’azione dell’agente in questione); ciò indipendentemente dal fatto se, nella popolazione animale di riferimento, l’incidenza spontanea della neoplasia in oggetto sia alta o bassa.
CANCEROGENI CHIMICI
Sostanze organiche: NATURALI
aflatossina
cicasina
alcaloidi senecio
etc.
ARTIFICIALI
catrame
amine aromatiche
etc.
Sostanze inorganiche:
REATTIVE
arsenico nichel
asbesto zinco
berillio piombo
cromo
INERTI
plastica
oro
platino
vetro
etc.
I cancerogeni chimici hanno una comune modalità di azione: allo
stato nativo o in seguito a trasformazioni metaboliche espongono
gruppi elettrofili altamente reattivi che stabiliscono legami
covalenti con siti nucleofili (ricchi di elettroni) di proteine e
acidi nucleici formando composti di addizione (ADDOTTI)
I cancerogeni chimici più importanti per la salute umana sono:
@ Gli idrocarburi policiclici benzenoidi
@ Le amine aromatiche
@ Le N-nitrosamine
@ Le aflatossine
BERSAGLI MOLECOLARI
Legame a DNA, RNA o proteine in modo covalente con formazione di Composti di addizione (addotti).
Il legame al DNA provoca alterazione trasmissibile alle cellule figlie
del codice genetico (mutazioni) che possono attivare proto-oncogeni (oncogeni silenti) o alterare geni regolatori. I gruppi piu’ reattivi sono quelli purinici.
Il legame a RNA o Proteine causa danni EPIGENETICI (es: istoni, sistemi di controllo dell’espressione genica, etc.)
I bersagli sono comunque geni regolatori della differenziazione
e proliferazione cellulare
MECCANISMO D’AZIONE
1) Azione DIRETTA: -sostanze elettrofile molto reattive possono essere inattivate chimicamente o enzimaticamente. Es: Sostanze ALCHILANTI (beta propionolattone, epossidi,iprite, antimitotici), alcune NITROSAMINE e NITROSAMIDI
2) PRECANCEROGENI: -vengono convertiti metabolicamente in CANCEROGENI TERMINALI. L’effetto massimo si ha nel tessuto capace di convertirli in composti
attivi (fegato).
Es: 2-acetoaminofluorene per os
idrossilazione epatica
EPATOCAR.
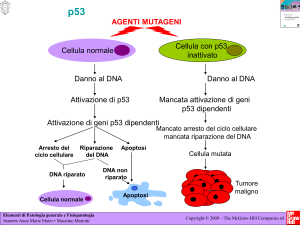
p53
LESIONE DEL DNA
p53 normale
p53 mutata
Arresto della crescita od apoptosi
No arresto della crescita
Efficiente riparazione del DNA
Inefficiente riparazione del DNA
Stabilita’ genetica
Instabilita’ genetica
Potenzialita’ maligna
Causa-effetto tra sostanze
chimiche e tumori umani
Sostanze e tipo di esposizione
●Industriale
Carbone fossile
Minerali di ferro
Asbesto
Asbesto + fumo di sigaretta
Ni, Cd, Cr
As
Benzene
Beta-naftilamina
Cloruro di vinile
●Medica
Clornafazina
Dietilstilbestrolo
Estrogeni
Idantoinici
Farmaci immunosoppressivi
Alchilanti, nitrosuree
●Sociale
Fumo di tabacco
Alcolici
Foglie di betel
Dieta
Sedi di neoplasia
Pelle, scroto, polmone
Polmone
Pleur, peritoneo
Pleura, peritoneo, polmone
Polmone
Pelle, fegato
Sistema linfatico
Vescica
Fegato
Vescica
Vagina
Mammella, fegato, utero
Tessuto linfatico
Sistema linfatico, midollo osseo
Midollo osseo, sistema linfatico
Polmoni, laringe, cavità orale,
vescica, forse pancreas
Esofago, fegato, cavità orale, laringe
Cavità orale
Colon, stomaco, mammella, prostata
ESPOSIZIONE AD AGENTI CANCEROGENI
ESEMPI DI ATTIVITA’ INDUSTRIALI CONTAMINANTI L’AMBIENTE E NOCIVE PER
L’UOMO TRASFERITE IN PAESI DEL “TERZO MONDO”
ATTIVITA’ INDUSTRIALE
LOCALITA’
FRANTUMAZIONE ASBESTO
SUD-AFRICA
MANIFATTURA TESSILE ASBESTO
MESSICO
MANIFATTURA CEMENTIERA ASBESTO
INDIA
PRODUZIONE PVC
MALAYSIA
MANIFATTURA α-NAFTILAMINA
PAKISTAN
PRODUZIONE COLORANTI
INDIA
MANIFATTURA CROMATI
MESSICO
IMPIANTI CLORO E MERCURIO
NICARAGUA
PRODUZIONE PESTICIDI
MALAYSIA, MESSICO
===============================================
LIMITI PER 1,3 BUTADIENE:
10 ppm
USA
780 ppm
BRASILE
1000 ppm
TAIWAN
Le linee guida epidemiologiche di
Evans e Mueller
• La distribuzione geografica dell’infezione virale dovrebbe
coincidere con quella del tumore, considerando eventuali co-fattori
noti
• La presenza del marcatore virale dovrebbe essere presente
prevalentemente nei soggetti neoplastici rispetto ai relativi
controlli
• I marcatori virali dovrebbero precedere il tumore, con una
prevalenza di soggetti malati positivi al marcatore rispetto ai
soggetti negativi
• La prevenzione dell’infezione virale dovrebbe diminuire l’incidenza
della neoplasia in oggetto
ASSOCIAZIONE VIRUSCANCRO
I virus sono necessari ma non sufficienti allo sviluppo di una neoplasia
(concetto di co-cancerogeno). Agiscono da fattori inizianti o promoventi,
infatti:
> 90% della popolazione è EBV+
neoplasia in ospite immunocompromesso
Solo 2-6% degli individui infetti con HTLV-1 e HPV sviluppano neoplasia
VIRUS UMANI SOSPETTATI DI SVOLGERE AZIONE ONCOGENA
VIRUS
PATOLOGIA
NEOPLASIA
COFATTORI
ASSOCIATA
------------------------------------------------------------------------------------------------------------
A DNA
EBV
Mononucl. Infett.
Linfoma di Burkitt
Ridotta immunocomp.
Cr. Nasofaringeo
Fattori genetici
Linfoma di Hodgkin
Linfomi T
Leiomiosarcomi
Adenok. Gastrico
HPV 16/18/33
Verruche e condil.
Cr. Ano-genitali
Contracc. orali
Cr Vescicali
Stato ormonale/
Immunol.
HBV
Epatite
Epatocarcinoma
Aflatossina, altri
Agenti epatot.Fattori
imm.
Herpesv. 8 o 9
Sconosciuta
Sarcoma di Kaposi
Ridotta immunoc.
HCV
Epatite
Epatocarcinoma
Sconosciuti
LEUCEMIA T
Paraparesi spastica
leucemia T, CD30+
Fattori immunogen.
(HTLV-1)
tropicale, uveite
anapl, Micosi
(HLA?)
A RNA
fungoide/Sezary
NELLA MAGGIOR PARTE DEI TUMORI SONO STATI INDIVIDUATI PRECISI FATTORI
AMBIENTALI COME FATTORI DI RISCHIO PER LA LORO INSORGENZA
RADIAZIONE
SOLARE
ALIMENTAZIONE
FUMO DI
SIGARETTA
VIRUS
CAUSE DI CANCRO
Impossibile visualizzare l'immagine. La memoria del computer potrebbe essere insufficiente per aprire l'immagine oppure l'immagine potrebbe essere danneggiata. Riavviare il computer e aprire di nuovo il file
potrebbe essere necessario eliminare l'immagine e inserirla di nuovo.
ATTIVITA'
INDUSTRIALI
4%
INQUINAMENTO
2%
NON NOTI
5%
VIRUS E
INFEZIONI
EREDITARIETA'
2%
10%
RADIAZIONI
1%
TABACCO
29%
ADDITIVI
1%
FATTORI SESSUALI
7%
ALCOOL
4%
LUCE SOLARE
1%
ALIMENTAZIONE
34%
LA PREVENZIONE
• ABITUDINI DI VITA: Evitare quei comportamenti che aumentano il
rischio diretto od indiretto di trasformazione neoplastica delle cellule
(fumo, dieta, vita sedentaria)
• SCREENING sulla popolazione maggiormante a rischio
(Mammografia, Pap Test, dosaggio PSA, Esame prostata,
Colonscopia)
• AMBIENTE: attivare politiche atte a ridurre la presenza di inquinanti
nell’ambiente (compresi i posti di lavoro) e nei cibi
• CHEMIOPREVENZIONE: Alcune sostanze chimiche potrebbero
avere effetto di prevenzione dei tumori (molecole presenti nel vino
rosso o inibitori dell’enzima COX-2). L’effetto comunque è incerto. Vincent Van Gogh: TESCHIO CON LA SIGARETTA (1886)
MUSEO VAN GOGH, AMSTERDAM
46)
r
r
r
r
r
Quale delle seguenti è causa di ipertrofia negli organi indicati
a)
Ipertensione arteriosa: ipertrofia del ventricolo sinistro.
b)
Ipertensione polmonare: ipertrofia del ventricolo sinistro.
c)
Insufficiente apporto di ossigeno ad un tessuto muscolare
d)
Stimolo meccanico: ipertrofia della prostata
e)
Nessuna delle precedenti