SISTEMA NERVOSO PERIFERICO
SINDROME DEL TUNNEL CARPALE
Definizione: compressione del nervo mediano nel canale osteofibroso del carpo
Eziopatogenesi:
•
•
•
Ipertrofia della guaina sinoviale in seguito a trauma o a qualsiasi altra causa che determini
alterazioni della guaina sinoviale dei flessori con loro ispessimento e conseguente
compressione del legamento sul legamento traverso anteriore (tetto del canale)
Lesioni del nervo: Possono essere traumatiche (lussazione del semilunare, esiti di frattura
dell’epifisi distale o radiale, neoformazioni sinoviali, lipomi, anomalie muscolari o tendinee
come l’ipertrofia congenita del tendine del palmare gracile).
Spontanee: eziologia ignota, ovvero casi in cui la guaina carpale venga stimolata da agenti
diversi con conseguente infiammazione ed ispessimento della stessa.
Teorie:
•
•
•
•
•
Teoria ormonale: l’alterazione del normale rapporto progesterone/estrogeni, che si verifica
in gravidanza e nel climaterio favorisce la ritenzione idrica con conseguente rigonfiamento
della guaina sinoviale ed irritazione del nervo
Teoria traumatica: macro o micro-traumi in pratiche sportive o lavori manuali con frequente
flesso-estensione del polso (tennis); traumi in iperestesnione del polso con o senza fratture
Teoria vascolare: alterazione della vascolarizzazione da prolungata compressione del nervo
Teoria dell’anossia tissutale: le alterazioni metaboliche a carico dei tendini e delle fasce,
conseguenti ad una circolazione sanguigna cronicamente deficitaria portano ad una
proliferazione fibroblastica con conseguente riduzione del canale osto-fibroso.
Teoria infiammatoria: infiltrazione di fibrina, materiale piastrinico e glicoproteine, che
stimolano una intensa produzione fibroblastica con crescente produzione di jaluronidasi e
maggiore imbibizione di acqua. Aumenta contemporaneamente il deposito di
mucopolisaccaridi. Si osserva in caso di fibrosità, tenosinoviti e fasciti.
Sintomi:
•
•
•
Turbe sensitive: dolore al polso. Irradiato alla mano, talvolta all’avambraccio sino alla
spalla. Violento, diurno ma frequentmente notturno ed è aumentato dallo sforzo.
Turbe motorie: riduzione della forza di opposizione (funzione di pinza): è un sintomo
tardivo
Turbe trofiche: a carico dell’eminenza tenar, soprattutto del muscolo opponente, talvolta al
primo interosseo; di solito è tardiva.
Fasi cliniche:
•
•
Fase irritativi: dolore, disestesia, in genere iperestesia sul territorio dal mediano alla mano.
Fase sensitiva: dolorte, con persistenti parestesie, ipoestesia termodolorifica sul territorio del
nervo mediano
•
Fase paralitica: riduzione o scomparsa del dolore con persistenza delle parestesie e segni di
paralisi motoria. Ipotrofia dell’opponente e adduttore breve del pollice con netta riduzione
della forza di opposizione e marcata ipoestesia, quasi anestesia termodolorifica nel territorio
del mediano.
Diagnosi:
•
•
•
Segno di Tinel: la percussione con il dito sul canale del carpo evoca dolore irradiato
distalmente lungo il percorso del ne4rvo
Segno di Phalen: la flessione dorsale, forzata del polso provoca parestesie sul territorio
d’innervazione del mediano
Elettromiografia
Terapia:
•
•
•
Medica con FANS, infiltrazioni con farmaci sterouidei
Può esserci guarigione spontanea com,e spessop avviene nelle donne in gravidanza: dopo il
parto si ha una regressione della sintomatologia algo-parestesica.
Terapia chirurgica: incisione cutanea, sezione del legamento traverso anteriore e liberazione
del nervo dal canale stretto.
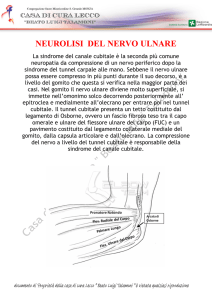
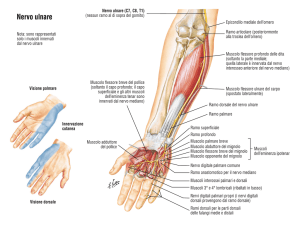
SINDROME DEL CANALE DI GUYON
Definizione: compressione del nervo ulnare al canale osteofibroso del polso
Eziopatogenesi:
•
•
Compressione intracanalare del nervo: da neoformazioni, anomalie vasali, alterazioni
strutturali dei vasi, cisti vasali, post-traumatismi
Compressione axtracanalare del nervo: neoformazioni cistiche o neoplastiche, alterazioni
infiammatorie e/o degenerative delle guaine sinoviali vicini al canale di Guyon spesso in
associazione a sindrome del tunnel carpale.
Sintomi:
•
•
•
Dolore: raramente presenta, si manifesta per lo più come dolenza al polso e nel territorio di
innervazione del nervo ulnare
Parestesie: persistenti. Ipoestesia o disestesia al quinto dito e metà ulnare del quarto dito
Ipotrofia: a carico degli interossei (soprattutto il primo) e dei muscoli dell’eminenza
ipotenar. Rapide insorgenza di quadro paralitico (in media 20 giorni) con aspetto della mano
di tipo benedicente
Diagnosi:
•
Esame della mano: ipovalidità dell’abduzione del quinto dito, ipotrofia del primo interosseo,
turbe sensitive (disestesie e/o parestesie nel territorio di innervazione), dolore
•
•
•
Aspetto della “mano benedicente”: atteggiamento ad artiglio incompleto della amno da
lesione completa del nervo ulnare; an ulare e mignolo con la prima falange iperestesia e le
altre flesse(paralisi degli interossei, del terzo e del quarto lombricali e prevalenza
dell’estensione comune delle dita. Indice e medio con la prima falange estesa e le altre
tendenzialmente semiflesse. Pollice con la seconda falange flessa o semiflessa
Atrofia dei muscoli interossei volari e dorsali
Elettromiografoa (EMG)
Terapia:
•
Da eseguire il più precocemente possibile la neurolisi e la decompressione del nervi intraextracanalare
SINDROME DEL TUNNEL RADIALE
Definizione: compressione del nervo radiale a livello del gomito
Eziopatogenesi:
•
•
•
Compressione a livello del tratto superiore del tunnel radiale: il connettivo lasso che avvolge
il nervo interosseo posteriore, ramo motore del nervo radiale, per azione di
microtraumatismi subisce la trasformazione in connettivo fibroso, scarsamente estensbile e
con azione compressiva diretta o da briglie aderenziali.
Compressione a livello del tratto medio del tunnel radiale: il muscolo estensore breve del
carpo può presentare, a livello dell’arcata che va dall’epicondilo alla facsia profonda
antibrachiale e all’aponeurosi dei muscoli flessori delle dita, margine fibrotico ed ispessito
con conseguente compressione del nervo interosseo posteriore
Compressione a livello del tratto inferiore del tunnel radiale: ispessimento fibrotico del
muscolo breve supinatore (arcata di Frohse) da attività lavorativa (in genere continui
movimenti di prono-supunazione)
Sintomi:
•
•
Dolore: localizzato alla regione epicondiloidea, spesso irradiato all’avambraccio in sede
postero-esterna, sino al polso. E’ diurno ma spesso permane anche durante le ore notturne,
con riacutizzazioni. Nel caso di compressione del nervo interosseo posteriore il dolore è
sordo, non bem delimitabile coinvolgendo la regione epicondiloidea simulando la patologia
muscolo-tendinea e osteoarticolare
Limitazione funzionale: diminuzione della forza alla estensione delle metacarpo falangee e
all’abduzione –estesnione del pollice. Nei casi avanzati, paresi dei muscoli innervati
distalmente alla compressione (estensori comuni e propri delle dita ed estensore del pollice)
con estensione conservata del polso ma con deviazione radiale per insufficienza
dell’estensore ulnare del carpo.
Diagnosi:
•
•
•
•
•
Cratteristiche del dolore: diurno e notturno, talora con riacutizzazioni notturne, irradiato
lungo il lato postero-esterno dell’avambraccio sino al polso.
Dolore evocato: la compressione sul canale supinatorio, distalmente al capitello radiale
evoca dolore
Dolore evocato dalla supinazione contro resistenza a gomito esteso: il nervo viene messo in
tensione dell’estensione del gomito e viene compresso dalla contrazione del muscolo breve
supinatore
Dolore alla pronazione passiva forzata dell’avam,braccio a gomito esteso
EMG
Terapia medica:
•
•
Nelle fasi iniziali riduzione o sospensione delle attività manuali, eventuale uso di ortesi,
FANS
Terapia chirurgica: neurolisi del nervo interosseo posteriore previa sezione dell’arcata
fibrosa del muscolo breve supinatore ed eventuale disinserrzione del muscolo breve
supinatore dall’epicondilo.ù
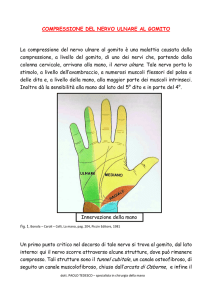
SINDROME DEL TUNNEL ULNARE AL GOMITO
Definizione: compressione del nervo ulnare nel canale osteofibroso al gomito
Eziopatogenesi:
•
•
Compressione del nervo ulnare a livello dell’arcata del flessore ulnare del carpo: da fibrosi
perineurai reattive, traumi, iperutilizzazione del comparto mediale del gomito con ipertrofia
del flessore ulnare del carpo ed ispessimento dell’arcata fibrosa di Osborne.
Compressione del nervo ulnare a livello della doccia epitrocleo-oleocranica: da traumi o
alterazioni degenerative che comportano deviazione in valgismo del gomito, da processi
sinovitici articolari in corso connettivopatie, da ipertrofia del legamento epitrocleooleocranico e nella iperlassità legamentosa congenita.
Sintomi:
•
•
•
Turbe sensitive: parestesie generalmente episodiche, al quinto dito e metà ulnare del quarto
dito con ipoestesia progressivamente ingravescente alla sola mano. Il dolore insorge
secondariamente con l’aggravamento delle parestesie, a livello periepitrocleare e sul
territorio di innervazione del nervo ulnare alla mano
Turbe motorie: diminuzione della funzione di presa della mano, specie nella presa polliceindice
Turbe trofiche: amiotrofia del primo spazio interosseo, lieve ipotrofia dei muscoli ipotecari
ed interossei. Con l’aggrvarsi atrofia muscolare con mano ulnare.
Diagnosi:
•
•
•
Comprasa di dolore e parestesie sul territorio del nervo ulnare alla flessione forzata del
gomito
Dolore alla percussione e palpazione sul tunnel cubitale
EMG
Terapia:
•
•
•
•
Riduzione e/o sospensione dei lavori manuali
Eventuale uso di ortesi
Terapia medica con FANS
Nella fase sensitivo-motoria e paralitica sezione dell’arcata del flessore ulnare del carpo e
del legamento epiotrocleo-oleocranico, neurolisi, epinerviotomia longitudinale anteriore,
epicondilectomia mediale ed eventuale trasposizione anteriore del nervo nei casi di gomito
valgo e salienze ossee post-traumatiche nella doccia epitrocleo-oleocranica.
NEUROCHIRURGIA PEDIATRICA
SPINA BIFIDA
La spina bifida è un difetto di chiusura dell’arco posteriore vertebrale (schisi) che si può associare
ad una mancata chiusura del rubo neurale e dei suoi involucri meningei. La più comune
localizzazione è quella lombare e sacrale, rara è l’estensione toracica.
L aspina bifida occulta è generalmente asintomatica e si caratterizza con il soito difetto di chiusura
ossea coinvolgente una o più vertebre. La
Presenza di un ciuffo di peli o di un angioma cutaneo in corrispondenza della schisi può essere
l’unico rilievo clinico.
La spine bifida cistica è un difettio di chiusura più complesso e si distingue:
•
•
•
Il meningocele che è una formazione cistica meningea a contenuto liquorale che occupa la
schisi ed è rivestita da un piano cutaneo integro. Raramente vi è una compromissione delle
radici nervose.
Il mielomeningocele è il classico e più comune difetto di chiusura del tubo neurale con
esposizione della placca neurale embrionale circondata da aracnoide e dura che si
impegnano nella schisi ossea, formando una cisti a contenuto liquorale. Il piano cutaneo è
interrotto e circonda la malformazione neurale esposta in superficie. La compromissione
delle radici nervose è costante.
Il lipomielomeningocele è una malfor,azione che si caratterizza con la presenza di un lipoma
aderente alle radici nervose (placide) che, attraverso una soluzione di continuo durale e la
schisi ossea, si continua con la voluminosa massa lipomatosa sottocutanea. Frequentemente
si manifesta una compromissione radicolare.
La spina bifida cistica ha una incidenza dello 0.6-4 per mille nati per anno e rappresenta il 30% di
tutte le malformazioni del sistema nervoso. Il mielomeningocele è la forma più frequente. La causa
che induce il difetto di chiusura non è nota. Attualmente viene somministrata nel primo trimestre di
gravidanza alla mamma acido folico. Questa terapia ha ridotto drasticamente ultimamente
l’incidenza di spina bifida.
Mielomeningocele
E’ la malformazione congenita più complessa perché al difetto spinale si associano quasi
costantemente (nel 90% dei casi) altre due malformazioni: la sindrome di Arnold Chiari e
l’idrocefalo.
Il difetto del tubo neurale condiziona nel neonato, in rapporto al livello e alla estensione della
lesione, un defcit motorio e senstivo agli arti inferiori di tipo radicolare con deficit sfinteriali
(vescica neurologica). A volte i neonatio affetti da tale malformazione presentano anomalie
osteoarticolari agli arti inferiori (piede torto) per alterati sviluppo da denervazione delle masse
muscolari. Il mielomeningocele costituito dalla placca neurale esposta e dall’esile membrana
aracnoidea può ulcerasi facilmente con fuoriuscita di liquor esponendo il piccolo paziente a gravi
complicanze infettive come la meningite o la meningo-encefalite. Si pone quindi la necessità di
riperare chirurgicamente il difetto neurale nelle prime ore di vita. L’idrocefalo triventricolare
concomitante deve essere tratta con un intervento di terzoventricolostomia o di derivazione
ventricolo peritoneale. L’ectopia dele tonsille cerebellari nel forame magno e nel canale rachideo
cervicale (S. di Arnold Chiari) condiziona raramente sintomi neurologici nel periodo neonatale. La
comparsa di una sintomatologia da compressione bulbo-midollare (deficit della suzione, difficoltà
respiratoria, ipostenia arti superiori) impone un intervento di decompressione craniospinale.
Lipomielomeningocele
I sintomi neurologici di questa affezione possono essere inizialmente modesti ma presentano una
notevole possibilità evolutiva. Possono comparire deficit radicolari per incremento della massa
lipomatosa intradurale con effetto compressivo e di trazione sulle radici lombari e sacrali. Inoltre
con l’accresxcimento possono comparire disturbi midollari dovuti ad uno stiramento del midollo
impedito nella sua risalita all’interno del canale spinale dall’aderenza con il lipoma (tethered cord).
Meningocele
E’ la forma più semplice di spina bifida cistica, libera da sintomi neurologici. L’intervento attuato
nei primi giorni di vita è finalizzato alla asportazione della cisti durale extrarachidea e alla
ricostruzione del piano durale meningeo.
SENO DERMICO
Il seno dermico, definito anche seno pilonidale, è un tragitto fistoloso rivestito da epitelio che dalla
superficie cutanea dorsale si approfondì fino a raggiungere o l’arco posteriore di una vertebra o
attraverso una schisi vertebrale il canale rachideo e lo spazio sottodurale. E’ dovuto ad una
incompleta separazione , nelle prime settimane di gestazione tra l’ectoderma che darà origine alla
placca nervosa dallo strato che svilupperà la cute ed i suoi annessi. La sua posizione è mediana e la
sede più frequente è a livello sacro-coccigeo (seno dermico spinale) e raramente a livello cranico.Si
caratterizza con un piccolo orifizio cutaneo, a volte ricoperta dapeli o cute iperpigmentata, che si
continua con un tragitto fistoloso rivestito da epitelio squamoso pliristratificato con annessi cutanei
(ghiandole sebacee, sudoripare, follicoli piliferi) E CHE TERMINA NELLO SPAZIO
SUBDURALE AVENDO RAPPORTI DI CONTINUITa’ CON LE STRUTTURE NERVOSE. Il
seno pilonidale può associarsi ad altre malformazioni quali il mielomeningocele ed il
lipomielomeningocele. La malformazione può terminare nella sua parte intradurale con una cisti a
contenuto cheratinico (cisti epidermoide) o contenente annessi cutanei e sebo (cisti dermoide) Il
seno dermico sacro-coccigeo può infettarsi e l’infezione propagarsi nello spazio intradurale e
causare una meningite. Se si associa la presenza di una cisti epidermoide o drermoide nellom spazio
endorachideo può manifestrsi una sindrome da compressione radicolare o midollare. La RMN
permette una diagnosi di certezza. L’intervento chirurgico da effettuarsi precocemente, consiste
nell’asportazione radicalòe del tragitto fistoloso e della eventuale cisti epidermoide o dermoide
concomitante.
DIASTEMATOMIELIA
Consiste nella divisione di un tratto del midollo spinale e dei suoi involucri meningei attorno ad
uno sperone osseo che origina dalla faccia posteriore di un corpo vertebrale e divide il canale
spinale in due metà, in ognuna delle quali è contenuto un emi-midollo circondato dalle meningi.E’
una malformazione rara e si può associare alla spina bifida occulta o cistica a lipomi e a fistole
dermiche. Di solito ha sede a livello toracico o lombare.
ENCEFALOCELE
Questa malformazione è riconducibile ad un difetto di chiusura del tubo neurale che coinvolge il
settore cranico; viene anche definita cranioschisi. E’ un difetto di chiusura delle ossa craniche e
attraverso la soluzione di continuo ossea, si realizza l’erniazione o della dura (meningocele) o di
una parte dell’encefalo rivestito dalle meningi (meningoencefalocele). Si sviluppano sulla linea
mediana a livello della volta cranica prevalentemente in sede occipitale e a livello del basicranio
anteriore nella regione fronto-etmoidale. Il classico encefalocele occipitale è già evidente alla
nascita come una massa, con dimensione comunemente di pochi cm di diametro, che sporge a
livello della fontanella occipitale, rivestita da cute a volte angiomatosa. A volte la grandezza della
malformazione è mostruosa. Il meningocele è una cisti a contenuto liquorale in comunicazione con
lo spazio subaracnoideo di solito molle riducibile che si tende con il pianto. Il meningoencefalocele
invece è una massa solida, non comprimibile che non varia la propria tensione con il pianto. Fli
encefaloceli fronto-etmoidali sono rilevabili alla nascita per la presenza di deformazioni della
piramide nasale o di ipertelorismo (aumentata distanza interoculare). Non infrequentemente si
rilevano durante l’età giovanile o per la comparsa di unam rinoliquorrea spontanea o per sintomi di
ostruzione rspiratoria. Nelle forme fronto-etmoidali l’encefalo è costantemente coinvolto
manifestandosi come meningoencefalocele. La diagnosi è ottenibile con studio TC ed RMN
encefalo. La correzione chirurgica del meningocele cranico prevede l’asportazione della sacca
meningea e la chiusura della breccia durale ed ossea. Il trattamento del menongoenecefaslocele
consiste nel riposizionamento del tessuto cerebrale erniato all’interno del cranio e nella
ricostruzione del piano durale ed osseo.
SINDROME DI DANDY-WALKER
Questa alterazione dello sviluppo cerebrale consiste nella atresia congenita dei forami di Magendie
e di Luschka. Questi forami mettno in comunicazione il quarto ventricolo con lo spazio
subaracnoideo (cisterne) di fossa posteriore. Ne consegue una enorme dilatazione del quarto
ventricolo con ipoplasia del verme e a volte degli emisferi cerebellari. L’ostacolato deflusso
liquorale causa un idrocefalo tetraventricolare no comunicante. Questa la formazione esordisce con
una sindrome di ipertensione endocranica che si manifesta nei primi mesi di vita del bambinio. La
Diagnosi è ottenuta con TC ed RMN encefalo ed il trattamento chirurgico prevede l’impianti di una
derivazione ventricolo-peritoneale.
CRANIOSINOSTOSI
La craniosinostosi o craniostenosi è un ridotto sviluppo del cranio per precoce saldatura di una o più
suture. Normalmente il volume dell’encefalo si raddoppia nei primi dodici mesi di vita, si triplica
nei primi 24 mesi e raggiunge l’80% del volume definitivo intorno ai tre anni di età.
La chiusura delle fontanelle si completa tra i 12 e i 18 mesi, ma il cranio può ancora espandersi
perché le suture non sono saldate definitivamente. Il cranio continua ad espandersi per la spinta che
riceve dall’encefalo. Quindi la precoce saldatura delle suture causa un impedimento al fisiologico
sviluppo accrescimento del cranio e l’encefalo rimane compresso nel suo contenitore in espandibile.
Si distinguono vari tipi di craniostenosi in rapporto alla sutura che riamne coinvolta dalla precoce
saldatura. Alcune forme associano alterazioni dello sviluppo facciale e si definiscono
malformazioni cranio-faciali complesse. La scafocefalia si caratterizza con un cranio lungo e stretto
ed è coinvolta la sutura saggitale. Nella brachicefalia il cranio è corto e largo e le suture colpite
sono la coronaria e la lambodoidea. La plagiocefalia si manifesta con un capo assimetrico
anteriormente da precoce saldatura di metà sutura coronaria. Nella trigonocefalia il cranio assume
un aspetto triangolare con apice anteriore per precoce chiusura dela sutura metodica. Quando tutte
le suture si saldano precocemente il cranio si sviluppa verso l’alto (turricefalia) o verso l’avanti
(oxicefalia). Le malformazioni cranio-faciali complesse, anche definite facio-craniostenosi,
coinvolgono il basicranio anteriore e le strutture osse del massiccio facciale. Il fulcro
dell’alterazione è a livello della regione etmoido-sfenoidale e della sutura coronaria. Si manifesta un
dimorfismo faciale con ipertelorismo, esoftalmo, ridotto sviluppo del basicranio e della volta
cranica anteriormente con asimmetria cranica in senso antero-posteriore (sindrome di Crouzon e
malattia di Apert se si associano malformazioni degli arti: sindattilia). Queste malformazioni
presenti alla nascita si accentuano nei primi mesi di vita quando si realizza il massimo sviluppo
cranico. Parallelamente al dimorfismo cranico si manifestano i sintomi neurologici da graduale
ipertensione endocranica: irrequietezza, vomito, strabismo convergente, papilla da stasi. Questi
sintomi iniziali a volte subdoli possono evolvere in atrofia ottica post-stasi, rallentamento dello
sviluppo psico-motorio, ipertono spastico e crisi epilettiche. La radiografia del cranio permette di
dimostrare l’assimmetrico sviluppo. La TC encefalo e le ricostruzioni tridimensionali permettono di
studiare in dettaglio l’alterato accrescimento. La terapia chirurgica si pone l’obbiettivo di favorire il
normale accrescimento del contenitore cranico.
SINDROME DI ARNOLD CHIARI
La sindrome di Arnold Chiari è una malformazione della giunzione cranio-vertebrale che si
caratterizza con uno spostamento (ectopia) delle tonsille cerebellari verso il basso, che si impegnano
nel forame magno e occupano la parte più alta del canale rachideo. Si distinguono diversi gradi di
ectopia tonsillare, nelle forme più gravi concomita lo spostamento verso il basso anche del verme
cerebellare e del quarto ventricolo. Questa affezione congenita si associa frequentemente con altre
malformazioni come il mielomeningocele, il lipomielomeningocele e la siringomialia. Raramente si
presenta in forma isolata. I sintomi clinici sono in relazione all’effetto compressivo che le strutture
erniate hanno sul passaggio bulbo-midollare costretto in uno spazio ristretto quale è il forame
magno. La sintomatologia raramente esordisce in epoca neonatale, più comunemente in età
giovanile ed adulta. Il Chiari associato alla spina bifida può causare raramente dei sintomi precoci
nel neonato quali: disturbi della deglutizione e della suzione, ipertono al tronco ed agli arti,
opistotono ed insufficienza respiratoria con fasi di apnea. Più comune,mente si manifesta in età
giovanile causando una siringomielia (cavità all’interno del midollo in vicinanza del canale
centrale).
La diagnosi è fatta con la RMN encefalo. La decompressiva chirurgica del forame magno con
rimozione dell’arco di C1 associata ad una plastica durale è la procedura chirurgica d’elezione.
TUMORI PEDIATRICI
Tra tutti i tumori dei bambini, i tumori cerebrali sono secondi come incidenza solo alle leucemie e
rappresentano il più comune tumore solido pediatrico (40-50% di tutti i tumori). L’incidenza è di 25 casi su 100.000 abitanti.
I tumori più comuni sono i glomi (del cervelletto, del tronco encefalico e del nervo ottico), i tumori
della pineale, i craniofaringiomi, i teratomi, i granulomi ed i tumori neuroectodermici primitivi
(PNET tra cui il più comune è il medulloblastoma). E’ comunem,ente ritenuto che la maggior parte
dei trumori pediatrici sia a sede sottotentoriale (60%). I tumori del cervello che si presentano
durante il primo anno di vita tendono ad avere un differente istitipo rispetto a quelli che si
presentano più tardivamente. Il 90% dei tumori nei neonati sono di origine neuroectodermica, con i
teratomi tra i più comuni. Alcuni di questi tumori possono essere congeniti.Altri tumori
sopratentoriali includono gli astrocitomi, i tumori del plesso corioideo, gli ependimomi ed i
craniofaringiomi. I tumori di fossa posteriore includono i medullobalstomi e gli astrocitomi
cerebellari. La maggior parte di questi tumori sfuggono alla diagnosi fino a che non hanno
raggiunto dimensioni ragguardevoli a causa della elevatissima elasticità della teca cranica. L
manifestazioni cliniche più frequenti sono il vomito, l’arresto o la regressione dello sviluppo
psicomotorio, la macrocefalia e l’arresto motorio.
Astrocitoma pilocitico
E’ benigno.Le sedi più frequenti sono nell’ordine gli emisferi cerebellari, la regione otticochiasmatica e gli emisferi cerebrali. Se sopratentoriali hanno una localizzazione mediana. Nella
forma cerebellare il tumore presenta una duplice componente, una solida ed una cistica la quale può
assumere notevole dimensione. Clinicamente si manifesta con il tipico quadro della sindrome da
ipertensione endocranica e disturbi cerebellari a graduale evoluzione.; condiziona costantemente un
idrocefalo triventricolare. E’ possibile l’asportazione totale della neoplasia ottenendo la completa
guarigione. La localizzazione ottico-chiasmatica condiziona la comparsa di deficit campimetriciche
gradualmente possono evolvere in amaurosi. La sede della neoplasia, il suo sviluppo verso
l’ipotalamo non permette l’asportazione totale della lesione configurandosi quindi una malignità di
sede. Il raro astrocitoma pilocitico a sede cerebrale presenta un comportamento biologico simile a
quello cerebellare: è asportabile radicalmente. Le crisi epilettiche focali sono i sintomi prevalenti di
esordioQuesto tumore è da considerarsi veramente l’unico benigno nell’ambito delle neoplasie
gliali. Rarissima è l’evoluzione maligna.
Medulloblastoma
E’ un tumore molto maligno a rapida crescita. Incide per l’11% nell’ambito dei glomi. Colpisce
particolarmente l’età pediatrica e meno frequentemente i giovani. Si sviluppa solo nel cervelletto di
solito nel verme invadendo il IV ventricolo, le tonsille cerebellari e a volte gli emisferi; quindi a
sede mediana-paramediana. La rapide crescita condizione una ingravescente ipertensione
endocranica (idrocefalo) e non infrequentemente ad un iniziale sindrome di impegno delle tonsille
cerebellari (ernia tonsillare, capo deviato e rigidità nucale). La chirurgia permette una apparente
radicalità, ma le recidive sono costanti. Inoltre è una neoplasia che favorisce una metastatizzazione
per via liquorale generalmente a livello spinale. E’ sensibile alla radioterapia che viene attuata sia su
tutto l’enecefalo che sul midollo spinale per limitare la diffusione liquorale. Comunque anche
associando la chemioterapia, la prognosi rimane severa con una sopravvivenza media di
3 anni.