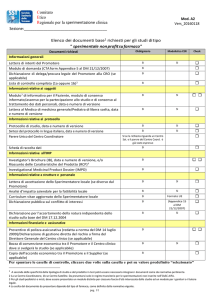
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 1/13
13/02/2009
Gestione degli studi clinici condotti presso l’ Azienda Ospedaliero
Universitaria S. Anna di Ferrara
1.
Lista di distribuzione ............................................................................................ 1
2.
Emissione ........................................................................................................... 1
3.
Scopo................................................................................................................. 2
4.
Campo di applicazione .......................................................................................... 2
5.
Riferimenti .......................................................................................................... 2
6.
Definizioni .......................................................................................................... 4
7.
Responsabilità e azioni ......................................................................................... 5
7.1 Processo complessivo .......................................................................................................... 5
7.2 Diffusione delle politiche e procedure aziendali in merito di studi clinici ...................................... 5
7.3 Autorizzazione di uno studio clinico........................................................................................ 6
7.4 Gestione di uno studio clinico approvato................................................................................. 9
7.5 Chiusura di uno studio clinico.............................................................................................. 11
7.6 Riesame del modello di gestione degli studi clinici ................................................................. 13
8.
Parametri di controllo ......................................................................................... 13
9.
Strumenti di registrazione................................................................................... 13
1.
Lista di distribuzione
La seguente procedura è messa a disposizione di tutto il personale coinvolto ed interessato attraverso la
pubblicazione su Intranet.
2.
Emissione
Rev.
Descrizione
modifiche
0
Prima
redazione
Data
Approvazione
13/02/09
Redazion
e
Gruppo di
redazione
Firma
Verifica
Responsabile
Qualità Aziendale
(Dott U.
Wienand)
Firma
Approvazione
Direttore Generale
(Dott R.Baldi)
Gruppo di redazione: Dott U.Wienand, Sig.ra F.Palara, Dott M.Voci, Dott.ssa A.Pozzati, Ing. A.Ferrozzi,
Dott. S.Bianchi, Dott.ssa L.Sighinolfi, Dott.ssa E.Raisi, Dott.ssa E.Vaccari, Avv. M.Uberti, Avv.
B.Paltrinieri, Dott.ssa R.Burattini, Ing. I.Braggion, Prof M.Simonato, Sig.ra C.Borsetti, Sig S.Boarini,
Dott.ssa A.Gualandi
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 2/13
13/02/2009
Questa procedura è stata sottoposta, prima della approvazione, al Comitato Etico in data 18 Dicembre
2008.
3.
Scopo
La procedura si prefigge i seguenti obiettivi:
concorrere ad agevolare tutti gli attori interessati al rispetto della vigente normativa in materia di
studi clinici, attraverso la esplosione del processo, la modulistica applicata e le eventuali
integrazioni in corso di utilizzo;
facilitare la comunicazione e l’integrazione fra i diversi Enti interni ed esterni all’Azienda coinvolti
nel processo;
ottimizzare i tempi di autorizzazione dello studio clinico e chiusura “amministrativa” della pratica;
chiarire gli ambiti di responsabilità dei diversi soggetti coinvolti;
garantire a Promotori e Sperimentatori trasparenza sulle modalità operative e facilitare la
collaborazione con l’Azienda;
dare evidenza e sostegno all’attività di ricerca sperimentale svolta nell’Azienda;
favorire la disponibilità in Azienda di dati ed informazioni su studi clinici condotti e risultati.
4.
Campo di applicazione
La procedura si applica in tutte le UU.OO. Sanitarie dell’Azienda Ospedaliero Universitaria S.Anna.
5.
Riferimenti
Esterni:
dlgs 507 del 14 dicembre 1992 - Attuazione della direttiva 90/385/CEE concernente il ravvicinamento
delle legislazioni degli Stati membri relative ai dispositivi medici impiantabili attivi
dlgs 46 del 24 febbraio 1997 - Attuazione della direttiva 93/42/CEE concernente i dispositivi medici
dm 15 luglio 1997 - Recepimento delle linee guida dell'Unione europea di buona pratica clinica per la
esecuzione delle sperimentazioni cliniche dei medicinali
dm 18 marzo 1998 sc - Modalità per l'esenzione dagli accertamenti sui medicinali utilizzati nelle
sperimentazioni cliniche
dm 18 marzo 1998 ce - Linee guida di riferimento per l'istituzione e il funzionamento dei comitati etici
dm 15 settembre 1998 - Integrazione al decreto ministeriale 18 marzo 1998, recante "modalità per
l'esenzione degli accertamenti sui medicinali utilizzati nelle sperimentazioni cliniche"
circ 6 del 8 aprile 1999 - Chiarimenti sui decreti ministeriali 18 marzo 1998 e 19 marzo 1998 pubblicati
nella Gazzetta Ufficiale n. 123 del 28 maggio 1998
dm 13 maggio 1999 - Integrazioni al decreto ministeriale 18 marzo 1998 recante: "Modalità per
l'esenzione dagli accertamenti sui medicinali utilizzati nelle sperimentazioni cliniche" e al decreto
ministeriale 19 marzo 1998 recante: "Riconoscimento della idoneità dei centri per la sperimentazione
clinica dei medicinali"
dm 23 novembre 1999 - Composizione e determinazione delle funzioni del Comitato etico nazionale per
le sperimentazioni cliniche dei medicinali, ai sensi del decreto legislativo n. 229 del 19 giugno 1999
dd 25 maggio 2000 - Trasmissione per via telematica dei dati inerenti le sperimentazioni cliniche dei
medicinali
circ 15 del 5 ottobre 2000 - Aggiornamento della circolare ministeriale n. 8 del 10 luglio 1997 relativa
alla sperimentazione clinica dei medicinali
circ 15 del 5 ottobre 2000 all 1-1quater
circ 15 del 5 ottobre 2000 all 6-10bis
dlgs integrati 178-44 - Testo integrato del decreto legislativo 178 del 29 maggio 1991 e del decreto
legislativo 44 del 18 febbraio 1997
dm 10 maggio 2001 - Sperimentazione clinica controllata in medicina generale ed in pediatria di libera
scelta
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 3/13
13/02/2009
dpr 439 del 21 settembre 2001 - Regolamento di semplificazione delle procedure per la verifica e il
controllo di nuovi sistemi e protocolli terapeutici sperimentali
diss 26 aprile 2002 - Accertamento della composizione e innocuità dei farmaci di nuova istituzione
prima della sperimentazione clinica sull'uomo. Individuazione della documentazione da sottoporre
all'Istituto superiore di sanità ai sensi dell'art. 4, comma 2, del decreto del Presidente della Repubblica 21
settembre 2001, n. 439
circ 6 del 2 settembre 2002 - Attività dei comitati etici istituiti ai sensi del decreto ministeriale 18
marzo 1998
dm 23 giugno 2004 - Individuazione degli uffici dirigenziali di livello non generale
dm 17 dicembre 2004 - Prescrizioni e condizioni di carattere generale, relative all’esecuzione delle
sperimentazioni cliniche dei medicinali, con particolare riferimento a quelle ai fini del miglioramento della
pratica clinica, quale parte integrante dell’assistenza sanitaria
dm 2 agosto 2005 - Modalità di presentazione della documentazione per notifica di indagine clinica con
dispositivi medici
dm 2 agosto 2005 - Allegati
dm 12 maggio 2006 - Requisiti minimi per l'istituzione, l'organizzazione e il funzionamento dei Comitati
etici per le sperimentazioni cliniche dei medicinali
dm 21 dicembre 2007 - Modalità di inoltro della richiesta di autorizzazione all’Autorità competente, per
la comunicazione di emendamenti sostanziali e la dichiarazione di conclusione della sperimentazione
clinica e per la richiesta di parere al comitato etico
determinazione aifa 20 marzo 2008 - Linee guida per la classificazione e conduzione degli studi
osservazionali sui farmaci
dm 22 dicembre 1997 - Tariffe residuali di cui al decreto ministeriale 19 luglio 1993, concernente le
tariffe e i diritti spettanti al Ministero della sanità, all'Istituto superiore di sanità e all'Istituto superiore per
la prevenzione e sicurezza del lavoro, per prestazioni rese a richiesta e ad utilità dei soggetti interessati
dm 19 marzo 1998 - Riconoscimento della idoneità dei centri per la sperimentazione clinica dei
medicinali
dm 7 ottobre 1998 - Integrazioni all'allegato al decreto 19 marzo 1998 recante "Riconoscimento della
idoneità dei centri per la sperimentazione clinica dei medicinali"
dm 20 gennaio 1999 - Misure relative all'immissione in commercio ed alla sperimentazione clinica di
medicinali contenenti materiali di origine bovina
circ 16 del 12 ottobre 1999 - Irregolarità nelle procedure autorizzative e nella esecuzione delle
sperimentazioni cliniche dei medicinali
dm 28 dicembre 2000 - Misure finalizzate alla minimizzazione del rischio di trasmissione all'uomo,
tramite farmaci, degli agenti che causano l'encefalopatia spongiforme animale
dm 30 maggio 2001 - Accertamenti ispettivi sulla osservanza delle norme di buona pratica di
fabbricazione e di buona pratica clinica
dm 8 maggio 2003 - Uso terapeutico di medicinale sottoposto a sperimentazione clinica
direttiva ce 20 del 2001 ita - Direttiva 2001/20/CE del Parlamento europeo e del Consiglio del 4 aprile
2001 concernente il ravvicinamento delle disposizioni legislative, regolamentari ed amministrative degli
Stati membri relative all'applicazione della buona pratica clinica nell'esecuzione della sperimentazione
clinica di medicinali ad uso umano
direttiva ce 20 del 2001 eng - Directive 2001/20/EC of the European Parliament and of the Council of
4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member
States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal
products for human use
dlgs 211 del 24 giugno 2003 - Attuazione della direttiva 2001/20/CE relativa all'applicazione della
buona pratica clinica nell'esecuzione delle sperimentazioni cliniche di medicinali per uso clinico
dlgs 200 del 6 novembre 2007 - Attuazione della direttiva 2005/28/CE recante principi e linee guida
dettagliate per la buona pratica clinica relativa ai medicinali in fase di sperimentazione a uso umano,
nonchè requisiti per l'autorizzazione alla fabbricazione o importazione di tali medicinali
Interni
Regolamento del Comitato Etico della Provincia di Ferrara con riferimento a Sperimentazioni
Cliniche, Farmacologiche ed Epidemiologiche
DOC-701-AZ “Documenti necessari per presentazione di una domanda di Studio Clinico”
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 4/13
13/02/2009
I-001-FARM “Ricevimento, verifica e consegna allo Sperimentatore di medicinali per studi
clinici”, del Servizio di Farmacia Ospedaliera
I-701-AcQuaRI “Archiviazione della documentazione relativa agli studi clinici presso l’Azienda
Ospedaliero Universitaria S.Anna di Ferrara”
6.
Definizioni
U.O.: Unità Operativa;
UU.OO.: Unità Operative;
AcQuaRI: Accreditamento Qualità Ricerca Innovazione;
Studio Clinico: qualsiasi sperimentazione o studio relativo a: farmaci, presidi, dispositivi medici,
qualsiasi altro materiale o prodotto (come ad esempio probiotici, cosmetici) ed a procedure o protocolli
diagnostico terapeutici;
Sperimentazione Clinica: qualsiasi studio sull'uomo finalizzato a scoprire o verificare gli effetti clinici,
farmacologici e/o altri effetti farmacodinamici di uno o più medicinali sperimentali, e/o a individuare
qualsiasi reazione avversa ad uno a più medicinali sperimentali, e/o a studiarne l'assorbimento, la
distribuzione, il metabolismo e l'eliminazione, con l'obiettivo di accertarne la sicurezza e/o l'efficacia.
Questa definizione include le sperimentazioni cliniche effettuate in un unico centro o in più centri, solo in
Italia o anche in altri Stati membri dell'Unione Europea;
Comitato Etico: un organismo indipendente, composto da personale sanitario e non, che ha la
responsabilità di garantire la tutela dei diritti, della sicurezza e del benessere dei soggetti in
sperimentazione e di fornire pubblica garanzia di tale tutela, esprimendo, ad esempio, un parere sul
protocollo di sperimentazione, sull'idoneità degli sperimentatori, sulla adeguatezza delle strutture e sui
metodi e documenti che verranno impiegati per informare i soggetti e per ottenerne il consenso
informato;
Promotori della Sperimentazione: una persona, società, istituzione oppure un organismo che si
assume la responsabilità di avviare, gestire e/o finanziare una sperimentazione clinica;
Sperimentatore: una persona responsabile della conduzione dello studio clinico presso un centro di
sperimentazione. Se uno studio viene condotto da un gruppo di persone in un centro di sperimentazione,
lo sperimentatore è il responsabile del gruppo e può essere chiamato «sperimentatore principale»
(Glossario Istituto Superiore Sanità 2008);
Studio Multicentrico: studio clinico effettuato seguendo un unico protocollo, ma in più centri e per
questa ragione condotto da più sperimentatori;
Centro Coordinatore: centro a cui viene attribuita la responsabilità del coordinamento degli
sperimentatori nei diversi centri che partecipano ad uno studio multicentrico;
Centri partecipanti: centri che partecipano ad uno studio multicentrico;
Monitor: soggetto responsabile dell’andamento di uno studio clinico per garantire che questo venga
effettuato, registrato e relazionato in osservanza del protocollo, delle Procedure Operative Standard
(SOP), della G.C.P. e delle disposizioni normative applicabili; il Monitor è nominato dal Promotore;
Good Clinical Practice (G.C.P.): lo standard a cui fare riferimento per la progettazione, la conduzione,
l’esecuzione, il monitoraggio, la verifica, la registrazione, le analisi ed i rapporti relativi agli studi clinici,
che garantisce che i dati ed i risultati riportati siano attendibili ed accurati, e che siano salvaguardati i
diritti, l’integrità e la riservatezza dei soggetti partecipanti allo studio;
Organizzazione di Ricerca a Contratto (C.R.O. Contract Research Organization): una persona o
un’ organizzazione (commerciale, accademica o di altro tipo) con cui il promotore ha stipulato un
contratto per assolvere ad una o più mansioni e funzioni del promotore relative allo studio;
Scheda Raccolta Dati (C.R.F. Case Report Form): un documento su supporto cartaceo, ottico, oppure
elettronico progettato per registrare tutte le informazioni richieste dal protocollo che devono essere
riferite al promotore relativamente a ciascun partecipante allo studio;
dm: decreto ministeriale;
dlgs: decreto legislativo;
circ: circolare;
diss: decreto istituto superiore della sanità;
dd: decreto delegato;
AIFA: Agenzia Italiana del Farmaco.
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
PROCEDURA
STAFF AcQuaRI
7.
P-701-AZ
Rev. 0
Pag. 5/13
13/02/2009
Responsabilità e azioni
7.1 Processo complessivo
Il processo di gestione degli studi clinici condotti presso l’Azienda Ospedaliero Universitaria di Ferrara
passa attraverso cinque macro fasi rappresentato del diagramma sottostante:
Diffusione delle
politiche e
procedure
aziendali
Riesame del
modello di
gestione degli
studi clinici
Chiusura di uno
studio clinico
Valutazione
delle proposte
e
autorizzazione
degli studi
clinici
Gestione di uno
studio clinico
Nei paragrafi seguenti vengono descritte in dettaglio le responsabilità e le attività (fasi del processo)
previste per garantire il conseguimento degli obiettivi stabiliti al paragrafo 3 “Scopo” della presente
procedura.
7.2 Diffusione delle politiche e procedure aziendali in merito di studi clinici
L'Azienda Ospedaliero Universitaria di Ferrara, attraverso la presente procedura e con la collaborazione
del Comitato Etico intende garantire la tutela del benessere e la sicurezza dei soggetti che partecipano
agli studi clinici svolti presso le proprie strutture,con lo scopo di proteggere e promuovere i valori della
persona umana;
Le modalità con cui un soggetto può promuovere l’avvio di uno studio clinico sono diffuse sul sito
aziendale www.ospfe.it nell’apposita sezione “Attività scientifica > Comitato etico > Sperimentazioni”.
L’aggiornamento di tale pagina informativa è assicurato dall’Ufficio Ricerca e Innovazione dello Staff
“Accreditamento Qualità Ricerca Innovazione” (AcQuaRI).
I principi generali sono contenuti nel “Regolamento del Comitato Etico della Provincia di Ferrara con
riferimento a Sperimentazioni Cliniche, Farmacologiche ed Epidemiologiche”.
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 6/13
13/02/2009
7.3 Autorizzazione di uno studio clinico
L’iter di valutazione delle proposte e autorizzazione degli studi clinici passa attraverso le fasi descritte
nella tabella seguente che portano alla predisposizione degli atti autorizzativi dello studio. Le note
riportate alle singole fasi dettagliano le modalità operative adottate.
N°
1
2
3
4
Fase
Presentazione di domanda di studio
clinico
Verifica completezza della
documentazione
Valutazione “etico-scientifica”
Valutazione impatto economicoorganizzativo con eventuale
predisposizione di piano di
monitoraggio
5
Redazione finale del contratto
6
Autorizzazione dello studio
7
Firma del contratto
Responsabilità
Promotore dello
studio
Ufficio Ricerca e
Innovazione
Comitato Etico
Ufficio Ricerca e
Innovazione
Ufficio Ricerca e
Innovazione
Direttore competente
Ufficio Ricerca e
Innovazione
Note alle fasi:
Fase 1: Presentazione di domanda di studio clinico
Il Promotore dello studio (vedi paragrafo 6 “Definizioni”) presenta domanda per l’autorizzazione di uno
studio clinico utilizzando il MOD-705-AZ (“Richiesta di autorizzazione allo svolgimento dello Studio
Clinico”).
La domanda deve essere inviata all’Ufficio Ricerca e Innovazione, in forma cartacea, e accompagnata da
quanto definito nel documento “Documenti necessari per presentazione di una domanda di Studio Clinico”
(DOC-701-AZ).
Fase 2: Verifica completezza della documentazione
L’Ufficio Ricerca e Innovazione verifica la completezza della documentazione inviata, controllando la
presenza di quanto elencato nel “DOC-701-AZ” (Documenti necessari per presentazione di una domanda
di Studio Clinico), contattando direttamente il “Promotore” in caso di incompletezze e/o incongruenze
(compresi quelli relativi agli aspetti di tutela dei dati personali) per richiedere eventuali integrazioni o
rettifiche e comunicando al Promotore che vengono sospesi i termini temporali previsti dalla legge per
l’emissione del parere di eticità.
Il modulo MOD-703-AZ “Dati per la fatturazione” viene inviato alla Direzione Amministrazione delle
Risorse Economiche Finanziarie per la fatturazione delle competenze del Comitato Etico.
Il Modulo MOD-706-AZ “Proposta di Contratto di Comodato” viene inviato alla Direzione
Approvvigionamento Beni e Servizi e Attività Economali per attivare la procedura operativa di
acquisizione del/i bene/i concesso/i in comodato d’uso gratuito.
Qualora l’Ufficio Ricerca e Innovazione rilevi l’esistenza di problematiche di natura assicurativa, inoltra la
documentazione relativa (comprensiva di polizza e certificato d’assicurazione nonché copia del protocollo)
all’Ufficio Legale che fornisce un proprio parere.
I dati degli studi vengono inseriti nel database aziendale e una selezione dei documenti (richiesta di
autorizzazione allo svolgimento dello studio “MOD-705-AZ”, sinossi “MOD-702-AZ” + eventuali altri
documenti di particolare interesse) viene pubblicata nell’area riservata ai componenti del Comitato Etico
del sito internet aziendale.
La trasmissione della documentazione alla Segreteria Scientifica del Comitato Etico attesta il superamento
delle verifica di completezza.
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 7/13
13/02/2009
Fase 3: Valutazione “etico-scientifica”
La Segreteria Scientifica del Comitato Etico effettua una valutazione tecnico-scientifica della proposta
verificando:
il disegno e la metodologia sperimentale per gli aspetti tecnico-scientifici, particolarmente in caso
di richiesta di parere unico come Centro Coordinatore;
l’istruttoria etico-deontologica, con riferimento specifico al diritto di protezione dei cittadini
coinvolti nello studio sperimentale:
valutazione dei criteri di inclusione ed esclusione nel piano sperimentale;
valutazione delle forme e dei contenuti per l’informazione ai probandi;
valutazione della forma e delle modalità di recepimento di un consenso valido;
le modalità di sorveglianza clinica;
le condizioni per la sospensione dello studio.
In caso di eventuali problemi riscontrati nel corso della verifica, la Segreteria Scientifica provvede a
contattare direttamente il Promotore per concordare le modifiche opportune. In caso di impossibilità di
trovare un accordo con il Promotore la Segreteria Scientifica comunica all’Ufficio Ricerca e Innovazione la
chiusura del procedimento autorizzativo.
Il superamento della verifica è attestato dalla redazione da parte della Segreteria Scientifica di una
“Relazione tecnica” con le caratteristiche previste dall’apposito allegato del Regolamento del Comitato
Etico.
Tale relazione viene inviata all’Ufficio Ricerca e Innovazione affinché l’Ufficio, in veste di Segreteria
Amministrativa del Comitato Etico, possa inserire la pratica nell’ordine del giorno del Comitato Etico.
Copia della Relazione Tecnica viene inviata a tutti i componenti del Comitato Etico.
Il Comitato Etico valuta i protocolli di studio, con particolare riferimento alla sicurezza ed al benessere dei
soggetti coinvolti, e può esprimere:
parere favorevole all’effettuazione dello studio;
parere favorevole all’effettuazione dello studio ma con richiesta di modifiche relative ai fogli di
informazione e di consenso;
sospensione con richiesta di chiarimenti e riesame nella successiva seduta;
richiesta di modifiche al piano clinico, nel caso in cui l’Azienda Ospedaliera di Ferrara sia Centro
Coordinatore;
parere negativo all’effettuazione dello studio.
Tutte le richieste di modifica e chiarimento vengono gestite dalla Segreteria Scientifica.
La Segreteria Scientifica rivede la “Relazione tecnica” sulla base delle decisioni prese dal Comitato Etico e
la invia alla Segreteria Amministrativa per la redazione del verbale che, una volta firmato dal Presidente,
viene inviato a:
Promotore dello Studio;
Sperimentatore;
Ministero.
L’Ufficio Ricerca e Innovazione provvede ad aggiornare, per quanto necessario, il database aziendale ed
inserisce i dati nel database dell’Osservatorio AIFA degli studi clinici.
Fase 4: Valutazione impatto economico-organizzativo con eventuale predisposizione di piano
di monitoraggio
Parallelamente alla valutazione etico-scientifica l’Ufficio Ricerca e Innovazione provvede, in apposite
riunioni periodiche interne, a valutare:
l’impatto economico generato dal protocollo rispetto alla normale gestione del paziente o del
normale follow up delle patologie in studio;
la congruità dell’analisi dei costi indicata dal Promotore;
per gli studi sponsorizzati, la copertura economica garantita dal promotore in base alla bozza di
contratto presentato;
per gli studi spontanei che comportano costi, le fonti di copertura degli stessi individuate dallo
Sperimentatore e indicate nell’apposito campo del modulo “Analisi dell’impatto economico ed
organizzativo per studi clinici” (MOD-704-AZ);
la presenza di eventuali problematiche di natura assicurativa, contattando l’Ufficio Legale per
quanto necessario;
l’impatto organizzativo dello studio, con riferimento all’impiego di attrezzature e coinvolgimento di
altre UU.OO., in particolare dei servizi diagnostici di supporto.
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 8/13
13/02/2009
La valutazione di impatto economico-organizzativo viene effettuata sulla base del modulo “Analisi
dell’impatto economico ed organizzativo per studi clinici” (MOD-704-AZ), compilato dallo Sperimentatore
e controfirmato dal Direttore dell’U.O. in cui si svolgerà lo studio. In tale modulo lo Sperimentatore indica
gli altri soggetti coinvolti (altre UU.OO. Sanitarie, Servizi Centrali, Direzione Medica) con cui ha
precedentemente concordato le modalità di collaborazione nell’ambito dello studio.
Nella valutazione dell’impatto economico-organizzativo l’Ufficio Ricerca e Innovazione si avvale della
collaborazione del Servizio di Farmacia Ospedaliera, della Direzione Programmazione e Controllo di
Gestione e della Direzione Medica di Presidio.
In caso di esito negativo della valutazione si provvede a contattare il Promotore per una ridefinizione
della modalità di copertura dei costi, compresa - per gli studi spontanei - la possibilità di accesso al Fondo
per la ricerca spontanea.
L’apposizione della firma sul modulo “Riepilogo delle valutazioni effettuate sullo studio clinico” (MOD-707AZ) da parte del Responsabile dell’Ufficio Ricerca e Innovazione attesta l’esito positivo della valutazione
compresi gli aspetti assicurativi.
L’Ufficio Ricerca e Innovazione, in caso di particolare complessità di uno studio, predispone un “Piano di
Monitoraggio” per verificare eventuali ricadute organizzative ed economiche non evidenziate
precedentemente.
Fase 5: Redazione finale del contratto
La stipula del contratto non è necessaria per gli studi spontanei.
L’Ufficio Ricerca e Innovazione valuta la bozza di contratto inviata dal Promotore e si rapporta con lo
stesso per concordare eventuali modifiche in vista dell’esame da parte del Responsabile della Direzione
Affari Giuridici e Attività Amministrative di Presidio - competente alla stipula dei contratti su delega del
Direttore Generale – che autorizza la predisposizione del provvedimento amministrativo di autorizzazione
allo studio.
L’utilizzo del format aziendale “Proposta di contratto” (MOD-701-AZ) da parte del Promotore garantisce
tempi più rapidi di approvazione.
Il contratto dovrà contenere i dati necessari per la fatturazione dei corrispettivi dello studio.
L’Ufficio Ricerca e Innovazione redige la proposta di Determina.
Fase 6: Autorizzazione dello studio
Il rilascio dell’autorizzazione allo svolgimento dello studio si formalizza:
per gli studi spontanei con lettera predisposta dall’Ufficio Ricerca e Innovazione, a firma del
Direttore Sanitario;
per gli studi sponsorizzati e no-profit attraverso una determina della Direzione Affari Giuridici e
Attività Amministrative di Presidio.
L’Ufficio Ricerca e Innovazione provvede:
a notificare l’avvenuto rilascio dell’autorizzazione ai seguenti soggetti: Direzione Medica di
Presidio, Servizio di Farmacia Ospedaliera, Direzione Amministrazione Risorse Economico
Finanziarie, Direzione Approvvigionamento Beni e Servizi e Attività Economali (per studi che
prevedono la fornitura di attrezzature da parte del Promotore), Ufficio Legale (per studi spontanei
e no profit che rientrano nella copertura assicurativa aziendale), Farmacologia Clinica (per avvio
monitoraggio), Sperimentatore;
ad aggiornare, per quanto necessario, il database aziendale e ad inserire lo studio nell’anagrafe
della ricerca della Regione Emilia Romagna.
Fase 7: Firma del contratto
L’Ufficio Ricerca e Innovazione riceve gli originali firmati dal Promotore, raccoglie le firme del
Responsabile della Direzione Affari Giuridici e Attività Amministrative di Presidio, del Responsabile del
procedimento amministrativo e dello Sperimentatore Principale. Quindi archivia una copia agli atti ed
inoltra le copie di competenza al Promotore unitamente alla Determina di autorizzazione.
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 9/13
13/02/2009
7.4 Gestione di uno Studio Clinico approvato
N°
Fase
8
Avvio fatturazione
9
10
11
Presa in carico di eventuali
attrezzature
Apertura del centro
Organizzazione dello studio clinico nell’
U.O.
12
Presa in carico di eventuali farmaci
13
18
Svolgimento dello studio
Gestione di eventuale richiesta di
interruzione dello studio
Verifiche periodiche del Promotore in
caso di studi sponsorizzati
Verifica del rispetto del protocollo
Effettuazione attività di verifica
previste dal piano di monitoraggio
Report annuale al Comitato Etico
19
Avanzamento della fatturazione
14
15
16
17
Responsabilità
Direzione
Amministrazione
delle Risorse
Economiche
Finanziarie
Direzioni Competenti
Promotore
Direttore Unità
Operativa
Servizio di Farmacia
Ospedaliera
Sperimentatore
Ufficio Ricerca e
Innovazione
Monitor
Farmacologia clinica
Ufficio Ricerca e
Innovazione
Promotore
Direzione
Amministrazione
delle Risorse
Economiche
Finanziarie
Note alle fasi:
Fase 8: Avvio fatturazione
La Direzione Amministrazione delle Risorse Economiche Finanziarie provvede all’emissione della fattura
relativa alla prima tranche di pagamento secondo le indicazioni contrattuali.
Fase 9: Presa in carico di eventuali attrezzature
Le eventuali attrezzature (sanitarie, non sanitarie e informatiche) necessarie verranno acquisite mediante
stipula di un contratto di comodato, secondo le procedure operative aziendali e previa acquisizione dei
pareri dei Servizi aziendali competenti (U.O. interessata, Direzione Medica di Presidio, Servizio di Fisica
Medica, Direzione Attività Tecniche e Patrimoniali, Sistema Informatico e Informativo).
Fase 10: Apertura del centro
Il Promotore valuta la necessità di effettuare una visita di sopralluogo presso il Centro e/o la Farmacia
Ospedaliera per favorire l’organizzazione operativa dello studio.
Il Promotore (anche qualora coincida con lo Sperimentatore) garantisce la presenza presso il Centro al
momento della sua apertura della seguente documentazione:
Protocollo e relativi emendamenti se presenti;
valutazioni etiche;
elenco Centri partecipanti;
eventuali recapiti del promotore e C.R.O.;
scheda di segnalazione di eventi avversi;
copia della documentazione assicurativa;
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 10/13
13/02/2009
modulistica per l’osservazione delle persone in studio (“diario”, “scale”, “questionari”, “CRF”,
etc.);
fogli di informazione e consenso;
eventuali fogli di contabilità del farmaco.
Il Promotore (anche qualora coincida con lo Sperimentatore) garantisce la diffusione e condivisione del
Protocollo di Studio con lo Sperimentatore Principale ed suoi collaboratori.
Il Monitor, su incarico del Promotore, raccoglie i documenti essenziali a firma dello Sperimentatore
Principale.
Per studi spontanei e no-profit, lo Sperimentatore comunica all’ Ufficio Legale l’apertura del centro per
ottenere la copertura assicurativa.
Fase 11: Organizzazione dello studio nella U.O.
Il Direttore dell’Unità Operativa sede dello studio provvede, con la collaborazione dello Sperimentatore e
del Coordinatore Infermieristico/Tecnico, a:
individuare e organizzare le risorse umane, tecnologiche e strutturali necessarie;
definire e formalizzare eventuali procedure o istruzioni operative necessarie per l’effettuazione
delle attività previste dallo studio;
istruire il personale coinvolto;
adeguare i piani di lavoro;
definire le modalità di documentazione delle prestazioni che verranno erogate nel corso dello
studio.
Fase 12: Presa in carico di eventuali farmaci
Per la presa in carico dei farmaci, si fa riferimento all’istruzione operativa (I-001-FARM) “Ricevimento,
verifica e consegna allo sperimentatore di medicinali per studi clinici”, del Servizio di Farmacia
Ospedaliera.
Le operazioni che vengono effettuate sono:
valutazione tecnico-farmaceutica: verifica della corrispondenza tra farmaco e G.C.P. (Good
Clinical Practice);
definizione del luogo di stoccaggio e formulazione di eventuali indicazioni per l’U.O. sulle
modalità di conservazione ed utilizzo del farmaco;
controlli di conformità dei prodotti consegnati e delle preparazioni galeniche;
distribuzione del farmaco all’U.O.
Fase 13: Svolgimento dello studio
Lo svolgimento dello studio prevede una serie di operazioni: l’informazione al paziente e l’acquisizione del
consenso, la fase di screening (selezione dei pazienti arruolabili secondo i criteri di inclusione previsti dal
protocollo), l’arruolamento dei pazienti e l’assegnazione ai bracci (gruppi) di trattamento, la visita di
baseline (prima visita, indagini clinico-laboratoristiche ed eventuale somministrazione del farmaco), le
visite programmate e le attività clinico-diagnostiche previste dal protocollo, l’eventuale uscita del paziente
dallo studio (in caso di: ritiro del consenso, eventi avversi seri, decesso, trasferimento del paziente,
decisione medica, violazione dei criteri del protocollo), la visita al paziente di fine studio e le visite di
follow-up.
La responsabilità di individuare e adottare modalità di documentazione delle operazioni previste al fine di
garantire puntuale identificazione e rintracciabilità lungo tutto il processo di gestione dello studio è del
Promotore.
Fase 14: Gestione di eventuale interruzione dello studio
Uno studio può subire una chiusura anticipata per motivi di sicurezza, per arruolamento competitivo, per
inadempienza contrattuale di una delle parti, per decisione diretta dello Sperimentatore o per una scelta
strategica del Promotore.
L’intenzione di interrompere lo studio è comunicata all’Ufficio Ricerca e Innovazione che provvede ad
individuare l’iter di gestione in base alle motivazioni presentate.
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 11/13
13/02/2009
Fase 15: Verifiche periodiche del Promotore in caso di studi sponsorizzati
Il Monitor effettua visite periodiche per verificare l’aderenza al protocollo, controllare la contabilità del
farmaco, monitorare la C.R.F. (scheda di raccolta dati) cartacea od elettronica, confermare la
corrispondenza tra C.R.F. e cartella clinica, verificare la completezza delle registrazioni e della
documentazione dello studio.
Fase 16: Verifica del rispetto del protocollo
La Farmacologia Clinica ha ricevuto il mandato dal Comitato Etico di vigilanza sul concreto procedere
degli studi approvati dal Comitato Etico. Essa effettua un monitoraggio interno svolgendo le seguenti
attività:
1. contattare il referente dello studio, designato nel modulo “Analisi dell’impatto economico ed
organizzativo per studi clinici” (MOD-704-AZ), per avere riscontro dell’effettivo avvio dello studio;
2. dall’avvio alla chiusura dello studio effettuare visite periodiche per il controllo del rispetto del
protocollo (ogni 3-6 mesi, a seconda della tipologia dello studio in oggetto), in collaborazione con
l’Ufficio Ricerca e Innovazione, con particolare riferimento a:
numero di pazienti reclutati/usciti e cause dell’uscita;
compliance ed eventuali rifiuti;
modalità di raccolta dei dati per gli studi spontanei;
obblighi di farmacovigilanza;
3. raccogliere un report di fine studio (sia per studi sponsorizzati, sia per i non sponsorizzati), che
contenga i risultati scientifici ottenuti.
Lo sperimentatore deve segnalare alla Segreteria Scientifica del Comitato Etico gli eventuali eventi
avversi verificatisi nel corso dello studio clinico. La Farmacologia Clinica ne cura la valutazione.
Fase 17: Effettuazione attività di verifica previste dal piano di monitoraggio
L’Ufficio Ricerca e Innovazione, durante lo svolgimento dello studio, potrà monitorare i dati dichiarati nel
modulo “Analisi dell’impatto economico ed organizzativo per studi clinici” (MOD-704-AZ), e verificare che
siano effettivamente rispettati, concordando con lo Sperimentatore periodiche visite di controllo.
Fase 19: Avanzamento della fatturazione
Lo Sperimentatore, qualora siano soddisfatte le condizioni per l’emissione delle successive fatture,
comunica i dati relativi all’avanzamento dello studio all’Ufficio Ricerca e Innovazione che li inoltra alla
Direzione Amministrazione delle Risorse Economiche Finanziarie per l’emissione delle fatture.
7.5 Chiusura di uno Studio Clinico
N°
20
Fase
Chiusura del centro
21
Chiusura dello studio
22
23
Valutazione del rispetto dei costi
approvati per lo studio
Raccolta e restituzione farmaci non
utilizzati
24
Distribuzione proventi
25
Archiviazione dello studio
Responsabilità
Promotore
Ufficio Ricerca e
Innovazione
Ufficio Ricerca e
Innovazione
Servizio di Farmacia
Ospedaliera
Direzione
Amministrazione
delle Risorse
Economiche
Finanziarie
Ufficio Ricerca e
Innovazione
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 12/13
13/02/2009
Note alle fasi:
Fase 20: Chiusura del centro
La chiusura del Centro da parte del Promotore avviene attraverso una visita di chiusura da parte del
Monitor il quale procede alla chiusura di contabilità del farmaco, alla chiusura delle C.R.F. cartacee od
elettroniche, al ritiro della copia della documentazione scientifica relativa allo studio ed alla stesura della
relazione finale.
Fase 21: Chiusura dello studio
Lo Sperimentatore comunica per iscritto i dati relativi alla chiusura dello studio (numero soggetti
arruolati, completati e usciti) all’Ufficio Ricerca e Innovazione che li verifica alla luce della comunicazione
di chiusura del centro del Promotore. Tali dati vengono quindi trasmessi alla Direzione Risorse Economico
Finanziarie e Contabilità, che emette fattura.
Fase 22: Valutazione del rispetto dei costi approvati per lo studio
L’Ufficio Ricerca e Innovazione verifica con lo Sperimentatore, utilizzando il modulo “Riepilogo delle
valutazioni effettuate sullo studio clinico” (MOD-707-AZ), le reali prestazioni aggiuntive effettuate
nell’ambito dello studio clinico (numerosità e tipologia) e comunica il dato alla Direzione Amministrazione
delle Risorse Economiche Finanziarie, che procede alla riscossione delle somme dovute attraverso:
emissione della fattura al Promotore, in caso di studi sponsorizzati;
emissione della fattura nei confronti di Enti Esterni (Università), qualora fossero indicati per la
copertura delle spese;
utilizzo di fondi accantonati dall’Unità Operativa;
addebito al centro di costo proprio dell’Unità Operativa.
Fase 23: Raccolta e restituzione farmaci non utilizzati
Alla chiusura del centro o in altri casi di non utilizzo dei medicinali oggetto dello studio (scadenza del
medicinale stoccato, ritiro di un lotto del medicinale precedentemente consegnato) il Promotore richiede
autorizzazione al Servizio di Farmacia Ospedaliera per il ritiro dei medicinali.
Fase 24: Distribuzione proventi
I proventi degli studi clinici sono distribuiti sulla base di quanto previsto dal Regolamento Aziendale.
Lo Sperimentatore ed il Direttore dell’U.O., se non coincidenti, ricevono comunicazione dell’avvenuto
incasso dei corrispettivi da parte della Direzione Amministrazione delle Risorse Economiche Finanziarie.
Si precisa che l’utilizzo delle risorse attribuite sulla base dei criteri da Regolamento alle Unità Operative
segue quanto previsto dalla normali procedure aziendali in tema di acquisizione di risorse, beni e servizi.
Fase 25: Archiviazione dello studio
L’Ufficio Ricerca e Innovazione provvede a:
registrare i dati di chiusura dello studio nel database aziendale;
trasmettere alla Regione le informazioni da questa richieste con le modalità stabilite;
archiviare la documentazione relativa allo studio sulla base di quanto previsto dall’Istruzione
Operativa “Archiviazione della documentazione relativa agli studi clinici presso l’Azienda
Ospedaliero Universitaria S.Anna di Ferrara” (I-701-AcQuaRI) che stabilisce responsabilità,
modalità e tempi di archiviazione sia della documentazione per esigenze di natura medico legale
che le copie di archivio corrente.
L’Ufficio Ricerca e Innovazione conserva un archivio corrente a fini operativi, fino alla conclusione di ogni
studio. L’archivio legale viene mantenuto in sede e successivamente stoccato in un archivio di deposito.
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]
STAFF AcQuaRI
PROCEDURA
P-701-AZ
Rev. 0
Pag. 13/13
13/02/2009
7.6 Riesame del modello di gestione degli studi clinici
L’Ufficio Ricerca e Innovazione redige annualmente un report di analisi dei seguenti dati:
dati di attività relativi agli studi clinici (n. studi attivati nell’anno, n. studi chiusi nell’anno, numero
di studi divisi per U.O., numero di studi con contratto, numero di studi suddivisi per tipologie)
verifica del rispetto dei protocolli (dati forniti da Farmacologia Clinica);
attività del Servizio di Farmacia Ospedaliera sugli studi clinici;
verifica del rispetto dei costi previsti a budget (Ufficio Ricerca e Innovazione con supporto del
Servizio di Farmacia Ospedaliera);
relazioni annuali a cura dei Promotori;
ammontare dei proventi derivanti da studi clinici e loro utilizzo (dati forniti dalla Direzione
Amministrazione delle Risorse Economiche Finanziarie);
dati statistici sull’attività del Comitato Etico;
andamento degli indicatori di monitoraggio sul processo di gestione degli studi clinici (vedi
paragrafo successivo)
Il report è inviato a:
Comitato Etico;
Comitato di Indirizzo;
Direttore Generale, Direttore Sanitario e Direttore Amministrativo;
Il report è inoltre base per sviluppare il capitolo sulla ricerca del Bilancio di Missione Aziendale.
Il report è presentato, a cura del Responsabile dell’Ufficio Ricerca e Innovazione, al Collegio di Direzione
che valuta la necessità di revisione del processo di gestione degli studi clinici.
Le eventuali indicazioni vengono gestite dall’Ufficio Ricerca e Innovazione con le modalità previste per
azioni preventive o correttive.
8.
9.
Parametri di controllo
% di proposte di studio che hanno parere motivato dal Comitato Etico entro i tempi di legge (3060 giorni)
% di proposte di studio sponsorizzato o no profit, presentate con contratto impostato come da
MOD-701-AZ “Proposta di Contratto”, con rilascio dell’autorizzazione entro 60/90 giorni dalla
presentazione della domanda
% di proposte di studio spontaneo con rilascio dell’autorizzazione entro 60 giorni dalla
presentazione della domanda
Strumenti di registrazione
MOD-701-AZ “Proposta di Contratto”
MOD-702-AZ “Sinossi riassuntiva delle caratteristiche dello studio”
MOD-703-AZ “Dati per la fatturazione”
MOD-704-AZ “Analisi dell’impatto economico ed organizzativo per studi clinici”
MOD-705-AZ “Richiesta di autorizzazione allo svolgimento dello studio clinico”
MOD-706-AZ “Proposta di contratto di comodato”
MOD-707-AZ “Riepilogo delle valutazioni effettuate sullo studio clinico”
Database Aziendale
Database AIFA
Anagrafe Regionale della Ricerca
AcQuaRI: Accreditamento Qualità Ricerca Innovazione
Corso Giovecca, 203 - 44100 Ferrara | Tel. 0532 236398; 0532236896 Fax. 0532 237718 | e-mail: [email protected];
[email protected]