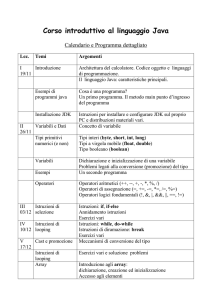
COSTRUZIONE
DI UN CDNA-MICROARRAY
ORIENTATO ALL’ANALISI DELL’ESPRESSIONE GENICA
DI CELLULE APOPTOTICHE UMANE
1.0 INTRODUZIONE 2
1.1 GENE EXPRESSION PROFILING 2
1.2 I MICROARRAY
1.2.1 GENE CHIP PER SINTESI DIRETTA SUL SUPPORTO 3
1.2.2 CDNA ARRAY 5
1.2.3 OLIGONUCLEOTIDE ARRAY
1.3 PROCEDURE
1.3.1 DISEGNO DEGLI OLIGO-SONDA (OLIGONUCLEOTIDE ARRAY)
1.3.2 PREPARAZIONE DEI CAMPIONI 6
1.3.3 RILEVAZIONE CON LASER-SCANNER 7
1.4 A COSA SERVE UN MICROARRAY ? 8
2.0 STRATEGIA ADOTTATA 8
2.1 OBIETTIVO 8
2.2 SELEZIONE DEI GENI PER L’ARRAY 9
2.3 SCELTA DEL SUPPORTO 9
2.4 SCELTA DEL PROBE 9
2.5 SCELTA DEI CDNA DA SPOTTARE 10
2.6 SCELTA DEI CONTROLLI 10
2.7 MARCATURA DEI PROBE 11
3.0 PROTOCOLLI SPERIMENTALI PER LA COSTRUZIONE DI CDNA
MICROARRAY 12
3.1 PREPARAZIONE DI UN SUPPORTO DI VETRO 12
3.2 PREPARAZIONE DEL CDNA DA SPOTTARE 13
3.3 PROTOCOLLO RT-PCR 13
3.4 PROTOCOLLO PCR 14
3.4.1 MIX PCR 14
3.4.2 Tempi di incubazione: 14
3.4.3 Controlli di efficienza per RT-PCR 14
3.5 DISEGNO DEI PRIMER * 14
3.6 SCELTE OPERATIVE PER PROTOCOLLO PCR 15
3.7 SPOTTING 16
3.8 PREPARAZIONE FINALE DELL’ARRAY 17
4.0 PROTOCOLLI PER L’UTENTE 17
4.1 SINTESI DOPPIO FILAMENTO CDNA MEDIANTE RT 17
4.1.1 Amounts of Reverse Transcriptase 18
4.1.2.First Strand cDNA Synthesis Components 18
1
4.1.3 Second strand synthesis 18
4.2 CLEANUP OF DOUBLE STRANDED CDNA 19
4.3 PREPARAZIONE DEL CRNA BIOTINILATO 19
MEDIANTE TRASCRIZIONE IN VITRO 19
4.5 CLEANUP DEL CRNA BIOTINILATO 20
4.6 PROTOCOLLO DI FRAMMENTAZIONE DEL CRNA 20
4.7 IBRIDAZIONE 21
4.7.1 Prehybridization of array 21
4.7.2 Wash array 21
4.7.3 Stripping e reprobing 22
4.8 ANALISI DATI 22
4.8.1 ANALISI PRELIMINARE ASSOLUTA 22
4.8.2 ANALISI COMPARATIVA 24
INTRODUZIONE
Gene expression profiling
Per "gene expression profiling" si intende l'espressione ordinata e temporale
dell'informazione genica1.
Lo studio delle differenze nel pattern di espressione genica é uno dei più
promettenti approcci per la comprensione dei meccanismi di differenziamento e
sviluppo. Infatti, essendo i processi cellulari governati dal repertorio dei geni
espressi e dai livelli ed i tempi di espressione, diviene importante avere degli
strumenti sperimentali per il diretto monitoraggio di numerosi geni in parallelo2.
Numerose sono le metodiche disponibili per lo studio della espressione genica,
molte delle quali si basano sulla rilevazione e sulla quantizzazione degli mRNA.
Tre sono i gruppi di metodiche comunemente usate per lo studio
dell'espressione genica 3:
1. Tecniche basate sull'ibridazione Northern Blotting 4RNase-protection assay
5
Differenzial plaque hybridization Subtraction cloning DNA microarray
2. Tecniche basate sulla PCR Differential display 6RDA (representational difference
analysis) 7
3. Tecniche basate sulla sequenza ESTs (expressed sequence tags) SAGE (serial
analysis of gene expression) 8
2
Il Northern Blotting e l'RNase protection hanno come limite quello di rilevare un
solo RNA per volta e di richiedere tempi molto lunghi oltre che grandi quantità di
campione9.
Il Differential display e l'RDA presentano lo svantaggio di dare risultati
generalmente non di tipo quantitativo e di non essere sempre affidabili, in
quanto frequente é l'incidenza di falsi positivi.
Il SAGE, invece, richiede procedure molto complicate per la preparazione dei
campioni, risulta quindi una metodica alquanto laboriosa e non particolarmente
sensibile.
Una metodica che cerca di superare tutti gli svantaggi sopra menzionati é
quella del DNA microarray. Principale vantaggio di tale approccio é quello di
consentire un' analisi comparativa e contemporanea, oltre che molto rapida,
dell'espressione di migliaia di geni10.
I Microarray11
Ideati nei primi anni novanta, consentono l'analisi contemporanea di un immenso numero
di geni mediante un'unica reazione d'ibridazione con migliaia di sonde immobilizzate su un
supporto solido,vetro o nylon12, chiamato chip o array.
A seconda del tipo di probe si distinguono in cDNA e Oligonucleotide Microarray. Un Gene
chip non è altro che un particolare tipo di oligo microarray che si differenzia per la modalità
con cui gli oligo vengono sintetizzati.
Gene chip
(ottenuti per sintesi diretta sul supporto)
Gli oligonucleotide array prodotti adattando la tecnologia fotolitografica dei
semiconduttori alla sintesi chimica degli oligonucleotidi sono propriamente detti
13
GeneChip.14
Sono costituiti da un supporto solido non poroso (vetro15) della grandezza 1.28
X 1.28 cm2 suddiviso in aree da 5 X 10 -2 x 5X10 -2 cm2 contenenti ognuna circa
10 7 oligo identici lunghi 20 basi.
La superficie del supporto solido viene modificata utilizzando gruppi MeNPOC
((R,S)-1-(3,4-(Metilendiossi)-6-Nitrofenil)Etil Cloroformio) che impediscono il
legame dei desossinucleosidi.
Tali gruppi sono fotolabili e, se illuminati da una luce UV che passa attraverso
dei fori presenti su una maschera fotolitografica, sono rimossi dando origine a
dei gruppi idrossili.
3
A questo punto un 5'-0-fosforamidite-deossinucleoside attivato viene presentato
alla superficie del chip e può avvenire il legame con i siti che erano stati esposti
alla luce.
Questo desossinucleoside è a sua volta protetto in 3' da un gruppo MeNPOC, la
superficie viene quindi esposta di nuovo alla luce che passa attraverso una
seconda maschera fotolitografica e nuovi gruppi idrossido vengono liberati per
una seconda reazione, e così via 16.
Il processo viene ripetuto fino a che non è sintetizzato l'intero numero di
oligonucleotidi sonda e di controllo che è stato programmato.
Ogni singolo gene è rappresentato da 20 aree distinte,ognuna contenente 107
copie di uno di 20 oligonucleotidi disegnati in base alla sequenza del gene in
studio in maniera tale da poter distinguere tra geni membri di famiglie
strettamente correlate e anche tra varianti di splicing di uno stesso gene.
Per minimizzare gli effetti di una ibridazione aspecifica ciascuna area è
fiancheggiata da un' area di controllo i cui oligonucleotidi differiscono per una
singola base nella zona centrale dagli oligo-sonda scelti per la caratterizzazione
del gene.
4
cDna array
Questi array vengono prodotti "spottando" sul supporto alcuni nanolitri di prodotti di PCR
purificati che rappresentano i geni di interesse. In questo caso ogni gene è rappresentato
dall’ unica area in cui il suo amplificato è stato spottato. In questo array i controlli non sono
rappresentati da sonde mismatched dello stesso gene ma sono controlli indipendenti che
utilizzano geni particolari ( vedi scelta dei controlli).
Il processo di spotting è eseguito da un robot "arrayer" che sfrutta il principio della
Deposizione Piezoelettrica:17
1. Un capillare di vetro, in cui è contenuto il probe , è inserito in un “collare” di
ceramica.
2. Una differenza di potenziale applicata alla ceramica causa la contrazione del
liquido contenuto nel capillare.
3. Tale contrazione permette il rilascio di una microgoccia di c.a. 200 picoL .
La differenza di potenziale applicata ad alta frequenza (1000 Hz) provoca il rilascio di
1000 gocce al secondo dispensando così
5
Se come supporto è utilizzato il vetro esso è rivestito da poli-lisina o amino-silani
che ne aumentano l'idrofobicità favorendo l'aderenza del DNA ; un ulteriore
trattamento con anidride succinica riduce l'adesione aspecifica del DNA .Il DNA
è poi cross-legato al supporto attraverso irradiazione ultravioletta.
Successivamente viene denaturato al calore o a pH 18alcalino.19
Oligonucleotide array
Questo array unisce l’ utilizzo degli oligo sintetizzati in vitro (e non direttamente sul
supporto!) con la metodica dello spotting mediante Arrayer.
Procedure
Disegno degli oligo-sonda (oligonucleotide array)20
La sequenza degli oligo-sonda (probe) che ibrida con i loci bersaglio del gene
d'interesse deve prendere in considerazione i seguenti parametri:21
composizione in basi per uniformare il Tm
presenza di appaiamenti G/C all'estremità 3' aumenta l'efficienza di ibridazione
usare almeno 200-300 basi rappresentative del gene di interesse ( da EST o
cDNA).
i 20 tipi di oligo , che rappresentano il gene, devono ,se possibile, contenere
sequenze non "overlapping"
sequenze specifiche che individuino inequivocabilmente il gene in studio.
preferenzialmente scelte al 3' dell'mRNA per ridurre problemi derivanti dalla
retrotrascrizione o dalla parziale degradazione dell'mRNA.
Preparazione dei campioni
Il campione che può essere costituito da DNA22 o da RNA viene estratto dalle
cellule di interesse, marcato ed infine ibridato sull’array.
L'mRNA viene retrotrascritto in vitro a cDNA e utilizzato come tale oppure
trascritto di nuovo in RNA (cRNA). La miscela di cRNA viene prima sottoposta a
frammentazione prima dell' ibridazione.
6
La marcatura può avvenire impiegando nucleotidi coniugati a fluorocromi o
biotina:
per il campione di DNA durante la fase di PCR
per il cRNA durante la ritrascrizione del cDNA
La marcatura con biotina sarà evidenziata mediante il legame con streptavidina
coniugata a un flurocromo.
A questo punto il campione può essere ibridato.
La resa dell'ibridazione aumenta notevolmente :
In tutti i tipi di array, frammentando i cRNA ad una lunghezza vicina a quella delle sonde
sull'array così da eliminare le strutture secondarie che potrebbero interferire con l'ibridazione.
Nei gene chip, introducendo tra la superficie e gli oligo-sonda degli "spacers"
costituiti da esaetilenglicol che diminuiscono l'ingombro sterico delle sonde.11,13
Rilevazione con Laser-scanner
Dopo che l'ibridazione è avvenuta, il cRNA marcato può essere individuato sull’array
mediante l'emissione di luce visibile quando il fluorocromo viene stimolato da un laser. La
luce emessa viene catturata da un microscopio confocale che ne registra l'intensità. I dati
sono quindi trasferiti ad un computer e con specifici software è possibile elaborare i dati di
fluorescenza per valutare la presenza di un particolare trascritto.
Un campione e il suo controllo possono essere marcati con lo stesso fluorocromo su slide differenti
oppure sulla medesima slide con fluorocromi diversi (come in figura sotto)
7
Inoltre, dato che l'intensità di fluorescenza è proporzionale alla concentrazione di un
particolare cRNA e quindi alla concentrazione di quel particolare messaggero all'interno
della cellula, il calcolo fornisce anche valutazioni quantitative e comparative sui livelli di
espressione in diversi campioni.
Nota: Non solo l’efficienza di trascrizione ma anche l’emivita del messaggero influenza l’intensità
del segnale.
E’ possibile quindi individuare RNA che hanno frequenze bassissime all'interno della
miscela di reazione, anche di 1/300.000.
A cosa serve un microarray ?
per valutare l'espressione genica in un pool di cDNA.
per un'analisi quantitativa dei livelli di specifici mRNA.
creare un nuovo genere di mappa genica
per identificare modificazioni dell'espressione genica in seguito a stimoli di diversa natura.
(es. agenti patogeni,farmaci etc.)
per l'analisi comparativa dell'espressione in condizioni normali e patologiche ad esempio in
cellule tumorali.
genotipizzazione
STRATEGIA ADOTTATA
Obiettivo
Lo scopo é quello di realizzare un microarray in grado di valutare qualitativamente e
quantitativamente l’espressione genica in cellule umane apoptotiche ed identificare eventuali
pathway non ancora conosciuti.
8
Il microarray proposto potrà essere utilizzato dall’utente per studiare la risposta apoptotica in
condizioni sperimentali diverse:
Stimoli apoptotici diversi
Stimoli applicati per tempi diversi
Estrazione di RNA a tempi diversi dall’applicazione dello stimolo
Stimoli applicati su diverse linee cellulari
Selezione dei geni per l’array
Secondo i seguenti criteri è stato selezionato un migliaio di geni:
Geni umani noti coinvolti in apoptosi
EST23 umane coinvolte in apoptosi
EST umane apparentemente non coinvolte24
Geni umani cardine del ciclo cellulare ed oncogeni
Geni non correlati di specie non umana
Scelta del supporto
E’ stato scelto come supporto il vetro poiché può garantire:
Il DNA può essere covalentemente legato alla superficie di vetro il vetro è un
materiale che non si deteriora con il tempo e con le alte temperature.
L'assenza di porosità limita i fenomeni di diffusione del probe25 spottato consentendo
l'utilizzo di un minimo volume di ibridazione.
Riduce al minimo il segnale di fluorescenza di fondo non specifico.
Migliore resa di ibridazione e rilevazione grazie alla sua rigidità .
Scelta del probe
Tre le scelte a disposizione:
Oligo sintetizzati sul supporto
Oligo spottati sul supporto
cDNA spottati
L’esclusione di oligo sintetizzati con la fotolitografia é stata dettata dal fatto che tale tecnica é sotto
brevetto dell’ AFFYMETRIX C.
L’opzione degli oligo presintetizzati presenta diversi vantaggi:
alta specificità
possibilità di controllo dell’ibridazione per ciascun oligo introducendo singoli mismatch in
oligo di controllo
9
velocità di sintesi in automatico
tale scelta, però, é stata scartata per i seguenti limiti operativi:
il disegno degli oligo prevede la disponibilità di un software specifico per la scelta di
sequenze specifiche non ridondanti
alto costo
difficoltà di deprobing26 per successivo uso.
La tecnica di spot dei cDNA è stata quindi valutata come quella più rispondente alle nostre esigenze
operative per:
facilità di disegno e disponibilità di software per la scelta dei primer
stime di specificità e sensibilità non di molto inferiori rispetto alla tecnologia degli oligo.
possibilità di deprobing.
Scelta dei cDNA da spottare
1. cDNA library
2. mRNA totale
La prima alternativa prevede lunghi tempi di esecuzione :
trasformazione
selezione dei cloni mediante sonde specifiche
ciclo di amplificazione
sequenziamento
quindi l’estratto totale di un mRNA é da preferire per i bassi costi e per la rapidità dell’estrazione e
del successivo processamento:
RT-PCR
Amplificazione con primer specifici
Sequenziamento
Scelta dei controlli
Dovendo garantire controllo e normalizzazione dell’efficienza di ibridazione sono stati inseriti i
seguenti controlli:
Controlli con geni housekeeping umani( G-3-PDH, actina). Rappresentano dei controlli
positivi dell’ibridazione. Inoltre, poiché questi geni presentano livelli di espressione simili in
cellule sottoposte a stimoli diversi, permettono di normalizzare 27i dati di fluorescenza ottenuti
da più slide (ad esempio ibridando su una slide il campione ricavato da cellule apoptotiche e su
un’altra il campione da cellule non stimolate).
10
Controlli con cDNA di geni non umani che presentano diversi gradi di
omologia con il gene umano prescelto.
Tutto questo permette di stimare grossolanamente il grado di
affidabilità dell’ ibridazione:
ad esempio, se si ottengono segnali positivi dal gene umano (100% di
omologia ) e da quello di Cercopiteco (97% di omologia ) ma non da
quello di cavallo (94% di omologia ), si può affermare che
indicativamente l’array riesce a distinguere fra loro 2 geni con grado di
omologia superiore a 94% ma inferiore a 97%.
Il gene prescelto è quello della -Actina umana confrontata con
quella di Cercopithecus aethiops (97% di omologia ), di Ovis aries
(94% di omologia ), di Equus caballus (94% di omologia ), di Mus
musculus (91% di omologia ).
Il software on line “Blast” ha permesso l’ analisi delle omologie di sequenze, tutte catalogate
in Gene Bank.
Clicca qui per vedere i risultati dell’ analisi di sequenza mediante Blast 2.0
Controlli con geni di piante (Arabidopsis).Possono avere una duplice funzione:
1)Controlli negativi per verificare ibridazione non specifica poiché tali mRNA non si
appaiano a geni umani.
2)Quantizzazione del campione.
Concentrazioni note di un certo gene vengono ibridate con sonde per lo stesso gene in un
esperimento indipendente. I segnali sono associati alle diverse diluizione così da ottenere
una “barra dei colori” con la quale i segnali di fluorescenza vengono confrontati per
quantizzare il campione.
Marcatura dei probe
Tecnica con radioattivo
Tecnica con fluorocromi
Tecnica immunoenzimatica
Metodi con rilevazione radioattiva non consentono alta risoluzione.
La fluorescenza presenta maggiori problemi di background e minore sensibilità rispetto al metodo
immunoenzimatico.
Il metodo immunoenzimatico consente sensibilità maggiore dovuta alla possibilità di amplificazione
del segnale e specificità grazie al legame antigene-anticorpo.
11
PROTOCOLLI SPERIMENTALI PER LA
COSTRUZIONE DI CDNA MICROARRAY
Preparazione di un supporto di vetro
E’ necessario rivestire il supporto con poli-lys amilosilani o silani aminoreattivi per intensificare sia
l’idrofobicità del supporto che l’aderenza del DNA al supporto, limitando la dispersione del DNA
quando spottato sul supporto.
Materials
Qty
Glass microscope slides 60
Slide rack 28
Slide chamber
2
29
30
6
ddH2O
~5 L
NaOH
70 g
95% Ethanol
420 mL
Poly-L-lysine
70 mL
Vacuum oven (45°C)
PBS tissue culture
70 mL
Slide box (plastic only) 1
1. Place slides in slide racks. Place racks in chambers.
2. Prepare CLEANING SOLUTION:
Dissolve 70 g NaOH in 280 mL ddH2O.
Add 420 mL 95% ethanol. Total volume is 700 mL (= 2 X 350 mL); stir until
completely mixed.
If solution remains cloudy, add ddH2O until clear.
3.
Pour solution into chambers with slides; cover chambers with glass lids. Mix for
2 hr.
Once slides are clean, they should be exposed to air as little as possible. Dust
particles will interfere with coating and printing.
4.
Quickly transfer racks to fresh chambers filled with ddH2O. Rinse vigorously by
plunging racks up and down.
Repeat rinses 4X with fresh ddH2O each time. It is critical to remove all
traces of NaOH-ethanol.
5. Prepare POLYLYSINE SOLUTION:
12
70 mL poly-L-lysine + 70 mL tissue culture PBS in 560 mL water.
6. Transfer slides to polylysine solution and shake 15 min. - 1 hr.
7.
Transfer rack to fresh chambers filled with ddH2O. Plunge up and down 5X to
rinse.
8.
Centrifuge slides on microtiter plate carriers (place paper towels below rack to
absorb liquid) for 5 min. at 500 rpm.
Transfer slide racks to empty chambers with covers for transport to vacuum
oven.
9. Dry slide racks in 45°C vacuum oven for 10 min. (Vacuum is optional.)
10. Store slides in closed slide box (plastic only, without rubber mat bottom)
11.
BEFORE PRINTING ARRAYS:
Check that polylysine coating is not opaque.
Test print, hyb and scan sample slides to determine slide batch quality.
Preparazione del cDNA da spottare
Si estrae mRNA totale dalle linee cellulari prescelte, contenenti i geni selezionati, utlizzando
QIAGEN’s Rneasy Total RNA isolation kit.
La qualità dell’RNA è essenziale per il successo dell’analisi dell’espressione genica, sia nella fase
di costruzione del cDNA microarray che nella fase di utilizzo dell’utente.
Nota: Giacché il protocollo più appropriato per l’isolamento dell’RNA può dipendere dalla sua
fonte, si raccomanda di utilizzare un protocollo che é normalmente usato per quel tessuto o tipo
cellulare.
Protocollo RT-PCR
Miscela di reazione RT:
Reagenti
MgCl2
TrisHCl
KCl
dNTP
Oligo d(T)
Conc. finale
5mM
10mM
50mM
1mM(each)
RNAsin
1U/L
RT
2,5U/L
RNA totale
1-2g/40-50L
2,5M
13
Temperature di incubazione:
10 min
30 min
10 min
25°C
42°C
95°C
Protocollo PCR
Prelevare 20L del prodotto RT e aggiungerli nei tubi eppendorf con la mix per la reazione PCR.
MIX PCR
Componente
Conc.finale
Conc.iniziale
buffer
dNTP
F.Primer*
10x
10mM
1x
200mM(each)
50M
50M
R.Primer*
50M
5U/mL
25mM
50M
25 U/mL
1mM
Taq
MgCl2
Tempi di incubazione:
Denaturation:
5min
95°C
20s
20s
30s
94°C
55°C
72°C
30 cycles:
denaturation:
annealing:
elongation:
Controlli di efficienza per RT-PCR
Amplificazione di un gene standard espresso in tutte le cellule
( mRNA actina) per controllare l’integrità dell’RNA dopo il processo di estrazione e come
controllo dell’efficienza di RT
Per ogni serie di campioni amplificati inserire sempre un controllo negativo costituito da
H2O. Ciò controlla la presenza di contaminanti.
Disegno dei primer *
14
I primer sono stati scelti con il programma Primer Express 4.0 che consente di settare la lunghezza
dell’amplificato a seconda delle proprie necessità e fornisce gli estremi per una reazione di PCR
multiplex.
A titolo esemplificativo i primer sono stati disegnati per il gene (BNIP3L) considerando il metodo
utilizzato come prototipo per la realizzazione dei primer per tutte le altre sequenze inserite nell’
elenco “Geni chip”.
Le sequenze di domini comuni a tutte le proteine BCL2-like sono state individuate facendo una
ricerca con Omni-blast e sottratte alla sequenza del cDNA di BNIP3L.
Le regioni restanti del cDNA sono state considerate potenziali target per la scelta delle coppie di
primer.
Questo tipo di analisi, esteso poi a tutti gli altri geni, permetterà dunque di minimizzare le
interferenze di segnale dovute all’ibridazione di sequenze appartenenti alla stessa famiglia genica.
Forward primer
Start:522
Lunghezza:20
GACCCGAAAACATTCCACCC
Reverse primer
Start:622
Lunghezza:21
AATACCCCCTTTCTTCATGGC
Forward primer
Start:133
Lunghezza:22
CAACTGCGAGGAAAATGAGCAG
Reverse primer
Start:252
Lunghezza:19
AGCCCCCCATTTTTCCCAT
Forward primer
Start:587
Lunghezza:22
ATGAGGAAAAGTGGAGCCATGA
Reverse primer
Start:705
Lunghezza:22
CCAATATAGATGCCTAGCCCCA
Tm:60°C
%GC:55
Tm:58°C
%GC:48
Tm:61°C
%GC:50
Tm:61°C
%GC:53
Tm:60°C
%GC:45
Tm:59°C
%GC:50
Scelte operative per protocollo PCR
Lunghezza dei primer: dovrebbe essere compresa tra 20-30 bp ( comunque non inferiore a
16 bp per assicurarsi la specificità del processo).
Contenuto di GC compreso tra 45-50%
Evitare distribuzioni di sequenze inusuali come serie di purine o pirimidine che potrebbero
dare appaiamento di primer
15
Le estremità 3’ dei primer non devono essere tra loro complementari per evitare i dimeri.
La concentrazione dei primer: 1M è sufficiente per almeno 30 cicli di amplificazione.
MgCl2 è il parametro più critico.
Il buffer usato ha un pH=8.3. Quando la reazione è a 72°C il pH diminuisce ad un valore
di 7.2. Per questo la presenza di cationi bivalenti rappresenta il punto cruciale( Mg++) così
come l’assenza di agenti chelanti.
Inoltre ricordare che Mg++ agisce attivando la Taq e favorendo l’annealing.
La presenza di dimeri può essere minimizzata ottimizzando la
concentrazione di
MgCl2
Concentrazioni troppo alte favoriscono annealing aspecifici.
La concentrazione dei dNTP di 200M è sufficiente a sintetizzare 12.5g DNA quando la
metà degi dNTP è incorporata. Inoltre dNTP sequestrano Mg++ e concentrazioni troppo elevate
potrebbero inibire la Taq.
Scelta DNA polimerasi: la Taq Pol è attiva a 72°C. A questa temperatura procede ad una
velocità di 60 nucleotidi per secondo.
Per realizzare una PCR multiplex: il disegno dei primer deve tenere conto di cinetiche di
reazione simili.
I prodotti post-PCR devono essere controllati mediante sequenziatore automatico.
Caratteristiche degli amplificati
Lunghezza di c.a. 100bp
Tm comparabile( tutti i geni devono ibridare nelle stesse condizioni ! )
Sequenza del gene selezionato che maggiormente possa distinguerlo dai suoi omologhi.
Protocollo di precipitazione per purificare i prodotti di PCR
1.
2.
3.
4.
5.
6.
7.
Aggiungere 1/10 volume 3 M NaAc, pH 5.2, and 2.5 volumi etanolo.
Mix e incubare a -20°C over night.
Centrifugare a ~12,000 x g in una microcentrifuga per 20 min a 4°C.
Aggiungere 100 L 70% etanolo tenuto in ghiaccio
Centrifugare 30 min come al punto 3.
Asciugare il pellet all’aria.
Risospendere il pellet in DEPC-treated H2O 31 over night.
Aggiunta di un Carrier :
Glicogeno
0.5 to 1 L of (5 mg/mL) di glicogeno aggiunti alla precipitazione possono favorire la
visualizzazione e il recupero del pellet. Il glicogeno sembra non influenzi i successivi passaggi.
Spotting
16
Sospendere i cDNA precipitati in SSC 2x.
La concentrazione finale dei campioni dev’essere stabilita in base alla grandezza degli spot
desiderati, tenendo conto anche della viscosità dei campioni stessi.
Qualora i campioni risultino troppo viscosi è opportuno diluirli per avere omogenea disposizione
sullo spot. Per altre informazioni consultare Aecom site.
Preparazione finale dell’array
Eseguita per:
fissare gli spots all’array ( fissaggio mediante UV)
disattivare i gruppi aminosilani rimasti scoperti dopo lo spotting
1.
2.
3.
4.
5.
6.
Orientare la slide segnandola con la matita
Idratare al vapore la slide con l’array rivolto verso l’acqua bollente.
crosslinkare il DNA al vetro mediante UV con Stratalinker a 65 mJ
Reidratare come al punto 2.
Riscaldare su piastra per 3’’
Immergere la slide per 15' agitando nella seguente soluzione:
0.56 g di Succinic anhydride in 36 ml di N-methyl-pyrrilidinone + 4 ml di 0.3 M Boric
acid pH 8.0
7. Immergere la slide in 0.1% SDS 10-20 secs
8. Immergere la slide in ddH2O x 10-20 secs
9. Bollire a 95 °C per 3-5 min
10. Porre la slide in etanolo freddo
Adesso l’array è pronto per essere ibridato.
PROTOCOLLI PER L’UTENTE
Sintesi doppio filamento cDNA mediante RT
Occorrono almeno 5 g di RNA totale, la cui qualità è essenziale per la riuscita del saggio,si
consiglia un rapporto 260/280 tra 1.9 e 2.1 (considerando 1 OD a 260 nm = 40 g /mL di RNA).
Se ci sono contaminanti tale rapporto sarà significativamente minore e l’efficienza della reazione
sarà diminuita.
Per la sintesi si utilizza come primer purificato un oligo dT(24).
La temperatura indicata nei protocolli deriva dalla constatazione sperimentale che l’optimum
d’azione per l’enzima RT è 42°C e che a 92-95°C la sua attività è bloccata.
17
Amounts of Reverse Transcriptase
Total RNA (g)
RT (L), 200U/L
5.0 to 8.0
1.0
8.1 to 16.0
2.0
16.1 to 24.0
24.1 to 32.0
32.1 to 40.0
3.0
4.0
5.0
First Strand cDNA Synthesis
Components
Step 1: primer hybridization
Incubate at 70° C for 10
minutes
Quick spin and put on ice
Reagents in reaction
Volume l
DEPC- H2O32 (variable)
final vol 20
RNA (variable, 5.0 -40,0g)
5.0 to 40 g
5.0 to 40
g
1l
(dT)24 primer (100
pmol/L)
100 pmol
5X First strand cDNA buffer
Step 2: temperature adjustment 0.1M DTT2
Incubate at 42° C for 2 minutes
10 mM dNTP mix1
Step 3: first strand synthesis
Mix well,
Incubate at 42° C for 1 hour
Final
Conc.
RT
4
1X
2
10 mM
DTT
1
See Table
RT
Total Volume
500 M
each
200 U to
1000 U
20
Second strand synthesis
Component
DEPC-treated water
5X Second Strand Reaction
Buffer
lOmM dATP, dCTP, dGTP,
dTTP
10 Ul / DNA Ligase
Volume l
Final concentration or
amount in reaction
91
30
1X
3
200 M each
1
10U
18
10 U/l DNA Polymerase I
4
40U
2 U/l RNase H
1
2U
Final Volume
150
Place first strand reactions on ice. Centrifuge briefly.
Add the reagents listed in the table
Gently tap tube to mix. Then, briefly spin in a microcentrifuge to remove
condensation
Incubate at 16°C for 2 hours in a cooling waterbath.
Add 2 ml[10 U] T4 DNA Polymerase.
Incubate for 5 minutes at 16°C
Add 10 ml 0.5 M EDTA
Proceed to cleanup procedure for cDNA or store at –20°C for later use.
Cleanup of double stranded cDNA
Precipitation of cDNA
1. Aggiungi 1/10 volume 3 M NH4Ac, pH 5.2, and 2.5 volumi etanolo.
2. Mix e incuba a -20°C over night.
3. Centrifuga a ~12,000 x g in una microcentrifuga per 20 min a 4°C.
4. Aggiungi 100 L 70% etanolo tenuto in ghiaccio
5. centrifuga 30 min
6. Asciuga il pellet all’aria.
7. Risospendi il pellet in 12 L DEPC-treated H2O over night.
Aggiunta di un Carrier : Glicogeno 0.5 - 1 L di (5 mg/mL)
Nota: La quantificazione del cDNA ds non può esser effettuata con lo spettrofotomentro perchè
l’assorbanza a 260 nm è influenzata dalla presenza dei primers.
Preparazione del cRNA biotinilato
mediante trascrizione in vitro
La preparazione consiste in una reazione di trascrizione in vitro a partire dal cDNA utilizzando una
miscela di dNTP contenente CTP e UTP biotinilati.
1. A temperatura ambiente miscelare i reagenti (vedi tabella)
Attenzione! Il DTT nel 10X transcription buffer può precipitare a freddo
2. Incubare 4-6 h a 37°C
Note:
19
l’incubazione overnight può ridurre la resa dei prodotti più grandi di 200 nt
è importante utilizzare come nucleotidi modificati biotin-11 CTP e biotin-16 UTP
.Nucleotidi modificati in altre posizioni possono non lavorare bene per questa applicazione
Reagents
14.5 l NTP labelling
mix
Mix:
7.5 mM ATP
7.5 mM GTP
5.625 mM cold UTP / 1.875 mM bio-UTP
5.265 mM cold CTP / 1.875 mM bio-CTP
2.0l of 10X T 7 transcription buffer
1.5 l of resuspended cDNA of the 12 l ds cDNA double strand synthesis
2.0 l 10X T7 enzyme mix
Cleanup del cRNA biotinilato
Dal cRNA così prodotto è essenziale rimuovere nucleotidi non incorporati cosicché gli RNA
marcati possano essere quantizzati mediante assorbanza a 260 nm.
I metodi utilizzati per la purificazione dei prodotti della IVT 33utilizzano colonnine
cromatografiche(Qiagen Rneasy columns) .
È consigliabile controllare l’integrità del cRNA mediante gel elettroforesi.
Nota:bisogna assolutamente escludere l’utilizzo di estrazione con fenolo/cloroformio che
implicherebbe perdita di RNA biotinilato a causa della partizione della biotina nella fase organica.
Protocollo di frammentazione del cRNA
Risospendere il cRNA così da ottenere un volume piccolo che durante la procedura di
frammentazione minimizzerà la quantità di magnesio nel cocktail di ibridazione.
Si otterranno frammenti tra 35-200 basi.
1) Aggiungere 2 l di fragmentation buffer 5X per ogni 8 l di RNA in acqua ( la
concentrazione finale di RNA può essere tra 0.5 g/l e 2 g/l).
2) Incubare a 94°C per 35 min. e mettere in ghiaccio.
3) Conservare a – 20 °C fino all’ibridazione.
5X fragmentation buffer :
200mM Tris-acetate pH 8.1 ; 500mM KOAc ; 150mM MgOAc
Nota:E’ necessario un controllo pre e post frammentazione su gel
(occorre 1 g di cRNA se si usa EtBr , meno con SYBR green).
20
Ibridazione
5 g del campione da ibridare devono essere denaturati e subito raffreddati in ghiaccio.
Si utilizza poi una soluzione di ibridazione contenente formamide : essa presenta una
cinetica di ibridazione più lenta rispetto ad una sol. acquosa, comporta però un minor
background; in alternativa si può usare una soluzione contenente PEG se target ha un
basso numero di copie.
Prehybridization of array
Place 15 µl prehybridization solution on top of array (use dummy slide)
Place coverslip over array
Place slide in hybridization chamber and incubate for at least two hours in a 50 °C
water bath
Add 10 µl dd H2O (or mix of 50% formamide/dd H2O) in each corner of chamber to
maintain humidity
Hybridization
Remove cover slide and wipe off excess solution from edge of slide
Add 20 µl hybridization solution over array
Place new coverslip on array
Place in hybridization chamber
Hybridize O/N in a 50°C water bath
Wash array
Place slide in 50 ml tube with 1x SSC/0.1% SDS
Shake gently so that coverslip falls off
Place slide in slide holder/glass dish with 200ml 0.2x SSC/0.1% SDS
Shake slide for 10 minutes
Place slide in holder with 200 ml 0.2x SSC and wash for 20 min
Repeat this step
Make sure all residual SDS is removed
(you may repeat last wash step with less SSC)
Place slide in 50 ml tube and spin for 5 minutes at 1000 rpm to dry slide (array
facing outwards)
21
Stripping e reprobing
Immergere l’array in dimetil-formamide a 65°C per 1 h
Lavato 2 volte per 5 min in SSC 2X contenente 0.1 %
porre in 40 ml di NaOH 0.4 M a 60° C per 90 min
incubare in 5 ml di soluzione neutralizzante (SSC 0.1X SDS 0.1% Tris HCl 0.2M pH 7.5) a
temperatura ambiente 15 min
Nota : la denaturazione e la neutralizzazione potrebbero essere ripetuti per
assicurare un completo stripping del DNA probe
Prehybridization solution
Hybridization solution
3.5 ml formamide
2 ml 20x SSPE
0.5 ml 10% SDS
0.5 ml 50x Denhardt's
0.2 ml ss salmon sperm
DNA
3.3 ml ddH20
10 ml total
700 µl formamide
(35%)
50 µl 20% SDS
(0.5%)
100 µl 50x
Denhardt's (2.5x)
400 µl 20x SSPE (4x)
1.25 ml total
ANALISI DATI
Nota: Per portare a termine l’elaborazione dei dati descritta in questa sezione l’utente deve
possedere uno array-scanner e un programma in grado di elaborarne i dati.
Se l’utente è sprovvisto del materiale suddetto si occuperà della preparazione del campione (fino
alla sez. 4.6 ) , affidando le procedure di ibridazione e rilevazione a compagnie che offrono servizi
biotecnologici avanzati 34
Sarà compito dell’utente far pervenire alla compagnia il campione secondo le modalità indicate
dalla stessa.
Analisi preliminare assoluta
Consente di determinare l’indice di fluorescenza assoluta per ciascuno spot presente su un cDNA
microarray.
22
1) scanning dei segnali di fluorescenza:
Uso di un microarray scanner .
Esempio :GenePix 4000 (Axon Instruments)
2) acquisizione dell’immagine risultante come file dati. DAT e
file immagine .BMP
3) Uso di un software per l’analisi dei dati per la disposizione automatica di una griglia sopra il
DAT file che demarca ciascuno spot e per la determinazione della intensita’ media di
fluorescenza per ciascuna area
4) raccolta dei dati e dell’ immagine per ulteriore analisi in opportuni database
Alcuni esempi di software per l’analisi dei dati sono :
ScanAlyze (scaricabile dal sito inserendo la propria mail)
OptiQuant (vedi in questa figura una grid che consente ad esempio di evidenziare i
singoli spot collegando le loro coordinate ad una flag).
Il software utilizzato :
assegna coordinate (X,Y) che corrispondono alla posizione di ciascun campione sull’array .
calcola l’ intensita’ media per ciascuno spot attraverso le intensita’ dei pixels contenuti in
ciascuna area.
Evidenzia l’area con l’intensita’ di fluorescenza piu’ bassa ,il 2% della quale sara’ sottratto
dall’ intensita’ media di tutte le aree poichè rappresenta il background .
Il background è il segnale causato dall’autofluorescenza della superficie
dell’array , dai legami aspecifici del campione nonche’ da molecole residue.
Calcola le variazioni nei segnali di intensità pixel per pixel per ciascuna area e ne determina
la deviazione standard stimando così il noise.
23
Il noise è rappresentato dalle piccole variazioni nel segnale di fluorescenza
acquisito dallo scanner entro ciascuna area .
Il software consente inoltre l’utilizzo di :
PROBE MASKS :
per “mascherare” aree di ibridazione non significative
(es: contenenti geni con regioni omologhe ridondanti ,ad esempio la BCL-2 family).
PROBE SET MASKS :
per selezionare quali aree di ibridazione possono essere utili nell’ interpretazione del
fenomeno biologico considerato (apoptosi).
Analisi comparativa
Consente di determinare il cambiamento relativo nella intensita’ di fluorescenza per ciascun
trascritto esaminato.
Utilizza i dati di due distinti esperimenti su cDNA microarray.
Il primo esperimento è condotto su un campione “basale”.
Esempio: cellula
normale
Il secondo su un campione “sperimentale”.
Esempio: cellula apoptotica
Questo tipo di analisi permette così una rilevazione più accurata dei cambiamenti di espressione
genica rilevanti da un punto di vista biologico.
Fattori non biologici possono contribuire alla variabilità dei dati ad esempio:
quantità e qualità del campione aggiunto all’array
quantità utilizzata di rilevatore (streptavidina-ficoeritrina)
variabilità generale nelle intensità di ibridazioni
Sono però necessarie una normalizzazione 35 ed uno scaling36 per minimizzare le differenze in tutti i
segnali di intensità tra i due array.
Normalizzazione
L’array sperimentale puo’ essere “normalizzato” cioè reso equivalente all’array basale .
L’intensità media del normalizzato si ottiene moltiplicando l’intensità media dell’array basale per
un fattore di normalizzazione.
L’ intensità media dell’intero array è la media di tutti i valori d’intensita’ media di fluorescenza per
ciascuna area –esclusi il 2% del valore piu’ alto e il 2% del valore piu’ basso-.
Scaling
Un singolo fattore di scaling per ogni area viene calcolato dal software assegnando il valore 1 alla
fluorescenza delle aree dei controlli basali su ciascun array.
Gli indici di fluorescenza vengono calibrati indipendentemente per ciascun array utilizzando questo
fattore.
24
Per ogni trascritto viene poi calcolato il fold change che è la differenza tra l’intensità media
sperimentale (normalized/scaled) e l’intensità media basale (normalized/scaled).
Questo ulteriore parametro consente di evidenziare una possibile espressione genica differenziale in
presenza di stimolo apoptotico.
Autori :
V.Gasperi
A.Livigni
M.Mazzone
N.Montesano Gesualdi
G.Prencipe
C.Quintarelli
Tutor: Prof. S. Cocozza
1
Velculescu, V.E., Zhang, Volgelstein, B. & Kinzler, K. W. Serial analysis of gene expression. Science 270, 484487 (1995)
2
Lockart, D.J., Dong, H., Byrne, Follettie, Gallo Expression monitoring by hybridation to high density
oligonucleotide arrays. Nat. Biotechnol. 14, 1675-1680 (1996)
25
3
Kozian & Kirschbaum;. Comparative gene expression analysis. TibTech 17, 73-78 (1999)
4
Alwine, Kemp, D.J. & Stark. Method for detection of specific RNAs in agarose gel by transfer to diazobenzylpaper and hybridization with DNA probes.PNAS 74,5350-5354(1977)
5
Berk ; Sizing and mapping of early adenovirus mRNAs by gel electrophoresis of S1 endonuclease-digested
hybrids.Cell 12,721-732(1977)
6
Liang . Differential display of eukariotic messenger RNA by means of the polymerase chain reaction .Science
257,957-971(1992)
7
Lisitsyn N., Wigler M. Science 259,946-951 (1993)
8
Velculescu, V.E., Zhang, Volgelstein, B. & Kinzler, K. W. Serial analysis of gene expression. Science 270, 484487 (1995)
9
Lockart, D.J., Dong, H., Byrne, Follettie, Gallo Expression monitoring by hybridation to high density
oligonucleotide arrays. Nat. Biotechnol. 14, 1675-1680 (1996)
10
. David J., Duggan , Bittner , Chen . Expression profiling using cDNA microarrays . Nature Genectics
(supplement) 21,10-14 (1999)
11
Array:l’ insieme del supporto (slide) e delle sonde geniche (probe).
12
Atlas array www.clonetech.com
13
. Brown P.O. & Botstein D. Exploring the new world of the genome with DNA microarray. Nat Genet. 1999
Jan;21(1 Suppl):33-37. Review
14
. Lipshutz R.J. et al. High density synthetic oligonucleotide arrays. Nat. Gen. suppl.1 ; ( 21) jan 99
15
Fodor Sp et al . Light directed spatially addressable parallel chemical synthesis. Science 1991; 251:767-73
www.affimetrix.com
16
Singh-Gasson S. et al. Maskless fabrication of light-directed oligonucleotide microarrays using a digital
micromirror array Nat.Biot. 17; oct 99 974-78
17
Cheung VG, Morley M, Aguilar F, Massimi A, Kucherlapati R, Childs G.Making and reading
microarrays.
Nat Genet. 1999 Jan;21(1 Suppl):15-19. Review.
18
Shalon D, Smith SJ, Brown PO.
A DNA microarray system for analyzing complex DNA samples using two-color
fluorescent probe hybridization.
Genome Res. 1996 Jul;6(7):639-45.
19
Bowtell DD.Options available--from start to finish--for obtaining expression data bymicroarray.Nat Genet.
1999 Jan;21(1 Suppl):25-32. Review.
20
Per il razionale della scelta dei primer per le sonde dei cDNA array si veda la sezione Disegno dei primer
Southern E. et al. Molecular interctions on microarrays. Nat. Gen. suppl.1 ; ( 21) jan 99
22
Nel caso del genotyping
23
EST : Expressed Sequence Tag
24
Inserite di routine per identificare nuovi geni eventualmente coinvolti in apoptosi.
25
Il cDNA sonda spottato sulla slide di vetro
26
procedura che permette di rimuovere il campione precedentemente ibridato per poter riutilizzare la stessa slide
27
Uniformare i dati di fluorescenza
28
supporto per la slide di vetro
29
cameretta per la slide
30
acqua Rnasi free
21
26
31
acqua Rnasi free
acqua Rnasi free
33
In Vitro Transcription
34
Vedi i link citati
35
Uniformare i dati di fluorescenza
36
Bilanciamento
32
Ciao a tutti! Buona lettura e buon lavoro!!! ( siate clementi….)
Bibliografia
Oltre alle references consultate integralmente ( vedi note e links) per questo lavoro sono stati
raccolti molti titoli, alcuni dei quali consultati parzialmente (abstract) .
Essendo molto numerosi non sono stati inseriti qui ma potete consultarli in microarray references
Links
http://www.affymetrix.com/
http://www.interactiva.de/vlab/xna.html
http://www.packardinstrument.com/
http://www.biol.rug.nl/lacto/bsmicroarray.html
http://www.brunel.ac.uk/depts/bl/project/biocomp/sequence/seqanal_guide/glossary.html
http://ruly70.medfac.leidenuniv.nl/~gtc/arrayNL.html
http://www.biodiscovery.com/
http://www.genelogic.com/
http://www.imagingresearch.com/
http://www.microarrays.com/
http://www.resgen.com/
http://www.biol.rug.nl/lacto/www.scienceonline.org
http://www.nhgri.nih.gov/DIR/CGB/TMA/index.html
http://cmgm.stanford.edu//pbrown/
http://genome-www.stanford.edu/
27
28