II LEZIONE
FARMACOLOGIA:
farmacocinetica
(distribuzione ed
eliminazione per
escrezione)
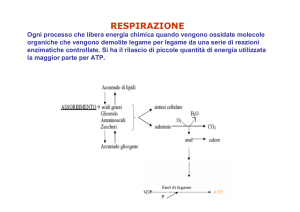
• Somministrazione farmaco
• Assorbimento
• Distribuzione (raggiungimento equilibrio tra conc. plasm. ed
extraplasm.)
• Eliminazione
(metabolizzazione o escrezione)
Distribuzione-eliminazione:
Caso lidocaina
fase iniziale rapida caduta
(caduta conc. plasm. x uscita da letto vascolare
e distribuzione ai tessuti)
fase tardiva più lenta
(caduta conc. plasm. x processi di
metabolizzazione ed escrezione)
Distribuzione
Insieme di fenomeni alla base del trasporto del F
tra i diversi compartimenti dell’organismo.
Importanti:
aspetto di equilibrio, relativo a concentrazione;
aspetto cinetico, relativo a velocità.
Dipendenza da:
•PM
•Idro-liposolubilità
(F idrosol. in spazi interstiziali;
F liposol. anche dentro cellule).
Concetto di AVIDITA’
(rapporto tra concentrazione tessutale totale del F
e
concentrazione plasmatica dello stesso)
(rimane costante fino a nuova somministrazione,
x cui è indicata con Kp)
La conc. di 1 F all’equil. dip. dalla Kp del tessuto.
Distribuzine F all’equilibrio:
volume effettivo diverso da quello reale (Vd apparente)
Volume di distribuzione
Parametro dal quale dipende
la concentrazione ematica di un F.
La massa dei liquidi dell’organismo può essere
suddivisa in tre distinti compartimenti:
plasmatico, interstiziale ed intracellulare.
L’acqua extracellulare è la somma di quella contenuta
nel plasma e nei liquidi interstiziali.
Una sostanza può avere un basso o un alto volume di
distribuzione, a seconda che si distribuisca
esclusivamente nei liquidi all’interno dei vasi o anche
negli spazi extracellulari e all’interno delle cellule.
Vd = Q / concentrazione plasmatica
(Q= quantità di F somministrata)
Ogni tessuto o organo ha un suo Vd dato da
Kp x Vreale
Vd molto elevato (maggiore volume acqua corporea),
Kp maggiore 1
1 + tessuti accumulano il F (deposito)
Le sedi principali di accumulo dei F sono:
- fegato e reni (grazie ad una proteina che
trasporta gli anioni organici dal plasma
all’epatocita, legando anche i corticosteroidi e i
coloranti azoici cancerogeni, ed un’altra, la
metallotioneina, che lega con varia affinità il Cd, Zn,
e altri metalli);
- tessuto adiposo (per incorporazione fisica nei
grassi neutri);
- tessuto osseo (x gli scambi tra la superficie
dell’osso, costituita da cristalli di idrossipatite, e i
fluidi interstiziali a contatto con essa.
Per ogni farmaco
Kp e Vd di ogni compartimento,
insieme al flusso ematico specifico ed alle caratteristiche di
permeabilità
del letto capillare regionale,
permettono di stimare
• la concentrazione del F all’equil.
• la velocità con cui tale equil. è raggiunto:
La concentrazione di equil. sarà Kp volte quella plasmatica
La velocità sarà data dal rapporto:
Flusso /Vapp= flusso/VxKp= flusso specifico/Kp
Farmaco nel sangue
• Libero
• Legato a proteine plasmatiche
• Legato a cellule circolanti
• È importante conoscere la quota libera
Legame F-proteine plasmatiche
Grado di legame = % concentrazione
F legato/ concentrazione totale F
Varia tra 0 ed 1:
Maggiore 0.9, F fortemente legato
Minore 0.2, F scarsamente o x nulla legato
Importante Albumina
(Alfa-1 globulina→molecole basiche)
L’entità del legame con le proteine
varia da un composto ad un altro.
Ad esempio:
- l’antipirina non si lega affatto;
- il secobarbitale per il 50%;
- il warfarin per il 99%.
Le proteine plasmatiche possono legare
tanto le sostanze acide (fenilbutazone)
quanto quelle basiche (imipramina) o
neutre (digitossina). La concentrazione
totale della sostanza è data dalla somma
della frazione legata e di quella libera.
La quota legata alle proteine può essere
quindi calcolata per differenza tra la
frazione legata e quella libera.
Il legame dei F alle proteine plasmatiche è legato
all’affinità di legame.
La quantità di f legata x mole di proteina sarà:
C Fleg/Cproteine = n x C Flib/K+C Flib
dove:
n è il n. di siti di legame su ogni proteina
C la concentrazione
K la costante di dissociazione del complesso
F o sostanze endogene possono
spiazzare altri F dal loro legame
alle proteine plasmatiche,
modificandone la quota libera
(Casi warfarina-F acidi,
sulfamidici-bilirubina)
La distribuzione in vari organi e tessuti dipende:
• dal flusso ematico distrettuale;
• dalla velocità di diffusione dai capillari agli spazi
interstiziali e alle cellule;
• dall’affinità della sostanza per i componenti dei tessuti.
Il passaggio nelle cellule avviene sia per diffusione
passiva che mediante processi
di trasporto specializzato.
Fattori importanti nel determinare la distribuzione sono
inoltre:
- formazioni di legami con cellule circolanti;
- Dissoluzione della sostanza in tessuti che sono sede
di accumulo o di deposito nell’organismo (fegato,
tessuto adiposo, tessuto osseo).
L’ accumulo può essere visto come un meccanismo
protettivo che fa diminuire livelli plasmatici e quindi la
concentrazione della sostanza nella sede d’azione.
Le dimensioni dell’organo
determinano il gradiente di
concentrazione tra sangue e tessuti
Casi
• muscolo (ampia superficie)
• cervello (organo più piccolo)
Flusso ematico importante nella
velocità di captazione
Organi ben perfusi raggiungono più
rapidamente elevate concentrazioni di F
rispetto a tessuti meno perfusi.
Permeabilità tessutale
• Un F può passare dal sangue al tessuto
per gli stessi processi attraverso cui è
assorbito
• Anche gli ostacoli (barriere) da superare
sono fli stessi
Una zona meno permeabile rispetto alle altre nell’organismo,
è la barriera emato-encefalica (BEE).
La ridotta permeabilità dipende da fattori anatomici o
fisiologici definiti:
1) presenza di giunzioni serrate delle cellule endoteliali nei
capillari del cervello ed assenza di pori;
2) presenza di carrier proteico ATP dipendente che trasporta
le sostanze in direzione del sangue;
3) i capillari del SNC sono avvolti da processi delle cellule
gliali (astrociti);
4) bassa concentrazione di proteine nel liquido
interstiziale rispetto agli altri liquidi fisiologici.
L’efficienza della BEE varia a seconda dell’area cerebrale.
Si pensa che i fattori limitanti l’ingresso di sostanze molto
lipofile nel cervello siano:
- un forte legame con le proteine del plasma o con le
lipoproteine;
- la particolare composizione del tessuto nervoso.
Alcuni F sembrano entrare nel
cervello attraverso carrier. Ad
esempio il metilmercurio si
combina con la cisteina formando
una struttura simile alla metionina.
La BEE non è completamente
sviluppata alla nascita ed è questa
una delle ragioni che determinano
la maggior tossicità di certe
sostanze nel neonato
rispetto allo adulto.
La velocità di distribuzione di un F
tra il sangue ed un tessuto
è limitata dalla
PERFUSIONE, quando il F
sufficientemente lipofilo da
attraversare le barriere, e/o dalle
permeabilità locali.
Distribuzione = cinetica di I ordine
• Emivita = tempo necessario xkè la
concentrazione tra i 2 compartimenti si dimezzi
T1/2 = 0.693 / Kt
• Kt = flusso / Vd = flusso / Kp x Vt
QUINDI
T1/2 = 0.693 x Kp x Vt / flusso
Se la C di F è mantenuta costante
La C tessutale del F continuerà a salire fino
a raggiungere un equilibrio x il quale tanto
F entra nel tessuto che altrettanto ne esce.
Il tempo necessario a raggiungere
l’equilibrio dipenderà dall’emivita: maggiori
sono il V e l’avidità del tessuto x il F, +
tempo ci vorrà a raggiungere l’equilibrio
La velocità di distribuzione del F
tra il sangue ed il tessuto
sarà in questo caso limitata dalla
PERMEABILITÀ, qualora il F
non fosse libero di diffondere dal
sangue al tessuto e viceversa.
Eliminazione dei farmaci
• Per metabolizzazione
• Per escrezione
Un F può esser metabolizzato x eliminazione molto prima che
venga escreto dall’organismo.
Per F con metaboliti attivi l’escrezione rappresenta, invece,
la modalità di eliminazione
Caso fenossibenzamina!
Anche l’eliminazione segue una
cinetica di ordine I
Maggiore sarà la quantità di F presente
nell’organismo, maggiore sarà la quantità
di F eliminata nell’unità di tempo.
Emivita di eliminazione
(quale sia la quantità di F presente nel
corpo, è il tempo necessario perché la
concentrazione plasmatica si dimezzi)
Vd / T1/2
• In linea di max, F con con grosso volume
di distribuzione hanno anche un’emivita
lunga perché il farmaco eliminato è
continuamente rimpiazzato da quello
accumulatosi nei depositi; il contrario si ha
con bassi Vd.
Alterazioni patologiche degli organi
di eliminazione aumentano l’emivita
fel F, prolungandone gli effetti.
Clearence
Se la quantità di F che si è distribuita è data da:
Vd x Cp
la quantità eliminata,
nell’unità di tempo,
sarà la quantità presente in un Vpl pari a Ke x Vd; quindi:
Qe = Vd x Cp X Ke
E’ come se Ke x Vd litri di plasma fossero ripuliti (cleared) del F nell’unità di t
La CL è il V di sangue virtualmente ripulito nell’unità di tempo dai processi di
eliminazione, quindi
CL = Ke x Vd
x cui
Qe = Cp x CL
ke è costante (dipende dall’efficienza dei processi di eliminazione)
Vd è costante e dipende dalla costituzione fisica del paziente)
T1/2
Vd
CL
Stretta correlazione
T1/2 = 0.693/Ke = 0.693 x Vd/CL
La CL totale sarà data dalla somma
delle CLs
• Si parla di CL renale (CLR) e CL non
renale (CLNR), quindi:
CLtot = CLR + CLNR
La CL di un F sufficientemente estratto
da un organo è limitata dal flusso: il
sangue è completamente depurato
dal F nel suo passaggio x l’organo
Escrezione
I F vengono eliminati dall’organismo attraverso
diverse vie:
-
renale (la più importante);
Epatica
intestinale (con le feci);
respiratoria (principalmente le sost. gassose);
Altre vie di minore importanza sono:
sudore, saliva, secrezioni lacrimali, latte, bile, liquido
cerebrospinale.
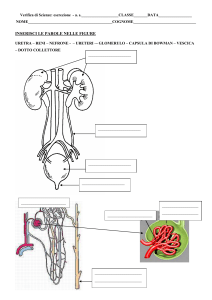
Escrezione urinaria
I reni eliminano i composti chimici esogeni con
gli stessi meccanismi che intervengono per
l’allontanamento dei prodotti finali del
catabolismo:
filtrazione glomerulare, secrezione tubulare,
per diffusione passiva o trasporto attivo.
Vai ad altra presentazione
• Come si deduce dall’equazione di
Handerson-Hasselbalch, le sostanze acide
sono escrete, cioè non riassorbite dal
tubulo, in maggiore misura ad alti valori di
pH urinario; l’opposto (limitata escrezione)
si ha a valori di pH acidi.
Ad esempio il grado di ionizzazione del fenobarbitale
(pKa=7.2) può essere notevolmente aumentato
modificando il pH urinario verso l’alcalinità; perciò in
caso di avvelenamento da barbiturici, la
somministrazione di una base (bicarbonato sodico)
riduce la quota di farmaco riassorbito nel tubulo con
conseguente aumento della sua
eliminazione urinaria.
Analogamente l’alcalinizzazione delle urine aumenta
l’eliminazione renale dei salicilati.
I diuretici possono perciò accelerare l’eliminazione di
acidi e basi organici/che deboli.
Le sostanze tossiche vengono eliminate nelle urine
anche tramite secrezione attiva.
Caso eliminazione morfina
• Una volta coniugata con l’acido glicuronico
a livello epatico, verrà escreta dal sistema
degli anioni anzicchè da quello degli
cationi organici (Es di F e metaboliti
escreti x diverse vie!).
La CL di un F è ridotta
• Da alterazioni del flusso renale o
glomerulare
• Da patologie che limitano la permeabilità
capillare o la funzionalità tubulare
Il fegato possiede almeno 4 sistemi di trasporto attivo
per le sostanze organiche escrete nella bile: due
specifici per gli acidi organici (es. la
sulfobromoftaleina, BSP), uno per le basi (es. la
procainamide etilbromuro) e uno per le sostanze
neutre (es. la ouabaina); infine ne esiste un altro per
l’escrezione dei metalli (il Pb viene escreto contro un
gradiente negativo di concentrazione bile/plasma
molto elevato e con un trasporto massimo).
In generale, i composti con basso pm sono poco
escreti con la bile; quelli con valori oltre 325 o i loro
prodotti di coniugazione (soprattutto con
ac.glicuronico) hanno una spiccata tendenza
all’escrezione biliare.
L’escrezione biliare può anche essere aumentata
somministrando determinati farmaci. Ad es. il
fenobarbi- tale induce gli enzimi di fase II e accentua
la capacità del fegato di coniugare le sostanze
chimiche aumentando la velocità con cui esse
vengono eliminate.
L’escrezione biliare può essere il fattore limitante o,
per alcuni versi, essere determinante per la tossicità
delle sostanze. Ad es.:
- il fenobarbitale aumenta l’escrezione biliare del
metilmercurio e ne favorisce l’allontanamento;
- lo spironolattone e il pregnenolone (induttori degli
enzimi microsomiali) 1) aumentano la produzione
della bile e incrementano l’escrezione biliare di BSP;
2) riducono la tossicità dei glicosidi cardioattivi
riducendone la concentrazione a livello cardiaco.
Per contro:
- l’indometacina può causare lesioni intestinali.
La suscettibilità a questo effetto è direttamente
correlata con la quantità di farmaco che viene
escreta con la bile.
In epoca neonatale, i sistemi fisiologici preposti
all’escrezione biliare degli xenobiotici sono
ancora poco sviluppati e per questa ragione
alcune sostanze possono risultare più tossiche
nel neonato che negli adulti. Lo sviluppo di
questi sistemi può essere stimolato mediante
somminis-trazione di induttori enzimatici
microsomiali.
Variazioni del flusso ematico possono alterare la CL
epatica
La variazione dell’attività enzimatica è importante
quando l’attivazione enzimatica è il fattore limitante
l’eliminazione
L’indice di estrazione è più elevato x i F con basso
legame F-Proteine plasmatiche
Eliminazione con le feci
Gran parte delle sostanze di sintesi ha un
certo grado di lipofilicità e perciò viene
riassorbita. Alcune eccezioni sono
rappresentate dalle macromolecole e dai
composti ionizzati ad alto peso molecolare
(polimeri o le basi di ammonio quaternario)
che presentano assorbimento molto limitato.
Perciò, dopo somministrazione orale di
colestiramina o di sostanze analoghe, la
maggior parte della dose si ritrova nelle feci in
maniera più o meno grande in rapporto alla
frazione della dose non assorbita.
Escrezione intestinale
Alcune sostanze vengono eliminate con le feci
passando direttamente dal sangue all’intestino,
forse con un meccanismo di diffusione passiva.
All’eliminazione fecale può anche contribuire la rapida
desfoliazione delle cellule epiteliali che tappezzano
la parete dell’intestino.
L’escrezione intestinale diretta è un processo
abbastanza lento, e ha importanza solo per quei
composti che vengono poco metabolizzati o poco
eliminati con il rene o la bile.
Per alcuni composti liposolubili, l’escrezione può esser
favorita aumentando la lipofilicità del contenuto
gastrointestinale.
Numerose sostanze vengono metabolizzate a livello
della mucosa intestinale e quindi riescrete all’interno
del lume. Circa il 30-40% del materiale fecale solido
deriva dall’azione dei batteri intestinali. Le sostanze
che originano dalla frazione non assorbita della dose
di uno xenobiotico assunto per via orale, oppure
dalla bile o dalle pareti intestinali, vengono
sequestrati da questi microrganismi secondo i
principi della permeabilità di membrana e da questi
possono venire anche biotrasformati (specie
nell’intestino crasso dove la flora batterica è più
abbondante) prima di essere eliminati.
Questo processo sembra favorire più il riassorbimento
che l’escrezione.
Escrezione per via respiratoria
Le sostanze che, alla normale temperatura
corporea, sono allo stato gassoso, vengono
eliminate attraverso l’apparato respiratorio.
Negli alveoli polmonari i liquidi volatili si
trovano in equilibrio con la loro fase gassosa
e la q.tà eliminata è proporzionale alla
tensione di vapore (teoria applicata nel “test
del palloncino”). Non esistono specifici
sistemi di trasporto per l’eliminazione delle
sostanze. Queste vengono eliminate per
semplice diffusione ad una velocità che è
inversamente proporzionale a quella con cui
vengono assorbite.
- I gas poco solubili nel sangue (es.l’etilene),
vengono eliminati con grande rapidità. Il fattore
critico che determina la velocità di eliminazione
è la perfusione.
- I gas molto solubili nel sangue (es. cloroformio)
vengono eliminati molto lentamente. Il fattore
determinante la velocità di eliminazione è la
ventilazione polmonare.
- Sostanze molto liposolubili (anestetici quali
fluotano, metossiflurano) si concentrano nel
tessuto adiposo e vengono eliminati molto
lentamente (2-3 settimane dopo un anestesia di
alcune ore).
Altre vie di eliminazione
• Liquido cerebrospinale: attraverso i villi aracnoidei;
quelle liposolubili attraverso la BEE;
• Latte: per semplice diffusione; passano con i lipidi del
plasma nella ghiandola mammaria e da qui vengono
escreti con il latte. Es.: DDT, bifenili policlorurati e
polibromurati, le dibenzo-p-diossine, i furani, analoghi
del Ca come il Pb, sostanze che formano complessi
con il Ca (certi chelanti);
• Sudore e saliva: avviene per diffusione del composto
non ionizzato (liposolubile). Il passaggio di sostanze
tossiche nel sudore può essere causa di dermatiti; le
sostanze escrete nella saliva possono essere deglutite
ed essere disponibili per l’assorbimento
gastrointestinale.
THE
END