Appunti di chimica
Edoardo Tomelleri
1 giugno 2014
1
Indice
0 Changelog
• 07/04/2014: corrette lezioni del 28/03 e del 01/04
• 08/04/2014: aggiunto changelog
• 09/04/2014: correzioni 03/04/2014, aggiunta tabella
• 13/04/2014: correzioni 13/04/2014, aggiunto grafico energia di legame 03/04/2014
• 04/05/2014: correzioni 02/04/2014 (completo), aggiunti un po’ grafici, corretto grafico 03/04/2014
• 05/05/2014: aggiunte un po’ di equazioni e sistemato elenco con graffe del 29/04/2014, aggiunto indice!!!
• 16-17/05/2014: correzioni per 08,09,13,15,16/05/2014, aggiunta tabella 15/05/2014 e equazioni varie, la lezione del 15 è sistemata, le parentesi in 13/05/2014 sono umane, le equazioni
in 09/05/2014 sono scritte decentemente, introdotto l’uso di siunitx per questi giorni, risolti
problemi di interlinea con sistemi e frazioni. In 16/05/2014 risistemate interlinee, resta da decidere se avviare nuova sezione per l’ultima osservazione. Aggiunto grafichino 15/05/2014. La
lezione del 9 è sistemata.
• 26/05/2014: correzioni per 20/05/2014
Indice
0 Changelog
2
1 Modelli atomici
1.1 18/03/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2 20/03/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.3 25/03/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3
3
7
10
2 Configurazioni elettroniche
2.1 27/03/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.2 28/03/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
14
14
16
3 Energia: ionizzazione e affinità
3.1 01/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
18
18
4 Legami chimici
4.1 01/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2 03/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
21
21
21
5 Legame ionico
5.1 03/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
23
23
6 Legame covalente
6.1 03/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
6.2 04/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
25
25
25
2
Indice
7 VB: polarità, elettronegatività
7.1 08/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.2 10/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.3 11/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
28
28
30
32
8 Preparazione alla nomenclatura
8.1 15/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
34
34
9 Ancora struttura delle molecole
9.1 15/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
35
35
10 Nomenclatura
10.1 17/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
38
38
11 Ossidoriduzioni
11.1 29/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
40
40
12 Interazioni deboli
12.1 29/04/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
12.2 02/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
44
44
45
13 Stati della materia
13.1 02/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
46
46
14 I Gas
14.1 06/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
49
49
15 Teoria cinetica
15.1 08/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
15.2 09/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
15.3 13/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
50
50
53
54
16 Soluzioni e proprietà colligative
16.1 15/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
16.2 16/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
16.3 20/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
57
57
62
63
17 Cenni di termochimica
17.1 20/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
66
66
18 Sistemi in equilibrio
18.1 22/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
18.2 27/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
69
69
71
19 pH
19.1 27/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
19.2 29/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
19.3 30/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
72
72
72
76
20 Pila
20.1 30/05/2014 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
80
80
3
Modelli atomici
18/03/2014
1 Modelli atomici
1.1 18/03/2014
La prima idea è quella di Thomson, ottenuta non conoscendo niente, senza conoscere fenomeni
particolari, del 1897. Egli sapeva che dentro l’atomo ci sono cariche elettriche positive e negative,
individuate mediante altri esperimenti elettrici.
Thomson pensò che gli elettroni fossero particelle disperse dentro una massa di carica positiva. Gli elettroni sono carichi negativamente e portano con sé una minima quantità di carica, il
modello è detto a panettone, la massa è data dalla sfera di carica positiva. La dimensione della
sfera di carica positiva era identificabile e misurabile ai tempi di Thomson.
Supponiamo di prendere l’oro, che ha Z = 79. Ai tempi di Thomson si sapeva che c’erano l’equivalente di 79 cariche positive, non si parlava ancora di protoni. Si pensava che la carica positiva
fosse diffusa uniformemente.
Oro: Z = 79 PA = 196,96654 , densità = 19,32
g
cm 3
Se si ipotizza che gli atomi siano sferici, tutti attaccati gli uni agli altri, rimangono dei vuoti quindi
il calcolo che si fa è di sicuro approssimato, ma si cerca l’ordine di grandezza della dimensione,
l’errore va bene. Si procede così: si prende 1 cm 3 di oro, che ha massa m = 19,32 g. Quante moli
19,32
sono? n =
= 19,32/196,97 = 9,81 · 10−2 moli (circa).
P A(Au)
N = n · N A = 9,81 · 10−2 · 6,02 · 1023 = 5,9 · 10−22 atomi. Ne sto prendendo 1 cm 3 . Supponendo che
gli atomi siano messi tutti così
<1.1>
Allora posso pensare che in 1 cm 3 ci siano quel numero di atomi.
Vat omo =
1
= 1,69 · 10−23 cm 3
5,9 · 1022
Dal momento che il volume della sfera è
diametro dell’atomo:
s
D=
3
6V
=
π
4
3
s
3
· π · R3 =
4
3
· π · ( D2 )3 =
π
6
· D 3 . Ho quindi una stima del
6 · 1,69 · 10−23
= 3,19 · 10−8
π
Non mi fido un granché del 3,19, visto che ho un errore introdotto dal fatto che gli atomi fra di loro
hanno dei vuoti, mentre nel conto che ho fatto questo non è previsto, ma mi posso fidare dell’ordine di grandezza, cioè 10−8 cm, cioè 10−10 m = 1 Å
Thomson sapeva quindi che il suo panetto aveva un’estensione di 10−8 cm, in cui era distribuita
tutta la massa dell’atomo. E’ una massa piuttosto diluita, occupa un volume relativamente grande. Questo vuol dire che se io prendo degli atomi di oro in una lamina sottile (un foglio) (l’oro è un
metallo che ha buone caratteristiche elettriche, termiche ed è malleabile, cioè può essere lavorato
in modo da ottenere lamine sottilissime, anche trasparenti, senza che si rompa)... riformula
Esperimento di Rutherford: si sapeva che degli atomi emettevano dei raggi (radioattività, Curie e
Becquerel). In realtà non erano solo raggi, e i due scienziati si accorsero che c’erano tre tipi di raggi, alfa beta e gamma. Furono individuati bene, i raggi alfa erano particelle con massa significativa
e carica positiva.
Particella alfa: carica +2, massa: 4u
Particella beta: carica -1, massa: <?>
4
Modelli atomici
18/03/2014
Particella gamma: né carica né massa.
Si accorsero che questi raggi venivano emessi dall’atomo, era attivo, da qui venne il nome di radioattività. (Studiavano il Radio...)
I raggi α quando incontravano la materia venivano assorbiti facilmente, difficilmente passavano
attraverso uno spessore di un qualsiasi materiale, mentre i raggi β e γ passano più facilmente. Allora Rutherford prese una lamina sottilissima di oro, ci sparò contro un fascio di raggi α tramite
un minerale che conteneva radio. La lamina era tanto sottile in modo che le particelle non ci si
potessero fermare. Ciò che si aspettava Rutherford era che se la particella α incontrava un elettrone, non sarebbe successo praticamente niente, vista la differenza di massa, ma avrebbe dovuto
subire un qualche effetto per via della sfera di carica, ma siccome la massa era distribuita su tutta
la lamina, si aspettava che le particelle la attraversassero.
Usando uno schermo fluorescente che quando viene colpito da un raggio α, emetteva luce. Rutherford per fortuna non mise uno schermo piatto dietro la lamina d’oro, ma uno schermo che ci
girava tutto intorno, perché così poté vedere che oltre ai segnali che si aspettava, ve ne erano altri
analoghi però in direzioni deviate molto, e questo era strano. Ciò che era ancora più strano era che
alcuni segnali erano addirittura dalla parte opposta a dove si aspettava, cioè dalla stessa del fascio,
come se alcuni raggi rimbalzassero. Il fatto che fossero pochi, non significava che non contassero,
e furono questi tipi di segnali a far venire in mente a Rutherford come fossero fatti gli atomi.
Si può calcolare, in base all’energia della particella α, quanto è grossa la particella che la ha respinta. Si sa che la carica della particella alfa è +2, quella <...> dell’oro è +79. Si sa che mano mano che
la particella α si avvicina, rallenta e poi viene respinta dalla carica dello stesso segno. Si trova così
un limite superiore al diametro di ciò che ha respinto la carica, che non è 10−8 cm, ma 10−13 cm.
Rutherford determinò che la particella era in grado di arrivare al massimo a 10−13 cm dal centro
dell’atomo, prima di tornare indietro. Quindi la massa dell’atomo era concentrata in una zona
decisamente più piccola, che chiamò nucleo. Tutto ciò che c’era intorno era vuoto, per questo
motivo tante particelle α passavano indisturbate, perché è difficile prendere il nucleo. Quelle che
lo incontrano, subiscono una deviazione significativa, quelle invece che sono praticamente allineate (pochissime) al nucleo, vengono respinte indietro. Questo fece capire che l’atomo era una
specie di sistema planetario, con un nucleo pesante nel centro e il resto erano particelle leggere
intorno, con una dimensione media di 10−8 cm e un nucleo di 10−13 cm.
Il problema che si poneva allora, era descrivere con le leggi della fisica un atomo fatto in quel
modo.
Ci sono degli elettroni che ruotano intorno ad un nucleo, quindi si suppone che le orbite siano ellittiche o circolari come quelle planetarie, ma c’è una piccola differenza: le cariche elettriche sono
cariche elettriche, non masse.
Quando le cariche elettriche si muovono in moto accelerato, emettono energia sotto forma di onde elettromagnetiche. Onde elettriche e magnetiche sono caratterizzate da direzione, lunghezza
d’onda (λ), velocità di propagazione (c, velocità della luce), frequenza ( ν = c/λ), e trasportano
energia in modo proporzionale a E 2 (E = campo elettrico) o al quadrato del campo magnetico.
Un elettrone in moto accelerato, come un atomo intorno ad un nucleo, emette una sua energia,
sotto forma di onde elettromagnetiche, quindi la sua energia diminuisce, quindi il nucleo lo attira
di più, quindi oscilla ancora di più, emette ancora più energia, e così via. Teoricamente l’elettrone dovrebbe cadere sul nucleo, secondo la fisica classica. Se si prova a fare una stima di quanto ci
mette a fare l’elettrone a fare la spirale su un atomo di idrogeno, si ottiene 10−9 secondi, ma questo
non spiega come mai la materia sia stabile da più di 15 miliardi di anni. Questa era la crisi della
fisica classica di allora.
La seconda notazione: (spettro elettromagnetico) le onde elettromagnetiche sono caratterizzate
da lunghezza d’onda e frequenza, che hanno prodotto costante (c, velocità). Se si prendono lun5
Modelli atomici
18/03/2014
ghezze d’onda molto grandi, centinaia di metri, si prendono le onde radio. Se si scende, intorno
a 10−2 m, quindi nell’ordine del cm, si ottengono le microonde, che sono particolari perché la loro frequenza è molto vicina alla frequenza di risonanza delle molecole d’acqua, che è presente in
tutti i tessuti e quando un’onda di elettromagnetica di opportuna frequenza la incontra, l’acqua
entra in risonanza, cioè è in grado di assorbire moltissima energia dall’onda, vanno in vibrazione,
ed è il motivo per cui i cibi cuociono.
Poi ci sono gli infrarossi, poi c’è un intervallo piccolissimo delle onde cui gli occhi sono sensibili.
Oltre ci sono gli ultravioletti, che non vediamo, poi i raggi X e infine i raggi gamma, un’estensione
molto grande di onde elettromagnetiche con frequenze elevatissime, emessi anche dalle sorgenti
radioattive e dalle radiazioni cosmiche.
Gli oggetti portati a temperatura di incandescenza, emettono luce di tutto lo spettro del visibile e
oltre, noi vediamo il bianco, ma si possono individuare le componenti. Osservando ciò che fa la
pioggia con la luce del sole, si prende un prisma e lo si colpisce con raggio di luce con un certo
angolo di incidenza.
L’angolo di uscita dipende dalla lunghezza d’onda. Quelle sul blu hanno un angolo più stretto di
quelle sul rosso, in questo modo si separano le varie lunghezze d’onda. Si vede così lo spettro continuo, dove ci sono tutte le lunghezze d’onda.
A quei tempi, quando si scoprì questa storia degli spettri, la si usò per identificare gli elementi dei
composti, in base a due tipi di spettri diversi, gli spettri di assorbimento e di emissione. La differenza sta solo nella modalità sperimentale: si prende un gas, lo si eccita, per esempio attraverso
una scarica elettrica, gli atomi di idrogeno emettono luce, si fa attraversare la luce da un prisma, e
la si scompone nelle varie componenti. Non si vede un arcobaleno, ma solo alcune righe luminose
ben precise. Ogni riga corrisponde con un certo angolo di deviazione, cioè una ben precisa lunghezza d’onda. Lo si può fare con tutti gli elementi gassosi, e ogni elemento ha il suo ben preciso
spettro. In questo modo gli spettroscopisti capivano che sostanza avevano di fronte.
L’altra tecnica è usare una luce bianca (lampada ad incadescenza), si interpone il gas da osservare,
si fa passare la luce attraverso il prisma e si osservano delle righe scure sullo spettro, che coincidono con le righe chiare nello spettro di emissione.
Non si sapeva perché, ma si era certi di questo comportamento.
Gli spettroscopisti avevano trovato una formula empirica che dava
¶
µ
1
1
1
=R 2 − 2
λ
n
m
L’atomo di idrogeno aveva uno spettro di assorbimento fatto così: <1.5>
Lo spettro di emissione così: <1.6>
Osservarono che la prima famiglia c’era in entrambi, le successive solo in emissione. (le famiglie
erano i raggruppamenti per n: n =1 prima famiglia, n=2 seconda famiglia e così via).
<1.7> Se l’onda elettromagnetica è molto intensa (cioè E è molto grande) e lo lascio per molto tempo, alla fine gli elettroni presenti sulla superficie del metallo possono aver assorbito tanta di quella
energia da venir via dall’atomo: effetto fotoelettrico, che è l’emissione di elettroni ad opera della
luce, che era già stato osservato per alcuni metalli particolari.
All’interpretazione semplice classica contemporanea, si opponeva questo esperimento: <.prendo
un certo metallo M..> se la frequenza dell’onda che uso ni è minore di ν0(M ) , posso lasciare il raggio
attivo per tutta la vita, anche a intensità bestiale, ma non esce nessun elettrone, in nessun modo.
Gli elettroni escono se la frequenza dell’onda è maggiore di questa soglia.
Se si traccia un grafico di energia cinetica degli elettroni in funzione della frequenza <1.8>, non si
trovava alcuna spiegazione al comportamento prima di ν0 .
La spiegazione a questo fenomeno venne data da Einstein, con questo esercizio:
E ond a = E el et t r one +E l avor oDi E st r azi one . L’energia dell’elettrone è l’energia cinetica (una volta estrat6
Modelli atomici
18/03/2014
to ha solo quella), l’energia dell’onda la indicò così: <1.9>
<...> nel mondo atomico queste quantità variano senza continuità. Planck chiamò queste quantità
Quanti. L’energia associata ad una particella è h · ν, è un risultato quantistico, non classico.
Einstein non usò un’espressione classica, ma quella dovuta a Planck.
<1.9>, che ha il grafico di <1.8>.
Anche l’effetto fotoelettrico non aveva spiegazione in fisica classica, il primo che si prese la briga
di spiegare questi fenomeni fu Bohr.
Il modello di Bohr dell’atomo di idrogeno è un modello semi-classico. Fece questo: prendiamo un
atomo di idrogeno, un neutrone ed un solo protone.<2.1>
e2
L’elettrone interagisce con il protone con una forza di tipo elettrico: F e = 2
r
Suppose che 1) le orbite che percorreva l’elettrone fossero circolari e stazionarie, motivo per cui
non fu creduto all’inizio, perché si prevede che l’elettrone non emetta energia nonostante sia sottoposto ad accelerazione.
e2
m · v2
Si ha questa relazione in fisica classica: 2 =
(forza centripeta), che permette di avere:
r
r
e2
r=
m · v2
Un’altra cosa che si sa è che l’energia cinetica E c = 12 · m · v 2 , e l’energia dovuta all’interazione elet−e 2
1
e2
trostatica è E p =
. Si sa quindi che E = E c + E p = · mv 2 −
=<2.2>
r
2
r
2) quantizzazione del momento angolare <2.3>. Classicamente può assumere valori qualunque,
Bohr invece ipotizzò che il momento angolare variasse solo per quantità discrete, ciò in modo
quantizzato, per multipli di un numero n intero di una certa quantità:
nh
h
= n · ħ. Ricavo v =
. Lo sostituisco in <2.4>
m ·r ·v = n ·
2π
2π · m · r
Viene fuori che i raggi permessi non sono qualunque, ma sono indicizzati da un indice n, il più
piccolo lo ottengo per n = 1.
La seconda orbita ha un raggio 4 volte più grande della prima, poi 9 volte e così via.
Se si calcola il primo raggio, si ottiene r 1 = 0,529Å, cioè il diametro di un atomo di idrogeno è grosso circa 1 Å, che è perfettamente in accordo con ciò che si sa già. Ciò che determina la grandezza
dell’atomo sono delle grandezze fisiche universali.
7
Modelli atomici
20/03/2014
1.2 20/03/2014
Bohr aveva dato due postulati: 1° postulato) orbite circolari stazionarie
2° postulato) quantizzazione del momento angolare
h2
rn = n2 · 2
= n 2 · a 0 con n = 1,2,3,..., dove n è detto numero quantico, a 0 = raggio di Bohr =
4π me 2
0,529 Å (raggio della prima orbita), è il raggio dell’orbita.
Questa dimensione ha coincidenza quasi esatta con la dimensione di un’atomo di idrogeno, che
era nota (1 Å), e veniva fuori dalle costanti fisiche universali.
e2
Un’altra cosa importante che segue è: E = − . La prima conclusione è che l’elettrone può stare
2r
solo sulle orbite calcolate, sono le uniche permesse. Ciò vuol dire che su ogni orbita c’è un’energia
precisa, che quindi anch’essa può assumere solo alcuni valori.
1 2π2 me 4
Se si sostituisce: E n = − 2
n
h2
che è solo un’altra combinazione di grandezze fisiche universali che ha un valore numeri preciso.
1
La si scrive così di solito: − 2 E 0 , con E 0 = 13,6 eV (1 eV = 1,602 · 10− 19 J). L’eV è più adatto a lavon
rare con le cariche atomiche.<...>
Cosa sono questi 13,6 eV?
n = 1 r 1 = a 0 = 0,529 Å E 1 = −E 0 = −13,6 eV
13,6
= −3,4 eV L’energia diminuisce con il quadrato di n.
n = 2 r 2 = 4a 0 = 2,13 Å E 2 = −
4
13,6
n = 3 r 3 = 9a 0 = 4,71 Å E 3 = −
= −1,51 eV
9
<1.1>
Il limite che si può ottenere per questa energia è 0, cioè E ∞ = 0, tutti gli altri stanno tra 0 e -13,6 eV.
A quelli con E = 0, corrisponde un raggio infinito, cioè che l’elettrone è stato portato via al protone,
cioè l’atomo di idrogeno è stato ionizzato.
H −−→ H+ + e
Questo processo si può fare sperimentalmente, e si può misurare l’energia necessaria. Si trova
esattamente -13,6eV. Normalmente si trovano tutti gli elettroni di idrogeno nella condizione E 1 ,
cui bisogna fornire proprio questa energia per estrarlo dall’atomo.
Quindi il modello di Bohr giustifica la misura sperimentale molto precisamente. Queste due prove
sperimentali (dimensione atomo e energia di estrazione) spiegate dalla sua teoria fecero sì che le
sue idee fossero prese in considerazione.
Bohr si domandò come l’atomo facesse ad assorbire ed emettere energia. Dal momento che lo
schema di energia è quello in <1.1>, allora l’elettrone non può fare altro che passare da un valore
all’altro di queste energie.
3° postulato) variazioni di energia = transizioni permesse
Questo secondo lui era l’unico modo per cui un atomo di idrogeno poteva emettere o ricevere
energia dell’esterno, solo spostando l’elettrone fra uno dei livelli di energia permessi.
Prendiamo atomi di idrogeno con gli elettroni tutti in E 1 e lo si colpisce con la luce. Esprimo la
quantità di energia dell’onda elettromagnetica così:
hν = E m − E 1 , dove m è un intero come n (parte da 2). Questa è l’energia che fa passare l’elettrone dall’orbita 1 a 2, ma se l’energia è inferiore, non succede nulla, cioè l’idrogeno non l’assorbe,
cioè appare trasparente a quella certa lunghezza d’onda. Se invece l’onda ha energia esattamente
coincidente con quella necessaria,
µ l’atomo
¶ l’assorbe.
1
1
hν = E m − E 1 = − 2 E 0 − E 0 = E 0 1 − 2 dove m = 2,3,4,...
m
m
µ
¶
µ
¶
hc
1
1 E0
1
Sostituisco ν :
= E 0 1 − 2 , cioè =
1− 2
λ
m
λ hc
m
8
Modelli atomici
20/03/2014
Questa cosa giustifica ciò che gli spettroscopisti conoscevano già, spiegando la costante di R... e
giustificava lo spettro dell’idrogeno.
E0
coincide con il valore sperimentale della costante di R.
hc
Quindi l’atomo di Bohr spiega perfettamente lo spettro di assorbimento dell’idrogeno, e anche il
motivo per cui lo spettro di assorbimento è solo un sottoinsieme dello spettro di emissione (che
ha una parte che coincide e una serie di righe che in assorbimento non ci sono).
Quando ho un’emissione? Per esempio appena dopo ho avuto un assorbimento. L’elettrone ci
sta pochissimo tempo (10− 9 s) nello stato E 2 , e la differenza di energia la emette sotto forma di
fotone, con energia precisamente del salto, cioè si vede l’atomo di idrogeno che emette luce. Cioè
l’atomo di idrogeno riemette la stessa lunghezza d’onda della luce che ha assorbito. Se ogni livello ritornasse sempre sul primo, avremmo uno spettro di assorbimento coincidente con quello di
emissione, ma in emissione <1.2> ha tanti modi per tornare giù. Se torna allo stato E 1 , si ha coincidenza degli spettri, mentre se invece fa un salto più
un’energia diversa. La formula
µ corto, emette
¶
E0
1
1
=
1/32 − 2
se sono coinvolti il quarto e terzo:
λemi ssi one hc
4
Si capisce che in assorbimento tutte le freccie partono da E 1 , mentre in emissione possono partire
da qualsiasi stato, in questo esempio
ci sono
µ
¶ almeno 3 lunghezze d’onda che prima non vedevo.
1
E0 1
1
La formula generale:
=
−
λem hc n 2 m 2
Se n 6= 1, si ottengono le altre bande, che si potevano misurare con estrema precisione.
Sommerfeld cercò di perfezionare il modello di Bohr, generalizzando il modello di Bohr, usando
orbite ellittiche, in cui compare un altro numero quantico, detto numero quantico principale, che
teneva conto della forma dell’ellisse, indicato con <l>.
Un’altra generalizzazione importante è prendere un atomo con Z protoni e un solo elettrone, detto
atomo idrogenoide (si prende cioè un atomo qualunque ionizzato Z-1 volte).<1.3>
Per esempio si può prendere He+ . Il modello di Bohr riesce a descriverlo, come anche con il Li2+ ,
per il tempo in cui si riesce a mantenere in vita questi ioni. La differenza che compare nelle formule:
1
r n = − n 2 a 0 che ha senso, perché se ci sono più protoni l’elettrone si trova più vicino al nucleo.
Z
E0
E n = −Z 2 2 Questo vuol dire che l’elio ha energia di ionizzazione 4 volte più alta, per via della
n
maggior carica del nucleo.
Un limite principale di questo modello è quando si cerca di inserire due elettroni, per descrivere per esempio l’elio normale, non funziona più in nessun modo, e le cose peggiorano se si vanno
a vedere gli atomi degli elementi successivi. Questo modello funziona solo se c’è un elettrone solo
all’esterno.
L’altra critica maggiore si basa sui postulati, in particolare sul primo, cioè che le orbite sono stazionarie, ed era il motivo principale per cui Bohr non era stato creduto. Il fatto è che secondo la
fisica classica, un moto circolare o ellittico non può essere stazionario.
De Broglie ipotizzò che la materia in realtà avesse una doppia natura, corpuscolare e ondulatoria.
C’era da capire quando un’oggetto mostrava un aspetto e quando l’altro (per esempio, l’effetto fotoelettrico della luce e la diffrazione con una fenditura piccola).
h
Trovò che λ =
legava i due aspetti.
mv
Una conseguenza importante era: <1.4> e <1.5>. Se l’elettrone è un onda, allora deve essere stazionaria se è stabile<?>, quindi occorre che 2πr = nλ, spiegando così cosa era il significato di
nh
h
stazionario di Bohr, trovando 2πr =
, che si può scrivere mvr = n , spiegando la quantizzamv
2π
9
Modelli atomici
20/03/2014
zione.
Heisenberg (fisico tedesco) dimostrò il principio di indeterminazione, fondamentale, che si traduce in:
<1.6> (è il prodotto tra due errori di misura)
Quando si deve descrivere un moto, serve sapere ~
r (t ) e ~
v (t ) nello stesso istante, ognuna con il loro relativo errore. In una dimensione per esempio: x ± ∆x e v x ± ∆v x . Riscrivendo il principio di
h
.
indeterminazione: ∆x∆v x >=
4πm
Significa che il prodotto dei due errori ha un limite inferiore, cioè che posso determinare per esempio con precisione elevata la posizione (ho ∆ x piccolo), ma non posso scegliere un valore a caso
di velocità, sono costretto a perdere informazione sulla velocità. Il concetto è che non si possono
misurare posizione e velocità contemporaneamente.
Quando funziona questo principio?
Esempio: <1.7>
<1.8>
Anche accontentandosi di un errore grosso sulla posizione, l’errore sulla velocità è enorme, quindi
non posso trovarne la traiettoria.
La difficoltà emerge perché sto usando una particella con una massa molto piccola, dove l’indeterminazione diventa evidente.
Quindi è assodato che non si può descrivere l’elettrone come un corpuscolo che si muove su una
traiettoria. A livello atomico, un elettrone ha un comportamento che somiglia di più a un onda,
che è una “cosa” distribuita che trasporta energia ma non si sa dove è.
Schroedinger riuscì a trovare un’equazione che lega onda e comportamento dell’elettrone: <1.9>
La risoluzione di questa equazione sono funzioni d’onda Ψ, il cui significato fisico è d P = |Ψ|2 dV
<1.10>
Se sommo la probabilità di trovarlo in tutti i punti dello spazio, ottengo 1 (cioè ce lo trovo di sicuro). <1.11>
Quando l’equazione di Schroedinger si applica all’atomo di idrogeno, si ottengono funzioni che
dipendono dalle tre coordinate spaziali:
Ψ(x,y,z; n,l ,m)
n = numero quantico principale, assume tutti i valori interi da 1 a infinito, cui è legato il valore
dell’energia dell’elettrone (E)
1 2π2 me 4
cioè proprio l’espressione di Bohr.
Quando si trova l’autofunzione, si ha che E = −1 2
n
n2
l = numero quantico orbitale (o angolare) è un numero intero che va da 0 a n - 1
m = numero quantico magnetico, i cui valori sono 0, ±1, ±2 fino a ±l .
La distribuzione nello spazio si chiama orbitale, m è legata alla sua orientazione nello spazio.
10
Modelli atomici
25/03/2014
1.3 25/03/2014
4πr 2 |Ψ1s |2
|Ψ1s |
1
q
πa 03
max
r
a0
r = a0
r
a0
Figura 1: Densità di probabilità e densità di probabilità radiale
Gli orbitali di tipo s (l = 0) sono caratterizzati da una
sferica. Gli orbitali s si potevano
¡ simmetria
¢
scrivere, in coordinate polari, come Ψ = R nl (r ) · A l m θ,ϕ .
Nel caso in cui l, il numero quantico secondario è 0, il secondo termine della moltiplicazione è una
costante. La funzione d’onda quindi assume sempre la stessa forma, e dipende solo dalla distanza.
Questo ha una conseguenza, perché l’orbitale s si presta meglio a individuare un punto nello spazio <1.3> (ricorda che il significato della funzione d’onda è quello di densità di probabilità) <...> Il
guscio ha tutto la stessa probabilità di individuare un elettrone.
Ñ
|Ψ1s |2 dV
Z 2a0 Z 2π Z
P (a 0 < r < 2a 0 ) =
=
=
1
π
e
πa 03
a0
1
Z
πa 03
0
2a 0
− a2r
0
0
4πr 2 e
−2r
a0
r 2 sin θd θd ϕd r =
dr
a0
Osserva il termine da integrare:
e
− a2r
0
(4πr 2 d r )
| {z }
è la superficie di una sfera di raggio r per lo spessore dr, cioè è l’elemento di volume di un guscio
sottile.
Il secondo grafico di Figura 1 rappresenta la somma delle probabilità in tutte le direzioni, è la densità di probabilità radiale, è lineare, in funzione della distanza. qui c’è qualcosa che non torna!!
Risponde alla domanda: a che distanza trovo l’elettrone?
Ha max in corrispondenza del raggio di Bohr, ma questo non significa che la probabilità max si ha
per r = a 0 , è max sul nucleo, il fatto che ce l’abbia è un fatto puramente matematico.
Un modo per fare il grafico di questa funzione <densità di probabilità?> in tre variabili (servirebbero 4 assi è questo):<1.6>
Dove i punti sono più fitti, è più alta la probabilità di trovare elettroni.
L’integrale per parti che dovevi fare:
P (a 0 < r < 2a 0 ) = −e
− a2r
0
· µ ¶2
¸¯2a0
¯
r
2r
2
+
+ 1 ¯¯ = 0,4386
a0
a0
a0
11
Modelli atomici
25/03/2014
cioè la probabilità di trovare l’elettrone nella corona circolare con raggio compreso tra a 0 e 2a 0 è
del 43,86%.
δ
= 10−3 se δ è molto piccolo, è il caso di un volumetto
L’altro esercizio: a 0 < r < a 0 + δ , dove
a0
infinitesimale, un dV . La probabilità, in questo caso si può calcolare anche direttamente, invece
che con l’integrale:
¡
¢
1 −2
∆P = |Ψ1s |2 ∆V = |Ψ1s |2 4πa 02 a 0 · 10−3 =
e 4πa 03 · 10−3 = 5,41 · 10−4
3
πa 0
che è una probabilità molto piccola.
Il succo della questione da capire è: quale è il significato di queste funzioni d’onda, la "roba"
radiale<...>
Serve comunque avere un’immagine anche se approssimata, ma significativa di come è fatta questa distribuzione nello spazio che sia più pratica della nuvola di punti sempre più rarefatti. Pur
perdendo alcune informazioni, una convenzione che si usa è quella di individuare una superficie
che ha un certo raggio che chiamo r ∗ che abbia una probabilità di trovare l’elettrone all’interno
della superficie del 95%, così che fuori da questa superficie, ci sia solo il 5% di avere elettroni. E’
solo una convenzione, che dice solo di delimitare la zona di spazio dove la probabilità è del 95%.
<1.9>
Nel caso di una funzione d’onda sferica, la superficie è quella di una sfera. Ricordati che questa è
solo una semplificazione, e che questa funzione d’onda ha un massimo che è dentro alla sfera, e
che la probabilità di trovarvi elettroni non è tutta uguale.
L’orbitale con n = 1 ha un solo orbitale, il successivo, con n = 2, ne ha 4. L’orbitale con i numeri
2,0,0, è l’orbitale 2s, anch’esso con simmetria sferica, ed ha questa espressione:
µ
¶
1
r
1
− r
2−
e 2a0
Ψ2s = p
3
a0
2 π 2
2a 0
L’equazione di Schroedinger per gli atomi di idrogeno e per tutti gli idrogenoidi, ha sempre la
stessa forma:
Ψ = R(r ) · A(θ,ϕ)
µ ¶
r
− r
dove R(r ) = e na0 · P n−1
(P n−1 è un polinomio di grado n-1)
a0
La conseguenza non è solo matematica, se si fa il grafico:
I nodi sono i punti a probabilità 0, dove non si troverà mai un elettrone. Il 2s si osserva che ha 2
massimi, proprietà di tutti gli orbitali s è di avere un massimo assoluto. Si può immaginare quindi
una nuvola di puntini molto fitta, poi ad un certo punto si annulla, poi aumenta, poi si dirada di
nuovo. Anche questa si può rappresentare con una superficie limite, si trova che è una superficie
molto più grossa di quella dell’1s.
2 1 1
Con n = 2, esistono altri tre orbitali, che sono 2 1 0 La forma di questi tre orbitali è identica,
2 1 -1
basta vedere come è fatto uno per sapere come sono fatti tutti e tre, perché differiscono solo per
l’orientamento spaziale (ognuno è orientato lungo un asse). Le loro equazioni sono: <2.3>
Si vede che con n = 2, l’angolo adesso compare nell’equazione. Un dettaglio, per vedere come è
fatta, si osserva l’equazione più semplice lungo la z. Queste funzioni hanno un nodo subito per r
= 0, cioè non troverò mai un elettrone nel nucleo. La superficie limite è fatta di due lobi, una sopra
e una sotto il piano xy. Il 95% di trovere un elettrone di questo orbitale è dentro questa coppia di
12
Modelli atomici
25/03/2014
|Ψ2s |2
A2
Ψ2s
A
nod o
2
r
a0
r
a0
2
4πr 2 |Ψ2s |2
A
|Ψ2s |2
ingrandimento del
secondo grafico per
vedere il massimo
2
r
a0
2
r
a0
Figura 2: Ψ, densità di probabilità e densità di probabilità radiale dell’orbitale 2s
lobi. Questa forma è determinante per capire il meccanismo di formazione dei legami. Inoltre la
funzione d’onda ha un segno (la distribuzione di probabilità è sempre positiva, è un quadrato).
Questi lobi hanno una simmetria assiale.
Con n = 3, si ha l’orbitale 3,0,0, che si indica con 3s, che ha 2 nodi. La parte radiale sarà un poli3 1 1
nomio di grado 2 e andrà a 0 più lentamente. Poi ci sono i 3 1 0 Tutti i nodi sono sul nucleo,
3 1 1
poi hanno un altro nodo all’interno, sono fatti di due lobi in simmetria assiale, ciascuno diretto
3 2 -2
3 2 -1
lungo un asse. Poi ci sono 3 2 0 In questo caso, non cambia solo l’orientazione, ma anche
3 2 1
3 2 2
la forma. <2.4>
La capacità di un atomo di legarsi o il modo in cui si lega dipende fortemente dagli orbitali in cui
gli elettroni si trovano.
<2.5> La coincidenza tra i risultati di Bohr <...>
Questo è solo un problema matematico, non concettuale. Quindi per trovare gli atomi degli elementi successivi, si usano approssimazioni matematiche e si trovano numericamente le funzioni
d’onda. Il risultato di questo calcolo è che tute le funzioni d’onda di tutti gli atomi sono sempre
classificate con 3 numeri quantici (n, l, m: principale, orbitale, magnetico), sempre con la stessa
dipendenza dai parametri già vista, con dipendenza analitica analoga a ciò che si è già visto, con
differenze minime. Ciò che si vede è che, con n maggiori, gli orbitali esterni sono molto più estesi.
Una delle varianti più significative rispetto all’atomo di idrogeno è questa: negli atomi a più elet13
Modelli atomici
25/03/2014
troni, l’energia comincia a dipendere anche da l. <2.6>
Quale è la differenza più vistosa tra l’atomo di idrogeno ( 1 protone ed 1 elettrone), e l’idrogenoide
dell’elio (2 protoni e 1 elettrone)? Le energie sono tutte più basse perché il nucleo attira di più gli
elettroni, ma i livelli energetici rimangono gli stessi. Se si mette un elettrone in più nel sistema,
cioè si aggiunge un qualcosa che aggiunge una forza di repulsione tra gli elettroni, che non c’era prima nell’idrogeno. Nell’equazione di Schroedinger, il termine della repulsione è ciò che non
consente di risolvere matematicamente l’equazione. Che cos’è la repulsione?
Pensa all’idrogenoide dell’elio. L’elettrone da solo è molto attratto, che vuol dire che l’energia di 1s
è molto basso, e l’energia che serve per ionizzare è più elevata, 4 volte quella dell’idrogeno. Se nello stesso sistema ho una carica negativa, mi aiuta a rimuovere un elettrone. Quindi la repulsione
sposta "tutto in su". C’è poi una certa probabiltà di avere i due elettroni vicini. Se i due elettroni
sono sullo stesso orbitale, c’è una certa probabilità che siano vicini. Complessivamente, tra un 2s
e un 2p, il secondo occupa un volume minore. Se si sommano le tre probabilità degli orbitali p si
trova una funzione radiale che ha simmetria sferica, che ha circa lo stesso volume del 2s. Questo
significa che 2 elettroni che stanno su uno stesso orbitale p, sono in un volume più piccolo, quindi
è più probabile che siano vicini, quindi l’effetto di repulsione si vede di più su un orbitale p che su
un orbitale s. Quindi è più facile togliere un elettrone da una p che da una s (guarda 2.6). Questa è
l’origine della rimozione della degenerazione. Se i due elettroni possono occupare uno spazio più
piccolo, in media si respigono più energicamente. Gli orbitali d, nonostante la loro complessità,
hanno tanti nodi, cioè un volume a disposizione minore del p, quindi l’effetto di repulsione è mediamente maggiore, infatti si "spostano ancora più un su".
Posso immaginare virtualmente di creare l’elio aggiungendo un protone, poi un elettrone. <2.7>
In generale, la repulsione tra gli elettroni è minore dell’attrazione verso i protoni. L’incremento da
p ed s, e tra d e p si riduce sempre di più mano mano che aumenta n. Man mano che si sale, le
variazioni sono sempre più ravvicinate. Questa scala porta a questa conseguenza: ci sono delle
inversioni, la prima è tra 4s e 3d, in cui 3d ha più energia di 4s. E salendo ne avvengono altre. Succede che mentre salgo i livelli energetici, trovo 1s, 2s, 2p, 3s, 3p, 4s, 3d, ...
Il trucco per impararseli è <2.8>
Va letta lungo le diagonali dei quadrati. L’ordinamento di questi livelli energetici spiega la regola
usata per ordinare la tabella periodica.
14
Configurazioni elettroniche
27/03/2014
2 Configurazioni elettroniche
2.1 27/03/2014
La sequenza vista ieri serve per capire la struttura della tavola periodica.
Per iniziare il discorso bisogna fare un’anticipazione. Occorre aggiungere un altro numero quantico magnetico, che non ha analogo classico, e si riferisce allo SPIN.
La prima cosa che differenzia lo spin dagli altri numeri quantistici, è che lo spin come grandezza
può assumere solo 2 valori, +1 e -1 o +1/2 o -1/2 a seconda di come si prende la costante di Plank,
noi lo rappresentiamo con <1.1>
Il principio di esclusione (di Pauli) è un teorema dimostrato che dice che in qualunque sistema
atomico non ci possono stare elettroni che hanno tutti e 4 i numeri quantici uguali. Questo in
termini pratici significa che siccome m s può avere 2 valori, per ogni terna n,l,m può avere solo 2
valori, cioè su un orbitale ci possono stare solo 2 elettroni, con spin opposto. Questo è importante
perché finora nessuno aveva detto quanti potevano esserci su un orbitale, adesso si sa che se ci
sono, sono 2 e uno ha spin in sù e l’altro in giù.
Si può fare quindi una costruzione sulla tabella periodica, detta “AUFBAU”.
z=1 H <1.2>
Per il litio: di sicuro 2 elettroni sono in 1s, ma poi non ce ne posso mettere più per il Principio di
esclusione, quindi lo devo sistemare nell’orbitale a più vicina energia, il 2s.
Già ora si nota che (quando s’era fatta la tabella?)<..> il guscio con n = 1 al massimo aveva 1 orbitale, l’1s, e avevamo detto anche che il guscio si completa, <...> l’indicazione di guscio completo
è tradotta con l’accapo nella tabella periodica. Le righe si chiamano periodi, dentro i riquadri di
ogni elemento è riportata la configurazione elettronica. Quando si passa al litio, per poterlo costruire devo aggiungere un protone e un elettrone. Il terzo elettrone ho appena visto che lo posso
sistemare solo ricorrendo ad un orbitale 2s, cioè vado ad utilizzare gli orbitale con n=2, perché
quelli con n=1 (uno solo) sono già pieni. Ed n significa iniziare una nuova riga, infatti il litio si
trova nella prima posizione della seconda riga.
<1.3>
Visto che la notazione diventerà pesante, introduciamo una notazione nuova: 1s 2 significa che il
guscio è completo, la si indica con [He] e la si porta avanti, quindi si possono riscrivere le configurazioni degli elementi successivi.
Per il carbonio: Regola di HUND (è la tendenza dei sistemi a raggiungere il minimo energetico): se
ho più elettroni da sistemare, essi preferiscono occupare orbitali diversi con spin parallelo piuttosto che orbitali identici ma con spin opposti. Questo succede perché quando comincio a riempire
gli orbitali con gli elettroni, cambia la loro energia di poco, per questo a gli elettroni normalmente
(cioè a riposo) si dispongono su orbitali diversi con spin paralleli.
Per l’ossigeno: ora gli orbitali sono di nuovo tutti degeneri, quindi non c’è scelta, l’elettrone si mette in un orbitale p con spin opposto.
Adesso non posso più aggiungere un elettrone in 2p, perché gli orbitali sono già pieni, quindi l’undicesimo elemento, deve avere un orbitale 3s, che vuol dire che sono andato a capo perché ho
dovuto usare il numero quantico successivo, infatti il sodio è il primo elemento della terza riga, e
inoltre la sua configurazione elettronica la si può scrivere meglio, perché anche il Neon ha il guscio
completo, quindi in realtà la si può scrivere [Ne] 3s 1 .
C’è una regolarità: l’idrogeno era il primo elemento del primo periodo, e ha 1 solo elettrone, il
litio (primo elemento del secondo periodo) ha guscio completo e 2s 1 , il sodio ha guscio completo
e 3s 1 , cioè il guscio più esterno hanno lo stesso numero di elettroni. Le colonne, dette gruppi, è
fatta di elementi la cui configurazione elettronica finice sempre con s 1 , hanno un solo elettrone
esterno, tutti gli altri sono distribuiti in gusci completi. Questa regolarità si ripete anche per gli
15
Configurazioni elettroniche
27/03/2014
altri elementi, perché dopo il litio c’è il berillio che ha 2s 2 , l’elemento successivo al sodio, il magnesio, ha 3s 2 . Sotto il magnesio c’è il Calcio, che è completo e 4s 2 , cioè nel secondo gruppo gli
elementi finiscono tutti con s 2 . Dal punto di vista chimico, il fatto di avere solo un elettrone, o due
o tre esterni, determina la capacità di un atomo di interagire con il mondo esterno, che avviene
infatti tramite gli elettroni più esterni. Gli elettroni nei gusci completi sono molto più schiacciati
sul nucleo, quindi avere un elettrone invece di 2 esterni cambia completamente il tipo di reazione
con gli altri atomi.
L’ultimo gruppo ha elementi con guscio completo: He, Ne, Ar, e così via; tutti gli elementi di questi
atomi hanno il guscio più stabile possibile perchè è completo, quindi questi atomi per modificare la loro configurazione esterna dovrebbero sacrificare energia, tradotto in termini pratici: sono
elementi che non si legano con nessuno (gas nobili). Il gruppo subito prima hanno tutti configurazione ns 2 np 5 con un solo elettrone <...> e hanno tendenza ad acquisire l’elettrone che manca loro
per completare il guscio, raggiungendo una configurazione molto stabile. Simile cosa succede per
esempio al Na, infatti lo ione Na+ è molto stabile, ancora più stabile del neon, perché ha anche
un protone in più, ma siccome Na+ ha una carica, e ipotizziamo il fluoro che diventa ione F-, si
legano perché sono cariche opposte (meccanismo di formazione dei legami ionici, ed è uno dei
legami più diffusi, che tipicamente coninvolge gli elementi del primo e penultimo gruppo).
16
Configurazioni elettroniche
28/03/2014
2.2 28/03/2014
<1.1>
Si arriva fino a Z=18, con l’argon. Ora, con n=3, ho anche l’orbitale 3d, quindi per Z=19 (K), lo
dovrei aggiungere alla configurazione elettronica che ho già. Ma dopo il 3p non incontro il 3d, avviene la prima inversione, trovo il 4s (il motivo è l’attrazione del nucleo per i livelli e la repulsione
degli elettroni che rialza il livello energetico a seconda di l, cioè se l’orbitale è s, p o d). Quindi dobbiamo cambiare n, infatti andiamo a capo. Dal punto di vista energetico o fisico, il riempimento
ottenuto con 3s 2 3p 6 è come un guscio stabile; il fatto che ci sia anche il 3d a disposizione, non
cambia questa natura di stabilità, perché comunque il 3d è abbastanza lontano (dal punto di vista
energetico), e questo costringe a incrementare n ed ad andare a capo. I gusci successivi ripartono
dalla configurazione dell’argon.
<1.2>
Appena arrivo a Z=21 con lo scanio, l’ultimo elettrone va in 3d , visivamente questo si traduce con
l’inizio della zona arancione, costituita da 10 elementi per riga, perché in 3d ci stanno 10 elettroni. Questo elemento è l’inizio della prima serie di transizione. Il blocco S è costituito dai primi 2
gruppi, il blocco celeste è il blocco P, caratterizzati dalle configurazioni s complete e p 1,2,3,4,5,6
Si arriva quindi fino a Z=30. Una volta finito il 3d, l’orbitale successivo a disposizione è di nuovo un
orbitale con n=4, il 4p. Con Z=31 (Ga) inizia il riempimento dell’orbitale 4p, che termina quando
si arriva a Z=36 (Kr).
Il 37-esimo elemento ripresenta la stessa inversione di prima, quindi l’elemento successivo è a capo, e comincia a riempire l’orbitale 5s.
Ci sono un paio di dettagli da osservare: se si prende la prima serie di transizioni e si seguono le
configurazioni, per esempio dallo scanio, si osserva che è 4s 2 3d 1 , poi si va avanti si trova il titanio
con 4s 2 3d 2 , poi il vanadio che è 4s 2 3d 3 , poi il cromo che ci si aspetterebbe 4s 2 3d 4 , mentre invece
è 4s 1 3d 5 . Questo fenomeno si rifà ancora alla regola di Hund. <1.3> Quando un atomo ha possibilità di avere tutti gli spin paralleli, allora lo fa. Quando si va a orbitali molto elevati (d e f), le
configurazioni diventano ancora più complesse, con particolari simmetrie. Più la configurazione
è simmetrica, più l’elettrone “preferisce starci”.
Continuando, con il manganese, si ha 4s 2 3d 5 cioè l’elettrone va di nuovo in s.
<1.4>
Il quindo periodo (che comincia con il rubidio) ha l’inversione anch’esso: con Z=39, ritorna 4d e
inizia la seconda serie di elementi di transizione.
Andiamo a due periodi dopo, che inizia con il cesio, Z=55. Accanto c’è il bario, con 6s 2 , con l’elemento successivo si vede un’ulteriore inversione, compare 4 f (con 7 orbitali, max 14 elettroni).
Per motivi di grafica, queste tabelle sarebbero dovute essere state inserite nel mezzo come le zone arancioni, ma viene messa sotto. Sono elementi di transizione anche questi, corrispondenti al
riempimento dell’orbitale f, detta transizione interna (transizione nella transizione). Dopo il bario
c’è il lantanio e da esso (che è un elemento di transizione) prendono nome quelli successivi della transizione interna, i lantanidi. Dopo 4 f , torna il 5d , in serie di transizione, poi il 6p. Questo
si replica nello stesso modo nel periodo successivo. Dopo l’attinio, ci sono altri 14 elementi (gli
attinidi) che riempono l’orbitale 5 f , con Z da 90 a 103. Gli attinidi sono tutti elementi radioattivi
instabili, caratterizzati da tempi di decadimento compresi fra i 100.000 e qualche milione di anni.
Dopo l’elemento 92, che è della seconda serie della transizione interna (uranio) i simboli di questi
elementi vengono rappresentati in modo diverso, per indicare che sono elementi che non esistono
in natura ma che si producono artificialmente (negli acceleratori o nei reattori nucleari). L’ultimo
elemento presente in natura è l’uranio, tutti i successivi non si trovano perché non stabili.
Riepilogo: i colori della tabella periodica sono fatti per facilitare l’interpretazione di alcune proprietà degli elementi, i primi due gruppi fanno parte del blocco S, perché riempono l’orbitale s più
17
Configurazioni elettroniche
28/03/2014
esterno, quellli a destra fanno parte del blocco P, che riempono via via l’orbitale p più esterno, ci si
riferisce agli elettroni esterni alle configurazioni interne complete perché gli elettroni più interni
(quindi più legati al nucleo) non intervengono mai nelle reazioni; poi c’è il blocco arancione che
rappresenta gli elementi di transizione e poi gli elementi in verde, di transizione interna (o terre
rare). Nel blocco S gli atomi hanno tutti tendenza a perdere il singoli o i due elettroni che hanno esternamente. Spostandosi verso destra, occorre sempre più energia per sottrarre un elettrone
al nucleo, si hanno comportamente intermedi, con elementi che tendono a perdere o ad acquistare elettroni a seconda di come conviene, fino ad arrivare ai penultimi elementi del blocco P,
tipo fluoro, bromo, iodio che hanno tutti gli orbitali p completi tranne uno, quindi con la tendenza a completare la configurazione catturando un elettrone in più. Questo si traduce in valori di
quantità fisiche misurabili (energia di ionizzazione). L’ultimo gruppo è caratterizzato dalle configurazioni più stabili del periodo, che richiedono moltissima energia per la ionizzazione, che di
fatto significa che questi elementi non si legano ad altri perché darebbero origine a strutture con
altri atomi e molecole con spesa energetica troppo alta rispetto a quello che ci si guadagna.
18
Energia: ionizzazione e affinità
01/04/2014
3 Energia: ionizzazione e affinità
3.1 01/04/2014
La numerazione dei gruppi della tabella periodica non è univoca, ce ne sono almeno 3, noi stiamo
usando quella semplice. Per noi il terzo gruppo è il primo gruppo del blocco p. Un altro sistema
è usare il numero di elettroni esterni utilizzabili per i legami (i gas nobili infatti hanno gruppo 0
secondo questa numerazione).
Colori della tabella periodica: il blocco giallo è indicato con il nome di blocco s, caratterizzati da
configurazioni elettroniche ns 1 o ns 2 . Gli elementi del primo gruppo tendono a ionizzarsi così:
X −−→ X+ + e, si chiamano metalli alcalini. Gli elmenti del secondo gruppo tendono a ionizzarsi
così: Y −−→ Y2+ + 2 e, e si chiamano alcalino-terrosi (perché compresi tra gli alcalini e le terre rare).
Nel blocco p, i cinque gruppi terminano con ns 1 np 5 ... ns 2 np 5 , ns 2 np 6 .
Gli elementi che terminano in ns 2 np 5 sono detti alogeni, sono caratterizzati da una configurazione elettronica quasi completa a meno di un atomo, e tendono a raggiungere una configurazione
più stabile quando prendono un elettrone, alll’opposto di ciò che fa un alcalini, e formano facilmente ioni negativi: X + e −−→ X–
Più elettroni mancano per completare il guscio, più si riduce la tendenza ad attirare elettroni, ad
esempio il boro si comporta in entrambi i modi, a seconda della situazione. Nell’ultimo gruppo
del blocco P ci sono i gas nobili, chiamati così perché non hanno tendenza a formare legami (sono
già stabili e hanno il guscio esterno completo), anche se in realtà è possibile avere composti anche
con essi, ma sono molto instabili <1.1>
<1.2>
Na+ è particolarmente stabile, di più dell’atomo di neon, quindi l’energia per la seconda ionizzazione è molto maggiore dell’energia della prima, in generale le ionizzazioni sono sempre più
costose mano mano che si procede. Questa energia si può misurare sperimentalmente e le energie composte nelle formazioni di legami derivano da questa quantità.
<1.3> Mi aspetto che l’energia di ionizzazione dell’elio sia più alta, non 4 volte più grande (deriva
dal quadrato di cosa??) perché ho elettroni sullo stesso orbitale, quindi si repellono tra loro.
Con il litio: l’elettrone che mediamente sta più lontano dagli altri due dal nucleo, vede il nucleo
schermato dai due elettroni più interni, percui sotto di sé vede una carica complessiva molto vicina a 3 - 2, circa 1, vede una carica netta simile a quella dell’idrogeno. Inoltre l’elettrone si trova
anche a distanza più grande, e sente sicuramente una forza minore, percui l’energia di ionizzazione è più bassa.
Con il berillio: ci sono ancora due elettroni interni che schermano la carica interna (+4), quindi i
due elettroni esterni vedono una carica +2 che li attrae, infatti l’energia di ionizzazione è un po’
più alta.
Con il boro: succede un qualcosa di particolare, passo a 5 elettroni nel nucleo (B [1s 2 ]2s 2 2p 1 ). Gli
orbitali s e p non si schermano tra di loro, hanno più o meno la stessa distanza dal nucleo (hanno stesso n). Deve ancora avvenire la rimozione della degenerazione, sta cominciando, questo
significa che l’elettrone in 2p ha ancora energia più alta degli elettroni nel 2s, quindi è più facile
ionizzarlo, nonostante il nucleo abbia un elettrone in più. Il motivo è perché la ionizzazione coinvolge l’orbitale 2p che per ora è ancora più alto.
Con il carbonio: vado a togliere uno dei due elettroni in 2p, ma la carica del nucleo è aumentata di
uno, la repulsione tra gli elettroni non contribuisce più di tanto.
Con l’azoto: 7 protoni schermati da 2 elettroni, gli elettroni in p vedono carica circa 5, quindi aumenta l’energia.
Con l’ossigeno ( [1s 2 ]2s 2 2p 4 ]): visto che è aumentata la carica del nucleo (+8) e che gli elettroni in
2p vedono carica risultato +4, mi aspetto un’energia di ionizzazione più alta, ma adesso un elet-
19
Energia: ionizzazione e affinità
01/04/2014
trone condivide un orbitale p, quindi in realtà l’energia diminuisce leggermente rispetto all’azoto,
nonostante aumenti la carica del nucleo, e il primo elettrone a venire via è uno dei due presenti
nello stesso orbitale.
Con il sodio: ([Ne]3s 1 ) ho un 3s molto grande, l’unico elettrone vede una carica +1, quindi l’energia di ionizzazione crolla giù e si ricomincia da capo.
Il risultato è una spezzata che ha nei vertici superiori tutti gli elementi a più alta energia di ionizzazione, che comunque tendono ad avere mano mano sempre valori più bassi. Tutti i vertici inferiori
sono gli elementi ad energia di ionizzazione più bassa, gli alcalini (litio, sodio...) e se si procede
lungo il gruppo, la loro energia si riduce sempre di più.
2) AFFINITA’ ELETTRONICA X + e −−→ X– è la grandezza opposta alla ionizzazione, e si misura
con la stessa unità. Tipicamente il risultato di questa reazione è più stabile del reagente, cioè
X– ha meno energia di X. Vista in valore assoluto che elementi come gli alcalini, che hanno un
solo elettrone nella loro configurazione esterna che è anche poco legato (erano tutti i punti di
minimo di <1.3>), hanno un’affinità molto bassa. Non solo, più l’alcalino è grosso, più l’energia di
ionizzazione è bassa, quindi aggiungergli un elettrone in più significherà una bassissima riduzione
di energia, quindi bassissima affinità. Viceversa, per gli elementi cui manca un solo elemento per
raggiungere una configurazione stabile, viene fuori tantissima energia, lo ione che si forma è molto
più stabile dell’atomo di partenza (esempio: Cl– è molto più stabile di Cl), per questo mi aspetto
affinità molto elevata per gli alogeni, e mi aspetto lo stesso comportamento che vedevo prima con
gli alcalini: mi aspetto di guadagnare di più aggiungendo un elettrone al fluoro, che a metterne
uno in più sullo iodio (perché vado ad aggiungere un elettrone ad un guscio molto più lontano),
vedo cioè che l’affinità diminuisce scendendo nei gruppi, e aumento se ci si sposta in un periodo.
<1.4>
Per quanto riguarda la dimensione dell’atomo, ci sono dei parametri importanti e più definizioni usate.
Intanto: esprimere la dimensione di un atomo isolato non ha senso, comincia ad averla quando
si forma un legame con un altro atomo, per esempio con H2 si prende come raggio dell’atomo la
metà della distanza fra i due atomi legati, è altresì detto raggio covalente per via del tipo di legame
presente fra i due atomi (esiste anche il raggio ionico).
<1.5> Quando ci sono raggi di elementi non gassosi, si usa un altro criterio: i metalli sono ordinati
nello spazio secondo un retino cristallino, in modo regolari. Gli atomi in un cristallo sono disposti
in modo preciso e ordinato, in questo caso si prende la distanza cristallina divisa per 2.
Si può osservare che la dimensione degli atomi varia in base al numero di protoni nel nucleo e di
elettroni esterni. Nella tabella periodica sono riportate le dimensioni, per esempio per l’idrogeno
è riportato il raggio covalente. Per il litio, che è un solido, la dimensione è calcolata con il raggio
cristallino, così è per il sodio, per il potassio e così via, ma nel reticolo si osserva una dimensione diversa del reticolo, e siccome l’unico atomo per legarsi che hanno è sempre più lontano dal
nucleo, la dimensione di questi atomi è sempre più grande. Se ci si muove lungo il periodo, con
atomi con lo stesso n, cambia la carica del nucleo, che via via aumenta, quindi gli elettroni via via
sono sempre più attratti dal nucleo, percui la dimensione atomica scende. Quelli più piccoli sono i
gas nobili, e c’è una coincidenza tra i più grandi (quelli a più bassa energia di ionizzazione) e quelli
più piccoli, i più legati (quelli a più alta energia di ionizzazione).
Il carattere metallico: qualitativamente un metallo è un elemento con buona conducibilità elettrica e termica, quasi tutti gli elementi della tabella sono metalli seguendo la definizione qualitativa, tranne gli elementi dopo la diagonale del blocco p. La zona adiancente alla diagonale, che
è il confine di separazione tra il carattere metallico e non metallico è popolata da elementi con
caratteristiche ambigue, tra cui il silicio e il germanio.
Tutto ciò che abbiamo discusso finora deriva dalla configurazione elettronica e da come sono mes20
Energia: ionizzazione e affinità
01/04/2014
si i livelli di energia.
Se ci guardiamo intorno, non vediamo gli elementi così come sono nella tabella periodica, a
parte i gas nobili, spesso vediamo composti o miscele. La presenza spontanea dei composti invece
che degli atomi singoli è la riprova della tendenza dei sistemi fisici e chimici di evolvere verso la
configurazione di minimo possibile.
Se prendiamo due atomi, uno di un elemento A e l’altro di un elemento B
<1.6> Nella maggior parte dei casi, è vero che la variazione di energia è negativa. Questo tipo di
reazioni sono dette esotermiche (in teoria sarebbe esoergoniche), avvengono spontaneamente in
un sistema isolato. Le trasformazioni in cui succede il contrario, le reazioni endotermiche, avvengono se si fornisce energia.
Atomi isolati e atomi legati formano sistemi diversi, con valori di energia minori per gli atomi legati.
21
Legami chimici
03/04/2014
4 Legami chimici
4.1 01/04/2014
I legami chimici sono i metodi con cui gli atomi si legano tramite gli atomi più esterni.
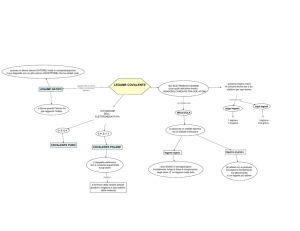
1) LEGAME IONICO <2.1>
Sto supponendo un passaggio fisico di un elettrone da un atomo all’altro, posto che siano abbastanza vicini. Si formano due atomi con cariche opposte, e si attraggono quindi elettrostaticamente.
2) LEGAME COVALENTE <2.2>
Se lascio le due cariche positive sulla retta così, si allontanano indefinitivamente. Se però aggiungo
un elettrone nel mezzo, ottengo un sistema stabile. L’idea del legame covalente è proprio questo,
cioè che l’elettrone sia condiviso e sia efficace per rendere il sistema stabile, e fa in modo che due
atomi vadano l’uno verso l’altro.
4.2 03/04/2014
2
La forza di attrazione tra due cariche di segno opposto è F = re 2 (in modulo), a parte dei fattori
moltiplicativi per le diverse unità di misura. Il lavoro che la forza fa è l’energia potenziale, e l’enerR 2
2
gia potenziale associata a questa forza è E = re 2 dr = − er (è l’integrale sulla distanza)
E
Cl–
Na+
r
+e
AB
energia di
legame
AB
A B
A
2
F = re 2
B
r
r0
-e
2
E = − er (caso ionico)
distanza di
legame
Energia potenziale associata alle forze di due atomi quando si legano. Quando i due atomi sono sufficientemente separati, l’energia potenziale è praticamente nulla (debolissima interazione).
Quando i due atomi cominciano ad essere sufficientemente vicini, si attraggono, e si avvicinano ancora di più, facendo diminuire l’energia potenziale (cioè il sistema diventa più stabile). Se
fossero due corpi puntiformi, l’energia divergerebbe a −∞, ma abbiamo a che fare con un sistema
fisico, e c’è un limite alla distanza che i due atomi possono avere. Quando A e B arrivano abbastanza vicini, comincia ad esserci la repulsione dovuta agli elettroni esterni dei due atomi, perché le
regioni dove si collocano intorno ai due nuclei si sovrappongono, e maggiore è la sovrapposizione,
maggiore è la repulsione degli elettroni. La repulsione è una forza cui si può associare un’energia
potenziale però positiva. La sovrapposizione dei due comportamenti riporta alla fine sul segno
positivo l’energia potenziale.
22
Legami chimici
03/04/2014
Questo comportamento è comune a tutti i legami, ciò che lo caratterizza è che c’è un punto r 0 di
minimo dell’energia potenziale, dove le due forze di attrazione e repulsione si bilanciano, che è il
valore energetico che il sistema raggiungerà in condizioni normali. Questo valore è detto energia
di legame, r 0 è detta distanza di legame, è la distanza a cui si trovano i due atomi nella molecola.
Se ai due atomi dall’esterno fornisco energia pari all’energia di legame dissocio la molecola. Nel
caso di molecole biatomiche semplici come H2 , Cl2 , N2 , O2 coincide con l’energia necessaria per
staccare i due atomi e portarli a distanza infinita (in questo caso si chiama energia di dissociazione).
<1.2>
Se si prende una molecola più complessa come quella dell’acqua, posso staccare un atomo di
idrogeno con un energia di legame pari a quella del legame che rompo, poi posso staccare anche il
secondo, e dissociare del tutta la molecola (nonostante si parta da un sistema simmetrico, le due
energie non sono uguali, perché dopo aver staccato il primo idrogeno gli elettroni si ridistribuiscono). In molecole complesse l’energia di legame si misura più difficilmente, per esempio con
H2 O gli si associa l’energia media per spezzare i due legami.
Angolo di legame: è una caratteristica geometrica che determina la forma spaziale delle molecole, e necessito di almeno 3 atomi per avere un angolo come in H2 O. Due legami consecutivi che
insistono sullo stesso atomo formano due angoli (quello compreso e quello esterno), la convenzione stabilisce di prendere quello minore di 180° (Ci sono casi in cui entrambi sono 180°, quindi
è indifferente prendere l’uno o l’altro). Gli angoli si misurano sperimentalmente con la diffrazione
dei raggi x. <1.3>
Questi angoli hanno conseguenze sulla geometria , quindi sulle proprietà di queste molecole.
E l eg ame
H2
O2
N2
F2
HF
³
kJ
mol
436
497
949
157
569
´
d ( Å)
0,79
1,21
1,10
1,42
0,92
omonucleari
ª
etero nucleare
La distanza di legame in realtà è una distanza media, perché i due atomi sono in continua vibrazione, così anche gli angoli sono valori medi.
Nota che ossigeno e azoto sono uno accanto all’altro nella tabella periodica, quindi le dimensioni
dei due differiscono di poco, nonostante questo c’è un’enorme differenza nell’energia di legame e
una più piccola distanza di legame; le variazioni in distanza di solito sono sempre piuttosto piccole, ma se si considera le energie che agiscono (e 2 /r 2 ) basta poco per avere una variazione di entità
grande.
Il fluoro ha energia che è circa un terzo di quella di ossigeno e i due atomi stanno un po’ più lontani: è molto più facile dissociare due atomi di fluoro che due di ossigeno o di azoto.
HF: mi aspetterei intuitivamente che HF abbia un’energia che sia una via di mezzo tra H2 e F2 , in
realtà è più alta ed è una molecola molto più piccola di F2 , c’è un meccanismo molto sottile che
regola la distanza di legame e l’energia di legame.
23
Legame ionico
03/04/2014
5 Legame ionico
5.1 03/04/2014
<1.5> Il meccanismo di formazione del legame ionico avviene quando c’è possibilità di far passare
un elettrone da un atomo all’altro, è un problema classico di elettrostatica. Se avessi i due elementi
A e B gassosi, loro si avvicinerebbero, raggiungerebbero il minimo energetico e si manterrebbero
in equilibrio ad una certa distanza. Di fatto non si ha mai un singolo atomo di A e un singolo di
B, si hanno quantità grosse di entrambi. Questo significa che se gli ioni A+ e B– si sono formati, ne
avrò molti tutti intorno. Gli ioni con la stessa carica si disporranno al massimo valore di angoli e
a distanza tale per non darsi fastidio l’un l’altro. Questo è il principio della creazione del reticolo
cristallino, la distanza è sempre quella r 0 di equilibrio. Ogni ione intorno a sé fa disporre gli altri
ioni in modo tale da ottenere una struttura spaziale cristallina. Nel caso del NaCl, i due ioni Na+
e Cl– si dispongono nello spazio secondo un reticolo cubico, cioè le posizioni di tutti gli ioni sono
vertici di cubi, che si propaga a livello macroscopico. Quando ho questo tipo di legami, si crea un
solido ionico, che ha struttura cristallina. La struttura geometrica può essere diversa a seconda
delle dimensioni, carica degli atomi di partenza.
Quale è allora il senso di scrivere PM(NaCl), se PM(H2 O) è una molecola e NaCl no?
HCl + NaOH −−→ NaCl + H2 O
la indico come se fosse una molecola biatomica, in
realtà è un cristallo ionico dove c’è un rapporto 1:1
tra Na e Cl.
Si tratta NaCl come se fosse una molecola (in realtà indica che all’interno del composto c’è la stessa quantità di ioni Na+ e Cl– ), allo stesso modo, non comporta nulla il fatto che sia fatto come un
cristallo o che sia una molecola biatomica. Talvolta per riferirsi a questo tipo di molecole si preferisce parlare di peso formula PF(NaCl).
Immaginiamo di voler ottenere NaCl direttamente partendo da sodio e cloro. <1.7>
Metto il sodio sotto vuoto e ci faccio passare del cloro gassoso. Inizia a formarsi uno strato di NaCl
sulla superficie di sodio metallico, dove arrivano gli atomi di Cl2 . Il legame si forma spontaneamente, basta che il cloro “veda” il sodio, è sufficiente che il ∆E della reazione sia negativo.
Come collego questa cosa reale alla formazione del reticolo ionico? Immagino di andare dalla
condizione inziale a quella finale usando un percorso alternativo: trasformo il sodio da solido a
gassoso (sublimazione), con una certa energia ∆E s (in KJ/mol). Prendo le molecole di Cl2 e le dissocio, con l’energia di dissociazione ∆E D di Cl2 . Trasformo il Na in Na+ ionizzandolo e Cl in Cl– . A
questo punto i due ioni si dispongono formando il reticolo, con un altro ∆E R , indicato con energia
reticolare, che è la somma di tutte le energie potenziali presenti nel reticolo cristallino.
Nel caso del sodio: ∆E S = +109 KJ/mole
∆E D = +123,5 KJ/mole
∆E I ON = +497 KJ/mole
∆E AF = −349 KJ/mole
Per il cloro e per gli alogeni, questo ultimo è un grosso guadagno di energia. Se si fa la somma viene
un numero positivo, è il bilancio complessivo di energia che servirebbe per avere tutti gli ioni separati. Se a questo punto, invece di ottenere un reticolo riuscissi ad ottenere molecole biatomiche
separate le une dalle altre, dovrei aggiungere come ultimo passaggio la formazione di una mole di
molecole biatomiche, ciascuna delle quali ha un’energia di legame che di -57 KJ/mole (è l’energia
di un singolo legame). Se si fa la somma, viene un numero positivo, che significa che il bilancio
complessivo per far avvenire la reazione porterebbe ad una reazione non spontanea. Quello che
24
Legame ionico
03/04/2014
realtà succede in natura è che in realtà non si formano molecole singole, avviene un processo che
è la formazione del reticolo tridimensionale spaziale macroscopico cui è associata l’energia reticolare, che per NaCl vale:
∆E R = - 767 KJ/mole
Molecole singole quindi non si formano, solidi cristallini sì. Il bilancio complessivo dà quindi un
∆E = −386 KJ/mole (grazie all’energia reticolare).
Il percorso che si è realizzato per costruire la reazione è virtuale, il perché l’energia necessaria
per far avvenire la reazione direttamente dipende da come si ottiene questa energia. Qualunque
strada si prenda per ottenere lo stesso stato, è uguale. Questo metodo si chiama CICLO DI BORNHABER e lo si usa spesso per studiare composti ionici. E’ un modo che dimostra come mai NaCl è
fatto così e non di NaCl2 .
25
Legame covalente
04/04/2014
6 Legame covalente
6.1 03/04/2014
Il legame ionico non è direzionale, il legame covalente è fortemente direzionale (l’elettrone deve
essere in una regione di spazio intermedia tra i due nuclei). Ci sono poi vari modelli con cui il
legame covalente è stato affrontato, noi trattiamo quello di Lewis e quello VB (legame di valenza).
La teoria di Lewis in realtà è un modello, che non ha basi teoriche ma nasce da un’intuizione: guidato dal fatto che elettroni ha spin opposto, che gli elettroni sono sempre due nello stesso orbitale,
allora immaginò che un legame tra due atomi coinvolge gli elettroni e che gli elettroni che si mettono in comune devono essere due, a coppie. Si inventò una rappresentazione grafica (simboli di
Lewis) fatta così: <1.8>
cioè due atomi di idrogeno condividono una coppia di elettroni.
Per capire come fa invece l’ossigeno, Lewis pensò che gli elettroni più interni non vengono mai
coninvolti, ma vengono coninvolti solo gli elettroni più esterni, detti di valenza, in questo caso
sono 6.
Sapendo come sono messi gli elettroni esterni, si ipotizza che solo due elettroni solitari si leghino.
Per Lewis il fatto che l’energia di legame dell’ossigeno fosse alto era dovuto al fatto che il legame
era doppio.
Prendiamo l’azoto: <1.9>
In generale, i due elettroni condivisi vengono uno da un atomo, uno dall’altro. <1.10> Già qui c’è
qualcosa che non torna, perché HF ha alta energia di legame.
Una delle caratteristiche che hanno condizionato la tradizione chimica è una regola nota come la
regola dell’ottetto, perché una conseguenza di questo meccanismo qui è che i due atomi si uniscono per fare una molecola e il risultato è (per molti ma non per tutti, soprattutto quelli del secondo
periodo) che ogni atomo vuole 8 elettroni. Allora se si guarda N2 , dal punto di vista dell’atomo di
azoto, ognuno vede 8 elettroni, così anche per O2 .
<1.11> La regola dell’ottetto qui non è rispettata, ma questa eccezione però conferma il fatto che
NO sia un radicale libero, cioè una molecola con forte tendenza a legarsi a qualcos’altro con cui
reagire.
<1.12> è un’altra eccezione alla regola dell’ottetto; il modello di Lewis non dice perché metto gli
elettroni in un modo piuttosto che in un altro.
<1.13> <1.14>
Il legame che si forma con H+ viene indicato con una freccetta perché gli elettroni vengono solo
da N, e viene chiamato legame dativo.
6.2 04/04/2014
<1.1> In questo esempio, siccome si sa a posteriori che la molecola esiste, si disaccoppiano gli
elettroni e si rappresenta il legame, che rispetta anche la regola dell’ottetto, anche se non ci sono
motivazioni teoriche per questo comporamento (nel modello di Lewis).
<1.2> Vedo che c’è una certa simmetria, mi aspetto che due atomi di idrogeno si leghino ad un carbonio, ci si aspetta tipo CH2 CH2 . Questo modello prevede che questa molecola abbia un doppio
legame carbonio-carbonio, che è vero. Anche per C2 H2 il modello funziona, perché è vero che c’è
un legame triplo tra i due atomi del carbonio.
VB: modello quantistico del modello di Lewis. Stesso principio, un legame si forma con una
coppia di elettroni, però su un orbitale che ha almeno due centri <...12:53>. Altra ipotesi: gli elet-
26
Legame covalente
04/04/2014
troni usati sono solo quelli di valenza, quelli più interni non vengono coinvolti mai, <...12:54>. Per
questi elettroni esterni il problema è trovare le funzioni d’onda che descrivono le coppie di elettroni. Dal punto di vista matematico è molto complesso, perché non è la funzione d’onda di un
elettrone, ma di due, bicentrica (tiene conto della presenza di due nuclei). Anche solo esprimere
l’equazione del potenziale è molto complesso. La soluzione che si può ottenere è approssimata
per passaggi iterativi.
<1.3> La prima approssimazione è questa: supponiamo di avere un atomo A con un nucleo e un
elettrone spaiato esterno (gli altri elettroni sono più interni), lo indico con e 1 , e supponiamo di
avere un atomo B con un altro elettrone spaiato, chiamato e 2 . Finché sono lontani e isolati, non
interagiscono.<...12:57>.
Se gli atomi sono vicini, quello che devo inserire come termine correttivo è qualcosa che tenga
conto del fenomeno che si verifica di interazione anche con l’altro nucleo (riformula), e così vale anche per l’altro elettrone. Si costruisce quindi sempre con le stesse funzioni d’onda iniziale la
funzione d’onda ΨI I , che prevede che i due elettroni si scambino di nucleo La due funzioni d’onda
quindi si sommano: ΨCOV = ΨI +ΨI I <...13:00>. Non si tiene conto però di un altra cosa, e cioè del
fatto che gli elettroni possono trovarsi contemporaneamente sullo stesso atomo. Viene aggiunto
un altro termine, ΨI ON = Ψ A (1)Ψ A (2)+ΨB (1)ΨB (2) che approssima la probabilità di trovare tutti e
due gli elettroni su A + la probabilità di trovarli tutti su B <...13:02>, quindi ha un aspetto che somiglia molto al carattere ionico. La funzione d’onda complessiva finale ha un’espressione id questo
tipo: Ψ = αΨCOV + βΨI ON , che è una combinazione lineare delle due funzioni d’onda trovate.
<...13:03> ed è ottenuta per costruzione matematica a partire dalle funzioni d’onda atomiche. I
due coefficienti α e β possono essere α >> β, cioè se nella funzione c’è una prevalenza della prima componente rispetto la seconda, significa che è bassa la probabilità che entrambi gli elettroni
siano sullo stesso atomo, cioè che è basso il carattere ionico, questo dà luogo a un legame covalente apolare, o covalente puro, è il legame che si forma tra atomi uguali come H2 , O2 , N2 , ma
è anche il legame che si forma in una molecola come C2 H6 in corrispondenza del legame fra i due
atomi del carbonio. <1.4>
Può avvenire anche il contrario: se α << β, si ha bassa probabilità di avere elettroni equamente
condivisi, questo è il legame ionico secondo questa formazione teorica, quindi appare come una
situazione limite del legame covalente, quindi in realtà tutti i legami sono descrivibili in questo
modo.
Nella maggior parte dei casi, α e β sono confrontabili, sono legami covalenti polari, non è una
condivisione simmetrica degli elettroni, cioè le funzioni d’onda danno una probabilità più alta di
trovare gli elettroni su un atomo piuttosto che sull’altro.
Quello che viene fuori è che la funzione d’onda dei due elettroni legati la si può <...13:10>
come soR
vrapposizione spaziale delle funzioni d’onda atomiche. Viene fuori un termine S = spazi o Ψ A ΨB dV
che è detto integrale di sovrapposizione <...13:11>.
<1.5> <...13:12> <1.6> <manca un monte di roba fino a 13:16>
Conclusioni: la molecola di H2 , si usano gli orbitali 1s, localizzati in posti diversi rispetto allo
spazio. Quando i due atomi sono distanti, i due orbitali 1s rappresentati con le superfici limite
sono sferici, sono lontane e non si intersecano. Se invece sono a distanza sufficientemente piccola
da creare una regione di sovrapposizione tra i due <...13:17>, si forma il legame, gli elettroni sono
prevalentemente nella zona intermedia, l’energia di legame corrisponde all’estensione della zona
intermedia. <1.7>
La forma di <...>
<1.8> <...> che è escluso dal principio di esclusione di Pauli. Lo stesso vale per i primi due orbitali
p, perché verrebbero fuori sovrapposizioni dove ci sono più di <due elettroni?>. L’unico che forma
legame è l’ultimo orbitale p. Quindi nella molecola F2 il legame è spiegato come legame di orbitali
27
Legame covalente
04/04/2014
p, nella molecola H2 il legame è spiegato come legame di orbitali s. Questa formulazione contiene
spiegazioni che nel modello di Lewis non ci sono, per esempio: nel caso che abbiamo preso in
considerazione, come si sceglie un orbitale, perché prendere proprio p x ? L’unica cosa che serve
è che gli atomi per legarsi presentino gli orbitali giusti, con direzione coassiale, altrimenti potrei
avere la situazione <1.9>. Ho lo stesso una sovrapposizione fatto da due pezzi, ma l’integrale di sovrapposizione viene nullo, perché le funzioni d’onda (ci sono le Ψ, non Ψ2 , quindi hanno segno)
si annullano. <..13:24> Questo modello quindi spiega perché i legami si formano con gli orbitali
collineari.
<1.10> <...13:28> Si vede che si formano due tipi diversi di legame, perché l’integrale di sovrapposizione collineare (detto legame σ) è leggermente maggiore rispetto alla sovrapposizione laterale
(detto legame π).
28
VB: polarità, elettronegatività
08/04/2014
7 VB: polarità, elettronegatività
7.1 08/04/2014
<...> <1.1>
Quando un legame si forma ed è singolo <...10:46> è sempre un legame σ, mentre gli altri sono
sempre π, ovviamente non è possibile avere un legame π senza un legame σ, e di legami σ ce ne
può essere solo uno, <10:46> conseguenze sul <...> fisico.
La teoria del legame di valenza è il modello di Lewis trattata in maniera più formale, in termini di
orbitale, funzione d’onda, eccetera.
<1.2> Nel modello di Lewis c’era il caso del berillio, in cui i due elettroni sono nel 2s, e ci si aspetterebbe che non formi legami. Lewis quindi separava i due elettroni e indica come si forma BeCl2 .
La teoria di valenza per dare una spiegazione fa un’ipotesi in termini energetici: nella formazione
di un legame gli atomi non usano gli elettroni della configurazione dello stato fondamentale (che
sarebbe il 2s), ma usano gli elettroni di una configurazione diversa, che si chiama eccitato di valenza. <1.3><10:50>
Una configurazione in cui uno dei due elettroni dello stato fondamentale viene portato ad un livello leggermente superiore (configurazione 2s 1 2p 1 ) è ipotizzato dalla teoria del legame di valenza.
In questo caso gli elettroni spaiati sono 2, quindi si spiega perché il Be si lega con due atomi di Cl.
Questo fenomeno va cominciato a osservare a partire dal gruppo 2, nel primo gruppo viene usato
l’elettrone già spaiato. <1.4>
Nel terzo gruppo ci si aspetterebbe di avere valenza 1, perché c’è un elettrone spaiato, in realtà non
lo si osserva mai, perché lo stato eccitato di valenza diventa ns 1 np 2 . Tieni conto: la formazione di
un legame è un processo in cui la differenza di energia del sistema è negativa. Se un atomo può
fare più legami, il sistema finale è più stabile, quindi è più stabile più è alta la valenza. Il passaggio
dallo stato fondamentale a quello eccitato di valenza consente di fare più legami. Il boro nel suo
stato fondamentale può fare un solo legame, nel suo stato eccitato ne può fare 3, quindi lo stato
legato risultante è più basso rispetto a quello isolato, e ci si guadagna tanta più energia (e stabilità) tanti sono i legami. Questa è l’unica giustificazione del passaggio allo stato eccitato (l’energia
rilasciata è più bassa). Questa è un’interpretazione teorica, gli atomi non fanno il passaggio intermedio.
<1.5> Per il boro, lo stato di energia minima è quello con cui riesce a fare 3 legami, percui spende
un po’ di energia per passare nello stato eccitato e poi ne ottiene molta di più dopo la formazione
dei 3 legami (invece che di uno solo).
<1.6>
Nel caso del secondo gruppo, il passaggio è forzato, altrimenti non ci sono legami. Nel caso del
terzo gruppo, il fattore di aumento è 3, nel caso del gruppo 4 il fattore è 2, quindi l’aumento è sempre minore. Ciò significa che gli elementi del quarto gruppo (come il carbonio) si comportano in
modo diverso a seconda dell’ambiente, infatti si può avere valenza 2 o 4.
Per il quinto gruppo (con n=2, azoto), anche se venisse mandato un elettrone in un altro orbitale,
la valenza rimarrebbe comunque tre. Però per gli elementi di questo gruppo esistono (a partire dal
periodo 3) gli orbitali d molto vicini energeticamente, per esempio per il fosforo (n=3) posso avere
valenza 3 o 5. L’azoto non lo può fare, perché per poter avere 5 legami dovrebbe avere un elettrone
in 3s, che è molto distante energeticamente, e non si riguadagna la spesa con 5 legami, mentre per
il fosforo è conveniente perché gli orbitali 3d sono vicini.
Per il sesto gruppo, se si prende l’ossigeno (n=2), anche se un elettrone si sposta in un altro orbitale, può fare comunque 2 legami, come si comporta quasi sempre. Se si prende n >= 3, per
esempio lo zolfo (S) già si osservano più possibilità. Può avere valenza 2, 4 o addirittura 6 (la spesa
per mandare un elettrone da 2s a 3d è grande, ma può fare molti legami)
29
VB: polarità, elettronegatività
08/04/2014
Per il settimo gruppo, se n = 2 (fluoro), si ha valenza 1. Se n >= 3 si possono avere anche valenze
3, 5 e 7.
<1.7> Schema di Lewis per i composti del cloro in esempio.
La teoria VB spiega in termini energetici il motivo per cui gli elettroni vengono spaiati e colma la
lacuna del modello di Lewis.
Una delle cose che abbiamo intuito introducendo la VB è che in realtà il legame che si forma tra
atomi è di natura unica, poi noi lo distinguiamo tra ionico e covalente, in base a <11:25>
Se prendiamo due atomi di idrogeno <1.8> <1.9> c’è maggiore probabilità di trovare un elettrone
sul fluoro che sul’idrogeno, quindi il baricentro delle cariche positive e quello delle cariche negative non coincidono più. Questa molecola presenta verso l’esterno un’eccesso di carica negativa
su F e un’eccesso di carica positiva su H. Questsa caratteristica si chiama POLARITA’, la molecola
è polare. Si crea un dipolo elettrico, che è una molecola che ha una carica positiva e una negativa
rigidamente collegate ad una certa distanza (di solito le cariche sono uguali in modulo)
<1.10,11>
Se si mette in un campo elettrico delle molecole di HCl gassoso, le molecole si orientano tutte.
ELETTRONEGATIVITA’: è una grandezza non misurabile<...>, è la capacità di un atomo di attrarre
su di sé gli elettroni coinvolti in un legame. Siccome la molecola HF ha una disposizione asimmetrica degli elettroni, prevalentemente spostati sul fluoro, allora il fluoro è più elettronegativo di H.
Nel caso di due atomi con la stessa capacità di attrarre, l’elettronegatività è uguale e gli elettroni si
trovano in orbitali simmetrici. <11:43> quindi il legame è polare. Questa caratteristica elettrica è
legata alla differenza di elettronegatività tra gli atomi.
Una delle definizioni è quella di Pauling: <...11:44> alla capacità che ho di dissociare le molecole,
quando un atomo di per sé riesce a <...>. Definì una formula pratica per determinare l’elettronegatività:
r
E A A + EB B
(in eV)
|χa − χb | = 0,102 E AB −
2
E A A : energia per separare due atomi A <11:46>
<11:47> e stabilì l’elettronegatività dell’idrogeno: χH = 2,1, per mantenere compatibilità con una
scala già usata precedentemente dai chimici. Con questa formula l’elettronegatività diventa una
sequenza di numeri, <riportata sulla tabella periodica>.
Prendiamo H2 , l’elettronegatività di H è 2,1. Le caratteristiche di polarità del legame sono legate
~ | = 0. Quindi se messa in un campo elettrico
alla differenza di elettronegatività: ∆χ = 0, quindi |P
non fa niente.
Prendiamo HF, l’elettronegatività di F è 4 (il valore più alto di tutti, si vede che è in correlazione
all’affinità elettronica, il fluoro è quello che la ha più alta di tutti). ∆χ = 1,9, allora <1.13>.
Con HCl <1.14>
<11:55> non è strano che i metalli alcalini abbiano bassa elettronegatività <...> Il cristallo ionico
non è carico, ma all’interno <sì> <...>.
<11:57> ∆χ = 0 -> cov. puro
0 <= ∆χ <= 0,4 si considera covalente
0,4 <= ∆χ <= 2 si considera covalente polare
∆χ > 2 si considera ionico, perché gli elettroni stanno quasi praticamente sull’atomo più elettronegativo. Sono numeri scelti per convenzione.
<1.16> si può fare una considerazione su quanto sia <polare 12:00> questo legame <?>
Si sa quanto vale il dipolo (quando lo metto in un condensatore e osservo il comportamento, lo
~ | = 1,08D, dove D è l’unità di misura Debye = 3,336·10−30C ·m, e si può misurare
posso misurare) |P
~ | = δ · p, allora <1.16>.
quanto è lunga la molecola: r = 1,27Å. Sapendo che |P
<12:06> se fosse totalmente ionico, δ dovrebbe valere e = 1,602·10−19 Coulomb, ma il valore che ho
30
VB: polarità, elettronegatività
10/04/2014
δ
· 100 =
e
17,7%. Quindi il legame in HCl è un covalente polare in cui la componente polare(o ionica) è solo
del 17,7 %, quindi è più covalente che ionico.
Il carattere ionico in una molecola ha un effetto particolare: in HF veniva fuori che l’energia di
legame è più grande di H2 e di F2 <12:10>. Viene fuori che il legame tra HF è più forte, perché H
è lo stess ed H è più elettronegativo del cloro, quindi lo sbilanciamento è maggiore, quindi ha un
carattere ionico più forte del cloro. <12:11> inoltre, se ci sono cariche intorno ad HF, si attraggono
elettrostaticamente. <...> questo è il contributo ionico al legame.
Se si chiedesse il carattere covalente, basta fare %cov al ent e = 100 − %i oni co.
ottenuto è minore. Quindi posso calcolare la percentuale di carattere ionico: %i oni co =
7.2 10/04/2014
C’è rimasto un punto da trattare sulla teoria del legame di valenza.
<1.1> Se si guarda come è la configurazione elettronica, si dovrebbe avere anche un’idea della geometria della molecola, che si può determinare sperimentalmente sempre tramite la diffrazione dei
raggi X.
Per dedurre la forma della molecola BF3 dalla configurazione eccitata di valenza del boro <1.2>.
Però visto che l’orbitale 2s è <12:10>, mi aspetterei una molecola fatta così <1.3>, mentre in realtà
è <1.4>, completamente simmetrica e planare, con energie di legame tutte identiche, e questo non
è previsto dalla teoria VB.
La molecola d’acqua H2 O <1.5> mi aspetterei una molecola planare, con angolo a 90°, mentre
invece l’angolo reale è 104,5°, che è una deviazione troppo grande da 90° per essere considerato
valido. Un’altra molecola che dà problemi simili è quella dell’ammoniaca NH3 <1.6>. Mi aspetterei una piramide con tre angoli di 90°, invece sono di 107,5°.
<1.7> <12.19> ci dovrebbe essere un s che si sovrappone <...>, mi aspetterei una molecola una molecola con <tre> angoli di 90° e il quarto per simmetria sull’angolo risultante rimanente, quindi tre
legami uguali e uno diverso, mentre invece è un tetraedro <12.22>, con angoli di 109,5°, che non
tornano con gli angoli teorizzati.
Il modello di Lewis non diceva nulla sulla geometria spaziale, mentre giustificare la forma delle
molecole è importante, perché ha un ruolo importante nella proprietà delle sostanze. Il fatto che
una molecola sia un tetraedro piuttosto che <angolata> ha conseguenze diverse <12:24>.
C’è un approccio puramente matematico, una giustificazione a posteriori, somiglia a trovare una
base di uno spazio vettoriale. Le funzioni d’onda in qualche modo descrivono uno spazio vettoriale.
<1.8><12:25>
Queste due sono una base, sono indipendenti. Questa è una coppia di funzioni che è una base
di uno spazio di funzioni, ma posso descriverle anche cambiando base. Una base alternativa la si
può costruire partendo da queste due, facendo somma e differenza (a meno di qualche coefficiente numerico).
Questa coppia di orbitali sono detti ibridi sp, infatti ottenuti dall’ibridazione di un orbitale s con
uno p. Diventano direzionali sulla stessa direzione delll’orbitale p che uso e i due lobi non sono
più simmetrici, c’è ne è uno molto più esteso dell’altro. Un orbitale più esteso nello spazio significa una grossa capacità di sovrapposizione, cioè energia di legame significativa (integrale di
sovrapposizione <12:31>). L’ibridizzazione sp è ciò che la VB usa per spiegare la formazione della
molecola BeH2 .<1.9>
Se si ammette questo passaggio, <...>. Nota: dall’orbitale eccitato di valenza non si capisce che
l’energia dei due legami è uguale.
Prendiamo i due orbitali p e s <1.10>. Creano uno spazio di dimensione 3, e posso ottenere alme-
31
VB: polarità, elettronegatività
10/04/2014
no 3 funzioni diverse dalle prime che sono una nuova base per le funzioni d’onda, il cui risultato è
più elaborato ma intuitivo. (Sono tutti e tre sullo stesso piano xy).
Questo spiega la struttura della molecola BH3 . <1.11>, planare con angoli di 120°. L’orbitale p z c’è
ancora, è vuoto non ibridizzato.
Il passo successivo è l’ibridizzazione che coinvolge tutti gli orbitali, cioè l’orbitale s e tutti e tre gli
orbitali p.
<1.12>
Questa combinazione porta alla formazione del tetraedro <12:42>, l’ibrido è sp 3 . <1.13>
<1.14> Si era giustificato questo passaggio dicendo che l’energia era più bassa se si formavano più
legami. L’ibridizzazione in teoria prevede ancora un altro aumento di energia (in realtà è virtuale,
perché non stiamo spostando gli elettroni, ma stiamo solo facendo una costruzione matematica.
La condizione che vale è che l’energia si conserva). Nel momento in cui si formano i legami è più
bassa, perché essendo gli orbitali ibridi più estesi c’è una sovrapposizione più grande. Siccome
però è un’operazione matematica, e una delle condizioni che si pone è che non ci deve essere una
variazione di energia dal nulla, significa che l’ibridizzazione deve portare a (per esempio) 4 orbitali
(sp 3 ) con la stessa energia degli orbitali di partenza. Se parto dai 4 orbitali px, py, pz e s, si piazzano in un punto che è il baricentro dell’energia dei 4. Quindi la posizione degli ibridi in realtà è nel
mezzo <12:50>, è il motivo per cui mi invento l’ibridizzazione.
Ma c’è sempre l’ibridizzazione? Se si deve spiegare la geometria del metano, c’è necessariamente.
Se però si pensa a HF, dove il fluoro è fatto così: <1.15>, non c’è nessuna necessità di pensare ai
legami ibridi, perché c’è un solo elettrone che si lega, sono due atomi quindi non c’è nemmeno
una geometria da giustificare. In realtà il fluoro fa un’ibridizzazione sp, talvolta anche sp 3 . Nella
geometria della molecola non cambia nulla, rimane sempre lineare. Se si sposta un elettrone, la
valenza rimane sempre 1, però se si fa una ibridizzazione sp, <1.16>, si ha uno degli orbitali sp
che ha già 2 elettroni dentro, l’altro ha un elettrone e farà sovrapposizione con l’s dell’idrogeno.
La differenza è che <12:54>, ma se l’orbitale è tutto da una parte, la sovrapposizione può superare il 50%, quindi il legame può essere molto forte. L’atomo fa il legame che è più conveniente
energeticamente. Non succederebbe niente se il fluoro facesse un’ibridizzazione sp 3 , ci sarebbe
un orbitale davanti e gli altri dietro a coda, con 3 sp 3 , ciascuno con 2 elettroni dentro, che è una
configurazione stabile. Non si riuscirebbe a vedere la differenza, ma il fluoro ha fatto la scelta più
conveniente energeticamente. Gli orbitali non coinvolti in un legame (in questo caso 3) non si
vedono, perché non ci sono molecole in quelle posizioni come con il metano.
<1.17> La molecola di NH3 non è un tetraedro regolare perché ci manca una quarta parte, il risultato è che i due elettroni che stanno nell’orbitale occupano un grosso volume, pensa ad un
palloncino gonfiato ad elio. Se li prendi con una mano (adimensionale) tutti e 4, si dispongono
secondo un tetraedro. Se si gonfia di più il palloncino che sta sopra, si espande e comprime quelli
di sotto. Questo orbitale che non è legato a nessun nucleo positivo è più espanso perché non c’è
un nucleo che trattiene gli elettroni. Quindi le cariche negative respingono gli elettroni degli altri
tre atomi, e il tetraedro si chiude di più, formando angoli di 107,5°.
Il passo successivo è l’acqua. <1.18> Ci sono due palloncini più grandi, l’angolo tra i due H si stringe di più, a 104,5°. La geometria dell’acqua è comunque planare angolata (anche se si usa l’orbitale
sp 3 ), la cui forma si spiega con i due orbitali che non si vedono che condizionano l’angolo di legame.
<1.19> Mi aspetto una molecola simmetrica in base a quello che avevo visto con Lewis <13:10?>,
fa un’ibridizzazione sp 2 (l’s c’è sempre), usa due soli orbitali degli orbitali p. Ciascun atomo di
carbonio fa 3 legami, ha ancora a disposizione un orbitale con un elettrone (entrambi gli atomi di
carbonio ce l’hanno), che è perpendicolare al piano. I due p z danno luogo ad un legame π, per
cui la molecola diventa <1.20>, questo legame funziona da lucchetto, impedisce alla molecola di
ruotare, costringe i 3 atomi di un lato a stare sullo stesso piano dell’altro (c’è anche il legame σ tra
32
VB: polarità, elettronegatività
11/04/2014
i due C).
7.3 11/04/2014
<1.1> Riprendiamo l’esempio della molecola C2 H4 . Può essere utile per capire la geometria della molecola usare anche il modello di Lewis. Anche se non c’è nessun riferimento preciso, c’è
un’indicazione su quanti legami si formano tra gli atomi di carbonio. Sappiamo che il doppio
legame è dato da un legame σ, collineare, che può provenire dall’ibridizzazione, e un legame π,
che proviene dalla sovrapposizione laterale di due orbitali perpendicolari all’altro legame, quindi
non può provenire da orbitali ibridizzati. Già con questo quindi si sa che un legame è ibridizzato
e uno no, che l’orbitale ibridizzato non è sp 3 , ma mi aspetto che faccia un ibrido inferiore, infatti
è sp 2 , e l’orbitale p rimanente è perpendicolare al piano (dei primi due, cioè degli sp 2 ). Quindi se
disegno la geometria della molecola, partendo da un atomo di carbonio, vedo che: dei tre legami
realizzabili con sp 2 , che sono disposti a 120°, due sono legati agli atomi di idrogeno, uno è legato
all’altro atomo di carbonio, l’altro carbonio fa la stessa cosa, visto che la molecola è simmetrica, e
poi perpendicolarmente al piano ci sono gli orbitali p dei due atomi del carbonio che fanno una
sovrapposizione laterale, cioè il pegame π.
<1.2> Prima la guardiamo con Lewis: mi aspetto un triplo legame tra i due atomi di carbonio. La
molecola è simmetrica, quindi quello che fa un atomo di carbonio lo fa anche l’altro. Nel triplo legame, due sono π e uno σ, quindi ci sono due legami che provengono da sovrapposizioni laterali.
Quindi gli orbitali p che devono essere coinvolti nella ionizzazione è solo uno, così da lasciare due
orbitali per la formazione dei due legami π. Ho dunque un orbitale sp e due p. Il legame sp forma
un angolo di 180°, così fa anche l’altro carbonio. Ci sono poi gli orbitali p che fanno sovrapposizione nel piano e gli altri due che fanno sovrapposizione sul piano perpendicolare.
<1.3> La molecola di C6 H6 è più interessante, che con Lewis si vede peggio. Anche qui tutti gli
atomi di carbonio fanno la stessa cosa, i due 6 mi dicono che su ogni atomo di carbonio c’è un
atomo di idrogeno. Ogni C fa un ibrido sp 2 , a 120°. Un legame viene fatto con H, due con altri
due C. Risulta una molecola ciclica (chiusa su sé stessa). Ciascun atomo di carbonio ha ancora un
orbitale p con un elettrone perpendicolare al piano (siccome l’orbitale ibrido è sp 2 , tutti i legami
sono complanari) che può fare sovrapposizione laterale con il carbonio adiacente. Ma ogni atomo di carbonio può fare un legame π. La difficoltà che c’è è nello stabilire tra quali C viene fatto
il legame π. Ci sono due possibilità, ma la molecola apparrebbe diversa. In realtà, questo ha un
significato semplice. Basta immaginare l’esagono sul piano, ogni atomo di C ha la stessa distanza
dall’altro, ed ha il suo orbitale p perpendicolare; per questo motivo non c’è nessuna preferenza tra
avere un legame π con un C piuttosto che con un altro. Inoltre, sperimentalmente risulta che la
molecola è esattamente esagonale, con tutti e sei i legami C-C uguali (sia per distanza ed energia).
Se invece ci fossero legami π messi come visto, la distanza e l’energia di legame non sarebbero
uguali. Questo perché in realtà gli orbitali p si sovrappongono non a coppie ma in sequenza, cioè
tutti i lobi superiori degli orbitali p danno un unico orbitale (risultato della sovrapposizione di 6
lobi) che percorrono l’esagono sopra e sotto. Quindi la sovrapposizione degli orbitali p non dà
luogo a doppi legami, da luogo ad un doppio legame ad anello su tutto l’esagono.
Questo processo è detto risonanza: la struttura reale è una “cosa” intermedia tra le due ipotizzate. La conseguenza è che si ha un legame delocalizzato, risulta che l’energia tra un carbonio e un
carbonio è più di quella di un legame singolo, ma meno di due, è la media ((1+2) / 2 = 1,5) (media
tra C-C σ e C-C σ + π). La molecola del benzene (liquido) usa il carbonio nello stesso modo in cui
il carbonio si comporta in un altro materiale solido, che si può estrae dal carbone delle miniere, la
grafite, che è carbonio puro legato con l’ibrido sp 2 .
<1.4> (tassellatura piana esagonale) In essa, ciascun C ibridizzato fa la struttura ad esagoni, ed ha
un orbitale p non ibridizzato. La sovrapposizione degli orbitali p (perpendicolari al piano) coin33
VB: polarità, elettronegatività
11/04/2014
volge tutta la struttura, segue la mappa degli esagoni, è una nuvola (fatta da tanti esagoni) sopra e
sotto, gli elettroni sono delocalizzati su tutto il cristallo, il che è responsabile delle proprietà elettriche della grafite, perché gli elettroni (degli orbitali p) si muovono sopra tutta la molecola, che
conduce solo in una direzione, ma non perpendicolarmente al piano.
Questo è il primo modo con cui si può avere il carbonio. Altrimenti lo si può trovare nei diamanti,
che sono fatti di carbonio puro, però con legami sp 3 , ed hanno proprietà completamente diverse,
in termini fisici, chimici, elettronici...
L’sp 3 ha tre orbitali diretti sui lati del tetraedro. La posizione spaziale di tutti gli atomi di carbonio
legati non è graduale, risulta in un reticolo detto “cubico a facce centrate”, cioè regolare, perché
si ripetono sempre nelle stesse posizioni, gli elettroni sono tutti coinvolti nei legami infatti il diamante è un isolante e cattivo conduttore di calore. Un elemento comune alla grafite è che se si
avvicina alla fiamma, brucia. La differenza sta nella quantità di energia necessaria per avviare la
reazione e nell’energia che rilascia successivamente.
La stessa struttura del diamante si vede anche nel silicio, anch’esso ha solo legami sp 3 .
Vediamo adesso legami con ibridi non solo sp.
<1.5> Avevamo usato il fosforo per fare esempi di valenza diversa: PCl3 e PCl5 . Nel primo caso, il
comportamento è lo stesso che si osserva con NH3 , ha valenza 3 e arriva ad un ibrido sp 3 analogo,
formando una molecola piramide con il fosforo nel vertice. In PCl5 vengono fatti 5 legami, quindi
si ha un’altro stato eccitato in cui viene usato anche l’orbitale d vuoto, sp 3 d , combinazione lineare di 5 orbitali(s, 3 p e 1 d). C’è da considerare che gli orbitali d non hanno tutti la stessa energia,
quindi c’è una differenza di energia a seconda di quale orbitale d viene ibridizzato.
La struttura geometrica è questa: il fosforo è centrale in un triangolo su un piano, nei tre vertici del
triangolo equilatero ci sono i Cl, e i due rimanenti sono perpendicolari al piano, uno sopra e uno
sotto. Di questi cinque orbitali, in realtà non sono equivalenti, ma 3+2, cioè i 3 sul piano hanno la
stessa energia, i 2 perpendicolari sono diversi dai primi tre e uguali tra loro. Con questo ibrido la
molecola di PCl5 viene interpretata come una bipiramide triangolare. Quando si stacca un atomo
di cloro, vengono via prima i due perpendicolari perché hanno energia minore.
<1.6> Anche qui l’ibrido è sp 3 d . Anche qui ci sono 5 direzioni, ma una è già presa da due elettroni,
ed è uno degli orbitali del triangolo. Gli elettroni liberi si mettono su un orbitale del piano, e l’orbitale è più espanso nello spazio, gli altri quindi tendono a schiacciarsi un po’. Infatti i due atomi
di fluoro non sono allineati ma appena appena angolati e gli altri due sono messi con un angolo
più piccolo di 60°. <1.7> In questo caso si hanno 6 elettroni disaccoppiati (valenza 6), l’ibrido corrispondente è sp 3 d 2 . I legami sono tutti uguali (sia come lunghezza che angoli che energia), è una
doppia piramide a base quadrata.
<1.8> Quando si era parlato di HF, avevamo detto che era un legame covalente polare (dove
polare si riferisce alla posizione delle cariche elettriche nella molecola), è un dipolo elettrico (è un
vettore) la cui intensità è proporzionale all’elettronegatività dei due atomi, e determina come la
molecola è influenzata dall’ambiente esterno (campo elettrico, corrente, campo magnetico...). In
BF3 , il boro fa un ibrido sp 2 (angoli a 120° con tre legami uguali), ciascuno dei legami è polare, sono
3 dipoli, perché fluoro e boro hanno elettronegatività diversa, quindi ogni legame è polarizzato. Il
dipolo elettrico totale è la somma dei tre vettori, che è nulla, quindi il dipolo totale è nullo.
34
Preparazione alla nomenclatura
15/04/2014
8 Preparazione alla nomenclatura
8.1 15/04/2014
Quando ci sono due atomi e si forma un legame, sappiamo che c’è un trasferimento di elettroni,
che si distribuiscono in maniera diversa. In generale, quando gli atomi sono di elementi diversi,
gli elettroni stanno preferenzialmente localizzati su uno dei due. Se per esempio riconsideriamo
HF, nella formazione del legame l’atomo di idrogeno complessivamente cede parzialmente il suo
elettrone esterno al fluoro (che è più elettronegativo, percui attrae su di sé di più gli elettroni di
legame). Per questo l’atomo di fluoro subisce un guadagno parziale, l’atomo di idrogeno una perdita parziale. In NaCl questa cessione e acquisto è totale, mentre in HF è un’acquisizione parziale.
Si danno dei nomi convenzionali a questo fenomeno:
• OSSIDAZIONE: cessione (anche parziale) di elettroni
• RIDUZIONE: acquisizione di elettroni
L’atomo che cede elettroni in un legame si dice che si ossida, l’elemento che acquisisce (anche
parzialmente) si dice che si riduce. Nella formazione di legame, chi cede l’elettrone lo dà ad un altro, cioè il processo di cessione e acquisizione avviene continuamente, cioè se c’è qualcuno che si
ossida, c’è qualcuno che si riduce, ci deve essere un bilancio totale nullo, non rimangono elettroni
<10:42>.
Per individuare numericamente se un elemento si trova in uno stato ossidato o ridotto, si usa il
numero di ossidazione, che si indica con n.o. = numero di elettroni ceduti o acquisiti.
<1.1>. E’ il valore numerico <10:46>.
Esempio: in HF abbiamo visto che l’idrogeno cede l’elettrone. <1.2> <10:47> si verifica con il fatto
che <...>
<10:48> si vede chi dei due cede o acquisisce preferenzialmente l’elettrone, e questo si determina tramite l’elettronegatività. In HF, il fluoro ha elettronegatività 4, l’idrogeno <...>, quindi F avrà
tendenza ad acquisire, H tendenza a perdere, quindi +1 e -1. Stesso discorso vale per NaCl.
Prendiamo H2 O. Scriviamo la molecola secondo il modello di Lewis e si vede cosa succede legame per legame. L’ossigeno è più elettronegativo di H, quindi nella formazione di un legame OH,
l’idrogeno cede il suo elettrone, quindi n.o(H) = +1 per entrambi gli H, l’ossigeno sta acquisendo 2
elettroni, quindi n.o.(O)=-2. La regola da somma 0 = −2 + 1 + 1.
Prendiamo NH3 : non è necessario sapere la struttura, basta conoscere la formula. So però che N
è più elettronegativo di H, quindi di sicuro H ha n.o.>0, N invece negativo. Di sicuro H ha n.o.(H)=
+1, ce ne sono 3, quindi n.o.(N) = -3.
Prendiamo H2 O2 : (acqua ossigenata / perossido di idrogeno) ci sono 2 legami OH, l’ossigeno è
più elettronegativo, quindi n.o.(H) = +1, ciascuno degli atomi di ossigeno acquisisce un elettrone.
Ma cosa fa tra un O e l’altro? E’ un legame tra due atomi uguali, quindi gli elettroni non hanno
nessuna preferenza, si ha un legame covalente puro, cioè ciascuno dei due atomi di ossigeno cede
e acquista la stessa quantità di elettroni, non c’è nessuna cessione o acquisizione netta quando
si forma un legame tra due atomi uguali, quindi non contribuisce alla variazione dei numeri di
ossidazione. Quindi n.o.(O) = -1.
Ci sono 5 regole: <1.3>
• 1) n.o. allo stato elementare = 0 (stato elementare significa che l’atomo o non è combinato
con nessuno, oppure è legato con un atomo dello stesso elemento, come in O2 , F2 , H2 , N2 ,
O, F, H, N)
35
Ancora struttura delle molecole
15/04/2014
• 2) n.o.(H) = +1 sempre tranne quando è legato con elementi meno elettronegativi di lui (elettronegatività(H) = 2,1. Gli elementi meno elettronegativi sono gran parte dei metalli, tutti gli
alcalini, tutti gli alcalino terrosi: in LiH n.o.(H) = -1, n.o.(Li)= +1)
• 3 o 2 bis) n.o.(alcalino) = +1 sempre, n.o.(alcalino terroso) = +2
• 4) n.o.(O) = -2 tranne nei perossidi (composti dove c’è un legame O-O) in cui n.o.(O) = -1;
n.o.(O) = +2 solo in OF2
P
• 5) n.o. = carica elettrica. Se la molecola fosse uno ione come CO32– , significa che ci sono 2
elettroni in più oltre alla configurazione neutra, quindi la somma dei numeri di ossidazione
non può dare 0, deve dare -2. Ossigeno è più elettronegativo, quindi n.o.(O) = -2, n.o.(C) =
P
(regoletta) C T OT − n.o.(O) = −2 − (−6) = +4.
Prendiamo H2 SO4 : non ci sono alcalini né alcalino terrosi, quindi sicuramente n.o.(H) = +1, n.o.(O)
= -2 (non c’è il fluoro), manca lo zolfo. Questa è una molecola neutra, quindi: 2 · [n.o.(H )] + x + 4 ·
[n.o.(O)] = 0. Quindi 2(+1) + x(+4) = 0, x = +6. Spesso i numeri di ossidazione si riportano sulla
formula quando servono (nelle reazioni di ossido reduzioni).<1.4>
Prendiamo CH4 : n.o.(H) = +1, n.o.(C) = -4.
C2 H4 : n.o.(H) = +1; n.o.(C) = -2
C2 H2 : n.o.(H) = +1; n.o.(C) = -1
Il numero di ossidazione non è in relazione al numero di legami che l’atomo fa, è in relazione al
numero di elettroni acquisiti e ceduti, infatti se si osserva come è fatto CH4 <1.5><11:14>
8
C3 H8 : n.o.(H) = +1; n.o.(C) = 8(+1) + 3x = 0; x = . Cosa significa un numero frazionario? Questo
3
deriva dall’applicazione delle regolette, che non presuppongono che si sappia dove sono i doppi
legami. In realtà, ogni atomo trasferisce un numero intero di elettroni, visto nel complesso non è
detto, perche per esempio per questa molecola, se scriviamo la formula di struttura <1.6> si vede
che non tutti gli atomi di carbonio si comportano allo stesso modo, quindi hanno sicuramente
stato di ossidazione diverso (il carbonio nel mezzo è legato a solo 2 H, che contribuiscono alla variazione di n.o.). Se si conosce la formula di struttura, si può dare il valore intero ad ogni atomo.
8
−3 − 3 − 2
= − . Il numero di ossidazione frazionario è comunque utilizzabile, non comInfatti:
3
3
porta alcuna complicazione.
12
C5 H12 : (<normal 11:24?>pentano) se si fa il conto, viene 12 · (+1) + 5x = 0, x = − . Però se si ve5
de la molecola così <1.7> si vede che i numeri di ossidazione sono interi. C’è però anche un’altra
struttura (isopentano). Qui il C centrale ha n.o.=0, perché fa 4 legami con atomi di C. La media è
(−3 · 4 + 0)/5.
<11:27>
9 Ancora struttura delle molecole
9.1 15/04/2014
CH4 : quale è la geometria molecolare (spaziale)? Bisogna vedere quale ibrido fa. La geometria
deriva da una ibridizzazione sp 3 del C, quindi vengono fuori 4 legami diretti secondi i vertici di un
tetraedro regolare <2.1>. Spesso si chiede anche di analizzare i dipoli elettrici, sono 4 la cui somma
vettoriale è nulla. CH3 Cl: (cloroformio) se la si guarda si può intuire che non è altro che una molecola proveniente da CH4 sostituendo un idrogeno con il cloro (che ha valenza 1 in questo caso,
36
Ancora struttura delle molecole
15/04/2014
quindi non può essere l’atomo centrale, le altre valenze non vanno bene perché ci vorrebero 3, 5
o 7 altri atomi). <2.2> Questa è una molecola polare, basta disegnare i tre dipoli <2.2> e si consida
l’elettronegatività. Adesso il dipolo totale non è più nullo, qui si può anche approssimativamente capire quanto è: <2.3><11:37>. Un ulteriore passo che si può fare è ragionare sul fatto che gli
angoli non sono più quelli tetraedrici (fra H e C), ci sono 2 meccanismi che agiscono spesso in
maniera opposta per far variare gli angoli. Metto un atomo di cloro dove prima c’era un idrogeno,
l’idrogeno è molto piccolo. Ci sto mettendo un atomo di cloro che usa uno dei suoi elettroni, poi
esterni ne ha altri 8, è un elemento del terzo periodo, quindi la regione di carica negativa potrebbe
funzionare come gli orbitali ibridi quando non sono legati (che si gonfiano di più e schiacciano gli
altri), quindi così si potrebbero stringere i legami di sotto. <11:39>, per cui gli elettroni tendono a
stare di più sul carbonio. Quindi sull’idrogeno c’è una carica positiva, negativa su C. Si riprende
CH4 , supponiamo di avere quindi tanti +δ in equilibrio sui quattro vertici del tetraedro. Prendo
un H e ci metto Cl, che <11:41>, questo significa che gli atomi si spostano di più verso il carbonio,
perché c’è il Cl che sta attirando. Quindi se prima in H c’era una carica +δ, adesso ce n’è una ancora maggiore. Quindi il cloro grosso spinge i tre legami, ma il fatto che il cloro attiri fa sì che restino
cariche positive in eccesso su H, quindi si respingono tra loro di più. Tendenzialmente prevale il
secondo effetto, però l’accordo tra noi e il prof è che se si dice che tendenzialmente l’angolo in
basso aumenta va bene.
BH3 , BH2 F, BHF2 , BF3 : prendi le tre molecole e disponile dalla più polare alla meno polare. Tutte
si ottengono da una molecola di partenza (qui BH3 e BF3 ). Bisogna dire che l’ibridizzazione del
boro è sp 2 , quindi gli orbitali sono planari a 120°, l’idrogeno ha elettronegatività 2,1, ci sono tre
dipoli la cui somma è 0. <2.3>. Al primo posto ci sono a pari merito BH3 , BF3 che hanno |~
p | = 0.
Adesso si prende BH2 F <2.4>. Poi si fa il ragionamento sugli angoli: ho sostituito H con F, su ogni H
avevo un δ−, con un F, che è ancora più elettronegativo, quindi la carica su F è −δ∗ più grande, che
risucchia gli elettroni dagli altri H, che se prima erano un po’ in eccesso, adesso lo sono di meno,
quindi la nuova carica sugli H è minore della precedente. Quindi ora che sono diminuite potrebbero respingersi di meno e l’angolo chiudersi. Questa volta gli effetti vanno nello stesso verso: il
fluoro è più grande (potrebbe schiacciare), e la carica adesso è minore della precedente sui 2 H,
quindi mi aspetto che l’angolo tra B e i due H si stringe. <11:52, dipolo finale?>.
Prendiamo BHF2 : <2.5>, vediamo gli angoli. H è molto meno elettronegativo di F, quindi <..> se ci
metto H vanno verso il b, se ci metto <..>, quindi l’angolo tra F aumenta. Quindi la somma dei loro
dipoli non è più 2 ma un po’ meno. Quindi |~
p | < 1,9.
Se voglio mettere le molecole in ordine, posso fare due categorie: una con dipolo nullo, una con
dipolo vicino (a 1,9) <2.6>
Prendiamo ora C2 H4 : <2.7>
Da questa possiamo immaginare C2 H2 Cl2 : qui bisogna ragionare sul fatto che ci sono 2 atomi di
cloro da sostituire, e ci sono 3 modi diversi per farlo. <2.8>. Si può discutere come sono fatte dal
punto di vista del dipolo elettrico. <12:09 finito di disegnare un po’ di roba....>
Appendice: immagina di avere tante cariche elettriche nello spazio, ogni carica ha il suo vettore
posizione in un sistema cartesiano <2.9> La definizione di dipolo elettrico esiste anche quando la
somma di carica non è 0. Il valore del dipolo dipende da dove scelgo il sistema di assi di riferimento, quindi non si può definire in termini assoluti il dipolo, se non è nullo. Quando si ha la stessa
carica positiva e negativa, allora <12:15>. Se si prende un altro sistema, traslato rispetto al primo,
si può provare che se la carica totale non è nulla, il dipolo varia, se è nulla, il dipolo non dipende
dal sistema di riferimento. Bisogna stare attenti a come si sceglie il sistema quando il dipolo è carico <...12:17 casino>.
Prendi NH3 <2.10> [...] NH4+ Una volta che si sono i formati, non riesco a distinguere i 4 legami
con l’H, quindi è tetraedrica perfetta come quella del metano, identica, soo che ha una carica +. E’
sbagliato dire che ha dipolo nullo, perché se si prende il sistema di riferimento su N risulta nullo,
37
Ancora struttura delle molecole
15/04/2014
altrimenti è diverso da 0. Non basta la simmetria per dire che il dipolo è nullo.
38
Nomenclatura
17/04/2014
10 Nomenclatura
10.1 17/04/2014
L’esigenza della nomenclatura esiste da molto tempo, ce ne sono diverse. Noi trattiamo una di
quelle in vigore, ma ce ne sono altre due attuali. Noi seguiamo quella della IUPAC molto sinteticamente.
<11:46>. Gli ioni positivi si chiamano cationi, quelli negativi anioni. Fra questi si distingue se sono
ioni monoatomici o poliatomici. Poi guarderemo le molecole neutre, fra cui si distinguono prima
le molecole biatomiche, poi fra quelle neutre importanti specifichiamo gli idrossidi, gli acidi e i
sali. I nomi delle molecole neutre attingono ai nomi degli ioni.
Il caso più semplice è quello che ioni positivi monoatomici: uno ione positivo è un atomo che ha
perso uno o più elettroni, cioè si è ossidato. Un’ossidazione corrisponde a numero d’ossidazione
positivo. Quindi gli ioni positivi coincidono con numero d’ossidazione positiva, che coincida anche con la loro carica. La costruzione del nome è “Ione” + nome dell’elemento: Na+ ione sodio, K+
ione potassio.
C’è da distinguere quando si possono avere ioni con diverso numero di ossidazioni, come nel caso
di Cu. La vecchia nomenclatura usava una desinenza sul nome (-oso per n.o. minore e -ico per n.o.
maggiore, infatti Cu+ ione rameoso, Cu2+ ione rameico). La nomenclatura IUPAC invece aggiunge
un numero romano fra parentesi per indicare il numero di ossidazione: Cu2+ ione rame (II), Fe3+
ione ferro (III).
Per gli ioni positivi poliatomici, noi indicheremo solo quelli che possono essere pensati come
unione di un composto neutro più uno ione H+ . Prendiamo PH3 + H+ −−→ PH4+ che è uno ione
poliatomico positivo. Si indica lo ione usando una desinenza sul nome dell’elemento principale.
In questo caso: PH4+ ione fosfonio; AsH4+ ione arsonio.
Esistono delle eccezioni per motivi storici: certi composti non hanno cambiato nome: NH3 +
H+ −−→ NH4+ è lo ione ammonio, non nitronio (nella nomenclatura prevale la parola da cui proviene il simbolo (è un problema per l’italiano). <11:55> Ammoniaca è il nome usuale, non quello
IUPAC. Stesso discorso vale per H2 O + H+ −−→ H3 O+ è la forma fisica con cui si trova un protone
quando è sciolto in acqua (legato con legame dativo a una molecola d’acqua), è un protone “idratato”, è detto ione idrossonio.
Per gli ioni negativi monoatomici (n.o. < 0), a “ione” + nome dell’elemento si aggiunge la desinenza -uro all’elemento: H– ione idruro; S2– ione solfuro, N3– ione nitruro, P3– ione fosfuro, C4– ione
carburo. Eccezioni: O2– ione ossido, O– ione perossido.
Per gli ioni negativi poliatomici discuteremo solo quelli che contengono un elemento e ossigeno,
detti ioni ossigenati. La regola è “ione” + nome dell’elemento + -ato. Se ci sono n.o. diversi, si
mette il numero d’ossidazione in cifre romane. SO32– ione solfato (IV), SO42– ione solfato (VI);
nella precedente nomenclatura si distinguevano per la desinenza, quello con n.o. minore aveva
la desinenza -oso, quello con n.o. maggiore aveva desinenza -ico, rispettivamente: ione solforoso,
ione solforico.
ClO– ione clorato(I), ClO2– ione clorato (III), ClO3– ione clorato (V), ClO4– ione clorato (VII). La
vecchia nomenclatura li chiamava: ione ipocloroso, cloroso, clorico, perclorico.
NO2– ione nitrato (III), NO3– ione nitrati (V), PO3– <12:04>
CO32– ione carbonato, HCO3– ione idrogenocarbonato (si aggiunge idrogeno al nome). Nella vecchia nomenclatura era lo ione bicarbonato.
Eccezioni: uno degli ioni negativi monoatomici S2– , legato ad H: HS– diventa ione idrogenosolfuro
(non è una vera eccezione). CN– ione cianuro, OH– è detto ione idrossido (è un’eccezione importante).
Molecole neutre: Composti binari
39
Nomenclatura
17/04/2014
Fra due elementi, si scrive per primo quello meno elettronegativo. Il nome si legge seguendolo al
contrario, cioè si legge da destra a sinistra. Se si ha una molecola A n B m , il nome è m - (nome ione
negativo) di n - (nome ione positivo). B è uno ione negativo perché è quello più elettronegativo,
quindi avrà desinenza -uro <12:11>
NaCl (Na+ Cl– ) cloruro di sodio. <...> Si toglie il nome ione a tutto, rimane cloruro di sodio.
KBr bromuro di potassio, K2 S (2K+ S2– ) i nomi dei due ioni lo conosciamo, si ottiene solfuro di dipotassio, perché ci sono 2 K.
FeCl2 (Fe2+ 2Cl– ) dicloruro di ferro; cloruro di ferro(III), FeCl3 tricloruro di ferro; cloruro di ferro
(III). Siccome l’unica cosa che cambia è il numero di ossidazione è quello del ferro, la nomenclatura IUPAC prevede anche che se ne scriva il numero di ossidazione per indicarlo univocamente.
Nella vecchia nomenclatura si chiamavano cloruro ferroso, cloruro ferrico.
MgO (Mg2+ O2– ) ossido di magnesio, Na2 O (2Na+ O2– ) ossido di disodio
SO2 (S4+ 2 O2– ) diossido di zolfo; ossido di zolfo(IV)
SO3 triossido di zolfo; ossido di zolfo(VI)
FeO (Fe2+ O2– ) mon-ossido di ferro: si specifica mono perchè <12:17>.
Fe2 O3 triossido di diferro; ossido di ferro(III)
H2 O è acqua, il nome è ossido di di-idrogeno
LiH l’idrogeno è scritto dopo, perché è meno elettronegativo (è legato a metalli alcalini e alcalinoterrosi), è l’idruro di litio; NaH idruro di sodio, MgH2 di-idruro di magnesio
HF fluoruro di idrogeno, o acido fluoridrico, HCl cloruro di idrogeno (acido cloridrico), HBr bromuro di idrogeno (acido bromidrico), HI idruro di idrogeno (acido iodidrico).
Eccezione: NH3 si dovrebbe chiamare tri-idruro di azoto<12:22>, e dovrebbe essere H3 N, perché
l’idrogeno è meno elettronegativo. Si chiama ammoniaca. Tutti i suoi composti presentano questa
eccezione (l’azoto scritto per <primo>).
Idrossidi: ione positivo + ione OH (ione idrossido). Quando c’è più di uno ione idrossido, ci si
mette una parentesi: NaOH, Fe(OH)2 , sono idrossido di sodio e di-idrossido di ferro oppure idrossido di ferro(III).
NH4 OH idrossido di ammoniaca
Acidi (ossigenati): sono ottenuti come uno ione negativo poliatomico più lo ione H+ , che naturalmente si scrive prima.
H2 SO4 (2 H+ SO42– ) solfato(VI) di idrogeno (acido solforico), oppure si usa il nome di tutto: tetraosso-solfato di di-idrogeno.
H2 SO3 tri-osso-solfato di di-idrogeno; solfato(IV) di idrogeno (acido solforoso)
HNO2 nitrato(III) di idrogeno oppure di-osso-nitrato di idrogeno, HNO3 tri-osso-nitrato di idrogeno
H2 ClO clorato(I) di idrogeno oppure mon-osso-corato di <di-idrogeno>.
Sali: unione di uno ione positivo e uno negativo, dove lo ione positivo non è H+ e quello negativo
non è OH– , altrimenti nel primo caso si avrebbero gli acidi, nel secondo gli idrossidi.
KNO2 (K+ NO2– ) nitrato (III) di potassio, K2 S solfuro di di-potassio, BaSO4 (Ba2+ SO42– ) solfato(VI)
di bario
Na2 CO3 carbonato(IV) di sodio, NaHCO3 idrogenocarbonato(IV) di sodio (bicarbonato di sodio)
(NH4 )2 SO4 (2 NH4+ SO42– ) solfato(VI) di ammonio.
40
Ossidoriduzioni
29/04/2014
11 Ossidoriduzioni
11.1 29/04/2014
+2
±0
+2
±0
Zn + Cu2+ −−→ Zn2+ + Cu
Questa è una reazione in cui compaiono anche degli ioni che si bilancia mentre si scrive, però è
un’ossidoriduzione, cioè una reazione in cui delle specie cambiano il loro numero di ossidazione.
Le reazioni di tipo acido-base non c’è nessuna specie che cambia n.o. (basta andare a riprendere quegli esercizi per verificare). Qui Zn all’inizio ha n.o.=0, è solido. Nei prodotti n.o.(Zn) = +2,
questo è sufficiente a dimostrare che è una reazione di tipo ossidoriduzione. Se c’è qualcuno che
perde elettroni (come fa lo zinco qui) ci deve essere qualcuno che li prende, cioè se c’è qualcuno che si ossida c’è qualcuno che si riduce, qui lo ione rame, che parte da +2 e arriva a 0 come
prodotto.
Zn cede 2e → si ossida, è un riducente
Cu2+ acquista 2e →si riduce, è un’ossidante
Ciò che si fa è scomporre la reazione iniziale in due semireazioni, per individuare separatamente il processo di riduzione e di ossidazione.
Zn −−→ Zn2+ + 2 e è la semireazione di ossidazione
Cu2+ + 2 e −−→ Cu è la semireazione di riduzione
Sono reazioni virtuali, l’atomo di zinco cede direttamente elettroni al rame, sono due processi che
avvengono contemporaneamente, e avvengono anche nello stesso posto (lo ione zinco e il rame si
avvicinano per far avvenire la reazione). Se si sommano le due semireazioni:
Zn + Cu2+ + 2 e −−→ Zn2+ + Cu + 2 e
<10:47 cosa si ottiene?>
Noi usiamo il metodo ionico-elettronico per bilanciare le reazioni di ossidoriduzioni.
CuS + HNO3 −−→ CUSO4 + NO + H2 O
E’ da bilanciare, ed è possibile che ci sia qualche specie chimica omessa per farlo, tipicamente è
l’acqua (quando si chiede di completare, spesso c’è da aggiungere l’acqua). E’ qui che la nomenclatura fa comodo: CuS è il solfuro di rame, lo si immagina come Cu2+ + S2– . Lo ione solfuro ha
n.o.=-2. HNO3 è il nitrato(V) di idrogeno, quindi è H+ + NO3– , n.o.(N)=-5.
Cu
2+
−2
+5
+6
+2
+ S2− + H+ + NO3− −−→ Cu2+ + SO42− + NO + H2 O
CuS + HNO3 −−→ CuSO4 + NO + H2 O
l’ultima è la forma molecolare. <10:54>
Il primo passaggio è scomporre le molecole che ci sono negli ioni che le compongono (passaggio “ionico”). Non si scompongono <10:57>:
• gas
• ossidi
• solidi
41
Ossidoriduzioni
29/04/2014
Poi bisogna individuare chi cambia il n.o., qui lo ione solfuro diventa ione solfato, n.o.(N) va da
-2 a +6, quindi perde 8 elettroni. Questo è un processo di ossidazione che noi abbreviamo così
<10:59>
−2
+5
+6
+2
Cu2+ + S2− + H+ + NO3 −−→ Cu2+ + SO42− + NO + H2 O
Ox
Rid
Adesso scriviamo le semireazioni (questo è il passaggio “elettronico”):
S2− −−→ SO42− + 8 e
NO3− + 3 e −−→ NO
e le bilanciamo come se fossero reazioni singole. Nella prima ci sono 2 cariche negative nei reagenti, 8 nei prodotti, quindi bisogna bilanciarle. Abbiamo a disposizione delle specie che si possono
usare sempre:
• H+
• OH–
• H2 O
+
−
−−
*
H2 O −
)
− H + OH
L’acqua si dissocia continuamente in una situazione di equilibrio. In soluzione acquosa, ci sono
sempre queste tre specie, questo è il motivo per cui si possono usare (queste reazioni avvengono
spesso in ambiente acquoso). Per far tornare il numero di cariche ho 2 modi: nella prima semireazioni posso aggiungere 8 carice negative a sinistra oppure 8 cariche positive nei prodotti, in ogni
caso sarebbe bilanciata <numeri non tornano! 11:04>
S2– −−→ SO42– + 8 e + 8 H+ adesso devo bilanciare le masse, basta aggiungere 8 H2 O e ho bilanciato
l’idrogeno. Questo significa aver messo 4 atomi di ossigeno a sinistra, e va bene perché è anche
nei prodotti.
Passiamo alla seconda semireazione: adesso si è obbligati ad usare di nuovo gli H+ , bisogna usare
lo stesso criterio per entrambe. Ci sono 4 cariche negative a sinistra e carica neutra nei prodotti,
quindi bisogna aggiungere 4:
H+ a sinistra: 4 H+ + NO3– + 3 e −−→ NO + 2 H2 O
Adesso vanno risommate per ottenere la reazione complessiva. Questo passo lo si fa tenendo conto che abbiamo aggiunto elettroni come se fossero una specie chimica, ma non lo sono, il numero
di elettroni deve essere lo stesso <11:08>... mi rimarrebbero 5 elettroni. Quindi moltiplico le due
semireazioni per due coefficienti per avere lo stesso coefficiente stechiometrico per gli elettroni.
Moltiplico la prima per 3 e la seconda per 8. Ottengo:
12H2 O + 3 S2− + 32 H+ + 8 NO3− + 24 e −−→ 3 SO42− + 24 e + 24 H+ + 8 NO + 16 H2 O
Per prima cosa verifico che non devono rimanere elettroni. Poi vedo che ci sono 32 H+ e 24 tra
i prodotti, quindi si semplificano e ne restano 8 a sinistra. Così anche le molecole d’acqua. Si
ottiene:
3 S2− + 8 H+ + 8 NO3− −−→ 3 SO42− + 24 e + 8 NO + 4 H2 O
42
Ossidoriduzioni
29/04/2014
Questa è la reazione nella forma ionica. Il passo successivo che si può fare (perché la reazione
parte da specie in forma molecolare) è ritrovare la forma molecolare bilanciata, e lo si fa semplicemente ricostruendo le molecole neutre da cui i vari ioni provenivano. Per esempio lo ione
solfuro provenivva da CuS, quindi se voglio ricostruirla devo aggiungere le specie mancanti. Ho
3 S2– , quindi mi servono 3 Cu2+ per poter costruire 3 molecole di CuS, li devo aggiungere anche tra
i prodotti (altrimenti non è più bilanciata la reazione) <1.5 11:15>
Non è obbligatorio passare dalla forma ionica, ci sono un po’ più di complicazioni ma poi si ottiene direttamente la forma molecolare, la forma ionica è un po’ più semplice. Facciamo un altro
esempio: KMnO4 + HCl −−→ MnCl2 + Cl2 + KCl
Si chiede quanti grammi di cloro gassoso si formano facendo reagire insieme 150g di KMnO4
(manganato(VII) di potassio) e 290g di HCl.
La prima cosa da fare è bilanciarla, si nota che è una reazione di ossidoriduzione perché n.o.(Cl) è
passato da -1 a 0 nel prodotto Cl2 Scompongo in ioni: K+ +MnO4– +H+ +Cl– −−→ Mn2+ +Cl– +Cl2 +K+
NB: non serve indicare quanti di ogni ione si trovano, non servono i coefficienti stechiometrici,
serve sapere quali sono gli ioni presenti.
Nei reagenti lo ione Cl– ha n.o.=-1, nei prodotti però oltre a Cl c’è anche Cl– , quindi ci si aspetta
che non tutti i Cl– di partenza si ossidino<o riducano? 11:25>.
<1.6>
Le semireazioni sono <1.7>
Un altro esempio è:
I3− + S2 O32− −−→ I− + S4 O62−
E’ una forma ionica, quindi alla fine otterremo una forma ionica. C’è da vedere chi si ossida e chi
1
si riduce. Fra i prodotti c’è lo ione ioduro, n.o.=-1. Nei reagenti I3– , che ha n.o.=− . Lo iodio si è
3
ridotto. <1.8> <11:44>. Lo zolfo si ossida. Quindi si scrivono le semireazioni:
I3− + 2 e −−→ 3 I−
2 S2 O32− −−→ S4 O62− + 2 e
Per la prima: mi accorgo che devo fare la stessa cosa dell’esempio precedente, ho bisogno di 3
2
atomi di iodio, aggiungo il coefficiente al prodotto. Ogni atomo di iodio acquista di elettrone,
3
quindi ci sono 2e fra i reagenti. Per la seconda: aggiungo il coefficiente stechiometrico al reagente.
1
e ∗ 4 = 2e. Le cariche sono bilanciate e lo sono anche le masse. Adesso si sommano:
2
I3− + 2 S2 O32− −−→ 3 I− + S4 O62−
Se capita che il numero di elettroni nelle semireazioni è uguale senza dover introdurre coefficienti,
allora la reazione probabilmente si poteva risolvere a occhio.
Prendiamo ora:
Cl2 + NaOH −−→ NaCl + NaClO
Di sicuro è un’ossidoriduzione perché c’è lo ione cloruro e lo ione clorato.
−1
+1
Cl2 + OH− −−→ Cl− + ClO−
riduzione
ossidazione
43
(1)
Ossidoriduzioni
29/04/2014
In Cl2 n.o.=0, n.o.(Cl-)=-1, in ClO- n.o.(Cl) = +1. Il cloro sia si ossida sia si riduce.
Quando la stessa specie chimica si ossida e si riduce, la reazione è chiamata DISMUTAZIONE.<11:55
rivedi>
Le semireazioni:
Cl2 + 2e −−→ 2Cl−
4OH− + Cl2 −−→ 2ClO− + 2e + 2H2 O
La seconda va bilanciata con le cariche: è consigliabile osservare cosa c’è un partenza, nella forma ionica tra i reagenti <11:57>, conviene usare lo ione che è presente (OH– ). Quindi introduco
4 OH– .
Anche questa reazione si poteva bilanciare ad occhio. In forma molecolare il risultato è: Cl2 +
2 NaOH −−→ NaCl + NaClO + H2 O.
44
Interazioni deboli
29/04/2014
12 Interazioni deboli
12.1 29/04/2014
<12:04>. In alcuni testi sono chiamate anche legami deboli. Queste sono forze tra molecole distinte, non all’interno delle molecole, sono responsabili delle proprietà fisiche, soprattutto dello stato
delle molecole (liquido, solido, gassoso).
Le interazioni deboli si possono classificare così <2.1>:
• interazioni dipolari
– ione-dipolo
– dipolo - dipolo
Van Der Waals
– dipolo - dipolo indotto
– dipolo istantaneo - dipolo indotto
forze di dispersione
(di London)
• legame (ponte) a idrogeno
<12:08>
Le interazioni ioni-dipolo sono tutte di natura elettrostatica: immagina uno ione positivo (Na+ )
in presenza di molecole d’acqua, che è una molecola angolata polare (con carica relativamente
forte). Se uno ione Na+ si trova in presenza di dipoli come quelli dell’acqua (o viceversa), si orienta
con la parte negativa verso la carica positiva. A seconda della grandezza dello ione <12:13>. La
struttura che si forma intorno allo ione si chiama strato di idratazione oppure di solvatazione se si
ha un liquido diverso dall’acqua che si comporta nello stesso modo. La stessa cosa succede se si ha
uno ione Cl– , che attira verso di sé le parti positive dei dipoli. Tanto maggiore è il numero di dipoli
che uno ione riesce a coinvolgere, più è stabile il sistema. Questo meccanismo è responsabile del
fatto che il sale da cucina quando è messo in acqua si scioglie.<2.2> NaCl è un cristallo ionico, che
ha una superficie su cui c’è un pavimento di ioni alternati, legati all’esterno a nessuno e all’interno ad altri ioni. L’ambiente contiene molecole d’acqua, che si orientano verso le molecole degli
ioni. Quando diventano un numero sufficientemente alto, l’energia del sistema idratato supera
l’energia di legame dello ione nel cristallo e lo ione preferisce staccarsi e circondarsi di molte molecole d’acqua piuttosto che restare nel cristallo. Ugualmente fanno gli altri e così mano a mamo
si espongono tutti gli ioni e il sale si scioglie. Questa è un esempio di interazione fra uno ione e un
dipolo. Se si prova a sciogliere il sale in un liquido non polare, come CCl4 , esso rimane un solido
cristallino.
Le interazioni dipolo-dipolo sono interazioni fra molecole che sono già dipoli, come le molecole
d’acqua, che interagiscono tra loro. I poli cercano, quando l’agitazione termica lo permette, di disporsi in modo di avere energia minima. Sono interazioni piuttosto forti, che si verificano anche
in HF o NH3 .
Il dipolo-dipolo indotto: prendiamo l’acqua gassata, cioè acqua in cui c’è CO2 . La CO2 non è polare, però riesce a sciogliersi nell’acqua, quindi si instaura una interazione come quella dell’Na+ , ma
più debole. Come fa una molecola apolare ad interagire con una polare? Immagina una molecola
come una <12:22> elissoidale con il centro di carica <...>. Se la distanza è sufficientemente piccola,
attira preferenzialmente gli elettroni più esterni, quelli di legame (quelli più polarizzabili) <...> e
dalla parte opposta rimane una piccola parte di carica positiva. La polarità dell’acqua induce un
dipolo sulla CO2 , che è tanto più facile da indurre quanto più gli elettroni sono mobili. <2.3>
<12:24> esiste anche la CO2 solida, quindi ci devono essere delle interazioni, ma quali se non ci
sono molecole polari? E’ vero che il baricentro delle cariche positive e negative coincide con il
centro <...>, ma è vero in media, istantaneamente no, gli elettroni sono ogni volta in punti diversi.
45
Interazioni deboli
02/05/2014
E’ come se questa molecola in realtà avesse in continuazione un dipolo che si muove molto velocemente, che se si guarda su un arco di tempo anche breve si vede 0. <2.4>
Quindi è vero che questa molecola è polare istante per istante. Se ho un’altra molecola che ha
questo stesso comportamento, e sono sufficientemente vicine, questo comportamento genera un
dipolo indotto nell’altra. Quindi in un piccolo istante c’è un’interazione con energia bassissima,
ma spiega perché molecole apolari in realtà si possono solidificare o liquefare. L’esempio classico
è l’elio, che è un atomo con 2 protoni nel nucleo 2 due elettroni nell’orbitale 1s, è sferico, difficilmente polarizzabile perché i due elettroni sono molto legati al nucleo. Se si va a temperature sufficientemente basse (-270°C) l’elio diventa liquido, quindi si riesce ad osservare il comportamento
dipolo istantaneo-dipolo indotto.
12.2 02/05/2014
E’ rimasto da trattare il LEGAME A IDROGENO (o ponte a idrogeno). L’energia messa in gioco in
questa interazione è superiore a quella di tutte le precedenti già viste, è una sorta di via di mezzo
tra le altre interazioni deboli e i legami covalenti.
Supponiamo di prendere la molecola HF <1.1>, in cui c’è un atomo di idrogeno e un atomo
di fluoro legati insieme con un legame covalente fortemente polarizzato (il fluoro ha una elettronegatività 4, l’idrogeno 2,1) quindi come prima conseguenza sappiamo che c’è un’interazione
dipolo-dipolo con altre molecole di HF piuttosto intensa. In <1.1> vediamo come intuitivamente
sono disposti gli elettroni di legame, per avere un’idea sulla distribuzione di carica: i due elettroni
sono prevalentemente sul fluoro e lasciano poca carica negativa intorno all’idrogeno.
<1.2> In due molecole di questo tipo, i dipoli si attraggono, cercando di avvicinare la zona negativa di una con la zona positiva dell’altra (interazione dipolo-dipolo). In generale le due estremità
si avvicinano fino ad una certa distanza, compatibile con l’equilibrio tra l’attrazione che c’è e la
repulsione degli elettroni esterni (come già visto con i legami). In questo caso, siccome l’idrogeno
è legato ad un elemento molto elettronegativo, e siccome intorno a lui rimane poco dell’elettrone
originario, quando c’è l’avvicinamento delle due molecole l’effetto di rimozione fa sì che l’effetto
di repulsione sia poco. Il risultato è che questi due dipoli riescono ad avvicinarsi molto (mediamente la metà di quello che si vede con dipoli meno intensi) e le energie diventano molto intense
(le forze di tipo elettrostatico spesso dipendono da r12 o r13 ). Questo è un fenomeno che si verifica
solo con l’idrogeno legato ad un altro atomo molto più elettronegativo di lui, ticamente F, O, N,
più raramente con Cl (il cloro è molto più grosso, quindi l’effetto è più debole), ad essere rigorosi
HCl non presenta questo fenomeno.
Nelle molecole d’acqua <1.3>, che sono dipoli, in cui l’ossigeno si riesce ad avvicinare all’idrogeno, che di fatto è un protone scoperto (quindi molto piccolo) legato ad un altra molecola,
si instaura il legame a idrogeno. Nelle molecole di ammoniaca (NH3 ), può interagire con un H di
un’altra molecola <1.4>
In generale, le interazioni deboli sono responsabili dello stato fisico delle sostanze, ed il legame
a idrogeno porta conseguenze importanti. Un esempio tipico è quello dell’acqua, che è solida sotto gli 0 °C e liquida a temperatura ambiente. Prendiamo H2 O e H2 S: sono geometricamente simili,
entrambe polari, ma hanno la temperature di ebollizione molto diverse: essa dipende dall’energia
che serve per rompere le interazioni che tengono insieme le molecole nel liquido, nell’acqua queste interazioni sono dipoli-dipoli sovrapposti a legami a idrogeno, quindi serve parecchia energia.
La Teb dell’acqua è 100 °C, in H2 S, che è un dipolo più debole (differenza di elettronegatività tra
i due atomi minore) e non presenta legami a idrogeno si ha che Teb = −62 °C, infatti è un gas in
condizioni in cui l’acqua è liquida.
46
Stati della materia
02/05/2014
13 Stati della materia
13.1 02/05/2014
ev
ap
or
az
ion
e
GASSOSO
i
az
il m
b
su
e
on
co
nd
en
s
o
nt
az
io
e
ne
am
in
r
b
LIQUIDO
zione
solidifica
SOLIDO
fusione
I tre stati della materia sono gassoso (o aereiforme), liquido, solido. I passaggi di stato da liquido a gassoso è detto evaporazione, l’inverso condensazione, da solido a liquido fusione, l’inverso solidificazione, da solido a gassoso sublimazione, l’inverso brinamento. L’esistenza di una
sostanza in uno di questi tre stati è dato da un bilancio d’energia. All’interno della sostanza, per
esempio dell’acqua, ogni molecola ha una sua energia che in generale è la somma di due contributi: E = E c + E p , un’energia cinetica (dove il quadrato della velocità dipende linearmente dalla
temperatura) e un’energia potenziale, associata alle interazioni che le molecole fanno con l’ambiente circostante, legata alle forze cui una molecola è sottoposta, che possono essere quelle delle
interazioni deboli, oppure le forze del reticolo ionico come nel caso di un solido (sono forze elettrostatiche); nel caso dell’acqua sono interazioni dipolo-dipolo e legame a idrogeno. Queste forze
in generale sono dette forze di coesione.
|E c | ¿ |E p |, stato solido
E
solido
intervallo di movimento delle molecole
d
E
Ec
Ep
Prendiamo in considerazione il caso in cui |E c | ¿ |E p |, cioè che nel calcolo dell’energia complessiva l’energia cinetica influisce poco. Nel grafico che rappresenta l’andamento dell’energia
potenziale, c’è un punto di minimo, in cui la forza di attrazione e quella repulsiva sono in equilibrio, è l’E p . L’energia complessiva è dunque di poco superiore, ed è negativa. Questo significa
che la molecola (o atomo) è vincolata a muoversi in un piccolo intervallo di distanza dall’altra molecola (o atomo), cioè se le entità di cui è costituita la sostanza si trovano nella condizione in cui
l’energia potenziale supera di gran lunga quella cinetica, di fatto la loro posizione è fissa, al massimo vibrano intorno alla posizione di equilibrio, ma non si allontanano le une dalle altre. Questa
47
Stati della materia
02/05/2014
è la situazione semplificata dello stato solido, in cui le molecole sono a contatto l’una con l’altra e
sono in posizioni definite. Lo stato solido è caratterizzato da un ordine a lungo raggio (somiglia un
sistema cristallino), di cui un esempio sono i cristalli, ma anche nell’acqua solida siamo in questa condizione. La caratteristica più evidente che deriva da questa condizione è che la sostanza è
incomprimibile (le molecole non si possono avvicinare di più) ha una forma propria (le molecole
sono vincolate a stare in una certa posizione) ed un volume proprio.
E
prevale l’energia di movimento
E
gas
|E c | À |E p |
|E c | ∼ |E p |
liquido
d
Prendiamo il caso in cui |E c | À |E p |, quindi di avere un’energia totale molto più grande di
prima <1.6>. E p di fatto non modifica l’energia del sistema, le molecole interagiscono un po’ tra
loro, ma forze legate alle interazioni sono talmente deboli che prevale l’energia di movimento e
non la alterano. Le molecole sono praticamente libere le une dalle altre, si muovono casualmente,
si urtano con urti quasi elastici. Questo è lo stato gassoso, il fatto che le molecole non siano legate
o vincolate in posizioni spaziali comporta che la sostanza non abbia volume proprio (muovendosi
le molecole sono vincolate solo dalle pareti del recipiente che le contiene, cambio il recipiente e la
sostanza lo riempie sempre tutto), non abbia forma e sia compribimile.
C’è poi la situazione intermedia, in cui |E c | ∼ |E p |, la situazione dello stato liquido. Le molecole
riescono a muoversi con energie cinetiche significative ma comparabili con quelle potenziali, cioè
di fatto le molecole si possono muovere ma rimangono a contatto le une con le altre. Quindi se
rimangono a contatto hanno un volume proprio e non sono comprimibili, però siccome sono
libere di muoversi non hanno forma.
Il termine E c lo posso far variare linearmente variando la temperatura (partendo per esempio da valori molto bassi per basse temperature e portarlo a valori molto alti con temperature
alte). L’energia potenziale, che dipende da come sono fatte le molecole, è quasi indipendente dalla temperatura (se una molecola è polare, rimane tale a qualunque temperatura). Quindi facendo
variare la temperatura faccio variare il bilancio energetico e posso permettere il passaggio di stato.
Mi posso mettere in condizioni di temperatura in cui E c è molto piccolo, allo stato solido, e poi
aumentare la tempereratura e passare a stato liquido, se continuo arrivo allo stato gassoso.
<1.7 Diagramma di stato di H2 O> Prendo una mole d’acqua (circa 18g), e la sottopongo ad un
pistone che la comprime in un contenitore che la può riscaldare. Quello che succede è che per certi valori di temperatura sufficientemente bassi e per pressioni da alte a basse l’acqua la vedo solida.
Poi c’è un’altra zona del piano in cui ci sono altri valori per le coppie temperatura-pressione in cui
l’acqua è liquida, e altri in cui è gassosa. Queste tre regioni del piano sono separate da tre curve
caratterizzate da valori di coppie pressioni-temperatura in cui sono presenti entrambi gli stati (vedo contemporaneamente per esempio vapore e ghiaccio, in equilibrio). Le tre curve si intersecano
in un solo punto, P T (punto triplo, si può dimostrare dal punto di vista termodinamico), corrispondente ad una coppia precisa di valori di pressione e temperatura, che per l’acqua si valgono
P = 0,006 atm e T = 0,01 °C, in cui c’è presenza contemporanea di vapor d’acqua, acqua liquida e
48
Stati della materia
02/05/2014
ghiaccio in equilibrio. La curva tra stato liquido e gas termina nel PC (punto critico). Ad 1 atm, la
retta orizzontale interseca le curve in T f (punto di fusione normale) e in Te b (punto di ebollizione
normale). Nel caso dell’acqua, T f = 0 °C, Te b = 100 °C. Se si va in montagna, si ha una temperatura
di fusione un po’ più alta e una di ebollizione un po’ più bassa.
L’andamento teorico delle tre curve può essere approssimata dall’equazione di CLAUSIUSCLAPERYRON (indica la pendenza di quelle curve):
∆Q
dP
=
d T T ∆V
dove ∆Q è l’energia necessaria per far fare a 1 mole di sostanza un cambiamento di stato, T è la
temperatura a cui avviene, ∆V è la variazione di volume di quella mole di sostanza in quel cambiamento di stato. Per esempio, prendiamo il ghiaccio che fonde, partendo a 1 atm e -5 °C <1.8>:
la pressione rimane costante e lo si riscalda, si arriva ad un certo punto e si incontra la retta oltre
la quale l’acqua inizia a fondere e si ha contemporanea presenza di acqua e liquido se ci si ferma
sulla retta. Se si va oltre, tutto il ghiaccio fonde e si ha solo liquido. ∆Q > 0 (si sta fornendo calore),
∆V < 0, perché il ghiaccio quando fonde si contrae (e l’acqua quando congela si dilata), infatti la
pendenza della retta è negativa. Questo ha conseguenze dirette: se si mette una bottiglia piena in
congelatore la bottiglia si spacca, e spiega come i ghiacciai possano scivolare a valle oltrepassando
gli ostacoli <1.9>: nel punto di contatto tra ghiaccio e roccia la pressione aumenta, quindi l’acqua
fonde, procede e poi si ricongela dopo.
Un’altro esempio è la CO2 , che ha P T a 5,2 atm e T = −56,6 ◦C <1.10>. La pendenza della retta
tra solido e liquido è opposta a quella dell’acqua: la CO2 mentre fonde si espande, mentre l’acqua
si contrae, il motivo è legato al legame idrogeno. Le molecole d’acqua quando è liquida interagiscono tra loro e sono mobili, se si riduce la temperatura i movimenti rallentano e prevalgono le
interazioni; le molecole si dispongono lungo i dipoli, in un reticolo esagonale (vuoti all’interno)
<1.11>, che quando è liquida non c’è. Le interazioni costringono quindi le molecole ad allinearsi
e a lasciare dei vuoti, quindi occupano volume maggiore, giustificando la pendenza della curva
negativa.
49
I Gas
06/05/2014
14 I Gas
14.1 06/05/2014
N
= P a (Pascal). Altre unità di misura correnti sono il Torr (Torm2
ricelli), o mmHg. La definizione di questa unità di misura parte da un cilindro di sezione S e altezza
1mm, riempito di mercurio. La pressione che questo cilindro esercita sulla base dovuta al suo peso
è 1 Torr (o 1 mmHg). <1.1 altre unità>. Si misura con il manometro.
V Volume misurato in m 3 e in l (1m 3 = 1000l ), è il volume del recipiente che contiene il gas,
non è una caratteristica intrinseca del gas, si misura geometricamente. T Temperatura misurata in
°C, in cui 0 °C è in corrispondenza della fusione del ghiaccio a P = 1atm e 100 °C in corrispondenza
all’ebollizione dell’acqua a P = 1 atm, oppure misurata in K (Kelvin), detta anche scala assoluta (si
indica senza pallino). L’ampiezza di un grado è la stessa nelle due scale. <1.2>
<10:54>Prendiamo un cilindro con un pistone in cima, <10:55...>, e la pressione si misura attaccando un manometro in fondo. Il volume si misura con l’altezza del pistone. Il primo esprerimento fu fatto da Boyle, usando un gas abbastanza rarefatto (come l’aria o meno). Lasciando
la temperatura costante, osservò che P · V = costante. Se si vuole fare un grafico <1.4>. Poi ripetè l’esperimento cambiando la temperatura, e trovò che la relazione era sempre la stessa ma era
cambiato il valore della costante, cioè otteneva un’iperbole più lontana dagli assi.
Successivamente, Charles fece una cosa analoga con lo stesso apparato sperimentale, però
tenne costante la pressione. <come si fa 11:01> Se faccio qualcosa per cui il gas riesce a sviluppare
pressione maggiore, il pistone si alza, se lo faccio contrarre si abbassa, ma in ogni caso esercita la
stessa pressione. Trovo la legge sperimentale <1.5>:
¶
µ
t
V = V0 1 +
273,16
<10:44> P pressione misurata in
dove t è la temperatura in °C. Il grafico di questa relazione è <1.6>. <11:05 da dove viene fuori
273,16>. Questa legge è detta anche prima legge di Gay-Lussac.
Gay-Lussac fece un altro esperimento, tenendo fermo il volume (bloccando il pistone), variando la temperatura e misurando la pressione. Osservò che:
¶
µ
t
P = P0 1 +
273,16
(t è la temperatura in °C). Il grafico è <1.7> <11:07>.
<temperatura assoluta> Da qui si vede che le due scale differiscono solo per la posizione dello
0, ma non c’è nessun fattore moltiplicativo. Inoltre la pressione è una grandezza fisica definita
positiva, non può essere mai negativa. Questo significa che
T ≥0
t ≥ −273,16◦C
e questo ha una motivazione a livello molecolare. Anche dalla legge di Charles si arriva a questa
stessa conclusione.
Dunque le tre leggi sono:
PV = c 1
Tcostante
V = c2 · T
Pcostante
P = c3 · T
Vcostante
50
Teoria cinetica
08/05/2014
Se si usa la relazione centigrada, la relazione è lineare, se si usa la temperatura assoluta è direttamente proporzionale. <questo è il motivo per cui si usa 11:16?>. Si possono scrivere in una sola
equazione che le contiene tutte e tre:
PV = c · T
se si tiene T costante si ottiene la legge di Boyle, se si tiene P costante si ottiene V = Pc ·T , se si tiene
V costante si ottiene P = Vc · T . Un’espressione più precisa è:
PV = nRT
detta anche equazione di stato dei gas perfetti (ideali). Se io uso una quantità doppia di gas, tutto
ciò che misuro raddoppia, cioè dipende dalla quantità di gas. Quindi l’espressione costante deve
contere la quantità di gas <detto male 11:19>. R è il residuo della costante di proporzionalità, detta
costante dei gas, determinata sperimentalmente. R = 8,315J mol K−1 = 0,08206l atm mol−1 K−1 . PV
ha la dimensione di un energia, questo è il motivo per cui R ha quell’unità di misura. Se si scrive il
volume in litri e la pressione in atm, e li si moltiplica, si ottiene un’energia.
R non è una costante fisica universale, non lo è nemmeno il numero di Avogadro: è il numero di
entità in una mole, quindi deriva dalla definizione di mole, che si ottiene partendo da pesi atomici,
è il rapporto tra la massa dell’atomo e la massa di riferimento, indica quante volte è più pesante
del riferimento. Possiamo scegliere qualsiasi cosa come atomo di riferimento, ma in base a quella
scelta il numero di Avogadro cambia. Questo condiziona anche R, perché è definito a partire da
R
= κ è una
come definisco il numero di moli (cioè come definisco il numero di Avogadro).
NA
costante fisica universale, è la costante di Boltzman, non dipende dalla massa di riferimento.
Un gas che verifica la relazione dell’equazione di stato è un gas perfetto. Quando facendo
delle misurazioni si trovano dati che coincidono con la relazione, si dice che il gas è ideale. Le
relazioni di Gay-Lussac, Boyle e Charles erano misurate in situazioni non estreme, pressioni vicine
a quella dell’atmosfera e temperature non molto elevate o molto basse. Ripetendo gli esperimenti
con vari gas (ossigeno, ammoniaca, metano) trovarono sempre lo stesso comportamento, questo
giustifica il fatto che nell’equazione sia riferito solo il numero di moli ma non la natura chimica
dell’elemento. All’inizio del corso <11:32>. L’ipotesi di Avogadro era che se si ha un gas A con un
certo n a di moli, e un gas B con un certo n b , a stesse condizioni di pressione volume e temperatura,
<11:34>. Per il gas ... <1.8>
15 Teoria cinetica
15.1 08/05/2014
Nell’ambito della fisica statistica è stato fatto uno sforzo per dedurre la legge di stato dei gas a
partire dalla teoria microscopica <boh 11:39>. La teoria cinetica si applica alla materia in tutti gli
stati, lo stato solido è il più semplice da trattare.
La teoria cinetica è un modello di descrizione di fenomeni mascroscopici attraverso <o microscopici 11:40>. In particolare, visto che parliamo di un gas <..11:40.> (la validità cade se abbiamo
un gas fatto di solo tre particelle)
1. numero grande di particelle puntiformi
2. le particelle sono in continuo movimento (non ci sono forze dissipative), urti elastici
3. non ci sono interazioni a distanza tra le particelle
4. energia cinetica media è proporzionale a T (temperatura assoluta)
51
Teoria cinetica
08/05/2014
puntiformi: le dimensioni delle particelle sono trascurabili rispetto a quelle del recipiente
elastici: l’energia cinetica si conserva (oltre alla quantità di moto)
Nota: il punto 4 si deduce termodinamicamente in realtà.
<1.1> <11:46> questa particella ha una certa velocità, la cui componente su x è v x . <11:47> urta
e viene respinta (la parete rimane ferma perché ha massa infinita): ∆p x = mv x − (−mv x ) = 2mv x .
Questa stessa quantità è stata acquisita dalla parete nell’urto. Dopo il primo urto la particella
ritorna indietro, urta contro l’altra parete, e poi ritorna nella condizione iniziale e riurta di nuovo
la prima parete. La particella continua così andando da una parte all’altra. La particella percorre
un tratto 2l con velocità v x . v2lx è il tempo che intercorre tra un urto e l’altro sulla stessa parete. In
un tempo ∆t il numero di urti è
vx
∆t
=
∆t
2l
2l
vx
Se ogni urto cede alla parete la stessa quantità di moto e il numero di urti è quello appena
trovata, la quantità di moto complessiva ceduta in un tempo ∆t è
∆P x = 2mv x
mv x2
vx
∆t =
∆t
2l
l
<11:54>.
mv x2
∆P x
= fx =
∆t
l
f x è la forza complessiva che la parete subisce <no>. <11:56 distribuzione di velocità> non tutte
hanno la stessa componente v x sulla velocità. Se lo spazio che sto considerando non ha differenza
v2
nelle varie direzioni <11:57> quello che posso considerare è la velocità media: f x = m lx . Moltiplico
quest’espressione per il numero di particelle e trovo la forza dovuta agli urti di tutte le particelle:
Fx = m
v x2
F x mv x2
N =⇒ 2 = 3 N = P (pressione)
l
l
l
Questo fa vedere come la pressione sia un’espressione del numero di urti <11:59 non inventare>. Siccome una delle nostre ipotesi è che lo spazio sia isotopo (le particelle si muovono casualmente in tutte le direzioni), si ha che
v x2 = v 2y = v z2
(isotopo: non ci sono campi che influenzano la direzione delle particelle). Allora la pressione
anche sulle faccie perpendicolari all’asse y e all’asse z è la stessa. E si ha anche quindi che v 2 = 3v x2 .
Si può quindi riscrivere l’espressione così:
P=
m v2
V 3
1
2
2
PV = mv 2 N = E c · N
3
|2 {z } 3
è l’energia cinetica delle particelle
Infine, siccome l’energia cinetica è proporzionale alla temperatura, si ha che:
costante di Boltzmann), <1.5 12:05>
23
PV =
κT N = nN A κT
32
PV = nRT
52
3
2 κT
(κ è la
Teoria cinetica
08/05/2014
Si può quindi dedurre <12:09 la legge macroscopica da un semplice modellino meccanico>.
L’energia cinetica è definita positiva, quindi, di nuovo, la temperatura assoluta non può essere
negativa. L’interpretazione microscopica di questo limite è che se T è 0, l’energia cinetica è 0, la
massa non si annulla, è la velocità che si annulla, cioè a T=0 intuitivamente non si muove più nulla.
Quindi temperature al di sotto di questa situazione non hanno senso.
PV = nRT Se siamo nelle condizioni in cui si verificano le 4 ipotesi, questa relazione vale, e
non importa la natura chimica. Quindi se io avessi due gas, A e B, con n a e n b moli, dovrei scrivere
PV = (n a + n b )RT indipendentemente dal fatto che i due gas sono diversi.
PV = n a RT + n b RT
RT
V
RT
P b = nb
V
P = P A + PB
Pa = na
dove P a e P b sono dette pressioni parziali. Sono le pressioni che misurerei se ci fosse solo il gas A
o solo il gas B. La pressione che misuro quando sono presenti entrambi è la somma. Questa è la
Legge di Dalton.
In maniera analoga si possono definire i volumi parziali.
V=
n a RT n b RT
+
= V a + Vb
P
P
Va = n a
RT
P
RT
P
Questa è la Legge di AMAGAT. Inoltre posso fare il rapporto:
Vb = n b
Pa
=
P
na
RT
V
(n a + n b )
RT
V
=
na
= Xa
n a + nb
X A è la frazione molare di A. Similmente:
Pb
nb
=
= Xb
P
n a + nb
X B è la frazione molare di B.
Pa = X a P
Pb = X b P
Quindi se si conosce la pressione totale si possono trovare le pressioni parziali, e X a + X b = 1.
Questo è valido per una miscela gassosa di un qualunque numero di gas:
P i = X i P T OT
N
X
Xi = 1
i =1
Vi = X i VT OT
C’è un esercizio relativo su quad.
53
Teoria cinetica
09/05/2014
15.2 09/05/2014
3
κT , che è legata a v 2 ,
2
quindi ogni particella ha una velocità non necessariamente uguale a quella delle altre. La media
è quella che dà il risultato macroscopico, ma è importante andare a vedere come sono distribuite
queste velocità. Questo problema è stato affrontato da Maxwell.
Si immagini di avere un contenitore con un numero N grande di particelle puntiformi gassose.
Di queste, ce ne saranno alcune con velocità molto bassa, altre con velocità un po’ maggiore, altre
a velocità molto elevata. Il fatto che le particelle si urtino tra loro garantisce che ogni particelle
cambi in continuazione velocità, possono assumere tutti i valori possibili di velocità ammessi dal
sistema (rivedi urti e quantità di moto se non ti torna). Maxwell cercò la distribuzione di velocità
di queste particelle, e trovò l’equazione:
Una delle cose che abbiamo detto è che l’energia cinetica media è E c =
mv 2
d N = A · Ne − 2κT v 2 d v
v, v + dv
(trovo d N particelle con velocità velocità compresa tra v e v + dv) A è una costante, N è il numv 2
mero totale di particelle, all’espontente
c’è l’energia cinetica, κT dà l’ordine di grandezza
2
dell’energia termica. Il rapporto tra energia cinetica e κT dice quanto è grande (o piccola) l’energia cinetica di una particella rispetto all’energia termica ad una certa temperatura. L’equazione si
può riscrivere
mv 2
dN
= Ae − 2κT v 2 d v
N
(trovo la frazione di particelle con velocità compressa tra v e v + dv) La costante A si può ricavare
³ m ´3
2
.
(sommando tutte le frazioni si deve ottenere 1), A = 4π
2πκT
mv 2
1 dN
f (v) =
= Ae − 2κT v 2
N N
Questa funzione è fatta così <1.1>. Questo ci dice che le particelle con velocità 0 sono pochissime,
così come sono pochissime quelle che hanno velocità molto elevate. La gran parte delle particelle
hanno velocità che coincidono con il massimo di questa campana. Possiamo calcolare la velocità
media facendo:
s
Z ∞
2κT
v=
f (v)vd v =
m
0
ma a noi interessa di più v 2 :
v2
Z
=
∞
0
nota:
f (v)v 2 d v =
3κT
m
1
3
mv 2 = κT
2
2
Svolgendo i conti, si vede che la velocità media non è la velocità più probabile. Questa curva qui è
il grafico di una funzione dove A è un coefficiente, la massa è fissa, κ è una costante e c’è un parametro T che compare all’esponente e dentro A. Se con lo stesso sistema di particelle si cambia la
temperatura, si cambiano i parametri della curva. Come cambia la forma se si abbassa la temperatura? La forma qualitativamente rimane la stessa, solo più stretta con il picco più alto. Il punto
di massimo si trova spostato a velocità più basse. Cioè a temperature più basse, la distribuzione di
velocità diventa più stretta, cioè sono ancora meno di prima le particelle con velocità molto basse, e ancora molto meno quelle con velocità molto alte, si stringono intorno ai valori medi. Se fa
54
Teoria cinetica
13/05/2014
tendere T → 0 si vede che la velocità, tutta la “campana” si schiaccia sull’asse verticale, cioè tutte
hanno velocità 0. Se prendo invece una temperatura T3 > T1 il punto di massimo si sposta a velocità più grandi, la curva si abbassa e si allarga, cioè aumenta l’intervallo di valori di velocità a
disposizione delle particelle. Questa curva descrive anche liquidi e solidi, tutti i sistemi costituiti
da un grande numero di particelle.
s
s
2κT
2κT
di un gas A e la v B =
di un gas B.
Prendiamo per esempio la velocità media v A =
mA
mB
r
va
mB
Se si fa il rapporto tra le due velocità medie dei due gas, si otiene
=
. Quindi una particella
vb
mA
più leggera mediamente va più veloce. Possiamo sostituire le masse con i pesi molecolari.
s
r
va
mB
P M (B ) uma
=
=
vb
mA
P M (A) uma
cioè che
vA
=
vB
s
P M (B )
P M (A)
legge di Graham
Per esempio, prendiamo i due isotopi di elio: 3He e 4He, uno ha PM=3 e l’altro PM=4. Quando si prende l’elio gassoso, in realtà è sempre una miscela costituita da questi due. Ci si pone il
problema di separare gli isotopi: siccome l’elio è gassoso, con atomi anche piuttosto veloci perché
sono piccoli, un modo per farlo è usare la legge di Graham <1.2>. Si prende un contenitore vuoto
sufficientemente, in cui in una prima sezione ci si mette l’elio, poi si apre la valvola e gli atomi di
elio viaggiano lungo il tubo. Se ci si pone ad una certa distanza d dalla prima apertura, in media gli
d
d
, l’elio-4 ci mette t 4 = v(4)
, cioè il rapporto dei tempi
atomi di elio-3 ci mettono un tempo t 3 = v(3)
q
t 3 v(4)
è =
. Il rappoto delle velocità va con la relazione vista prima. 43 ' 1,15, quindi c’è un 16%
t 4 v(3)
di differenza nei due tempi, cioè se faccio il tubo sufficientemente lungo e l’elio-3 ci mette 100
minuti ad arrivare alla distanza d, l’elio-4 impiega 116 minuti, cioè è 16 minuti indietro rispetto
al primo atomo. Quindi se calcolo quanto tempo ci vuole per far arrivare l’isotopo più pesante,
chiudo la seconda valvola un attimo prima e tutto quello che arriva in fondo è elio-3, in media. In
realtà non si è separato tutto l’elio-3 dall’elio-4 (per via della distribuzione), però si è arricchita la
miscela a destra con l’elio-3. Questo processo è usato anche per l’uranio nelle centrali nucleari.
15.3 13/05/2014
PV = nRT
è l’equazione di stato dei gas perfetti che vale nelle 4 ipotesi della volta scorsa. Il comportamento
di un gas vero in alcune situazioni è approssimato bene (a pressione basse e temperature sufficientemente alte), mentre in altre come con l’ossigeno o l’azoto, il vapor d’acqua, in certe zone di
pressione e temperature questa relazione non è verificata.
Prendiamo una mole di gas, all’ora l’equazione diventa PV = RT . Prendiamo il rapporto Z =
PV
, detto fattore di comprimibilità, dove P è la pressione misurata con uno strumento (manomeRT
tro), V è il volume del recipiente (misurato geometricamente), T è la temperatura misurata (con un
termometro). Posso fare quindi un grafico in funzione della pressione, cioè fisso la temperatura
e vedo cosa succede <1.1>. Se il gas avesse un comportamento ideale, Z varrebbe sempre 1. In
realtà, per pressioni sufficientemente basse, il gas reale è più comprimibile di quello ideale (infatti
la curva è sotto 1), poi ad un certo punto l’andamento si inverte e il gas appare meno comprimibile
di uno ideale. Si osserva che a temperature molto elevate, un gas si comporta come un gas ideale.
55
Teoria cinetica
13/05/2014
Ci sono più modelli per giustificare questo comportamento, noi vediamo quello storicamente
più significativo e più semplice, il modello di Van der Waals. Le ipotesi che vengono riviste sono
1. dimensioni puntiformi delle molecole
2. interazioni a distanza delle molecole
P i Vi = nRT è l’equazione di stato se avessi un comportamento ideale (i sta per ideale). Se prendo
un gas reale, ho PV 6= nRT . Van der Waals ragionò così: come modificare P e V in modo che il loro
prodotto torni uguale a nRT ?
La prima correzione la ipotizzò sul volume:<1.1> immaginiamo di avere un contenitore e di avere
molecole puntiformi come se fossero punti luminosi. Quello che is vede è che le tracce di due
particelle si incontrano e da un punto in poi c’è un urto, si vedono le tracce che si incrociano. Ora
immaginiamo la stessa cosa però con particelle sferiche di dimensione finita trasparenti, con il
centro luminoso. Si seguono le tracce, di diverso si vede che ad un certo punto le due tracce cambiano direzione prima di incrociarsi, perché chi entra in contatto è la superficie della molecola,
non i due centri. Se descrivo le traiettorie possibili delle molecole a partire dai centri, <10:55> un
effetto di questo tipo è un effetto di esclusione <10:57>,<1.3> anche se arriva un po’ decentrata,
la superficie comunque urta l’altra e quindi il primo centro non può proseguire rettilineamente.
Quindi ad una molecola è escluso l’accesso ad un certo volume, che è una sfera di raggio R = diametro delle molecole. Van der Waals disse che quindi ogni sfera si sottrae volume. Se le molecole
fossero tutte sferiche, si può esprimere il volume totale escluso come 43 πd 3 · N2 (diviso due perché
se si conta il numero di particelle si contano due volte le <11:01?>, da cui si ottiene <1.4>, quindi
il volume si corregge con (V − nb). Consideriamo adesso quando le molecole interagiscono tra
loro: prendiamo il vapor d’acqua, che ha molecole polari, presenta interazioni dipolo-dipolo. Il
campo elettrico di una carica puntiforme varia con l’inverso del quadrato della distanza. Se invece si ha un dipolo, a distanza sufficientemente grande (più della molecola in sé) il campo elettrico
varia con l’inverso del cubo della distanza. <1.5> La molecola oltre alla sua velocità ha tantissime
forze di attrazione da tutte le direzioni. La media di queste interazioni è 0. Quindi la velocità rimane costante, come se non ci fossero interazioni. Quindi la particella viaggia a velocità costante,
e apparentemente il fatto che la molecola sia dipolare non ha effetto. Quando la molecola arriva
molto vicino alla parete, il sistema non ha più la stessa simmetria. Nel centro del contenitore la
media è nulla, ma quando si avvicina alla parete, lo spazio non è più isotopo, perché le particelle
sono solo da una parte. La risultante di queste interazioni è una forza diretta in direzione oppposta alla velocità, quindi nel momento in cui urta, la velocità è diminuita, quindi viene ceduta
una quantità di moto minore di quella che verrebbe ceduta se non ci fossero interazioni. Quinid
la pressione che misuro è dovuta a particelle che hanno perso un po’ di quantità di moto, cioè è
¶
µ
n2
minore di quella che avrei se il gas fosse ideale. Quindi la pressione viene corretta con P + a 2 ,
V
dove a è il coefficiente di interazione, legato al fatto che le molecole abbiano una struttura capace
di interagire a distanza, dipende da come sono fatte, moltiplicata per la concentrazione di moli
n
per volume al quadrato. Il primo
viene da <11:12>, e l’altro fattore viene dal fatto che sentono
V
un effetto di frenata dovuta alle molecole che sono presenti nel volume, <che porta al quadrato
11:12>. La forma corretta secondo Wan der Waals è quindi:
µ
¶
n2
P + a 2 (V − nb) = nRT
V
detta anche equazione di Wan der Waals, <coefficienti di ? 11:13>. Qui la natura chimica compare
in a e b, perché molecole con dipoli diversi hanno parametri a diversi <11:14>.
56
Teoria cinetica
13/05/2014
Se si svolgono i prodotti, viene fuori:
µ
¶
a 2
b
PV = nRT + nbP − n 1 − n
V
V
e se prendo una mole di volume
µ
¶
a
b
1−
PV = RT + bP −
V
V
da cui ricavo
µ
¶
P
a
b
PV
= 1+b
−
1−
Z=
RT
RT RT V
V
µ
¶
a
b
Se io mi metto in condizioni di pressione bassa, Z ' 1 −
1−
, che giustifica la comprimiRT V
V
bilità al di sotto di 1. Se invece si va a pressioni alte, il termine positivo diventa prevalente rispetto
P
a quello negativo, quindi Z ' 1 + b
.
RT
Nella nuova espressione di Z, T è al denominatore, quindi per temperature molto elevate, i termini diventano insignificanti rispetto a 1, quindi il gas comincia a comportarsi come un gas ideale.
Inoltre, a e b sono parametri che dipendono dal gas. Se ho a grande (molecole con dipoli molto intensi), per rendere trascurabile l’effetto del termine negativo occorre andare a temperature molto
alte. Quindi molecole molto polari cominciano a mostrare comportamento ideale a temperature
elevate. Se a è più piccolo, come nel caso di O2 o per l’azoto, bastano temperature più basse.
<1.6> Voglio disegnare un grafico per un gas reale. Intanto, ad alta temperatura ho un comportamento pressoché ideale, quindi ho un iperbole lontana dagli assi. Mano a meno che riduco la
temperatura, l’iperbole perde la sua “regolarità”. Se continuo ad abbassare la temperatura (posso
usare in contenitore con un pistone per fare questo grafico, misurando pressione e volume mentre
abbasso il pistone). <cosa succede 11:27>. L’evidenza fisica è questa: se riprendo l’isoterma a più
bassa, arrivo ad un certo punto in cui c’è un cambiamento di stato, cioè ho portato le molecole del
gas in condizioni tali per cui le molecole del gas hanno velocità sufficientemente basse e distanze
sufficientemente piccole da farle interagire come in un liquido, perché le interazioni prevalgono.
Il primo punto è quello in cui la prima coppia di molecole si “uniscono” diventando il primo “nucleo” liquido. Continuando a comprimere, sempre più parte del gas è diventato liquido, arrivo alla
fine del segmento in cui tutte le molecole si sono agganciate tra loro, non c’è più nessuna molecola
in fase gassosa, a questo punto tento di comprimere un liquido, quindi la curva di pressione sale
quasi in verticale, perché il liquido di fatto è incomprimibile. Ciò che succede nel tratto orizzontale è la presenza contemporanea di gas e liquido. Da qui si vede la differenza tra vapore e gas: il
vapore è gas sotto la temperatura critica. La definizione precisa di temperatura critica è temperatura per cui un gas non può essere liquefatto con una compressione isoterma. Non è vero che
sopra la Tc non si può liquefare più un gas, basta aumentare la pressione e contemporaneamente raffreddare (finendo sotto la Tc ) <11:38 ha disegnato un tratto orizzontale nel graf 1.6 in alto a
sinistra...> <1.7> <11:42>.
<11:45 domandina non scritta da copiare> <1.8> Per liquefare l’elio, devo rallentare tantissimo
le sue molecole, bastano 5 gradi sopra lo zero assoluto e l’elio non si liquefà più.
57
Soluzioni e proprietà colligative
15/05/2014
16 Soluzioni e proprietà colligative
16.1 15/05/2014
E’ esperienza comune che se si prende un bicchiere con dell’acqua e la si lascia all’aria, dopo un
tempo sufficientemente lungo l’acqua evapora. Il motivo è che le molecole d’acqua hanno una
distribuzione di velocità come quella teorizzata da Maxwell, c’è una coda di molecole che hanno
una velocità alta, con energia cinetica significativamente più elevata dell’energia potenziale dei legami dipolo-dipolo e idrogeno, quindi lasciano il liquido. Se il liquido è tenuto in ambiente aperto,
le molecole vengono “rimosse”. Le molecole rimanenti si ridistribuiscono e continuano ad avere
una distribuzione di Maxwell, quindi ci saranno ancora molecole ad energia elevata. Propagando
questo comportamento si ha l’evaporazione di tutto il bicchiere d’acqua.
Mettiamo l’acqua in un contenitore chiuso vuoto. <1.1> Sulla superficie dell’acqua ci saranno
alcune molecole con energia cinetica sufficiente da passare a stato gassoso. Piano piano la parte
vuota continua ad arricchirsi di molecole d’acqua gassosa, che si distribuiscono secondo la distribuzione di velocità di Maxwell a loro volta. Quindi alcune di esse, quando cominciano ad essere in
numero abbastanza grande, avranno velocità molto basse, quando urtano la superficie con energia cinetica bassa (confrontabile o minore alle energie potenziali nel liquido) vengono ricatturate
dal liquido. Man mano che osservo questo fenomeno vedo molecole che da liquido passano a
vapore, con una certa velocità, e delle molecole che da fase vapore ritornano in fase liquida, con
velocità diverse. All’inizio la velocità di evaporazione è maggiore, quando il numero di molecole
in fase vapore aumenta aumenta anche il numero di quelle che tornano in fase liquida, quindi
aumenta la probabilità di questo fenomeno e il numero di urti, quindi il numeo di molecole che
tornano liquide. Allora la velocità di condensazione da 0 piano piano aumenta fino ad arrivare
al punto in cui le due velocità si bilanciano. A questo punto vedo che la quantità di molecole in
fase vapore non cambia più, rimane costante. Non sono sempre le stesse molecole, ma il numero
è lo stesso. E’ un equilibrio dinamico (liquido-vapore). <1.2> La pressione nel tratto costante è
determinata dalle molecole in fase vapore. Quando le molecole in fase vapore hanno la velocità di
condensazione uguale alla velocità di evaporazione del liquido, il sistema è in equilibrio. ,La velocità di condensazione, cioè molecole per unità di tempo che tornano in liquido è proporzionale la
numero di urti che fanno sulla superificie del liquido, e anche a quello sulle pareti. Siccome il numero di urti è proporzionale a questa velocità, e questa velocità rimane fissa all’equilibrio (uguale
a quella di evaporazione), il numero di urti sulla superficie e le pareti è costante (nell’unità di
tempo), e siccome il numero di urti sulle pareti è legato alla pressione, la pressione è costante.
<1.3> Supponiamo ora che il recipiente abbia un pistone per variare il volume. Se diminuisco
il volume, il numero di urti in media sulle pareti aumenta, cioè aumenta la pressione, e quindi
la probabilità che le molecole urtando la superficie vengano ricatturate dal liquido. Quindi sto
creando un aumento della velocità di condensazione, quindi il sistema si sbilancia. Il numero di
molecole in fase vapore diminuisce, il numero di urti pure, fino a che la velocità di condensazione
non ritorna uguale all’altra, con una pressione identica a prima, quindi la stessa pressione, ma
meno molecole in volume più piccolo. Accade il viceversa se si aumenta il volume: diminuisce
la frequenza degli urti, prevale l’evaporazione, le molecole in fase vapore aumentano, aumenta la
frequenza degli urti, finché la velocità di condensazione non ritorna uguale a quella di evaporazione, con la stessa pressione di prima ma più molecole. Questo spiega perché la pressione rimane
costante in <1.2 diagramma di Andrews>. L’unica cosa da cui dipende il valore della pressione è
la temperatura, cioè spostandosi su un altra curva del diagramma, in qui l’equilibrio si instaura
a pressione maggiore. La pressione esercitata in fase vapore si chiama pressione (o tensione) di
vapore: è la pressione che la fase vapore esercita sul proprio liquido quando sono all’equilibrio
(quindi devono essere presenti entrambi). Ogni liquido ha la sua pressione di vapore, e dipende
58
Soluzioni e proprietà colligative
15/05/2014
solo dalla temperatura.
dP
Q
=
d T ∆V · T
E’ la legge di Clausius-Clapeyron, che descrive in maniera approssimata l’andamento dei tre tratti
di curve che delimitano gli equilibri fra le varie fasi. In questo momento, stiamo parlando dell’equilibrio liquido-vapore. Se parliamo dell’acqua ci stiamo riferendo a <1.4>. Nel diagramma di
stato, la curva tra queste due fasi è l’insieme dei punti in cui ad ogni temperatura trovo la pressione di equilibrio, cioè la pressione di vapore di equilibrio alla data temperatura. Per descrivere in
maniera un po’ più precisa quel tratto si riscrivere l’equazione di Clausius-Clapeyron:
Q ev
dP
=
d T (Vv ap − Vl )T
Vv ap ÀVl
≈
Q ev
Q ev * Q ev
≈
=
P
RT
Vv ap T
RT 2
·T
P
dove Q ev è la quantità di calore che serve per far passare 1 mol di H2 O da liquido a vapore (è
un’energia molare). T è la temperatura assoluta, ∆V è la variazione di volume che osservo quando
una mole di acqua passa da liquido a vapore. Il volume molare dell’acqua lo posso definire bene:
1 mol di H2 O è 18 g, la sua densità è 1 g cm−3 , quindi il volume di una mole d’acqua è 18 cm3 . Una
mole d’acqua in fase vapore non la posso definire a priori perché essendo in fase vapore non ha un
volume proprio, ha il volume del recipiente. Qui si fa l’ipotesi che il recipiente sia sufficientemente
grande da poter trascurare il volume dell’acqua liquida (in equilibrio). Quindi ∆V è la differenza
tra il volume finale (vapore) e il volume iniziale (acqua liquida) di una mole. Quindi Vl = 18 cm3 .
Se ci mettiamo nella condizione in cui Vr eci pi ent e = Vv ap À Vl (per esempio 50 l), allora Vl si può
trascurare e si può fare la prima approssimazione. Poi se ne fa un’altra (indicata con *): Vv ap ≈ RT
P
lo approssimo con l’equazione di stato dei gas perfetti con n = 1. Lo sto considerando quindi un
gas ideale, come se fossi lontano dalla liquefazione, invece sono sull’equilibrio tra liquido e vapore.
L’andamento è pressoché corretto, ma numericamente approssimato. Si ottiene quindi:
d P Q ev d T
=
P
R T2
Si integra:
Q ev
Q ev 1
+ costante =⇒ P = Ae − RT
R T
dove A è una costante, è l’andamento approssimato della pressione di vapore in base alla temperatura. A è determinata da una misura della pressione di vapore ad una certa temperatura, ed
è valida per tutte le temperatura tra punto triplo e il punto critico (cioè dove esiste l’equilibrio
liquido-vapore).
<1.6> Bisogna ridefinire alcuni elementi: il punto in cui la curva di equilibrio liquido-vapore
interseca il valore di pressione esterna al sistema, cioè l’equilibrio avviene al valore di pressione
esterno al sistema (quindi la pressione del vapore dentro è identica a quella esterna), individua la
Tebol l i zi one del liquido. Questo significa che le molecole del liquido sono in grado di formare la fase vapore al suo interno, non hanno bisogno di arrivare in superficie, quindi si vede la formazione
delle bolle. Quando la pressione esterna vale 1 atm, la Teb è detta temperatura di ebollizione normale. Quella dell’acqua è 100 ◦C (che si individua dall’intersezione della curva di equilibrio con 1
atm di tensione di vapore). Riformulando: alla temperatura di ebollizione normale, la tensione di
vapore dell’acqua è 1 atm.
Prendiamo ora un liquido differente, come l’alcool etilico, che è fatto così: CH3 CH2 OH. E’ sicuramento meno polare dell’acqua (ha un solo legame OH), fa legami a idrogeno (il legame OH
ne prevede l’esistenza), le interazioni sono meno forti perché c’è una grossa parte di molecola che
ln P = −
59
Soluzioni e proprietà colligative
15/05/2014
ostacola l’instaurazione di interazioni. Le interazioni sono dipolo-dipolo più deboli di quelle dell’acqua. Questo significa che basta meno energia per far passare l’alcool da liquido a fase vapore,
bisogna rompere legami meno intensi, cioè il Q v ap per l’alcool etilico è più piccolo rispetto a quello dell’acqua, infatti il suo grafico <1.6> è spostato più su. Questo vuol dire che l Teb (CH3 CH2 OH)
è più bassa, oppure che a parità di temperatura, l’alcool ha tensione di vapore più alta, cioè è in
grado di mandare in fase vapore un numero di molecole maggiore dell’acqua (per raggiungere il
suo equilibrio). Un liquido con tensione di vapore più alto rispetto ad un altro è detto più volatile.
Se un liquido è il doppio più volatile di un altro, allora un liquido ha tensione di vapore doppia
rispetto all’altro alla stessa temperatura.
Molte reazioni chimiche avvengono in soluzioni acquose, e le soluzioni sono miscele (dipendono dalle quantità che si mescolano insieme). Le proprietà del liquido cambiano e influenzano
le reazioni, dobbiamo quindi vedere cosa succede quando facciamo una miscela liquida. In generale si distinguono un solvente e i soluti. Ciò che si scioglie si chiama soluto (o soluti se sono
più di uno). Il solvente è, fra le varie sostanze mescolate, quella che ha la stessa fase della soluzione. Per esempio, quando si scioglie il sale nell’acqua, la soluzione finale è liquida, come lo era
l’acqua iniziale, cioè la fase dell’acqua è identica, mentre il solido non c’è più, ha cambiato fase.
Allora l’acqua è il solvente, gli altri elementi sono i soluti. Stessa cosa quando si scioglie la CO2
nell’acqua, l’acqua rimane nella stessa fase, quindi è il solvente, allora CO2 è il soluto. Quando
si mescolano due liquidi, per esempio acqua e alcool, ci sono due sostanze con la stessa fase (liquida) della soluzione finale. In questo caso si dice solvente quello che è contenuto in maggior
quantità fra i due (se sono uguali si sceglie uno dei due). Siccome le soluzioni sono miscele, la
prima cosa da conoscere è la sua composizione (quanto c’è di ogni componente). Introduciamo
alcuni modi diversi per misurarla:
frazione molare:
Xi =
ni
n t ot
frazione ponderale:
Wi =
mi
m t ot
g/l:
mi
Vsol uzi one
Molarità:
Mi =
Molalità:
mi =
=
ni
Vsol uzi one
mol
l
ni
· 1000 mol
=
g
massa di solvente in g
ni
mol
massa di solvente in kg kg
(i è l’i-esimo componente)
Per i gas avevamo usato le frazioni molari in una miscela omogenea. Anche una soluzione è
una miscela omogenea, percui la si può definire anche per le soluzioni, dove si prendono il numero
di moli di solvente sull numero di moli totali di solvente e soluti disciolti. Esistono poi la frazione
ponderale, il g/l, usato spesso in pratica. Altri due meno usuali ma che a noi ci interessano di più
sono la Molarità (indicata con M) e la Molalità (indicata con m). Osservazione: la frazione molare,
la frazione ponderale e la molalità, sono tre unità per esprimere la composizione di una miscela
che non dipendono dalla temperatura. I g/l e la molarità dipendono dalla temperatura, perché
il volume cambia per dilatazione. Quindi quando si esprime una concentrazione con questi due
modi andrebbe espressa anche la temperatura. Di solito si prende la temperatura a 25 ◦C. Un altro
modo per indicare la molarità è con [ ].
60
Soluzioni e proprietà colligative
15/05/2014
Riprendiamo l’esempio dell’acqua e del sale. <1.7> Se si mette una quantità abbastanza piccola di sale, esso si scioglie completamente. Se si continua ad aggiungerlo, si osserva che ad un
certo punto ne rimane una parte solida sul fondo. Questa è una soluzione satura, cioè nel solvente è sciolta la massima quantità di soluto che ci può stare. Se si prova a metterne un po’ di
più, quella quantità non si scioglie e rimane insoluto. La solubilità indica la massima quantità di
soluto che si può sciogliere nel solvente, la si determina sperimentalmente aggiungendo piccole
quantità successive finché da una certa quantità in poi si vede che si forma il solido sul fondo indisciolto. Sotto il valore è ancora solubile, sopra no, si ha un precipitato. La solubilità è la massima
concentrazione, quindi esprimibile in una delle unità viste.
La solubilità dipende dalla temperatura. Prendiamo l’esempio dell’acqua e il sale: all’inizio
abbiamo l’acqua liquida e NaCl solido ionico cristallino separati. Questo sistema ha una certa
energia, dato dalle interazioni delle molecole dell’acqua tra loro e dalle interazioni del reticolo
ionico. Le interazioni ione-dipolo (la più forte tra quelle deboli) sono quelle responsabili del fatto
che un reticolo cristallino ionico può passare in soluzione in un solvente polare (come l’acqua).
Nel passare da questi due separati a NaClsol uzi one (che è uno stato fisico completamente differente
<1.8>), con ioni Na+ e Cl– separati e staccati dal reticolo, intorno a ciascuno dei quali c’è un numero
di molecole d’acqua (di dipoli) che formano la struttura di idratazione (stato che si indica con il
pedice (aq)), ho una variazione di energia che è:
(
< 0 S↓ se T↑
∆E = ∆E r et + ∆E i d r at azi one
| {z } |
{z
}
> 0 S↑ se T↑
>0
<0
dove ∆E r et è l’energia che serve staccare ioni dal reticolo, per vincere l’attrazione che ogni molecola ha nel reticolo cristallino, che è grande, ∆E i d r at azi one è l’energia legata al fatto che ogni
ione isolato coordina intorno a sé dipoli dell’acqua e crea la struttura di idratazione. La prima è
positiva, la seconda è negativa perché le interazioni dei dipoli con lo ione abbassano l’energia,
rendono il sistema più stabile, viene restituita energia all’esterno. Il segno complessivo dipende
dalla natura chimica della specie che sto considerando, dalle interazioni che fanno tra loro, da come sono gli ioni... Potrei quindi avere un risultato > 0 o < 0, associato al passaggio NaCl e H2 O
da soli a NaClsoluzione . Se l’energia è negativa, cioè lo stato finale è più stabile (cioè la soluzione è
più stabile), il sistema sta restituendo energia (sotto forma termica), si vede che si riscalda. Se si
aumenta la temperatura si inibisce il fenomeno, si fornisce calore ma già la reazione fornirebbe
calore. Quindi se ∆E < 0 la solubilità diminuisce se la temperatura aumenta e viceversa. Un esempio di questo caso è l’acqua gassata, in cui c’è CO2 gassoso, H2 O e CO2(aq) in acqua. La CO2(g) non
ha praticamente interazione tra sé, è apolare, mentre CO2(aq) presenta interazioni dipoli-dipoli,
sicuramente è un sistema a un’energia più bassa. Un gas che si scioglie nell’acqua tipicamente ha
un ∆E < 0. Se invece il ∆E > 0, cioè nel passaggio da componenti separati a soluzione, trovo che
ho bisogno di fornire energia per facilitare il passaggio in soluzione, se aumento la temperatura
facilito il processo, cioè aumento la solubilità (è il caso del NaCl).
Ci sono casi che non si verificano quasi mai, quelli in cui ∆E = 0 (si chiama entalpia di mescolamento, è il calore coinvolto nel mescolamento). Quando ∆E = 0, c’è un bilanciamento preciso
tra quello che si deve consumare e quello che si riguadagna con le strutture di idratazione. Si parla
di soluzioni ideali. Il primo ragionamento che ci si può fare è quello della legge di RAOULT. Si
applica a soluzioni ideali:
P i = X i P i0
immaginiamo di avere una soluzione liquida con un certo numero di componenti: i è il componente, X i è la sua frazione molare in fase liquida (in soluzione), P i0 è la sua tensione di vapore (se ce
l’ha) quando è puro (indicato dallo 0). Esempio: miscela acqua-alcool, si ha una frazione molare
0
di acqua e una di alcool. Se considero l’acqua, X ac è la sua frazione molare, P ac
è la sua tensione di
61
Soluzioni e proprietà colligative
15/05/2014
vapore come acqua pura (a quella temperatura), P ac è la pressione parziale di acqua in fase vapore
in equilibrio con la miscela.
<1.10> Questo è un liquido fatto di due componenti, acqua e alcool. In basso abbiamo una
frazione molare di A e una di B (la composizione della soluzione). A ha una sua tensione di vapore
come liquido puro ad una certa temperatura, come anche il liquido B. Questa miscela di liquidi
sarà in equilibrio con una fase vapore. Ciascuno dei due manderà delle molecole in fase vapore.
Quindi anche la fase vapore è una miscela (gassosa), fatta di molecole di A e di B, in cui avrò una
pressione parziale di A è una pressione parziale di B (in fase vapore). La legge di Rault lega la
composizione della fase vapore con quella liquida. OCCHIO: questa legge non va confusa con
P i = X i P T , questa riguarda la pressione parziale in fase gassosa omogenea.
Facciamo un esempio: prendiamo di nuovo una miscela di liquidi A e B. Ho X A , X B che è la
composizione liquida. Applico la legge di Raoult e ottengo P A = X A P A0 che è la pressione parziale
di A e P B = X B P B0 che è la pressione parziale di B. Possiamo scrivere P A = X A P A0 = (1 − X B )P A0 . La
pressione totale è P = P A + P B .
P
P B0
P to
t
PB
P A0
PA
1
XB
Supponiamo di sapere la composizione della fase liquida. Quale è la composizione della fase
vapore?
Chiamo X A0 la frazione molare di A in fase vapore e X B0 la frazione di B in fase vapore, che sono
incognite. Posso scrivere (per definizione di frazione molare, poi con la legge di Raoult):
X A0
X A P A0
PA
=
=
PT
X A P A0 + X B P B0
X B0
X B P B0
PB
=
=
PT
X A P A0 + X B P B0
Supponiamo che X A = X B = 0,5 mol e che P A0 = 2P B0 (A è il doppio più volatile di B). La composi2P B0
2
1
0
=
,
e
X
=
. Ho ottenuto una fase vapore più ricca
zione della fase vapore viene X A0 =
B
3
2P B0 + P B0 3
di A rispetto a B, cioè più ricca della specie più volatile.
Supponiamo ora di aver raggiunto l’equilibrio, con fase vapore fatta come prima e una liquida
come prima, e di separare la fase vapore dal liquido, rimetterla in un contenitore. Essa ricondenserà in liquido. Supponiamo di riuscire a condensare tutta la fase vapore, si ottiene un liquido che
è una miscela di A e B (liquida) con la stessa composizione del gas (X A = 23 e X B = 13 ). Prova a calcolare le moli di composizione del vapore che si riforma. Si osserva che X A0 è diventato più grande
e X B0 più piccolo. Se si immagina di ripetere il procedimento, X A0 → 1 e X B → 0, si ottiene un gas
che è solo A e il liquido solo B: è la teoria alla base della distillazione.
62
Soluzioni e proprietà colligative
16/05/2014
16.2 16/05/2014
Vediamo adesso miscele liquide dove uno dei componenti non è volatile (per esempio un solido in
acqua). Abbiamo due componenti, A e B, A è il solvente, dotato di P A0 e B solido (cioè non volatile),
cioè che P B0 ¿ P A0 seguendo il modello di Maxwell. La frazione di molecole con energia sufficiente
a passare dallo stato solido a gas è praticamente nulla. Quindi il componente B ha una tensione di vapore pressoché nulla o comunque molto più piccola di quella del liquido. La pressione
in equilibrio è P = P A + P B = X A P A0 + X B P B0 (applico la legge di Dalton) che diventa praticamente
X A P A0 . Quando dentro A sciolgo un soluto non volatile, nella fase vapore sono presenti solo molecole di A, però non misuro la pressione di A puro, ma misuro quella di A per la sua frazione molare.
X A P A0 = (1 − X B )P A0 .
P = P A0 − X B P A0
∆P = P A0 − P = X B P A0
Se sciolgo qualcosa dentro A, vedo un suo abbassamento di tensione di vapore, con una variazione
direttamente proporzionale al contenuto di soluto.
XB =
nB
n A + nB
per definizione
ma noi abbiamo qui che n A ¿ n A . Ma cosa significa effettivamente scrivere questa cosa? Per
A
(questo significa accettare una precisione
utilità pratica utilizzeremo un criterio: x ¿ A se x ≤
50
del 2%). Quindi, se n B è 50 volte minore di A, allora lo posso trascurare. Quindi si può scrivere
XB =
La molalità è m B =
nB
nB
nB
nB
'
=
=
P M (A)
M A (g )
n A + nB n A
MA
P M (A)
nB
nB
mB
· 1000 →
=
. Allora la frazione molare di B diventa:
MA
M A 1000
X B ' mB
P M (A)
1000
∆P = X B P A0 = m B
P A0 · P M (A)
1000
∆P = k a · m B
in cui il delta dipende da una costante per la molalità del soluto <1.1> Sciolgo in A il soluto B, e os∗
servo che la pressione di vapore si riduce <12:52>. Teb
è la temperatura di ebollizione della soluzione, cioè vedo che aggiungendo soluto la temperatura di ebollizione aumenta. Finché gli intervalli
di variazione sono piccoli, posso considerare rettilinei i tratti di curva. Quindi mi posso disegnare
il triangolo <1.2>, dove α è fisso, il che vuol dire che ∆Teb ∝ ∆P , cioè che ∆T = k eb m B , dove k eb
è la costante ebullioscopica (per l’acqua vale k eb (H2 O) = 0,513◦C kg mol−1 . Quindi se si aggiunge
<12:58> bolle a 100.113°. Succedono cose analoghe per ogni passaggio di stato. Anche nel passaggio tra liquido e solido si ha un abbassamento della curva. L’altro fenomeno che si verifica è che
∆Tcr = k cr m B , dove k cr è detta costante crioscopica (per l’acqua vale k cr (H2 O) = 1,86◦C kg mol−1 .
Quindi quando si scioglie qualcosa nell’acqua in un liquido di non volatile, la temperatura di
ebollizione aumenta (innalzamento ebullioscopico) e la temperatura di congelamento si abbassa
(abbassamento crioscopico). <Come si getta il sale in terra quando la strada è ghiacciata? 13:04>
63
Soluzioni e proprietà colligative
20/05/2014
Questi fenomeni sono detti proprietà colligative: <13:25> la variazione della temperatura di
ebollizione, di congelamento, pressione osmotica (prossima volta).
C’è un’osservazione da fare: consideriamo CO2(g) −−→ CO2(aq) . I dipoli dell’acqua fanno un
dipolo-dipolo indotto e le molecole di CO2 rimangono nell’acqua <13:27 a corsa>. Prendiamo
C6 H12 O6(s) −−→ C6 H12 O6(aq) (saccarosio) quando la si mette nell’acqua riesce a coordinare i dipoli
dell’acqua e si scioglie. Specie chimiche come queste due sono dette non elettroliti, perché quando passano da stato solido a liquido non cambiano forma della loro molecola, cioè rimangono
intatte. Quando si calcola la molalità o la frazione molare, le moli sono relative a molecole fatte in
questo modo. Ci sono specie invece come NaCl(s) −−→ Na+(aq) + Cl–(aq) passa in soluzione attraverso la distruzione del reticolo cristallino e rimane in soluzione dell’acqua <così 13:30>. Quindi la
molecola cambia completamente in soluzione in acqua. Questi si chiamano elettroliti, cioè specie
che quando si sciolgono in acqua danno origine a ioni che componevano la molecola. Gli elettroliti si dividono in forti (NaCl) e deboli. NaCl è detto forti perché si dissocia in ioni, non c’è una
parte che riesce a stare in soluzione in forma di reticolo cristallino, altrimenti sarebbe solido e non
sciolto. Un elettrolita debole come HI(g) −−→ HI(aq) lo si può vedere in due fasi: si vedono molecole
di HI intere (è polare) ma una volta che si trova in soluzione si decompone come fa un elettrolita,
ma non tutto, solo una parte degli HI si decompone come NaCl, una parte rimane come molecola
−−
*
intera, e sono presenti tutti e tre nella soluzione: HI(g) −−→ HI(aq) −
)
− H+(aq) I–(aq)
La grandezza α, detta grado di dissociazione, è definito come
α=
numero di moli dissociate
numero di moli iniziale
Se si prendono i tre casi di prima, abbiamo un numero di moli iniziali di CO2 , rimane tutta indissociata. α = 0 per un non elettrolita. Prendiamo NaCl, di quelle moli si dissociano tutte: α = 1 per
un elettrolita forte. Se prendiamo HI, abbiamo 0 ≤ α ≤ 1, cioè un elettrolita debole, una parte si
dissocia e una parte rimane in soluzione indissociato.
16.3 20/05/2014
inizio
fine
Ap Bq
n0
n 0 − αn 0
−−−
*
)
−
pAq+
—
pαn 0
+
qBp–
—
qαn 0
Completiamo il discorso sulle proprietà colligative: consideriamo un composto generico, inizio con n 0 del primo composto, che non ha ancora fatto nessuna reazione. Quando la reazione è avvenua, ho che parte delle moli iniziali si sono dissociate. Uso il grado di dissociazione
dissociate
= moli dissociate
. Quindi avrò pαn 0 del primo prodotto e qαn 0 del secondo, n 0 −αn 0
α = moli
moli iniziali
n0
di reagente. La molalità è la molalità di tutto ciò che non è solvente, perché la devo calcolare guardando quante moli di soluto ci sono in 1000 g di solvente, dove il soluto è sia pA sia qB, che hanno
struttura di idratazione diverse, dimensioni diverse e interazioni diverse con l’acqua, si comportano come se fossero due soluti diversi presenti nella stessa soluzione, e anche il reagente se è in
soluzione con delle moli si comporta in maniera diversa dai due precedenti, quindi per calcolare
m, ho bisogno di sapere n t ot di moli di soluto:
n t ot = (n 0 − αn 0 ) + pαn 0 + qαn 0 = (n 0 − αn 0 ) + ναn 0
dove ν = p + q.
n t ot = n 0 [1 − α + αν] = n 0 [1 + α(ν − 1)]
64
Soluzioni e proprietà colligative
20/05/2014
il coefficiente è detto binomio di van ‘t Hoff ν è il numero totale di ioni che vengono fuori dalla
dissociazione. Se α = 0 =⇒ n t ot = n 0 (è il caso di un non elettrolita, cioè che non si dissocia in ioni,
tute le ioni di soluto sono uguali a quelle di sostanza iniziale). Se si ha α = 1, si ottiene solo prodotti
e nessuna mole di reagente di partenza rimasto (è un elettrolita forte) α = 1 =⇒ n t ot = νn 0 . Se
devo calcolare la molalità qui:
CO2(g) −−→ CO2(aq)
α = 0 n t ot = n 0
NaCl(s) −−→ Na+ + Cl−
α = 1ν = 2 n t ot = 2n 0
Quindi una mole di NaCl messa in un litro acqua per sapere che effetto ha sull’abbassamento
crioscopico o sull’innalzamento ebullioscopico produce un effetto pari a due moli di soluto, una
mole di uno ione e una mole dell’altro. Prendiamo
+
−
−−
*
HF(aq) −
)
− H(aq) + F(aq)
0 < α < 1 supponiamo α = 0,1 ν = 2 n t ot = n 0 [1 + 0,1(2 − 1)] = 1,1n 0
Quindi la sua efficacia in acqua è 1,1 volte di più.
Rimane ora la pressione osmotica: supponiamo di prendere una campana di vetro al cui interno si mettono due contenitori (bicchieri), in uno ci si mette H2 O, nell’altro H2 O + Zucchero (o
sale). Si mettono nello stesso ambiente. Se ci fosse stata solo l’acqua, se all’inizio non c’era acqua
in fase vapore nella campana, l’acqua comincia a creare la sua tensione vapore per arrivare all’equilibrio, quindi dopo un po’ di tempo si trova che nel bicchiere di acqua un po’ di liquido se ne
0
è andato, e il <vapore> eserciterà una pressione di vapore P H
O . La stessa cosa fa il liquido nel se2
0
condo bicchiere, che seguirà la legge di Raoult: P H2 O = X H2 O P H
. Cosa succede se li ho insieme?
2O
<1.1>Il primo bicchiere manderà molecole in fase vapore per raggiungere la sua tensione vapore, il
secondo bicchiere ne manda meno (legge di Raoult), quindi succede che il secondo bicchiere vede
un eccesso di molecole d’acqua in fase vapore, le riporta in fase liquida, sottraendola all’ambiente
0
della campana. Allora il primo bicchiere ne manda delle altre, perché vuole avere P H
. Quello che
2O
si vede dopo un po’ di tempo è che nel secondo bicchiere si ha acqua zuccherata un po’ più diluita. Questo processo non si ferma fino a che non viene consumata tutta l’acqua, la concentrazione
nella soluzione diminuisce, ma resta comunque una soluzione, in cui X H2 O < 1 sempre. Questo è
il meccanismo alla base della pressione osmotica.
<1.2> Il classico esperimento che si fa è quello con il tubo a U, dove nel punto più basso c’è
un diaframma, che è una membrana compatta non chiusa, fatta di tanti piccoli canali (pori) la cui
dimensione è a livello molecolare, tale per cui ci può passare una molecola d’acqua in entrambi i
versi (è piccola), ma non ci può passare per esempio uno ione Na+(aq per via della sua struttura di
idratazione, come non ci passa Cl–(aq) , oppure CO2 . Immaginiamo di mettere nei due tubi la stessa
acqua e acqua e zucchero di prima, nella stessa quantità (arrivando allo steso livello di altezza).
Le molecole di acqua si muovono in continuazione, ci sono delle molecole d’acqua che urtano la
parete della membrana, quindi in un secondo un certo numero di molecole urta la membrana.
Fra quelle ce ne è una frazione che urtano proprio dove ci sono i pori, quindi passano. La stessa
cosa accade dall’altro lato, dove ci sono molecole d’acqua con la stessa velocità media (siamo alla
stessa temperatura), per cui le molecole d’acqua possono attraversare quando trovano il foro. Però
il numero di urti che ho sul lato sinistro è dovuto solo alle molecole d’acqua, sul lato destro, che
in media è lo stesso, è dovuto a acqua e soluto, cioè non tutti gli urti sono di molecole d’acqua,
una parte proviene dai soluti, che però non passano. Quindi se sul lato sinistro su 100 urti per
65
Soluzioni e proprietà colligative
20/05/2014
unità di tempo, 100 passano, sul lato destro su 100 urti passano solo quelli che sono di acqua
(mettiamo 80). Quindi nell’unità di tempo ho 100 che vanno da sinistra a destra, 80 da destra a
sinistra, flusso netto di 20 da sinistra a destra. Dopo un tempo sufficiente si vede il livello sinistro
che scende e quello destro che sale. La differenza rispetto all’altro esperimento è che il primo non
finisce mai (il secondo bicchiere non arriva a diluizione infinita), mentre il secondo trova il punto
di equilibrio. La differenza di pressione idrostatica si manifesta sulla membrana con un aumento
della frequenza degli urti, quindi le 80 molecole d’acqua a sinistra ora urtano più frequentemente,
si raggiunge un punto in cui l’altezza a sinistra è tale per cui il numero di molecole d’acqua che
passano in un verso e nell’altro è uguale, e dalla parte dove la soluzione è più diluita la frequenza
è maggiore per via della pressione maggiore. Spesso la differenza di altezza è usata proprio per
misurare la pressione osmotica. E’ definita come Π = ρg h(ρ è la densità). La pressione osmotica si
osserva quando dopo un periodo sufficientemente lungo con le mani nell’acqua di mare. La pelle
è una membrana semipermeabile, quando siamo nell’acqua siamo in una soluzione acquosa con
una certa concentrazione di sali, e anche i fluidi cellulari o extracellulari hanno una concetrazione,
più bassa. Quindi i fluidi all’interno della pelle tendono a diluire la soluzione esterna, sparando
acqua fuori della pelle.
La definizione teorica di pressione osmotica è
Π = M RT
dove M è molarità totale di tutte le specie presenti
Si può riscrivere:
n t ot
RT
V
(somiglia un po’ all’equazione dei gas perfetti)
Π=
ΠV = n t ot RT
C’è un esercizino su quad.
Altro esempio: nel corpo umano ci sono circa 7 l di sangue, che è fatto da più componenti.
I globuli rossi hanno dimensioni nell’ordine delle decine di µm. Nei boccioni delle flebo, se si
prende la soluzione fisiologica normale, ci si trova un numero molto vicino a 320 mOsM (milli-OsMolare). Quell’Os dice che conta il numero totali di moli in soluzione. Per esempio: NaCl, 0,1 mol
in 1l. M (NaCl) = 0,1 M. OsM (N aC l ) = 0,2 M. E’ cioè la concentrazione in mol l−1 di tutto ciò che
produce NaCl in soluzione. Siccome produce due specie (Na+ e Cl– ), la concentrazione efficace
è il doppio, e per specificare questo si usa la dicitura os-molarità. Sul boccione c’è scritta questa
unità, quindi questo significa che c’è stata disciolta per esempio una quantità di NaCl per raggiungere quella concentrazione, ed è la stessa concentrazione che c’è dentro le cellule, come quelle dei
globuli rossi. Siccome le membrane dei globuli rossi sono semipremeabili, se il globulo rosso vede
una soluzione che ha la stessa concentrazione osmolare presente al suo interno, è in equilibrio, se
invece fuori per qualche motivo la concentrazione dovesse diminuire perché si diluisce l’ambiente, allora si avrebbe un flusso netto di molecole d’acqua che entrano nel globulo rosso, che rigonfia
<1.3>. Se la differenza di concentrazione tra dentro e fuori è sufficientemente grande, la quantità
d’acqua che entra è tale da spaccare il globulo rosso che libera tutto quello che ha dentro e muore.
Effettivamente, basta iniettare 40cc di acqua distillata nel sangue per spaccare la maggior parte
dei globuli rossi, ed è il motivo per cui non è bene bere l’acqua distillata.
66
Cenni di termochimica
20/05/2014
17 Cenni di termochimica
17.1 20/05/2014
Quando si scrive una reazione generica
∆HR
aA + bB −−−→ cC + dD
Se l’enegia di stato dei prodotti è più alto di quella dei reagenti, a questa reazione è associata una
variazione d’energia positiva, è detta endotermica, cioè devo fornire un’energia per passare da
reagenti a prodotti. Se invece <11:32> la differenza di energia è liberata verso l’esterno e si ha una
reazione esotermica. In termodinamica c’è una grandezza precisa indicata per indicare l’energia
scambiata nelle reazioni chimiche, chiamata Entalpia, indicata con H: è il calore associato ad una
reazione chimica che avviene a pressione costante. ∆H può essere positivo o negativo, il |∆H | è
detto calore di reazione. Supponiamo di avere questa reazione
C(s) + O2g −−→ CO2g
∆H = −394kJ mol−1
25 ◦C 1at m
<11:36> quindi quando si fa reagire <11:36>. Supponiamo di averne un’altra:
H2g + 21 O2g −−→ H2 Og
∆H = −286kJ mol−1
25 ◦C 1at m
anche questa è una combustione, questa fornisce <11:38 energia>. Prendiamo ora:
1
1
2 O2g + 2 N2g
−−→ NOg
∆H = +90,4kJ mol−1
25 ◦C 1at m
questa reazione è endoterma <11:39>.
La quantità di energia necessaria è proporzionale alla mole che reagisce:
O2 + N2 −−→ 2 NO
∆H = 2 · (90,4) = 180,8kJ mol−1
25 ◦C 1at m
Se è scritta così, mi produce il doppio di energia <invento 11:41>. Questa è l’energia associata alla
produzione di 2 moli di NO. Quindi ∆H dipende dai coefficienti stechiometrici, se si moltiplicano
i coefficienti stechiometrici per un numero, così deve essere anche ∆H . Questa discorso si può
generalizzare:
aA + bB −−→ cC + dD
naA + nbB −−→ ncC + ndD
cC + d D− > a A + bB
∆HR
n∆HR
− ∆HR
Se si inverte il senso di reazione, se la prima era endoterma, l’inversa è esoterma e viceversa.
<manca 11:43>. Riprendiamo le prime due combustioni di prima. I due dati di entalpia che abbiamo scritto così dice che la combustione del carbonio sviluppa più calore di quella dell’idrogeno.
Se misuro la combustione del carbonio ad un’altra pressione, l’entalpia sarebbe maggiore. Quindi
se voglio scegliere quella che dà più energia, devo confrontare questi due valori in modo che avvengano nelle stesse condizioni, soprattutto alla stessa pressione. Mi devo mettere in condizioni
standard in base alla quale definire tutte le specie e il ∆H .
Lo Stato standard è per un gas lo stato in cui la specie si considera ideale a P = 1 atm; per un
liquido puro è quello in cui si trova alla pressione di 1 atm. Per un solido (sostanza pura) è lo stato
più stabile alla temperatura che mi interessa e alla pressione di 1 atm. Un esempio di questo è
il carbonio, lo si può trovare come grafite oppure come diamante. A 25 ° lo stato più stabile è la
grafite <11:49>. Per una specie in soluzione, cioè per un soluto, si definisce lo stato standard quello
67
Cenni di termochimica
20/05/2014
in cui [] = 1M, P = 1 atm. Esiste uno stato standard per ogni temperatura. I valori di grandezze che
si riferiscono allo stato standard di solito si riferiscono sempre a 25 ◦C. <11:51> questo è un errore
teorico, sono i valori dello stato standard a 25 °.
Per specificare che una reazione avviene nello stato standard
C(s) + O2g −−→ CO2g
∆H 0
si indica <l’entalpia? 11:54> con ∆H 0 . Normalmente useremo entalpie nello stato standard, altrimenti specificheremo.
<Qui c’è un salto, aveva cominciato a dire prima la reazione dopo> Facciamo un esempio:
C(s) + 2 H2g −−→ CH4g
∆HR0
è la reazione di <formazione del metano= 12:01>. Posso aggiungere 2 ossigeni a entrambe e la
reazione rimane bilanciata. C(s) + 2 H2g + O2 −−→ CH4g + O2 , e immagino di seguire un’altra strada:
<1.6>. Quello che avevamo fatto con il circlo di <Born-Haber> <12:05> passando anche da un’altra
parte, basta che arrivi nello stesso punto. Ho:
∆HR0 = ∆H10 + ∆H20 − ∆H30
questa è una proprietà fondamentale dell’entalpia, che è una funzione di stato, cioè dipende solo
dall’energia del sistema, non dall’energia per andare dallo stato iniziale a quello finale. (Questa è
una proprietà delle funzioni di stato) <boh riscrivi un po’>. Questa relazione è detta Legge di Hess
(o somma dei calori). Si può usare la legge di Hess per ridurre <12:09>.
<aveva cominciato con questa>
CH4g + 2 O2g −−→ CO2(g) + H2 O(l)
∆HR0
quando abbiamo fatto il legame ionico, parlammo del ciclo di <Born-Haber>, in cui si passava da
∆H
Nas + 21 Cl2g −−→ NaCl usando un trucco per andare da uno stato all’altro. <forse manca qualcosa
11:58>. Questo ∆HR0 lo si può costruire facendo un percorso alternativo, purché si parta e si arrivi
sempre negli stessi stati.
<ripresa> <1.7> La reazione di formazione di un composto è la reazione che si scrive a partire dai
<composti elementari? 12:11> per ottenere quel composto. Nel caso del metano, prendo carbonio
e idrogeno e vedo quale reazione mi dà metano.
<1.8> Prendiamo H2 SO4 : H2 + S + 2 O2 −−→ H2 SO4 è la sua reazione di formazione. Solo che
<1.8> è fisicamente possibile, come l’idrogenazione del carbonio (l’esempio precedente). Questo
esempio appena fatto invece non avverrà mai. Non importa che la reazione non sia possibile, può
essere anche virtuale.
Torniamo alla reazione di formazione del metano. <12:15> mi immagino l’acqua <12:15>. Ad
essa associo l’entalpia di formazione dell’acqua. E’ il doppio perché mi si formano due moli di
acqua. <12:17 rivelazione> il che vuol dire che ∆HR0 la posso calcolare anche facendo quest’altro
percorso e ottengo:
∆HR0 = −∆H 0f (CH4 ) + 2∆H 0f (H2 O) + ∆0f (CO2 )∆HR0 = 2∆H 0f (H2 O) + ∆0f (CO2 ) − ∆H 0f (CH4 )
questo significa che a me interessano solo le entalpie di formazione, che possono essere reazioni
vere o virtuali (H2 SO4 ), la sua generalizzazione è che:
X
X
∆HR0 =
νi ∆H 0f (i ) −
ν j ∆H 0f ( j )
r eag ent i
pr od ot t i
68
Cenni di termochimica
20/05/2014
dove si prende come definizione che ∆H 0f (elementare) ≡ 0.
Questo significa che si può <12:22>
C(s) + 2 H2 g −−→ CH4g
<12:24> in realtà c’è una difficoltà pratica a misurare l’energia <12:24>, perché si formano tutti gli
idrocarburi possibili, non solo il metano, si forma una miscela, quindi misuriamo in realtà una
media di tutti i calori delle reazioni che sono avvenute. Però si può determinare l’energia usando
di nuovo la legge di Hess, facendo avvenire un’altra reazione:
∆HC0 = −394kJ mol−1
C(s) + O2g −−→ CO2g
H2g + 21 O2g −−→ H2 Ol
∆C0 (CH4 ) = ∆0f (H2 O = −286kJ mol−1
CH4g + 2O2g −−→ CO2g + 2 H2 Ol
∆C0 (CH4 ) = −891kJ mol−1
<12:28 perso> <1.9>. Prova a vedere quale è <cosa 12:31?>
69
Sistemi in equilibrio
22/05/2014
18 Sistemi in equilibrio
18.1 22/05/2014
Prendiamo una reazione qualunque
∆H < 0 esotermica
aA + bB −−→ cC + dD
∆H > 0 endotermica
Siccome tutti i sistemi tendono verso lo stato di minima energia possibile, emettendo verso l’esterno tutta l’energia, sembra che <11:44> siano solo quelle ad energia esotermica, sembra che
avvengano solo le prime, non le altre. Quindi se considero solo le reazioni spontanee, mi aspetterei solo le reazioni esotermiche. In realtà quello che si osserva sperimentalmente (o si dimostra
termodinamicamente) è che anche le reazioni endotermiche possono avvenire spontaneamente.
Il criterio della minima energia non è sufficiente a spiegare questo fenomeno. In effetti non è sufficiente tenere conto del calore per vedere se una reazione è spontanea o meno, serve un’altra grandezza, indicata con S, detta Entropia. Intuitivamente, mentre l’entalpia è una misura dell’energia
<11:48 di che?>, l’entropia è una misura del contenuto di disordine. Dal punto di vista entropico
(cioè del disordine) i sistemi tendono allo stato di massimo disordine, per cui viene fuori <1.1>.
Supponiamo di avere un contenitore con un isolante che non fa passare calore, con un diaframma
che lo divide in due parti. In una metà c’è un gas, nell’altra il vuoto. Se si toglie il diaframma, il gas
occupa tutto il volume a disposizione, spontaneamente. Questa è una trasformazione fisica, ma
avviene comunque senza scambio di energia, cioè con ∆H = 0. Allora dal punto di vista energetico
sembra che al gas non interessi essere in uno o in un altro stato. In realtà, lo stato preferito è quello
in cui si è occupato tutto, perché quando il gas occupa metà ha un certo grado di disordine, mentre se si aumenta il volume il grado di disordine aumenta. Questa trasformazione fisica ha una
variazione positiva di entropia, che avviene spontaneamente. Per tener conto di questi due effetti,
viene costruita una funzione, indicata con G = H − T · S, che è una funzione di stato (<11:53> anche S è una funzione di stato, quindi la combinazione di funzioni di stato è una funzione di stato).
Questa è detta energia libera di GIBBS. Tutti i sistemi evolvono per minimizzare G. In ogni trasformazione abbiamo un ∆G = ∆H −T ∆S. <11:55> la tendenza è legata a minimi di energia. Ma se
si prende per esempio l’espansione del gas, il ∆G negativo si ha per ∆S positivo. Questa grandezza
consente di studiare se una reazione chimica si svolge da reagenti verso prodotti oppure no.
Se ∆G < 0 evolve →
Se ∆G > 0 evolve ←
Se ∆G = 0 equilibrio
<reazione spontanea 11:58>. C’è un caso in cui ∆G = 0, e si ha una situazione di equilibrio,
che corrisponde a fenomeni opposti che avvengono con la stessa velocità (come il fenomeno della
tensione vapore, dal punto di vista microscopico <12:00>, ha ∆G = 0). Prendiamo la reazione generica: avrà una certa velocità in mol min−1 . <12:00> più se ne forma, più l’altra reazione diventa
efficace, e ad un certo punto non vedo più variazione di quantità tra reagenti e prodotti. Se evolvesse in un verso o nell’altro, una di queste due trasformazioni avrebbe un ∆G < 0, ma se non vedo
cambiare niente significa che le due trasformazioni stanno avvenendo in un verso e nell’altro con
la stessa velocità:
−−−
*
aA + bB )
− cC + dD
Consideriamo la reazione generica tutta gassosa:
−−−
*
aA(g) + bB(g) )
− cC(g) + dD(g)
70
Sistemi in equilibrio
22/05/2014
Per una specie gassosa, c’è un’espressione analitica dell’energia libera:
G = G 0 + RT ln
P
P0
<12:04> dove P 0 è la pressione a stato standard. Se la pressione cui si prova il gas è 1 atm, il secondo termine vale 0. In base a questa definizione, possiamo generalizzare l’espressione (è una def.
operativa):
P
G = G 0 + RT ln a
a = 0 attività
P
Avrò un’energia per ogni specie. Il ∆G della reazione è:
£
¤ £
0
∆G = G(prodotti)−G(reagenti) = (cGC +dG D )−(aG A +bG B ) = c(GC0 + RT ln aC ) + d (G D
RT ln a D ) − a(G 0A + RT
0
l’energia libera è un’energia su mole (kJ mol−1 ), ∆G 0 = cGC0 +dG D
−aG 0A −bG B0 . Quindi metto in un
contenitore una certa quantità di un gas A, B, C e D. Faccio i conti e trovo quanto vale la variazione
di energia libera, se è negativa vedo che si formano più prodotti, se invece è positiva vedo più
reagenti. Se non vedo cambiare nulla, allora ∆G = 0, cioè le quantità dei 4 gas sono in equilibrio,
perché le reazioni nelle due direzioni avvengono entrambe con la stessa velocità. Si ha cioè che
−
ac ad
∆G 0
= ln Ca D
= k costante
RT
a A a Bb
dove il primo membro dell’equazione è una costante (se faccio tutto a temperatura costante), che
vale quanto il secondo. k è la costante di equilibrio di questa reazione. Questo significa che se
metto quantità casuali di gas nel contenitore, finché ∆G < 0 vedo consumare i reagenti (A e B) e
la formazione dei prodotti (C e D). <1.2> Ogni reazione ha una sua costante ad una certa temperatura. Usando questa relazione, e trovo che ∆G < 0, vedo che aumenta il numeratore e diminuisce il denominatore, finché Q non diventa uguale a k. Se vedo invece un sistema che <12:21>, Q
diminuisce finché non diventa uguale a k.
Q
Q
< 1 →, ∆G > 0
> 1 ←, oppuUn sistema ha quindi sempre tre possibilità: ∆G < 0
k
k
Q
−−−
*
re ∆G = 0
= 1)
− . Q è detto quoziente di reazione, determinato con i valori delle quantità
k
iniziali. Facciamo un esempio:
P
a = 0 = P atm
P
Se uso atm come unità di misura, posso scrivere così. C’è una corrispondenza tra a che è un
numero puro e l’unità atm. Se uso questà unità, posso riscrivere k:
kP =
d
PCc P D
P Aa P Bb
Posso cambiare tutto quello che voglio, ma se lo faccio a temperatura costante, questo valore
numerico non cambia. <esercizino su quad.> NOTA: k P non ha una dimensione univoca. k è
adimensionale.
<13:12> il sistema reagisce diminuendo il numero di moli. Se si suppone di avere una mole
iniziale di reagente, e niente prodotti, e dopo la reazione si dissociasse tutto, avremmo 2 moli di
prodotti. Quindi le moli oscillano tra 1 e 2, cioè il sistema può cambiare il numero di moli. Se
riduco il volume, aumento la pressione, il sistema cambia il numero di moli diminuendolo. Il
sistema cerca di compensare la perturbazione. Questo risponde alla prima domanda dell’ultimo
esercizio.
71
Sistemi in equilibrio
27/05/2014
18.2 27/05/2014
−−
*
aA + bB −
)
− cC + dD
k=
d
aCc a D
a aA a Bb
eq. omogeneo gassoso
kp =
d
PCc P D
P Aa P Bb
¶
d µ
nCc n D
RT ∆ν
=
(dipende solo dalla temperatura) = a b
n A nB V
∆ν = c + d − (a + b)
nC
X Cc X Dd ∆ν
V
=
=
P
< copi ar ed aquad > X Aa X Bb t ot
<manca spiegazioni delle eq 10:43> Un aumento di V significa avere un aumento della prima frazione, l’equilibrio si sposta favorendo un <reagenti>, viceversa <perché ∆ν < 0>, quindi l’equilibrio
si sposta in un verso o nell’altro in base al segno di ∆ν. <10:48>
∆ν = 0 =⇒ k p = k c = k n = k X
−−
*
2HI −
)
− H2 + I2
<10:51> Se ∆ν = 0, posso variare il volume, ma non cambia il comportamento perché <gnamooooo>
viene sempre 1.
Immaginiamo ora che non tutte le specie siano nella stessa fase, supponiamo di avere:
−−−
*
aA(s) + bBg )
− cCg + dDg
Come abbiamo definito l’attività di una specie gassosa? ai =
ad altre specie non gassose.
ai =
Pi
P i0
=
ni
RT
V
ni
V 0 RT
=
mi
P M (i ) 1/V RT
mi
1/V 0 RT
P M (i )
Pi
.
P i0
Estendiamo questa definizione
= 1 solido, liquido
<10:55 un monte di roba> Siccome il solido per definizione è incomprimibile (può cambiare la
forma ma non il volume), V0 coincide con V , quindi per un solido l’attività è sempre 1. Ma anche
i liquidi sono incomprimibili, quindi vale lo stesso discorso.
[]
Come cambia l’attivitità di un <10:57>. n i /V 0 è la concentrazione molare = []0i = []i unità molai
ri. La definizione di stato standard è <pressione 1 atm 10:58> quindi se si esprime in unità <10:58>,
l’attività coincide numericamente con la concentrazione molare.
Torniamo all’esempio <11:00>.
d
PCc P D
k=
1 · P Bb
1 è l’attività di A, cioè non interviene nella costante di equilibrio, però non significa che non ha
effetto, perché se non ci fosse il solido l’equilibrio non ci sarebbe. Un esempio concreto è:
19,2g NH4 NO3 nitrato (V) di ammonio
◦
−−
*
NH4 NO3(s) −
)
− N2 Og + 2 H2 Og T = 250 CV = 10,0l raggiunto l’equil
72
pH
29/05/2014
19 pH
19.1 27/05/2014
−−−
*
H2 O va incontro a un fenomeno di dissociazione spontanea: H2 O )
− H+ +OH– . In realtà, siccome
H+ è effettivamente un protone che può formare legame con due elettroni, si forma un legame
−−
*
dativo con una molecola d’acqua: 2 H2 O −
)
− H3 O+ + OH– (ione idrossonio e ossidrile). Si ha
k=
aH3 O+ aOH−
a2H
=
2O
[H3 O+ ][OH− ]
a2H
2O
<12:14> nell’ipotesi che le concentrazioni siano molto basse, cioè che sia quasi acqua pura, posso
approssimare l’attività di H2 O: k · a2H O ' K W = [H3 O+ ][OH− ] = 10−14 . Ogni volta che ho a che fare
2
con l’acqua, gli ioni H3O+ e OH- hanno le concentrazioni legate da questa relazione, non possono
stare in acqua con la concentrazione che pare loro, il prodotto cambia solo se cambia la temperatura. Siccome la stechiometria della reazione è 1:1, le concentrazioni dei due ioni nell’acqua pura
sono uguali, si ha quindi
[H3 O+ ] = [OH− ] =
p
k w = 10−7 M condizione di neutralità
La parola neutralità si riferisce ad una carattaristica tipica delle soluzioni acquose: l’acidità. L’acqua è neutra per definizione. <12:19> p H = − log[10][H3 O+ ]. Quindi nell’acqua p H = 7. Il pOH
è definito così: pOH = − log[10][OH− ]. Nell’acqua pOH = 7. Se invece la soluzione è acida,
[H3 O+ ] > [OH− ], cioè [H3 O+ ] > 10−7 M =⇒ p H < 7, [OH− ] < 10−7 M =⇒ pOH > 7, se la soluzione è basica [H3 O+ ] < [OH− ], cioè [H3 O+ ] < 10−7 M =⇒ p H > 7, [OH− ] > 10−7 M =⇒ pOH < 7
<manca roba alla fine.. 12:20>
19.2 29/05/2014
<manca roba all’inizio, arrivato tardi 11:41>. <1.1>
Supponiamo di prendere HCl. Sappiamo che è un elettrolita forte, cioè che se lo si mette in
acqua si ha che HClaq −−→ H+aq +Cl–aq , per essere precisi bisognerebbe scrivere H+ +H2 O −−→ H3 O+ ,
combinando le reazioni:
HCl(g) + H2 O −−→ H3 O+ + Cl−
Supponiamo di avere HCl in soluzione acquosa, con una concentrazione generica Ca (M) in mol l−1 .
Io in soluzione quindi ho:
HCl(g) + H2 O −−→ H3 O+ + Cl−
+
−
−−−
*
2 H2 O )
− H3 O + OH
<11:46> quindi <...> saranno dati da un contributo <11:46>
[H3 O+ −[H3 O+ ]HCl + [H3 O+ ]H2O = C a + x
Questa reazione adesso non produce più la stessa quantità di ioni, perché non ho più solo acqua
ma ho anche HCl. Quindi non so quanto è la quantità ora. So però che la relazione di Kw è comunque valida. Se ho qualcosa che produce altri H3 O+ oltre quelli che fa l’acqua, è come se aumentassi
il suo numero, <diminuire questo 11:50>, cioè ciò significa che la reazione che produce OH– ne dovrà produrre meno, cioè l’acqua si dissocerà meno, è lo stesso concetto <dei gas? 11:50>. So quindi
73
pH
29/05/2014
che di sicuro [OH– ] dovrà avere molarità inferiore a 10− 7. Quindi posso scrivere:
K w = (C a + x) · x
x 2 +C ax − K w = 0
p
−C a + C a 2 + 4K w
x=
2 p
C a + C a 2 + 4K w
[H3 O+ ] = C a + x =
2
Se si studia questa funzione, si ottiene il grafico <1.2> <11:55> cioè ad un certo punto <11:55>
diventa insignificante. Già 10− 7 può essere più piccolo di C a , quindi il contributo dell’acqua non
lo vedo più. Questo è come si calcola il pH per un acido forte abbastanza concentrato (cioè C a À
2K w). Ricorda: noi consideriamo una cosa molto più piccola di un’altra se è pari a 50 volte meno
l’altra.
Se siamo invece in condizioni p
opposte, cioè se C a 2 ¿ 4K w, ossia c’è pochissimo
acido nell’acqua,
p
p
4K
w
4K
w
C
a
+
e se questa è vera si ha che '
= K w = 10− 7, cioè
di fatto si ha: [H3 O+ ] ≈
2
2
l’acido non interferisce con l’equilibrio dell’acqua.
Da qui deriva una regola pratica: se si è in presenza di un acido forte con concentrazione Ca
<sufficientemente elevata> <come è? 12:01>, si considera direttamente p H = − log[10]C a . Per
esempio, se si ha C a = 10−1 M , p H = 1, oppure C a = 10−3 M , p H = 3. Ma questa regola non è
sempre valida, perché se si ha C a = 10−8 M , p H = 8 non è vero, significa continuare ad usare l’approssimazione lineare dove non è più valida (si sta ottenendo una soluzione basica aggiungendo
acido? <12:05>).
p
p
C a + C a 2 + 4K w 10−8 + 10−16 + 4 · 10−14
+
=
= 1,05 · 10−7 M =⇒ p H = 6,98
[H3 O ] =
2
2
Si può fare il complementare di questo ragionamento, usando NaOH(s) −−→ Na+ + OH– , con
concentrazione C b (M), si trova che
[OH− ] = [OH− ]NaOH + [OH− ]H2 O
<12:09>.
Facciamo un passo indietro per vedere meglio cosa è un acido e cosa una base. Storicamente
la parola acido viene <12:11>, un modo per classificare le sostanze allora era “aspro” o no. In realtà
questa cosa ha un valore teorico: una sostanza acida ha un sapore “aspro” e c’erano sostanze in
grado di annullare questa caratteristica, dette alcali, <12:12> cioè basi. Dal punto di vista chimico,
fu abbastanza veloce individuare che il carattere acido è legato alla presenza di ioni H+ , mentre il
carattere basico è legato alla presenza di ioni OH– . Da qui venne la prima definizione di Arrhenius:
è un acido una sostanza che se disciolta in acqua libera ioni H+ , è una base una sostanza che
libera ioni OH– . Questa definizione va ancora bene per tanti composti, per esempio per HCl −−→
H+ +Cl– , oppure per NaOH −−→ Na+ +OH– . Dove queste relazioni sono evidenti, questa definizione
si può usare. Non faremo la definizione di Lewis, ma quella successiva a a quella di Arrhenius:
l’ammoniaca NH3 se messa in acqua si comporta da base, cioè si vedono gli OH– che aumentano,
anche se non sono presenti nella sostanza di partenza. Per risolvere questo problema Bronsted
- Lowry generalizzarono la definizione: l’acido è una sostanza che si comporta da donatore di
protoni, una base è una sostanza che è un accettore di protoni.
−−−
*
Supponiamo di mettere insieme HF+NH3 )
− NH4+ +F– HF cede un protone all’ammoniaca e
si osserva la reazione. Un protone è passato dal primo composto al secondo, quindi HF è un acido
74
pH
29/05/2014
e NH3 è una base perché l’accetta. Solitamente questo tipo di reazione è di equilibrio, cioè NH4+
cede un protone e diventa NH3 , F– lo acquista e diventa HF. Il comportamento acido base non
viene quindi più visto come una caratteristica di una singola sostanza ma come una caratteristica
di una coppia di sostanze: <1.3>. Sott’inteso in questo ragionamento è che non ci possa essere
un comportamento di acido o base se non c’è un altro elemento che accetta o cede un protone.
Nel caso di HCl −−→ H+ + Cl– , per questa nuova definizione si ha che HCl + H2 O −−→ H3 O+ + Cl– ,
cioè acqua e HCl si comportano come una coppia acido-base, dove l’acqua fa da base. Con l’am−−
*
moniaca: NH3 + H2 O −
)
− OH– + NH4+ , gli ioni H+ provengono dall’acqua, non dall’ammoniaca. Il
problema che si posero Bronsted e Lowry è che siccome queste reazioni sono spesso di equilibrio,
si ha sempre una certa quantità di reagenti e prodotti presenti. A seconda del valore della k di
equilibrio, si hanno comportamenti diversi <invenzioni 12:26>:
+
−
−−−
*
HF + NH3 )
− NH4 + F
+
−
−−−
*
HF + H2 O )
− H3 O + F
<...> non si riesce a capire se posso considerare in assoluto un acido più o meno forte rispetto ad
un altro, non posso ogni volta pensare a quale altra sostanza uso per farla reagire. I due fecero una
considerazione:
+
−
−−
*
H2 O + H2 O −
)
− H3 O + OH
<1.4> è la ionizzazione dell’acqua, che esiste in condizioni neutre, cioè di acqua da sola. Leggendola secondo lo schema di Bronsted e Lowry, si osserva che la prima molecola cede un protone
alla seconda, e si ottengono i due prodotti. Quindi una molecola fa da acido e una da base, c’è
comunque uno scambio di un protone tra due molecole identiche, e il risultato è la neutralità.
Questa reazione qui dice che l’acqua ha la stessa identica capacità di cedere e accettare protoni,
quindi è una sostanza unica da questo punto di vista. Bronsted pensò quindi di misurare questa
capacità facendo reagire la sostanza con l’acqua, cioè usare l’acqua come sostanza di riferimento:
+
−
−−
*
HF + H2 O −
)
− H3 O + F
di questa reazione di equilibrio possiamo scrivere l’espressione della costante di equilibrio: k =
a H3 O+ a F− [H3 O+ ][F− ]
=
per cui a H2 O è <cosa 12:34>, si scrive
a HF a H2 O
[HF− ]a H2 O
Ka =
[H 3O + ][F − ]
[H F ]
è la costante di dissociazione <acida 12:35>.<12:36> Il confronto numerico dei vari K a dà quindi
<cosa 12:36>.
Per esempio, K a di HF è 6,8 · 10−4 , per HCN 9,1 · 10−10 . Qui si capisce bene che nei confronti
con l’acqua, la prima ha una costante con due ordini di grandezze diversi. Cioè si ha indicazione
che HCN ha una capacità molto più debole di HF di cedere protoni. Si può prendere anche H2 S
9,5 · 10−8 , oppure CH3 COOH 1,8 · 10−5 <metti questa roba in una tabella 1.5>.
–
−−−
*
HCl + H2 O )
− H3 O+ + Cl anche se è un equilibrio, in soluzione sono presenti tutti tranne
HCl indissociata. Quindi nella reazione di K a <1.6>, allora si considera HCl ∞ <12:41>. Questo
dettaglio ha una controndicazione: tipici acidi forti sono HCl, H2 SO4 , HClO4 , HClO3 , HNO3 , che
hanno tutti K a = ∞. Non c’è modo di individuare quale sia più forte dell’altro. Questo è detto
effetto livellante. Mentre quelli deboli si distinguono, quelli forti hanno K a tutti ugale.
−−−
*
Adesso possiamo rifare tutto questo ragionamento, però sulle basi: NH3 + H2 O )
− NH4+ +
OH– . Questo è un equilibrio regolato da una costante
Kb =
[NH4+ ][OH − ]
[N H3 ]
75
pH
29/05/2014
<12:44> Dal confronto della K b riesco a fare una scala di basi come per gli acidi. In realtà <non
ci sono? 12:45> <1.6> nota: un acido o una base non devono essere molecole neutre per forza. <12:48> Il problema che c’è ora è come fare per calcolare il pH della base debole <o ha detto
acido?>. Prendo un acido generico:
+
−
−−
*
HA + H2 O −
)
− H3 O + A
Ka =
[H3 O+ ][A − ]
[H A]
Ca
K a li conosco
Nel caso dell’acido forte mi bastava sapere C a e K w , adesso invece servono C a e K a . Se l’acido è
più o meno in grado di cedere protoni <12:51>. Seguiamo le stesse <12:51>:
[H3 O+ ] = [H3 O+ ]H A + [H3 O+ ]H2 O
Quando avevamo un acido forte, si sapeva quanto valeva la molarità degli ioni H3 O+ di H A. Si
possono impostare delle equazioni che si risolvono matematicamente, oppure si può ragionare
così:
1a ipotesi: <12:54> [H3 O+ ]H2 O ¿ [H3 O+ ]H A
=⇒ [H3 O+ ] ' [H3 O+ ]H A =⇒ [A − ] = [H3 O+ ]
[H3 O+ ]2
Ka =
C a − [H3 O+ ]
questo sarebbe già sufficiente a risolvere il problema. Il denominatore è la differenza tra la concentrazione iniziale dell’acido e la concentrazione della parte che si è dissociata.
[H O+ ]2
2a ipotesi: [H3 O+ ] ¿ C a , quindi K a = 3
, cioè
Ca
p
[H3 O+ ] = K a C a
<13:00> gli H3O+ erano proporzionali a C a , adesso sono <13:01>. Se si prende 1 l di HCl a concentrazione C a (HC l ) = 0,1M , quindi p H = 1, e si diluisce portando tutto a 2l, la concentrazione
diventa C a (HC l ) = 0,05M , mentre invece <13:02> l’effetto di diminuzione è più forte con un acido
forte che non con un acido debole. Facciamo un esempio numerico:
H A C a = 0,100M
K a = 1,80 · 10−5
[H3 O+ ] =
p
KaCa =
p
1,8010−5 10−1 = 1,34 · 10−3 M
devo saper verificare che il numero che ho scritto in fondo abbia un senso: la prima ipotesi è
quella in cui si trascura il contributo dell’acqua. Come faccio a stimare il suo contributo? Se è
Kw
10−14
vero che ho calcolato tutti gli H3 O+ , so anche quanti sono [OH+ ] =
=
' 10−11 M .
[H3 O+ ] 1,3410−3
Nella reazione dell’acqua, i due ioni sono preseni con la stessa concentrazione. <13:08> quindi
posso dire che [H3 O+ ]H2 O = [OH− ], quindi la prima ipotesi è corretta. La seconda ipotesi diceva
che <13:10>, quindi devo vedere se 1,3410−3 ¿ 0,100. Usando il criterio che abbiamo stabilito:
0,100
1,3410−3 ≤
= 0,002, che è vero. Sono dentro l’approssimazione. Allora il numero che si è
50
calcolato è corretto. Allora calcolo p H = − log[10]1,3410−3 = 2,87.
76
pH
30/05/2014
Cosa succede se le ipotesi non sono verificate? Tipicamente, quella che non risulta verificata
è la seconda, cioè quella in cui [H3 O+ ] non è più piccolo di C a . Quello che devo fare è fermarmi
[H3 O+ ]2
e fare l’equazione di secondo
al passo precedente a questa ipotesi, cioè qui: K a =
C a − [H3 O+ ]
grado. Se non è verificata la prima ipotesi, tipicamente non lo è nemmeno la seconda. In ogni
caso se non è verifica la prima siamo in condizioni di acido molto debole molto diluito. Si risolve
impostando una serie di equazioni generali:
Conservazione della massa :C a = [H A] + [A − ]
Conservazione della carica elettrica (elettroneutralità) :[H3 O+ ] = [A − ] + [OH − ]
Prodotto ionico :K w = [H3 O+ ][OH− ]
Costante acida :K a =
[H3 O+ ][A− ]
[H A]
<13:19 manca roba qua dentro>
19.3 30/05/2014
+
−
−−−
*
HA + H2 O )
− H3 O + OH
<08:42 manca roba all’inizio>. Immaginate di avere in soluzione non l’acido ma direttamente lo
ione A − che è l’anione dell’acido. L’equilibrio con l’acido l’abbiamo <vista... 08:43>, questo si
comporta da acido, la specie coniugata da base. Mi aspetto che A − si faccia protonare dall’acqua:
−
+
−−−
*
A− + H2 O )
− A + H3 O K i =
[H A][OH − ] [H3 O+ ] K w
·
=
[A − ]
[H3 O+ ] K a
che evidenzia un comportamento come base. Questa è regolata dalla costante di equilibrio, che dipende dalla costante dell’acido di provenienza di A − . La i indica che siamo partiti da una molecola
non neutra, questa reazione si chiama idrolisi.
Esiste il complementare di questo discorso con le basi. Prendiamo una base generica:
+
−
−−
*
B + H2 O −
)
− BH + O K b =
[B H + ][OH − ]
[B ]
E prendiamo B H + :
B H + + H 2O <=> B + H3 O+
Ki =
[B ][H3 O+ ] [OH − ] K w
·
=
[B H + ]
[OH − ] K b
questa è un’idrolisi acida, perché produce H3 O+ . Questa relazione è interpretata qualitativamente
anche dicendo che se la base è abbastanza forte (non infinito) allora K b <? 08:51> moltiplicata per
<?>.
−
NaCls −−→ Na+
aq + Claq
Riconoscere per Na+ e Cl– da quale composto acido-base provengano. Per esempio, Na+ è il catione della base NaOH. Quale acido dissociandosi produce Cl– ? HCl. Allora se metto in acqua il sale,
ho la possibilità che avvengano le reazioni
+
−−
*
Na+ + H2 O −
)
− NaOH + H
−
−−
*
Cl − + H2 O −
)
− HCl + OH
Ki =
77
Ki =
Kw
→0
Kb
Kw
→ 0 dove K w è quella di HCl
Ka
pH
30/05/2014
Di fatto è solo un modo diverso per scrivere che Na+ viene protonato. Dal momento che K w (HC l ) =
∞, allora la seconda reazione significa che non avviene. <08:56> non succedono reazioni che
possano modificare il pH. Questo è il motivo per cui il sale non modifica l’acidità dell’acqua.
Supponiamo di fare un’altra cosa:
NH4 Cl −−→ NH4+ + Cl−
è un elettrolita forte che si dissocia in due ioni. NH4+ è il catione coniugato di NH3 , quindi in acqua
mi aspetto che si comporti da acido, cioè riesca a protonare l’acqua:
+
−−
*
NH4+ + H2 O −
)
− NH3 + H3 O
Ki =
Kw
10−14
=
= 5,5610−10
K b (NH3 ) 1,810−5
Se metto in soluzione per esempio C s = 0,1M di cloruro di ammonio (NH4 Cl) mi produce <09:02>,
p
per cui se lo considero acido debole normale, posso scrivere [H3 O+ ] = K i C s = 7,4510−6 M, quindi
non è neutro, p H = 5,13. <Se si prende un acido o base provieniente da ... debole il pH cambia
09:04>.
HC l
0,1M
N aOH
0,1M
+
HC l + N aOH = H +C l − + N a + + OH −
I due ioni C l − e N a + non fanno nulla. Siamo in acqua, deve essere verificato che K w = [H3 O+ ][OH− ],
ma ho qui 0,10,1 = 10−2 À 10−14 . Questo è come aver scritto la reazione di equilibrio
H + +C l − + N a + + OH − <=> H 2O + N a + +C l −
1
= 10−14
K=
Kw
1
1
Q = −2 = 102 ¿
10
Kw
quindi questa reazione è fortemente spostata verso destra, ottengo quindi acqua salata neutra.
Questa è una reazione di neutralizzazione: da una sostanza acida e una neutra messe insieme in
acqua si ottiene acqua neutra <09:11>. In questo caso, la neutralizzazione è totale. Cambiamo i
valori:
HC l
0,1M
N aOH
0,05M
Rimangono nell’acqua 0,05 M di HCl sopravvissute alla neutralizzazione, quindi è parziale.
0,1M
HC l
0,1M
NH3
+
HCl + NH3 −−→ Cl ↑ − + NH4 HCl cede un protone a NH3
78
pH
30/05/2014
Siccome sono partito da quantità iniziali uguali, mi si producono 0,1 M di NH4 Cl e rispetto al
caso precedente ho lo ione NH4+ la cui idrolisi può cambiare il pH. Quindi ho una completa
neutralizzazione, ma il pH non sarà neutro perché questo ione farà idrolisi.
+
−−
*
NH4+ + H2 O −
)
− NH3 + H3 O
Prendiamo ora
N aOH
0,1M
HF
0,1M
+
N aF = N a + F
−
−
F + H2 O> −>HF + OH−
<09:20> Quando si ha di fronte un sale o un acido, bisogna vedere se i suoi ioni provengono da
acidi o basi forti, o se l’acido o base sono forti o deboli.
Altro esempio pratico:
NaHCO3 = Na+ + HCO3− questo è l’idrogeno carbonato di sodio, come fa a far digerire?
Quando si scioglie produce i due ioni. HCO3– è l’anione dell’acido carbonico
+
−
−−−
*
H2 CO3 + H2 O )
− H3 O + HCO3
K a = 4,510−7
−
−−−
*
HCO3− + H2 O )
− H2 CO3 + OH
Kb =
Kw
< nonsonosi cur oper ni ent e09 : 25 >
Ka
Quando facciamo indigestione, si stimolano le mucose a produrre HCl in eccesso, quindi ci si
butta un qualcosa di basico e si prova a neutralizzare l’eccesso di acidità. Vengono fuori gas perché
l’acido carbonico si decompone: H2 CO3 −−→ H2 O + CO2 .
−−
*
AB(s) −
)
− AB(aq)
K1 =
+
−
−−
*
ABaq −
)
− Aaq + Baq
a AB (aq)
a AB (aq)
a A+ aB −
K2 =
a AB (aq)
+
+
−−
*
AB(s) −
)
− Aaq + Baq
K = K1K2
Se AB è un elettrolita forte, a AB (aq) → 0 K 1 → 0 K 2 → ∞, quindi K è indeterminato. Questo è
quello che succede con NaCl:
+
−
−−−
*
NaCl(s) )
− Na + Cl
s = 358g l
−1
K ' 36
≈ 6mol l−1
<09:42> s è la solubilità, il massimo numero di moli che posso sciogliere. Questo valore lo possiamo considerare enorme rispetto ai valori che usiamo di solito delle soluzioni diluite.
+
−
−−−
*
AgCl(s) )
− Ag + Cl
a Ag+ a Cl−
= [Ag+ ][Cl− ] = 1,7710−10
K ps =
a AgC l
79
pH
30/05/2014
K ps è il prodotto di solubilità. Supponiamo di avere <1.1 esercizio>
20,0g
1,00l
AgCl
acqua
K ps = 1,7710−10
+
−
−−−
*
AgCls )
− Ag + Cl
80
Pila
30/05/2014
20 Pila
20.1 30/05/2014
[Cu2+ + Zn −−→ Cu + Zn2+
Cu2+ + 2 e −−→ Cu
Zn −−→ Zn2+ + 2 e
<2.1> <manca 12:46> si lascia andare avanti la reazione e alla fine si è ricoperto di rame la superficie dello <zinco>. <12:47> a livello microscopico c’è la corrente, che può essere usata per far
funzionare un qualche dispositivo, l’idea è non farla dissipare all’interno del sistema chimico. Il
metodo usato è far diventare reali le due semireazioni, separandole fisicamente, costringendo gli
elettroni a non passare direttamente da una specie all’altra ma <12:48> <2.2> Ogni semicella è formata da <12:49>. Quello che succede è che finché la situazione è questa non cambia nulla, prendo
un filo conduttore (con una resistenza per esempio) e collego i due elettrodi. Gli atomi di zinco
nel metallo diventano ioni Zn2+ liberando due elettroni, che non si idratano, viaggiano nel conduttore, seguono il circuito e raggiungono l’altro elettrodo, sulla barra di rame si crea un eccesso
di 2 elettroni rispetto alla neutralità, incontrano uno atomo di rame sulla superficie della barretta
e si riduce. <12:52> si vede la barretta che si ingrossa. Qualce decimo di secondo dopo si ferma
tutto, perché all’inizio la soluzioneZnSO4 è neutra all’inizio, e la carica +2e di cui si libera lo zinco non viene bilanciata nella soluzione. Questo impedisce agli elettroni (negativi) di andarsene,
e l’inverso fa anche l’altra semicella <forse manca qualcosa 12:53>. Bisogna chiudere il circuito,
collegando fra loro le due soluzioni attraverso un oggetto (per esempio un tubo a U pieno di un
elettrolita molto concentrato, con due tappi porosi che permettono il passaggio degli ioni senza
far passare la soluzione). Così il sistema continua a funzionare, perché se a sinistra si crea un eccesso di carica positiva, dall’altra parte arrivano gli ioni carichi negativi <invento 12:54>. L’eccesso
di carica negativa viene compensato da uno ione SO42– che <12:55> e il ciclo continua perché la
corrente è composita, fatta di elettroni che circolano nella parte metallica e ioni nella <soluzione
12:55>. <12:56>.
Questa cosa si schematizza così:
Z n|....
<2.3> La prima semireazione (di riduzione) avviene sulla superficie di rame, l’elettrodo dove avviene la reazione di chiama catodo, è l’elettrodo con potenziale elettrico più alto, sullo zinco vengono
liberati elettroni, è detto anodo, è quello con potenziale elettrico più basso <2.4>. Posso misurare la differenza di potenziale tra anodo e catodo, e si indica con f.e.m., che per definizione è la
differenza di potenziale quando il circuito è aperto.
f .e.m. = EC − E A < 2.5 >
(è sempre potenziale più alto meno più basso, è sempre positiva per definizione)
La relazione che lega ∆G della reazione chimica al potenziale che si sviluppa nella pila è:
∆G = −nF ∆E
F = 96 485 C mol−1
dove F è la costante di Farady <13:03> in valore assoluto è <13:03>.
∆G = ∆G 0 + RT ln
81
aCu a Zn2+
a Cu2+ a Z n
Pila
30/05/2014
Le attività sono le concentrazioni delle varie specie nella condizione reale. Se la concentrazione
dei due ioni nelle due semicelle fossero 1 M, le due semireazioni <13:07> sarebbero a condizioni
standard. Il termine con il logaritmo è quindi la correzione da apportare al sistema quando le
concentrazioni sono qualunque.
∆E = −
aCu a Zn2+
aCu a Zn2+
∆G 0
RT
∆G
=−
− RT ln
= ∆E 0 −
ln
Equazione di NERNST
nF
nF
a Cu2+ a Z n
nF
a Cu2+ a Z n
∆E 0 = −
∆G 0
nF
∆E 0 è la differenza tra potenziale di catodo e anodo a condizione standard, il secondo termine è
<13:12>, quindi si può separare il tutto e raggruppare da una parte tutto quello che ha a che fare
con il catodo da una parte e tutto ciò che riguarda l’anodo da un’altra, ottenendo
µ
¶ µ
¶
RT
aCu
RT
aZ n
0
0
∆E = EC −
ln
− EA −
ln
< 2.6 >
nF
aCu 2+
nF
a Z n 2+
<13:16>
forma ridotta
RT
ln
nF forma ossidata
0,0591
Rid
E =E0−
log
n
Ox
E =E0−
quest’ultima è <13:18, e poi 13:20>. Scrivere un’espressione come potenziale assoluto pare non
avere senso, in realtà abbiamo scelto un riferimento rispetto al quale si sta misurando il potenziale. Il riferimento preso è un elettrodo particolare <2.7>, <13:21> è un platino spugnoso a livello molecolare, con un’elevatissima superficie di scambio a disposizione con la soluzione, e poi
perché il platino è un metallo affine all’idrogeno. <13:22 mancano bollicine> questo si chiama
elettrodo standard a H2 (tutto il sistema). L’unica reazione che può avvenire è
2 H+ + 2 e −−→ H2
o l’inversa. Allora un elettrodo fatto così ha tutte le specie in condizioni standard. Il potenziale
di questo elettrodo che si indica così <2.8> è 0 Volt. <13:25> uso per costruirmi una pila, quindi
una semicella è quella dell’elettrodo standard a idrogeno, e dall’altra metto <13:25> e misuro la
differenza di potenziale tra le due placche di metallo <siamo a condizioni standard perché...>.
∆E 0 = EC0 − E A0 < 2.9 >
Si può ripetere la stessa cosa per la placca di zinco <2.10>
Come si usano questi potenziali? <2.11 manca un sacco di roba scritta solo su quad fino alla
fine>
82