Scienze dei minerali
Il minerale può essere inteso come una sostanza cristallina con una disposizione ordinata
nelle tre direzioni dello spazio a uno stato di più bassa energia rispetto a una disposizione
casuale.
Una disposizione ordinata di oggetti è dotata di simmetria: infatti il concetto globale di
simmetria nasce dall’associazione di due concetti: la molteplicità e l’ordine. La simmetria è
una ripetizione ordinata di unità, o un ordinamento di oggetti identici
Ogni movimento che porta alla ripetizione di un motivo originale si chiama: operazione di
simmetria.
L’ente geometrico rispetto al quale si effettua l’operazione si chiama elemento di simmetria od
operatore di simmetria.
Sono elementi di simmetria tutti gli operatori che fanno sovrapporre idealmente un oggetto a
un altro identico. Sicuramente lo sono i VETTORI DI TRASLAZIONE e gli ASSI DI
ROTAZIONE(1,2,3,4,6);. Vi sono però oggetti identici, ma non sovrapponibili con una
semplice rototraslazione (ad es., mano destra e sinistra), in questo caso si parla di oggetti
ENANTIMORFI.
La loro giustapposizione richiede un’ulteriore operazione: lo “specchiamento” su un PIANO DI
RIFLESSIONE o il ribaltamento su un CENTRO DI INVERSIONE. Quindi le operazioni di
simmetria sono le seguenti:
traslazione: la traslazione di nodi reticolari porta alla costruzione di reticoli infiniti nelle
tre direzioni dello spazio;
rotazione: rotazione intorno a un asse con ordine dell’asse = 360°/n con n =
ricoprimenti in 360°
inversione: si inverte rispetto a un punto;
riflessione: la riflessione rispetto a un piano è equivalente alla rotoinversione su di un
asse di ordine 2(asse binario)
rotazione con traslazione (elicogire)
riflessione con traslazione (slittopiani)
rotoinversione: (1,2,3,4,6 soprasegnati);
Se uniamo questi elementi di simmetria con la traslazione ricaviamo 230 “gruppi spaziali”, se,
invece, non consideriamo la traslazione si scende a 32 classi cristalline o “gruppi puntuali”, in
analogia con quelli spaziali, in quanto gli elementi di simmetria passano (assi e piani) o
giacciono (centro) su un punto (normalmente il baricentro del cristallo).
Le 32 classi cristalline sono dovute a tutte le possibili combinazioni degli elementi di
simmetria.
Non esistono assi di ordine 5 o superiori a 7, in quanto con questi assi verrebbe meno la
traslazione di un uguale periodo.
I cristalli, quindi, sono la ripetizione nelle tre direzioni dello spazio di unità strutturali e le
superfici che li determinano (le facce del cristallo) dipendono da queste unità e dalle
condizioni di crescita: temperatura, pressione, ambiente chimico, direzione di flusso delle
soluzioni, spazio a disposizione per crescere, ecc.
Le relazioni angolari, lo sviluppo e la forma delle facce fanno parte della cristallografia
morfologica.
In un cristallo comunemente avremo poche facce (questo è stato trovato
sperimentalmente) e queste sono parallele a piani reticolari ad alta densità di punti
(nodi), legge di Bravais.
Sperimentalmente è stata anche ricavata la legge della costanza degli angoli diedri tra le
facce (legge di Hauy): se nei cristalli di un minerale si misurano angoli diedri tra coppie
di facce essi avranno valori uguali in tutti i cristalli dello stesso minerale, purché
misurate a temperatura costante.
Il miglior modo di rappresentare la morfologia di un cristallo è quello di riferire facce e spigoli
a una terna di assi (che si può pensare con l’origine nel baricentro del cristallo, anche se si
potrebbe prendere dove si vuole). Fissata la terna di riferimento ne possiamo individuare i
parametri: le distanze dall’origine dell’intersezione di una faccia sugli assi.
Se scegliamo una faccia in posizione generale, quindi staccherà tre parametri, come
riferimento (faccia fondamentale) e tutte le altre facce le riferiamo a questa avremo:
a/a’:b/b’:c/c’ = h:k:l (indici di Miller) dove a,b,c sono i parametri della faccia fondamentale e
a’,b’,c’ quelli dell’altra faccia.
Se si sceglie in maniera opportuna la faccia fondamentale e la terna di riferimento secondo
direzioni cristallografiche appropriate (direzioni di simmetria, spigoli reali o possibili, ecc.) gli
indici di Miller saranno generalmente piccoli e razionali.
Gli assi di riferimento possono avere:
3 angoli diversi da 90° (simmetria 1; sistema triclino)
1 angolo diverso da 90° (simmetria 2; sistema monoclino)
3 angoli uguali a 90° (simmetria 222; sistema rombico)
3 angoli uguali a 90° (simmetria 4: due assi equivalenti; sistema tetragonale)
3 angoli uguali a 90° e uno di 120° (simmetria 3,6: riferimento a 4 assi con 3
equivalenti; sistemi trigonale ed esagonale)
3 angoli uguali a 90° (simmetria 333: tre assi equivalenti; sistema cubico).
PROIEZIONE STEREOGRAFICA
La proiezione di un cristallo è un modo per rappresentare un cristallo, che è tridimensionale,
su di un piano. Nella proiezione stereografica si agisce nel modo seguente:
si pone il cristallo con il baricentro nel centro di una sfera, si tracciano le perpendicolari alle
facce e si determinano i punti di incontro delle perpendicolari stesse con la sfera. Individuati i
punti, si uniscono gli stessi con il polo dell’emisfero opposto: l’incontro con il piano equatoriale
determina la posizione della faccia. Se si va dall’emisfero nord al polo sud, si segna con una
crocetta o con pallino pieno; se si va dall’emisfero sud al polo nord, si segna con cerchietto o
con un pallino vuoti.
Simboli usati in proiezione stereografica:
roto‐riflessioni e roto‐inversioni
Centro di simmetria 1 nessuno
Piani di simmetria m ( linea continua)
Equivalenze
Rotazioni compatibili con i reticoli
cristallini
BB’ = BC+CC’+C’B’= AA’ + 2 t cos(180-) = t + 2 t cos(180-)
Sia alfa l’angolo generico di un punto di rotazione applicato nel nodo A.
I punti B e B’ devono trovarsi sullo stesso filare parallelo ad AA’.
Inoltre deve essere BB’ = m AA’ = m t segnato
dove m è un numero intero, positivo o negativo, 0 compreso.
BB’ = mt BB’ = t + 2 t cos(180-)
m = - 2cos
m = 1- 2cos- cos alfa = (1-m)/2 cos alfa : M/2
Questa dimostrazione impone che solo
cinque tipi di rotazione siano possibili nei cristalli, questo vale anche per le roto‐riflessioni e le
roto‐inversioni. Si noti, però, che gli ordini di rotazione non accettati per i cristalli sono però
possibili per le molecole isolate
REGOLE DI COESISTENZA DEGLI ELEMENTI DI SIMMETRIA
Possono coesistere solo queste combinazioni di assi di simmetria:
Tre assi binari ortogonali 222
Un asse ternario con tra assi binari ortogonali 32
Un asse quaternario con 4 assi binari ortogonali 422
Un asse senario con 6 assi binari ortogonali 622
Tre assi binari ortogonali e 4 assi ternari ciascuno dei quali forma lo stasso angolo con gli
assi binari 23
Tre assi quaternari ortogonali, 4 assi ternari ugualmente
inclinati rispetto ai quaternari e 6 assi binari bisettori dell angolo tra i quaternari 432
RAGGI X
L’applicazione dei raggi X allo studio dei minerali ha dato un grande impulso alla mineralogia.
Bragg (padre e figlio) nel 1914 risolsero la prima struttura, che fu quella del salgemma.
La scoperta dei raggi X è avvenuta per caso: Roentgen (1895) durante un esperimento per la
produzione di raggi catodici si accorse di aver causato fluorescenza in un minerale e la
imputò a una nuova radiazione e la chiamò X, perché non ne conosceva la natura.
La natura ondulatoria delle radiazioni luminose era stata dimostrata dal fatto che queste
davano fenomeni di diffrazione, fenomeni non erano stati riscontrati per i raggi X. Nel 1912
Von Laue, Sommerfeld, Ewald ed altri, supponendo che i raggi X avessero lunghezza d’onda
dello stesso ordine di grandezza del reticolo cristallino, fecero esperimenti di diffrazione con i
raggi X usando i minerali come reticolo di diffrazione e dimostrarono il reticolo ordinato e
periodico dei minerali e la natura ondulatoria dei raggi X, cioè che questa radiazione faceva
parte dello spettro elettromagnetico.
Le onde elettromagnetiche formano un spettro continuo, hanno proprietà comuni (rifrazione
riflessione,ecc.) e la relazione che lega energia e lunghezza d’onda è quella di Planck:
e=hη=hc/λ (e = energia, η = frequenza, c = velocità di propagazione, λ = lunghezza d’onda, h
= costante).
I raggi X nello spettro elettromagnetico appartengono a quella porzione compresa fra
l’ultravioletto e i raggi gamma; oltre che essere emessi naturalmente da alcuni isotopi
radioattivi essi sono normalmente prodotti da un improvviso rallentamento di elettroni
accelerati con trasformazione della loro energia cinetica in quanti di radiazione.
I raggi X vengono, quindi, prodotti da un tubo, dove è stato fatto un vuoto quasi completo, con
un filamento di wolframio (catodo) che riscaldato col passaggio di corrente emette elettroni
per effetto termoionico (raggi catodici) che vengono accelerati verso una targhetta di metallo
(anodo o anticatodo) da una grande differenza di potenziale : gli elettroni emessi dal filamento
del wolframio colpiscono gli elettroni degli atomi dell’anticatodo ed è in queste collisioni che
rallentano e perdono energia. Il tipo di radiazione dipende dalla targhetta e dalla differenza di
potenziale
Raggiunto un valore limite (a causa della perdita di energia degli elettroni frenati nel colpire
l’anticatodo) si ha uno spettro continuo o radiazione bianca o radiazione di frenamento.
Aumentando il voltaggio fino a un valore critico (che dipende dal materiale dello stesso
anticatodo) allo spettro continuo se ne sovrappone uno caratteristico .Quest’ultimo spettro si
produce quando gli elettroni che colpiscono l’anticatodo hanno energia sufficiente per
strappare (espellere) elettroni dagli strati più interni degli atomi del materiale dell’ anticatodo
stesso: rimangono dei vuoti che vengono riempiti a cascata dagli elettroni degli strati più
esterni e si accompagna a questo una emissione X con specifiche lunghezze d’onda. Le
transizioni da strati L a strati K sono le Kα, quelle da strati M a strati K sono le Kβ, ecc.
DIFFRAZIONE
Quando i raggi X colpiscono un ostacolo cristallino subiscono non solo il fenomeno
dell’assorbimento, ma anche quello della diffusione (gli atomi emettono raggi X della stessa
lunghezza d’onda di quelli incidenti. Ogni atomo diffonde la radiazione incidente in tutte le
direzioni e se si considera un filare di atomi investito da un fronte d’onda piano, ognuno degli
atomi diviene centro di propagazione di nuove onde che interferiranno fra di loro e, in alcune
determinate direzioni, produrranno un rafforzamento dando luogo al fenomeno conosciuto
come diffrazione.
La maniera più semplice per spiegare la diffrazione dei raggi X è quello di Bragg che
considera la diffrazione come una riflessione che riguarda non solo il primo strato superficiale,
ma anche quelli più interni. Adoperando una radiazione monocromatica (cioè di una sola
lunghezza d’onda), Bragg stabilì che si poteva avere effetti di diffrazione solo quando il
cammino dei raggi X incidenti e diffratti o riflessi differiva di un numero intero di lunghezze
d’onda, giungendo alla seguente equazione: nλ = 2dsenθ.
Trattazione di Bragg
Un omogeneo periodico tridimensionale si può interpretare in termini di fasci di piani reticolari
identici tra loro e ugualmente spaziati. Ogni terna di numeri hkl interi definisce un fascio di tali
piani, con distanza reticolare dhkl.
Consideriamo un fascio di raggi X paralleli e monocromatici che incide con un angolo su
un fascio di piani reticolari.
Il fenomeno è però assimilabile ad una riflessione selettiva: i piani reticolari riflettono i raggi X
SOLO quando l’angolo di incidenza dei raggi X sul piano reticolare (θ) soddisfa la relazione di
Bragg.La direzione di riflessione forma con il piano reticolare un angolo di riflessione uguale a
quello di incidenza. La direzione di riflessione forma con la direzione dei raggi X incidenti un
angolo doppio.
L’effetto di diffrazione del secondo ordine per la famiglia di piani hkl è equivalente all’effetto di
diffrazione del primo ordine della famiglia di piani 2h 2k 2l aventi distanza interplanare dhkl/2.
Significato di dhkl/n
Nella pratica, non essendo possibile riconoscere l’ordine di una riflessione, tutte le riflessioni
di ordine n superiore al primo sono considerate come riflessioni di primo ordine per la famiglia
di piani nh nk nl A livello cristallografico hanno significato anche i piani aventi indici hkl non
primi fra loro.
L’equidistanza dei piani reticolari (dhkl) – e quindi LA POSIZIONE DEI
MASSIMI DI DIFFRAZIONE – dipende esclusivamente dalla geometria della cella elementare
del minerale, cioè dal valore dei parametri di cella (a, b, c, , , ) e (ovviamente) dai valori
di h, k, l.
DIFFRAZIONE RAGGI X
MASSIMO DI DIFFRAZIONE POSIZIONE
INTENSITA’
Esistono, per ciascun sistema cristallino, delle formule per ricavare le dhkl di ciascuna
famiglia di piani reticolari (hkl) noti i parametri di cella. La formula è molto complicata per i
sistemi a bassa simmetria (p. es. triclino) e si semplifica via via che la simmetria cresce (fino
al cubico):
d(hkl)2 = a2/(h2+k2+l2)
(per sistema cubico)
L’INTENSITA’ di un massimo di diffrazione relativo ad una data famiglia di PIANI
RETICOLARI (hkl) si ottiene sommando i contributi dei vari atomi presenti nella cella
elementare. Essa dipende, per ciascun atomo, dal:
• fattore di scattering(fj);
• posizione dell’atomo nella struttura (x,y,z).
Fhkl è il Fattore di Struttura. Rappresenta in ampiezza e fase l’onda diffratta da tutti gli atomi
della cella elementare per il piano (hkl) (cioè nella direzione di diffrazione del piano di indici
hkl). L’ INTENSITA’ del riflesso (hkl) è direttamente proporzionale al quadrato del modulo del
FATTORE DI STRUTTURA.
LE POSIZIONI ATOMICHE SI POSSONO DETERMINARE DALLA MISURA DELLE
INTENSITA’ DIFFRATTE
I = F2 Fattore di struttura
Fhkl= Σfne2πi(hun+kvn+lwn) Σ estesa a tutti gli atomi della cella elementare fn= fattore di
diffusione atomico u, v, w = coordinate frazionarie dell’atomo ennesimo
Obiettivo dell’esperimento: portare in condizioni di diffrazione (riflessione) più piani reticolari
possibile.
Questo obiettivo può essere raggiunto:
a) Orientando sotto il fascio di raggi X in tutti i modi possibili un cristallo singolo
b) Operando su una polvere microcristallina, cioè su un vasto insieme di piccoli xlli orientati
statisticamente in tutte le direzioni.
Le varie tecniche di indagine diffrattometrica si differenziano per:
1‐ tipologia del campione
2 ‐ uso di radiazione poli‐ o mono‐cromatica
3‐ geometria dell’apparecchiatura
4‐ mezzo di rivelazione
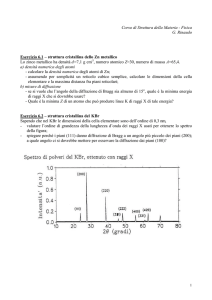
1)_Diffrattometro per polveri. E’ un’apparecchiatura costosa che costituisce una delle
tecniche di analisi mineralogica più moderne e fa parte dei metodi di diffrazione su polveri .
Se si colpisce con un fascio di raggi X monocromatici una sostanza cristallina finemente essa
li diffrange per determinati angoli,caratteristici di ogni singolo minerale, dipendenti dalla
disposizione interna del reticolo cristallino che la costituiscono.Un contatore misura l’intensità
dei raggi X diffratti che, insieme all’angolo sotto cui si verifica la diffrazione, viene
automaticamente registrata. Per far sì che tutti i possibili angoli di diffrazione vengano
esplorati, il portacampione (una lastrina su cui è pressata la polvere del campione) viene fatta
ruotare con continuità di fronte al fascio dei raggi X. Perché il contatore possa registrare i
raggi diffratti, deve ruotare con velocità doppia della lastrina portacampione
Il riconoscimento dei minerali per diffrattometria X è possibile sia in quanto tutte le sostanze
cristalline presentano uno spettro caratteristico, sia perché sono stati eseguiti numerosissimi
diffrattogrammi relativi a sostanze cristalline note. Una volta ottenuti gli angoli e le intensità di
diffrazione di una sostanza incognita, il confronto con quelli riportati nelle tabelle ci permette
di identificarla.
In altre parole:
Ogni cristallino possiede in sé tutte le serie di piani reticolari con distanze d1, d2,d3….dn
caratteristiche del reticolo di traslazione della sostanza in esame.
Ad ogni valore di d corrisponde un certo valore di che soddisfa le condizioni di Bragg.
Dato che nel campione di polveri sono rappresentate tutte le possibili orientazioni, vi sarà
sempre un certo numero di cristalli che presentano al raggio incidente una serie di piani
reticolari con intervallo di ripetizione d1 sotto l’angolo 1; altri granuli presentano altre serie
di piani reticolari con distanza d2, d3….dn, sotto angoli 2, 3… n.
L’angolo 1 (e analogamente 2, 3… n) è in realtà verificato da
innumerevoli piani
analoghi, contenuti in altrettanti granuli. In particolare, sono valide tutte le direzioni di raggi
diffratti appartenenti ad un cono di apertura angolare 4 1, 4 2… 4 n. Ciascuna serie di
piani con diversa distanza interplanare darà origine ad un proprio cono di diffrazione di
apertura crescente via via che d diminuisce.
Dalla misura degli angoli si ricavano i valori delle distanze interplanari dhkl, che dipendono
solo dai parametri di cella. Da database cristallografici si ricava la fase in esame.
Recenti simulazioni al computer hanno prospettato la possibilità di costruire anche nanotubi di
atomi di boro: al centro di alcuni esagoni sarebbe necessario inserire un atomo extra di boro.
Il boro è un elemento utilizzato per drogare microcristalli di diamante impiegati nella
ostruzione di fotocelle sensibili alla radiazione ultravioletta. Inoltre questi cristalli sono
superconduttori a temperatura inferiore a 20 K.
La formula di Scherrer è utilizzata per la determinazione,tramite diffrazione di polveri, della
dimensione media deicristalliti ( t)
t = / B cosθB
B = larghezza a metà altezza di un picco di diffrazione.Ipotizziamo il caso di un cristallo
costituito da due soli piani reticolari, chiamati 1 e 2, posti a distanza d.
Per ogni valore p dell’angolo di incidenza, tale che l’espressione 2d sen passuma un
valore uguale a un numero intero di lunghezze d’onda, i raggi riflessi dal piano 1 e dal piano 2
sono in interferenza positiva piena mentre per ogni valore n tale che l’espressione 2d sen
n assuma un valore uguale a un numero dispari di mezze lunghezze d’onda, i due raggi
sono in interferenza negativa piena. Per ogni valore di intermedio tra p e n si
realizzeranno condizioni di parziale interferenza positiva.
Quanto ora detto è illustrato graficamente dalla seguente figura nella qual in ordinata è
riportata l’ampiezza dell’onda diffratta dai due piani in funzione dell’angolo di incidenza.
Passiamo ora al caso di un cristallo costituito da una serie infinita di piani
reticolari.
Supponiamo che il valore dell’angolo sia tale da portare a una differenza di cammino, tra il
raggio riflesso dal piano 1 e quello riflesso dal piano 2, uguale a + (1/100) . La differenza
di cammino tra i raggi riflessi dal 1° e 3° piano sarà uguale a 2 + (2/100) , fra quelli riflessi
dal 1° e dal 4° piano sarà uguale a 3 + (3/100) , fra quelli riflessi dal 1° e dal 51° piano
sarà uguale a 50 +(50/100) , cioè 50 + (1/2) . Concludiamo quindi che il 51° piano
annulla il raggio riflesso dal 1°, il<52° quello del 2° e così via.E’ evidente che se il cristallo è
infinito, qualunque differenza di cammino, per quanto piccola, porterà ad una interferenza
negativa tra il primo e un n‐esimo piano. Si avrà quindi diffrazione solo nel caso che l’angolo
sia esattamente quello previsto dalla legge di Bragg.
A creare una situazione del genere contribuiscono il non perfetto parallelismo dei raggi X e la
non perfetta monocromaticità, ma soprattutto la struttura “reale” del cristallo. I cristalli reali,
infatti, hanno sempre delle imperfezioni reticolari, che fanno cadere in difetto l’omogeneo
periodico tridimensionale. In pratica il cristallo reale è costituito da una serie di porzioni
perfette, ma orientate in modo leggermente diversa l’una rispetto alle altre STRUTTURA A
MOSAICO
I dati di diffrazione a raggi X di un campione policristallino permettono di determinare:
• la composizione delle fasi cristalline (analisi qualitativa e quantitativa);
• gli indici dei picchi di Bragg, le loro intensità integrate e parametri di cella molto precisi;
• la distribuzione degli atomi in cella, ovvero la struttura cristallina e, ove pertinente,
molecolare;
• diverse caratteristiche (micro)strutturali.
Lo spettro di diffrazione di una fase cristallina è un fingerprint univoco che permette di
riconoscere una fase se se ne conosce già lo spettro. Condizioni necessarie per l’analisi
qualitativa:
• Avere a disposizione fasi pure di riferimento o:
• Avere a disposizione banche dati d/I o:
• Avere a disposizione banche dati di coordinate cristallografiche
CRISTALLOCHIMICA
Relazioni fra composizione chimica struttura dei minerali
Per determinate condizioni chimico-fisiche ambientali (m, P, T) struttura più stabile di un certo
insieme di atomi è quella a cui compete il valore minimo di ENERGIA LIBERA G.
Se variano le condizioni ambientali, possono invertirsi i rapporti di stabilità tra differenti
configurazioni e aversi FENOMENI DI POLIMORFISMO.
Dal momento in cui un minerale cristallizza da un magma o da una soluzione idrotermale,
esso è soggetto a cambiamenti sia nel suo intorno chimico che fisico.
Uno degli argomenti fondamentali della mineralogia riguarda il grado con cui i minerali
rispondono a tali cambiamenti, adattando la loro struttura e la loro composizione al nuovo
ambiente, possono essere piccoli cambiamenti nelle lunghezze o negli angoli di legame o
maggiori trasformazioni strutturali; essi possono comportare cambiamenti chimici su scala
atomica o vere e proprie reazioni con formazione di nuove specie. Tutti questi processi,
comunque, hanno un obiettivo comune:
LA DIMINUZIONE DELL’ENERGIA LIBERA DEL SISTEMA MINERALE SOTTO LE NUOVE
CONDIZIONI AMBIENTALI.
La termodinamica tratta degli stati iniziali e finali dei processi mineralogici, e non dei
meccanismi di trasformazione tra uno stato e l'altro.
La termodinamica descrive il comportamento ideale ed assume che i minerali siano in
equilibrio con l’intorno. Non vi è però nessuna garanzia che in realtà tale equilibrio si realizzi.
Es. velocità di raffreddamento superiore alla velocità intrinseca delprocesso; il minerale in tali
condizioni non è in grado di adattare la propria struttura al cambiamento di T e quindi si
hanno forti deviazioni dalle condizioni di equilibrio.
Il comportamento REALE dei minerali in fase di trasformazione è principalmente determinato
dalle velocità dei processi, cioè da FATTORI CINETICI
RAGGI IONICI
Il raggio ionico esprime le dimensioni di uno ione considerato come una sfera rigida.
Il modello approssimato che meglio e più semplicemente spiega il comportamento dei
minerali è il modello ionico a forza di campo centrale.
In questo modello i componenti la struttura cristallina sono rappresentati da ioni rigidi, aventi
un determinato raggio e una determinata carica
PRINCIPIO DEL MASSIMO RIEMPIMENTO:
Unità strutturali semplici (atomi o ioni) o complesse (gruppi di atomi o ioni) sulle quali
agiscono forze attrattive a campo centrale (o approssimativamente a campo centrale)
tendono ad aumentare al massimo i contatti reciproci, riducendo al minimo le loro distanze.
COORDINAZIONE
Nel caso in cui ioni di segno opposto ciascun ione tende ad attrarre il maggior numero
possibile di ioni di segno opposto, quindi ogni ione tende a legare (coordinare) ioni di segno
opposto in funzione della sua dimensione. Quando gli atomi sono legati da semplici legami
elettrostatici si possono supporre come sfere a contatto. La coordinazione si esprime con un
numero (a seconda degli ioni che vengono coordinati) e con il nome del poliedro su cui sono
disposti gli ioni coordinati (ad es. coordinazione 4 tetraedrica, coordinazione 6 ottaedrica,
ecc.)
Il numero e il tipo di coordinazione è funzione del rapporto dei raggi degli ioni (Rc:Ra, con
Rc=raggio del catione e Ra=raggiodell’anione). Se gli ioni coordinanti e quelli coordinati sono
uguali il rapporto fra i raggi sarà uguale a 1; in questo caso avremo delle coordinazioni
compatte (n. di coordinazione 12). Il massimo ricoprimento dello spazio sul piano si ha con
una sfera circondata da altre 6, tutte a contatto. Gli spazi vuoti tra le sfere (B e C) possono
essere riempiti da altre sfere; se la sequenza è: ABABAB avremo un impaccamento
esagonale compatto, se è ABCABCABC avremo un impaccamento cubico compatto.
Quando il catione è più piccolo dell’anione, cioè il rapporto fra i raggi è inferiore a 1, avremo
numeri di coordinazione più piccoli; Definiamo COEFFICIENTE DI IMPACCHETTAMENTO Ci
il seguente rapporto:
Ci = S (Va/Vcella)
dove SVa = volume occupato dagli anioni contenuti nella cella
elementare Vcella
In accordo con il principio di massimo riempimento:Ci max
In un insieme di sfere di uguale raggio, disposte secondo un impacchettamento compatto, si
formano due tipi di lacune:
- LACUNE TETRAEDRICHE (T)
- LACUNE OTTAEDRICHE (O)
Si dimostra che per ogni sfera (S) di un impacchettamento compatto si formano 2 lacune
tetraedriche e una ottaedrica:
T : O : S = 2 : 1 :1
Ciascun tipo di lacuna sarà occupato da cationi di raggio ionico appropriato.
Le lacune NON devono necessariamente essere tutte occupate da cationi, ma possono
anche rimanere vacanti.
ESEMPI
NaCl : impacchettamento cubico compatto di ioni cloro in cui tutte le lacune ottaedriche sono
occupate da Na
MgO : impacchettamento cubico compatto di ossigeni in cui tutte le lacune ottaedriche sono
occupate da Mg
NiAs : impacchettamento esagonale compatto di ioni As in cui tutte le lacune ottaedriche sono
occupate da Ni
Halite (NaCl): Impacchettamento CCP di anioni Cl-1 con i cationi Na+1 negli interstizi
ottaedrici.
Nickelite (NiAs): Impacchettamento HCP di ioni As3- con i cationi Ni3+ negli interstizi
ottaedrici.
Al2O3 : impacchettamento esagonale compatto di ossigeni in cui i 2/3 delle lacune
ottaedriche sono occupate da Al
CdI2 : impacchettamento esagonale compatto di anioni iodio in cui metà delle lacune
ottaedriche sono occupate da Cd
CRITERI DI STABILITA’ DELLE STRUTTURE AD IMPALCATURA POLIEDRICA
1) Quando due ioni di carica opposta si avvicinano, la loro distanza di equilibrio è determinata
dal bilanciamento di forze elettrostatiche attrattive o repulsive;
2) Nelle tre dimensioni, ioni collocati secondo i principi del legame ionico tendono a formare
poliedri di coordinazione altamente simmetrici; tetraedri e ottaedri sono i più comuni, ma
anche i triangoli e i cubi sono importanti;
3) Questi poliedri di coordinazione si collegano in vari modi per formare strutture ad
impalcatura poliedrica; queste includono minerali delle rocce come silicati, borati, solfati,
ossidi e idrossidi.
Regole di Pauling
I principi che si possono seguire per capire queste strutture sono stati forniti alla fine degli
anni ’20 da Pauling e Bragg e sono noti come le 5 regole diPauling.
Regola 1 – Distanze interatomiche
Attorno a ciascun catione si forma un poliedro di coordinazione di anioni. La distanza cationeanione è pari alla somma dei rispettivi raggi ionici; la forma del poliedro e il numero di
coordinazione dipendono dal rapporto dei raggi
Dc-a= rc+ ra
N.C. rc/ra
Regola 2_Principio della valenza elettrostatica
In una struttura ionica stabile, la forza del legame elettrostatico totale che converge su un
anione da tutti i cationi posti al centro dei poliedri di cui l’anione è vertice, è pari alla carica di
tale anione.
Questa regola è una diretta conseguenza del legame ionico. Poichè le interazioni sono
elettrostatiche, la capacità totale di legame di un catione è proporzionale alla sua carica.
Una misura della forza elettrostatica di un singolo legame è semplicemente la carica totale Z
del catione divisa per il numero totale di anioni a cui il catione si lega, cioè il numero di
coordinazione.
Regola 3-Condivisione di elementi tra poliedri - I.
La presenza di spigoli, e soprattutto di facce, in comune tra i poliedri di una struttura ionica,
ne decresce la stabilità. Questo effetto è maggiore per i cationi di alta valenza e basso
numero di coordinazione (es. Si4+).
Questa regola è ancora una volta una diretta conseguenza delle forze elettrostatiche che
tengono insieme le strutture ioniche. La configurazione più stabile si ha quando due poliedri
condividono solo un vertice perchè in tal modo I due cationi centrali, che tendono a
respingersi, sono il più lontano possibile l’uno dall’altro.
Regola 4-Condivisione di elementi tra poliedri - II.
In un cristallo contenente diversi cationi, quelli con valenza alta e numero di coordinazione
basso tenderanno a non mettere in comune nessun elemento del proprio poliedro di
coordinazione.
I cationi con alta carica tenderanno ad essere distribuiti alla massima distanza possibile nella
struttura cristallina. L’effetto di tale regola è che, a parità di carica, un catione tenderà a
privilegiare le coordinazioni più alte.
La terza e la quarta regola di Pauling spiegano come mai i poliedri tendono a non condividere
né facce né spigoli.
Regola 5-Principio di parsimonia
Il numero di costituenti di un cristallo che differiscono tra loro in modo essenziale tende ad
essere piccolo.
Il numero di diversi ioni e di diversi poliedri di coordinazione che possono formare un cristallo
è limitato e piccolo. Generalmente non troviamo più di 2 o 3 diversi poliedri in una struttura
cristallina. Il numero di diversi siti cristallografici è perciò piccolo ed I loro numeri stanno in
rapporto piccolo ed intero l’uno rispetto all’altro.
Questo è il motivo per cui i vari cationi e anioni nelle formule chimiche dei minerali stanno in
rapporti piccoli ed interi tra loro.
POLIMORFISMO
Il polimorfismo è il fenomeno a seguito del quale una sostanza di composizione chimica
costante può cristallizzare con strutture diverse.
Ogni polimorfo ha un suo campo di stabilità termodinamico definito da T, P.
Le trasformazioni polimorfiche sono esempi di TRASFORMAZIONI ISOCHIMICHE, cioè
avvengono con conservazione della composizione chimica
Diagramma di fase dei diversi polimorfi di Al2SiO5
Lungo ogni linea che separa due campi, sono stabili due polimorfi.
I tre polimorfi sono contemporaneamente stabili solo in corrispondenza del punto triplo
DINAMICA DEL POLIMORFISMO
Le trasformazioni polimorfe sono governate, oltre che da aspetti termodinamici, anche da
aspetti cinetici: affinchè una trasformazione avvenga è necessaria una certa ENERGIA DI
ATTIVAZIONE, che è tanto maggiore quanto più sono diverse le due strutture.
CLASSIFICAZIONE DEL POLIMORFISMO IN BASE AL MECCANISMO DELLA
TRASFORMAZIONE
1-POLIMORFISMO DISTORSIVO
La trasformazione da una modificazione all’altra avviene senza cambiamento del numero di
primi vicini e senza rottura di legami, ma solo con una distorsione degli angoli di legame.
-Basso assorbimento di energia
-Alta velocità di reazione
-Stretto intervallo di temperatura
-Completa reversibilità
Es. Quarzo a Quarzo b
g.s P312 (trigonale) g.s P622 (esagonale)
2-POLIMORFISMO RICOSTRUTTIVO
Il passaggio da un polimorfo ad un altro richiede almeno due stadi:
a. Rottura di un certo numero di legami e collasso della vecchia struttura
b. Diffusione allo stato solido degli atomi e loro riorganizzazione secondo un nuovo motivo
strutturale
-Notevole dispendio di energia
-Bassa velocità di reazione
-Vasto intervallo di temperatura
-Frequente irreversibilità
Es. Calcite ad Aragonite
3-POLIMORFISMO ORDINE – DISORDINE
Fasi con distribuzione ordinata o disordinata di cationi di raggio ionico e carica simili in una
serie di posizioni cristallografiche. Si può avere tra coppie di fasi con strutture molto simili: in
quella disordinata esiste una serie di posizioni cristallograficamente equivalenti che sono
occupate statisticamente da due ioni simili per raggio e carica.
Nella forma ordinata i due ioni sono distribuiti non più statisticamente, ma secondo una
determinata regola e le due posizioni, di conseguenza, non sono più equivalenti per
simmetria.
LA FASE ORDINATA HA SEMPRE SIMMETRIA INFERIORE A QUELLA DISORDINATA LA
FASE ORDINATA E’ IN GENERE STABILE A BASSA TEMPERATURA
Esempio: Feldspato potassico KAlSi3O8
Sanidino (g.s. C2/m) Microclino (C-1)
4-POLITIPISMO
Fenomeno per cui una sostanza può cristallizzare in un certo numero di modificazioni
strutturali diverse, le quali sono tutte costituite da strati atomici ISOSTRUTTURALI,
PARALLELI ed EQUIDISTANTI, ma DIVERSAMENTE ORIENTATI O TRASLATI l’uno
rispetto all’altro. Le celle elementari dei politipi avranno due vettori uguali (paralleli agli strati)
mentre il terzo vettore può essere variabile nei vari politipi.
5-ALLOTROPIA
Se la sostanza che dà origine a cristalli diversi è una specie allo stato elementare, si parla più
correttamente di ALLOTROPIA, cioè di polimorfismo per cambiamento di legame chimico.
Esempi
C diamante (cubica a facce centrate)
C grafite (struttura a strati).
ISOMORFISMO
Due composti isostrutturali possono presentare il fenomeno dell’ISOMORFISMO, cioè
possono cristallizzare dando relazioni di miscibilità allo stato solido, così da formare SERIE
ISOMORFE, di cui essi costituiscono gli end-members.
La formazione di soluzioni solide deriva dall’estensione di un fenomeno molto generale tra i
minerali: la vicarianza.
VICARIANZA = possibilità di un atomo di sostituirne un altro di specie diversa, provocando
un difetto puntuale sostituzionale che, ripetuto traslazionalmente, arriva a modificare la
composizione chimica della struttura nel suo insieme.
VICARIANZA Þ DIFETTO CHIMICO Þ RIPETIZIONE REGOLARE DEL DIFETTO Þ
ISOMORFISMO
La possibilità di vicarianza dipende dai seguenti fattori:
-natura della struttura in cui si realizza la soluzione solida
-corrispondenza dei raggi ionici
-mantenimento della elettroneutralità
-temperatura di formazione della soluzione solida
Esempi molto comuni di sostituzioni isomorfe nei minerali sono i seguenti:
Classificazione dei vari tipi di isomorfismo
I° specie: componenti miscibili con formula chimica simile, ugual numero di atomi,
valenza uguale e raggi ionici simili fra i cationi che si sostituiscono.
Es: Mg2SiO4-Fe2SiO4 (stabile a tutte le T) NaCl-KCl (stabile ad alta T)
II° specie: ioni vicarianti con raggio ionico vicino, ma cariche diverse (di 1 o
eccezionalmente di 2 unità); l’elettroneutralità è mantenuta con una seconda
sostituzione accoppiata.
Es: NaAlSi3O8-CaAl2Si2O8 (feldspato sodico - feldspato calcico = plagioclasi)
III° specie: l’aumento o la diminuzione di carica che si ha in una posizione reticolare
viene compensata dalla contemporanea sostituzione di uno ione di carica opposta.
Es: Ca2(Mg,Fe’’)5(Si4O11)2(OH)2 orneblenda
Ca2(Mg,Fe’’)4Fe’’’(Si4O11)2 O(OH) ossiorneblenda
Interstiziale: una posizione reticolare può essere occupata da un certo catione o
rimanere vuota, con formazione diuna lacuna reticolare.
Es: NaAlSiO4- SiSiO4 (Nefelina-Tridimite)
CaCa(Mg,Fe’’)5[(Si4O11)2(OH)2]-NaNaCa(Mg,Fe’’)5[(Si4O11)2(OH)2]
CLASSIFICAZIONE DEI MINERALI
Abbondanza e grande raggio ionico dell’O2- l’arrangiamento geometrico degli ossigeni
determina in grande misura la struttura della maggior parte dei minerali contenenti ossigeno.
• Altri anioni comuni: S2-, Cl-, F- dominano la struttura di solfuri e alogenuri.
• I cationi si combinano con anioni formando gruppi anionici che a loro volta dominano la
struttura di altri gruppi di minerali.
• Classificazione chimica basata sui cationi Þ NO nessuna somiglianza fra tutti i minerali
contenenti ad es. Fe.
Classificazione cristallochimica: basata sull’anione ogruppoanionico predominante
• Classificazione chimico-strutturale
• ogni gruppo di minerali ha certe proprietà comuni.
Dalla metà del diciannovesimo secolo i minerali vengono classificati in funzione dell’anione o
del gruppo anionici dominante. Questo metodo, però, non caratterizza completamente un
minerale, perché non tiene conto della sua struttura ed è per questo che, con l’applicazione
dei raggi X alla mineralogia, siamo passati a classificazioni cristallochimiche. Sulla base di
quanto detto i minerali vengono suddivisi nelle seguenti classi:
Elementi (leghe, carburi, nitruri)
Silicati
Solfuri (selenuri, arsenuri, tellururi, solfosali)
Ossidi (idrossidi)
Carbonati (nitrati, arseniti, seleniti, tellurati,iodati)
Borati
Solfati (tellurati, cromati, molibdati, wolframati)
Fosfati (arsenati, vanadati)
I minerali sono un numero (circa 3000-3500) molto inferiore a quello che ci si aspetterebbe
dalla combinazione degli elementi chimici costituenti la materia. Ciò è dovuto in parte alla
difficoltà di individuarli e maggiormente alla problematica della formazione e della stabilità
della crosta terrestre. Il numero è limitato per la distribuzione e concentrazione degli elementi
chimici e per i processi geologici relativamente monotoni in quanto, sulla crosta si hanno
specifiche temperature e pressioni pressoché stabili.
ELEMENTI
Escludendo le sostanze gassose dell’atmosfera, allo stato naturale si trovano solo circa 20
elementi che possono essere suddivisi in: metalli, semimetalli, non metalli.
Elementi metallici. Sono caratterizzati da malleabilità, conduttività, lucentezza metallica,
sono abbastanza teneri e, avendo delle strutture compatte (c f.c., hcp) o quasi-compatte
(ccc.), hanno alta densità. Si dividono in tre gruppi: gruppo dell’oro, gruppo del platino, gruppo
del ferro.
Oro (Au, cubico). Caratteristica distintiva è il colore unito al suo alto pesi specifico (19.3)e alla
malleabilità. Si trova in vene idrotermali quarzifere insieme con pirite e altri solfuri e in depositi
alluvionali. Contiene quasi sempre argento, con cui forma una soluzione solida completa.
Argento (Ag, cubico). Molto raro in cristalli, si trova in dendriti, filamenti, laminette, ecc. Si
distingue per il colore, la malleabilità e il peso specifico (10.5). Si deposita da soluzioni
idrotermali primarie.
Rame (Cu, cubico). Raro in cristalli è comune in masse compatte, spugnose in dendriti e in
filamenti. Si deposita da soluzioni idrotermali per azione di minerali di ferro. I depositi primari
sono associati a basalti.
Platino (Pt, cubico). Si trova in rocce basiche e ultrabasiche e in depositi alluvionali.
Ferro (Fe, cubico). Quasi introvabile come ferro terrestre, è costituente fondamentale di
alcune meteoriti (sideroliti) in associazione con il nichel (kamacite e taenite). E’
ferromagnetico, ed la sua più importante caratteristica diagnostica.
Elementi semimetallici. Vengono raggruppati gli elementi del V gruppo (As, Sb, Bi). Sono
fragili con un forte potere riflettente e legame metallico e covalente. Sono tutti e tre trigonali
con perfetta sfaldatura basale e si trovano in vene idrotermali associati a minerali di Ag, Co,
Ni, ecc.
Elementi non metallici. Zolfo(S, rombico). Esistono due modificazione monocline rarissime in
natura, ma semplici da produrre in laboratorio. Si trova in cristalli bipiramidali , ma spesso in
masse sferoidali, mammellonati, in incrostazioni, ecc. Colore giallo (con tonalità dal verde al
rosso per presenza di impurità), trasparente o traslucido è fragile con frattura concoide. Si
distingue per il colore e per la sua infiammabilità.
Genesi: sedimentaria (solfare) associato a gesso, celestina, anidrite, ecc.; connessa con le
ultime fasi di attività vulcanica (solfare, famosa quella dei Campi Flegrei) o come prodotto di
sublimazione di vulcani attivi; alterazione di depositi a solfati per azione di solfobatteri, ecc. Lo
zolfo è abbondantissimo in Italia dove sono molto sviluppati i terreni noti come “formazione
gessoso-solfifera”. La regione più ricca di zolfo è la Sicilia dove sono stati trovati bellissimi
cristalli, per forma e dimensione, specie nelle solfare in provincia di Agrigento solfatare di
Porto Empedocle figghiu miu.
SILICATI
E’ la classe più importante: circa il 25% dei minerali conosciuti e il 40% dei più comuni
appartengono a questa classe e, in più, i silicati costituiscono oltre il 90% dei minerali della
crosta terrestre, che è formata, principalmente, oltre dai silicati dagli ossidi e da altri composti
contenenti ossigeno come i carbonati.L’unità fondamentale di tutti i silicati è il tetraedro SiO4.
L’ ossigeno di un tetraedro può essere condiviso con quello di un altro tetraedro
indefinitamente.
Avremo una suddivisione dei silicati in:
nesosilicati (tetraedri isolati); Silicati isolati
SiO4 olivine,Granati
sorosilicati (gruppi di due tetraedri uniti per un vertice); Silicati a isola
Si/O = 2/7 (es. epidoti)
3/9 (es. benitoite)
6/18 (es. tormalina)
ciclosilicati (quando si uniscono più tetraedri);
inososilicati (quando si uniscono a formare catene sia semplici che doppie);Silicati a
catena
Catena singola: Si/O = 1/3 = 0.33 (es pirosseni, wollastonite)
Catena doppia: Si/O = 4/11 = 0.36 (es. anfiboli, sillimanite)
fillosilicati (quando si uniscono a formare strati);
Silicati a strati (fillosilicati):Si/O = 2/5 = 0.40 (es. miche, talco, pirofillite)
tectosilicati (quando i tetraedri costituiscono un’ossatura tridimensionalmente).
Silicati a framework (tettosilicati): Si/O = 1/2 = 0.50 (es. fasi della silice, feldspati,
feldspatoidi, zeoliti)
I tetraedri condividono tutti e 4 i propri vertici con altri tetraedri
Le fasi della silice sono caratterizzate da diffuse relazioni di: POLIMORFISMO
DISTORSIVO e POLIMORFISMO RICOSTRUTTIVO
Diagramma di fase dei polimorfi della silice
Il quarzo a è la forma stabile sulla crosta terrestre. All’aumentare
della T esso si trasforma nella fase b, che a sua volta si trasforma in tridimite a poi in
cristobalite. All’aumentare della P, il quarzo a si trasforma in coesite e poi stishovite.Nella
stishovite il Si non è più coordinato 4, ma 6.
Il quarzo a è trigonale, con catene elicoidali che corrono lungo l’asse c. Le eliche sono
collegate tra loro a formare una impalcatura tetraedrica priva di centro di inversione. Ciascun
tetraedro in una catena è ruotato di 120° rispetto al tetraedro sottostante ed èanche traslato di
c/3 lungo la direzione c. Questo corrisponde ad una elicogira ternaria 31. Ciascun tetraedro
condivide i propri 4 ossigeni con tetraedri adiacenti.
Ogni ossigeno è legato a due tetraedri e quindi cede ad ogni Si una carica negativa; ogni Si è
legato a 4 ossigeni e quindi è compensato elettricamente.
Nel quarzo b ci sono ancora elicogire ternarie, ma nel cristallo compaiono assi senari, mentre
nella fase a ci sono solo assi ternari.
La trasformazione a b (573°C) è distorsiva, non quenchabile, con nessuna discontinuità
nell’entropia o nel volume molare lungo la transizione.
Le strutture di quarzo a e b sono legate da polimorfismo distorsivo : le spirali di tetraedri, che
nel Qb hanno una simmetria esagonale, assumono una simmetria ternaria nel Qa quando la
T si abbassa sotto i 573°C a P ambiente. La tridimite è il più semplice polimorfo della silice.
E’ composta di strati di tetraedri legati a formare anelli a 6 in cui metà dei tetraedri puntano in
alto e metà in basso. Questi strati sono poi legati tramite gli ossigeni apicali che puntano
alternativamente in alto e in basso, secondo una sequenza ABAB…
La cristobalite ha un arrangiamento più distorto di tetraedri legati per i vertici. I vari strati si
succedono secondo una sequenza tipo ABCABC, simile a quella degli strati di anelli
esagonali di tetraedri di carbonio nel diamante.
SILICE IDRATA, AMORFA, MICROCRISTALLINA
Come abbiamo visto finora, i minerali sono quasi tutti cristallini; tra i pochi che non lo sono,
alcuni sono vetri (es. ossidiane), altri sono gel. Tra questi ultimi, abbastanza frequente in
natura è il gel di silice, cioè una sostanza sostanzialmente amorfa di composizione SiO2 +
H2O. La fase mineralogica corrispondente è l’OPALE, che contiene una quantità di acqua
variabile dal 3 al 10%, e ha una durezza poco inferiore a quella del quarzo .
Feldspati
Tettoalluminosilicati di formula generale: AT4O8
T= Si,Al
A = K, Na, Ca, Ba
Presentano una cristallochimica complessa:
-fenomeni di isomorfismo con formazione di soluzionisolide
-essoluzioni
-polimorfismo ordine-disordine e displacivo
-geminazioni
Anelli di 4 tetraedri che formano doppi colli d’oca.
Nelle cavità formate dai doppi collo d’oca trovano posto cationi di metalli alcalini e alcalino
terrosi
PLAGIOCLASI
Albite NaAlSi3O8 Anortite CaAl2Si2O8
C’è una soluzione solida completa tra albite e anortite ad alta T. I plagioclasi sono i minerali
più comuni della crosta terrestre.
Presentano un polimorfismo O-D che coinvolge Si e Al. Tutti i feldspati sono tettosilicati e tutti
hanno la stessa impalcatura di tetraedri (Si,Al)O4. In questa impalcatura tra un tetraedro e
l’altro ci sono degli spazi liberi nei quali trovano posto i cationi alcalini o alcalino-terrosi più
grandi. Tale impalcatura può essere schematizzata come un’interconnessione di catene
doppie note come “doppio collo d’oca”; i vari cationi extratetraedrici Na, K, Ca, Ba trovano
posto nelle cavità libere. La massima simmetria compatibile con tale impalcatura è quella del
gruppo spaziale C2/m monoclino, che è anche la simmetria effettiva di alcuni feldspati con
distribuzione disordinata di Si ed Al.
La simmetria reale della maggior parte dei feldspati è però inferiore, e ciò per due cause, che
possono anche coesistere:
1) Distribuzione ordinata di Si e Al nei tetraedri
2) Schiacciamento delle cavità ospitanti i cationi extraimpalcatura più piccoli delle
cavità stesse, provocando un collasso dell’impalcatura alluminosilicatica.
Questo collasso può risolversi o in una diminuzione di simmetria da monoclina a triclina,
oppure in una semplice maggior deviazione da 90 gradi degli angoli a e g della cellatriclina.
POLIMORFISMO DISTORSIVO
Il feldspato potassico presenta un polimorfismo O-D dovuto al fatto che la distribuzione di Si e
Al al centro dei tetraedri può essere disordinata o ordinata come nell’albite. Tale
cambiamento della distribuzione è associato quindi ad una diffusione di atomi nel reticolo e a
un cambiamento dell’assetto di tutta la struttura.
A T elevata, sopra 850°C, è stabile la fase disordinata, monoclina C2/m (SANIDINO), con 4
unità KAlSi3O4 nella cella e cioè con 16 tetraedri (Si,Al)O4 distribuiti su due posizioni di
molteplicità 8.
A T inferiore è stabile una fase a ordinamento intermedio (ORTOSE) ancora C2/m, nella
quale 8 Si sono disposti sul primo gruppo di 8 posizioni equivalenti e 4Si+4Al nel secondo
gruppo.
La separazione completa di Si e Al si raggiunge pressappoco sotto i 450°C, con
l’ordinamento tipo albite e la riduzione della simmetria al gruppo spaziale C-1
(MICROCLINO), ma con angoli a e g molto prossimi a 90°.
In tale gruppo spaziale le posizioni generali hanno molteplicità 4, e quindi i 16 tetraedri si
distribuiscono su 4 gruppi di 4 posizioni equivalenti: in un gruppo trovano posto 4 Al, negli
altri tre gruppi 12 Si.
PERTITE
Isole di feldspato sodico immerse in una matrice prevalentemente potassica, formatesi a
seguito dello smescolamento di un feldspato alcalino precedentemente stabile ad alta T
Feldspatoidi
I feldspatoidi sono tettoalluminosilicati abbastanza comuni, ma non frequenti come i feldspati
FELDSPATOIDI CUBICI
2/3 dei tetraedri sono occupati da Si e 1/3 da Al
Forma esterna: icositetraedro
Leucite e pollucite sono minerali comuni delle rocce vulcaniche ricche di alcali. La pollucite si
trova in alcune pegmatite
Tettosilicati idrati: ZEOLITI
FILLOSILICATI E MINERALI ARGILLOSI
Sono materiali nei quali si alternano fogli ottaedrici a fogli tetraedrici (Si2O5). Si classificano
in due gruppi:
1)Mg(OH)2 brucite,triottaedrico
2)Al(OH)3 Gibbisite, diottaedrico
SOLFURI
Lo strato ottaedrico deifillosilicati si può comprendere per analogia con la struttura degli
idrossidi
Strato tipo brucite: Mg3(OH)6
Nella brucite abbiamo strati di Mg coordinati a 6 ossidrili. Glistrati sono separati lungo c e
interagiscono solo con deboli forze di van der Waals. Gli ottaedri condividono spigoli. Tutti gli
ottaedri sono occupati da Mg.
Gibbsite: Al(OH)3 or Al2(OH)6
Strati di Al coordinati ottaedricamente a 6 (OH). Poiché Al è trivalente (Al3+), il bilanciamento
di carica impone che solo 2/3 dei siti ottaedrici possa essere occupato. I siti vacanti
provocano una deformazione dello strato rispetto a quello della brucite. La simmetria rimane
comunque esagonale. Lo strato tipo brucite è detto triottaedrico, quello tipo gibbsite è detto
diottaedrico.
Si può combinare il foglio ottaedrico con quello tetraedrico per ottenere una condensazione di
fogli. Può avvenire T-O oppure T-O-T:
1) T-O Asse c, varia la dimensione del parametro c 7 ampere
I pacchetti TO sono uniti da ponti H fra gli OH dello strato ottaedrico e gli O di quello
tetraedrico.
SERPENTINO: Mg3Si2O5(OH)4
(crisotilo, fibroso = Amianto)----> il foglio O è più lungo di quello T, dunque tende ad
incurvarsi e le fibre si arrotolano
CAOLINITE: Al2Si2O5(OH)4----> sfaldature parallele agli strati
(con gradi diversi di disordine planare)
2) T-O-T c= 10 ampere
1- Strati neutri
Talco: Mg3(OH)2Si4O10(Triottaedrico)
Pirofillite: Al2(OH)2Si4O10(Triottaedrico)
Considerando la successione di piani T-O .. i vertici sono ossidriliuniti da forze di Van der
Waals, quindi i vari piani tendono a sfaldarsi. Abbiamo diversi tipi di fogli e impalcature.
Cambiandoli avremo i fillosilicati.
La presenza di eventuali sostituzioni determinerà una diversa famiglia di composti.
Si individuano diversi tipi di sostituzione:
a - sostituzione di cationi tetraedrici con Al3+, Fe3+---> perdita di una carica positiva
b - sostituzione di cationi monovalenti o bivalenti al posto di bivalenti e trivalenti in ottaedro
c - vacanze in ottaedro
d - deidrossilazione di OH- a O2a_ esempio, la muscovite (KAl2(OH)2(AlSi3O10) diottaedrica,può avere K nell'interspazio. Si
presenta con un colore scuro, ottenendo una MICA
Non sostituisco nell'ottaedro, ma bensi nell' ossigeno, con Na, K, Ca, S, ovvero cationi che si
circondano di molecole d'acqua,csi si idratano e disidratano----> SMECTITI
b_Smectiti: gruppo di fillosilicati caratterizzati dalla capacità di gonfiarsi (assorbendo acqua),
disidratarsi e scambiare gli ioni interstrato (e anche molecole chimiche complesse) venendo a
contatto con soluzioni.
Tra i termini più comuni troviamo: Montmorillonite: dalla pirofillite (Al2(OH)2Si4O10) per
sostituzione dell’Al con Mg in ottaedro metalli alcalini e alcalinoterrosi in interstrato, al centro
di doppie maglie esagonali formate da molecole
d’acqua.(Na,K,Ca0.5)0.4(Al1.6Mg0.4)(OH)2Si4O10 4H2O
Distanza basale variabile con la quantità d’acqua o liquidi polari da 10 (disidratata) a 28 Å.
Tra i minerali a strati misti, troviamo strati di talco e brucite,
Clorite (14Å): successione di strati tipo talco e tipo brucite. Pacchetti neutri legati fra loro da
ponti idrogeno. Parziali sostituzioni in ottaedro.
Strati di illite e montmorillonite alternati senza regola
Altri interstratificati:illite-clorite, illite-vermiculite, caolinite-smectite, illite-smectite
QUALCHE DEFINIZIONE RELATIVA ALLE ARGILLE
Minerali argillosi
Fillosilicati con cristalli di dimensioni < 2μ.
( es: caolinite, illite, clorite, montmorillonite )
Argilla
Roccia incoerente costituita di minerali argillosi e altri minerali quali: sericite, talco, quarzo,
calcite, feldspato,miche, ossidi e idrossidi di Fe, solfati.
Composizione dell’ “argilla”
Minerali prevalenti: Fillosilicati del tipo “minerali argillosi” con cristalli di dimensioni < 2μ ( es:
caolinite, illite, clorite,montmorillonite )
Minerali secondari:
- altri fillosilicati (muscovite, biotite)
- quarzo
- feldspati
- carbonati
- ossidi e idrossidi (goethite, ematite, gibbsite)
- solfati (gesso)
- solfuri (pirite)
- sostanze organiche, ecc
Tipi di “argille”
Argille grasse - sono costituite da un’elevata % di minerali argillosi;
-trattengono una forte quantità di acqua e la perdono lentamente per evaporazione;
-subiscono un forte ritiro in essiccazione;
-sono molto plastiche;
Argille magre - contengono una % rilevante di frazione sabbiosa;
- trattengono poca acqua e la perdono più rapidamente
- hanno un basso ritiro;
- sono poco plastiche;
Argille caolinitiche - costituite prevalentemente da caolinite;
- colore bianco o giallastro;
- si impiegano per la realizzazione della porcellana
Argille refrattarie - contengono solo in piccola quantità i composti che
favoriscono la fusione (feldspati, carbonati di Ca, Mg, Fe ox)
PIROSSENI
Gli inosilicati sono minerali delle rocce molto importanti in cui i tetraedri SiO4 si legano in
catene semplici (pirosseni) o doppie (anfiboli). I pirosseni e gli anfiboli, ambedue rombici e
monoclini, hanno proprietà fisiche, chimiche e cristallografiche simili.. Tranne che per la
mancanza di (OH) nei pirosseni, i cationi presenti nei due tipi di catene sono gli stessi. La
presenza di (OH) negli anfiboli determina in questi un peso specifico leggermente più basso.
La differenza evidente tra i due gruppi riguarda l’abito, prismatico tozzo nei pirosseni e
prismatico allungato o aciculare negli anfiboli, e la sfaldatura .I pirosseni cristallizzano a
temperature più alte degli anfiboli e , quindi, nel raffreddamento di un magma si formano
prima; in alcuni casi, se c’è presenza di acqua, possono, al diminuire della temperatura,
reagire e formare gli anfiboli. In condizioni di metamorfismo, all’aumentare della temperatura
(metamorfismo progrado) gli anfiboli passano a pirosseni.
Pirosseni. XYZ2O6 è la formula chimica generale dei pirosseni, dove Na+, Ca2+, Mn2+,
Fe2+, Mg2+ e Li+, Y sono i cationi di X e Mn2+, Fe2+, Fe3+, Al3+, Cr3+, Ti4+ sono quelli di
Y. Z rappresenta Si4+ e Al3+ nel centro dei tetraedri. I pirosseni più comuni possono essere
rappresentati nel sistema di Pirosseni rombici. Sono minerali comuni di molte rocce,
principalmente gabbri, peridotiti, noriti e basalti.
Pirosseni monoclini. I principali sono diopside (CaMgSi2O6), hedenbergite (CaFeSi2O6), che
formano una soluzione solida completa, e augite, (Ca, Na)(Mg, Fe,Al)(Si.,Al)2O6, dove, come
si vede, un po’ di Na sostituisce il Ca e un po’ di Al, il Fe,il Mg e il Si. I cristalli sono prismatici
e frequentemente geminati, ma si possono trovare anche massivi, lamellari, colonnari, ecc.
L’augite è il pirosseno più comune e un minerale delle rocce molto importante. I pirossenoidi
sono inosilicati, triclini, con lo stesso rapporto Si/O dei pirosseni, ma con struttura diversa.
ANFIBOLI
Anfiboli. W0-1X2Y5Z8O22(OH,F)2 è la formula chimica generale degli anfiboli, dove W può
essere sodio e potassio; X sta per Ca2+, Na+, Mn2+, Fe2+, Mg2+ e Li+; Y rappresenta
Mn2+, Fe2+, Mg2+, Fe3+, Al3+, e Ti4+, mentre Z sta per Si4+ e Al3+. Gli anfiboli possono
esser rappresentati nel sistema di figura.Anfiboli rombici.Sono molto rari; il principale è
l’antofillite (Mg,Fe)7Si8O22(OH)2, che si trova raramente in cristalli, mentre è comune in
forma fibrosa. E’ un minerale di rocce metamorfiche. Anfiboli monoclini.
Cummingtonite, (Mg,Fe)7Si8O22(OH)2 e grunerite, Fe7Si8O22(OH)2 costituiscono una serie
di minerali per la quale non si conoscono termini completamente magnesiaci, che raramente i
trovano in individui distinti, ma che sono comunemente fibrosi raggiati. Hanno una sfaldatura
perfetta, una lucentezza sericea e un colore con tonalità di marrone, che insieme all’abito li
caratterizza. Sono minerali di rocce metamorfiche.
Tremolite, Ca2Mg5Si8O22(OH)2 e actinolite, Ca2(Mg,Fe)5Si8O22(OH)2 costituiscono una
serie, analoga a quella diopside-hedenbergite dei pirosseni, con caratteristiche e ritrovamenti
più o meno identiche ai minerali della serie cummingtonite-grunerite. Gli anfiboli con
composizioni variazionali molto complesse vengono chiamati orneblende:si trovano in cristalli
prismatici, ma anche con abito colonnare o fibroso,hanno sfaldatura perfetta, lucentezza
vitrea, colori verde scuro o nero, e sono minerali comuni di rocce ignee e fibrose. Un’altra
serie è quella degli anfiboli alcalini, dove c’è la presenza costante di sodio e, più raramente,
potassio.
NESOSILICATI
Nei nesosilicati, come abbiamo detto, i tetraedri SiO4 sono isolati e legati fra di loro da cationi
interstiziali e le loro strutture dipendono principalmente dalla dimensione e dalla carica di
questi cationi.
L’impaccamento è abbastanza compatto per cui questi minerali sono generalmente duri e con
alto peso specifico. L’abito dei nesosilicati è molto spesso equidimensionali, non essendo i
tetraedri legati a catene, a strati, ecc.
Olivine. Sono la completa soluzione solida di forsterite (Mg2SiO4 e fayalite
(Fe2SiO4)(rombiche).
Generalmente si trovano in masse granulari e i cristalli sono la combinazione di diverse forme
semplici (prismi, pinacoidi, bipoiremide).
la dunite è una roccia olivinica, la peridotite è una roccia olivinica con pirosseni.
La forsterite, che si altera facilmente ad antigorite, un polimorfo del serpentino, non è stabile
in presenza di silice libera: Mg2SiO4 + SiO2 = 2MgSiO3 (enstatite, un pirosseno) La varietà
trasparente (peridoto) è usata come gemma, mentre la normale olivina per il suo alto punto di
fusione è coltivata per l’utilizzo come refrattario in fonderia.
GRANATI
Granati. Sono un gruppo di minerali molto comuni nelle rocce, specialmente in quelle
metamorfiche, che comprende dei minerali cubici isostrutturali. suddivisi in due serie, quella
della pyralspite e quella dell’ugrandite. I nomi derivano dall’iniziali dei minerali di ciascuna
serie. Per la prima: piropo (Mg3Al2(SiO4)3; almandino (Fe3Al2(siO4)3; spessartite
(Mn3Al2(SiO4)3. e, per la seconda:
uvarovite (Ca3Cr2(SiO4)3; grossularia (Ca3Al2(SiO4)3; andradite (Ca3Fe2(SiO4)3. Sono
generalmente in cristalli romboedrici o trapezoedrici o in combinazione delle due forme
semplici.
L’andradite, di colore scuro, molto comune all’Isola d’ Elba, può essere il risultato di un
metamorfismo di contatto di calcari con silice e minerali di ferro: 3CaCO3 + SiO2 + Fe2O3 =
Ca3Fe2(SiO4)3 + 3CO2.
Zircone (ZrSiO4, tetragonale). Si trova comunemente in cristalli prismatici terminati da
bipiramidi, ma si rinviene anche in granuli di diverse dimensioni e forme. Ha lucentezza
adamantina e colore vario con predominanza di tonalità brune. La struttura consiste di
tetraedri isolati e cubi distorti.
I NON SILICATI
CARBONATI
Sono prevalenti nelle rocce sedimentarie nelle quali possono costituire anche il cemento. I
travertini sono generalmente di origine chimica, i calcari a lumachelle sono organogeni , le
calcareniti di origine clastica e i marmi metamorfica. Il complesso anionici (CO3)2- in
coordinazione triangolare planare costituisce l’ ossatura dei carbonati ed è, in massima parte,
responsabile delle proprietà di questa classe di minerali.
Il calcio può assumere due coordinazioni ed per questo che esistono due serie di carbonati
quella della calcite e quella dell’aragonite, anche se in quest’ultima la coordinazione del calcio
non è 8, ma 9.
Tra i carbonati esistono soluzioni solide, ma poiché in certi casi la differenza tra i raggi ionici
degli elementi è troppa grande si hanno composti intermedi, come la dolomite.
I carbonati anidri della serie della calcite, dell’aragonite e della dolomite, unitamente a quelli
contenenti l’ossidrile (azzurrite e malachite) sono gli unici carbonati importanti.
Calcite (CaCO3, trigonale). I cristalli presentano forme diverse, ma può trovarsi anche in
masse concrezionate, granulari e in masse spatiche limpide e trasparenti (varietà spato
d’Islanda). Può a causa di impurità assumere svariati colori. E’ costituente fondamentale di
molte rocce. La sua struttura si può pensare come derivata da quella del salgemma con il
calcio al posto del sodio e il gruppo CO3 del cloro: poiché nel caso della calcite il gruppo CO3
è triangolare e non sferico abbiamo una distorsione del cubo che passa a romboedro con tutti
i triangoli orientati nello stesso senso e su un piano strutturale.
Aragonite (CaCO3, rombica). E la modificazione polimorfa di alta pressione del carbonato di
calcio. Si trova anche a pressione ambiente (forse stabilizzata dalla presenza di stronzio), pur
essendo meno stabile e comune della calcite. Si rinviene in aggregati coralloidi e in cristalli
allungati prismatici e aciculari. Frequenti i geminati di tre individui a simulare un prisma
esagonale. Se pure è trasparente e bianca come la polvere. Spesso è colorata per impurità.
La struttura è con i triangoli CO3 su due strati a livello diverso e ruotati di 30° a destra e a
sinistra. Coordinazione 9
Dolomite (CaMg(CO3)2, trigonale). E’ un sale doppio, in quanto Ca e Mg possono sostituirsi
solo in piccola quantità, ma, data la differenza tra i raggi, occupano posizioni proprie. I cristalli
hanno forma romboedrica, talvolta con facce curve o raggruppati in aggregati selliformi. La
sua struttura differisce da quella della calcite, ma con Ca e Mg che occupano piani diversi e
che portano a una diminuzione di simmetria, in quanto rimane solo l’asse di ordine 3.
SOLFATI
Nei solfuri lo ione S2- è il risultato del riempimento con due elettroni “catturati” dello strato
esterno: in questo caso avremo uno ione negativo grande e bivalente. I sei elettroni presenti
nello strato esterno dello zolfo possono anche essere persi dando così luogo a uno ione
positivo esavalente con raggio tale da assumere con l’ossigeno una coordinazione
tetraedrica. I gruppi anionici (SO4)2- sono le unità base dei solfati, la cui genesi può essere:
sedimentaria evaporitica, idrotermale, di alterazione superficiale, fumarolica.
I solfati più importanti e comuni sono quelli anidri e quelli idrati. Al gruppo degli anidri
appartengono i minerali del gruppo della barite, rombici, isostrutturali e l’anidrite.
Gruppo della barite. Celestite (SrSO4), barite (BaSO4), anglesite (PbSO4). Hanno cristalli
incolori o con deboli colorazioni, normalmente tabulari con perfetta sfaldatura basale. La
lucentezza è vitrea o
perlacea, per celestite e barite, perlacea o adamantina per l’anglesite. Non si scalfiscono con
l’unghia.
Anidrite (CaSO4, rombica). Non è isostrutturale con i minerali del gruppo della barite (il calcio
è in coordinazione 8, mentre il bario 12). E’ rara in cristalli tabulari o prismatici, comunemente
è massiva.
Gesso (CaSO4.2H2O, monoclino).E’ il più importante solfato idrato e ha una struttura a strati
di (SO4)2- (paralleli alla forma semplice 010) fortemente legati a Ca2+ intervallati da molecole
di H2O che hanno legami con gli strati più deboli ed è per questo che il gesso ha una
eccellente sfaldatura. Si ritrova in bei cristalli tabulari o prismatici che possono raggiungere
anche dimensioni molto grandi.
Frequenti sono i geminati, in particolare a coda di rondine. Tra le sue varietà citiamo
l’alabastro (microcristallino compatto) utilizzato per sculture e oggetti di arredamento, la rosa
del deserto (gesso ricoperto da sabbia), la selenite (grandi masse limpide e trasparenti), la
sericolite (gesso fibroso con lucentezza sericea).
SOLFURI
I solfuri, che hanno un impaccamento compatto di atomi di zolfo tra i quali si distribuiscono i
metalli, possono essere suddivisi in piccoli gruppi sulla base delle strutture, ma una
classificazione di tipo cristallografico non si adatta molto bene. La suddivisione, quindi, è in
base al rapporto metallo/zolfo (Me/S). All’interno di questa teniamo conto del tipo di
coordinazione: coordinazione regolare tetraedrica (sfalerite), ottaedrica (galena), in poliedri
distorti come nei solfuri più complessi.
La genesi principale è di tipo idrotermale;. si hanno anche solfuri formatisi per sublimazione
da fumarole, per alterazione di giacimenti metalliferi (cappellacci) e per azione biochimica.
Solfuri con Me/S >1.
Calcosina (Cu2S, rombica). Massiva e granulare, raramente in cristalli a contorno esagonale.
Ha colore grigio piombo con riflessi bluastri (quando è esposta) e il colore della polvere è
grigio nerastro. Le caratteristiche distintive sono il colore e la buona sottilità. E’ uno dei più
importanti minerali di rame.
Poiché il rame si ossida con facilità (v. soluzioni solide con lacune)esistono molti minerali non
stechiometrici simili come, ad esempio, djurleite (Cu1.97S), digenite (Cu1.78S), ecc.
Solfuri con Me/S = 1.
Sfalerite (ZnS, cubica). Conosciuta anche con il vecchio nome di blenda ha una struttura
come quella del diamante in cui il carbonio è stato sostituito da uguali quantità di Zn e S e si
può trovare sia massiva sia in cristalli (cubici, tetraedrici, rombo- dodecaedrici); ha una
perfetta sfaldatura tetraedrica, anche se molte volte ha una grana troppo fina per poterla
osservare. Quando è pura è incolore, ma può contenere oltre il 50% moli di FeS (varietà
marmatite) e in tal caso diviene nera.
Calcopirite (CuFeS2, tetragonale). Ha una struttura derivata dalla sfalerite Massiva o in
cristalli bisfenoidici. E’ conosciuta come “l’oro degli stolti”, termine che vale anche per la pirite.
E’ uno dei più importanti minerali di rame ed è il più diffuso.
Galena (PbS, cubica). Ha una struttura tipo salgemma con il Pb al posto del sodio e lo S al
posto del cloro. Massiva o in cristalli cubici o combinati con l’ottaedro. E’ l’unico minerale di
piombo economicamente importante, è molto comune e ha la stessa genesi e gli stessi
ritrovamenti della sfalerite.
Solfuri con Me/S < 1.
Pirite (FeS2, cubica). Struttura (fig.9.3) tipo salgemma con gruppi S2 e Fe coordinati
ottaedricamente. Massiva, granulare, reniforme, stalattitica, ecc., anche in cristalli
pentagonoedrici, cubici, ottaedrico.
OSSIDI e Idrossidi
Nella classe degli ossidi e idrossidi (circa 250minerali) ve ne sono molti di grande interesse
economico.
Con il termine ossidi intendiamo i solidi con legame generalmente fortemente ionico di ioni O
e cationi metallici. La maggior parte degli ossidi può venire schematizzata secondo un
modello ionico e quindi viene spontaneo descrivere questi composti in termini di poliedri di
coordinazione catione-ossigeno.
Un altro modo di considerare la cristallochimica degli ossidi parte dall’ impiccamento
compatto dei grossi anione dell’ossigeno, con i cationi metallici che si pongono nelle cavità
ottaedriche e tetraedriche di questo tipo di impiccamento. Tuttavia un simile inquadramento
cristallochimica presenta notevoli difficoltà ai fini sistematici e, quindi, anche per gli ossidi si
segue una classificazione basata sul rapporto
metallo/ossigeno: Me/O >1; =3/4; =2/3; =1/2
GRUPPO DELL EMATITE
l quarzo (SiO2), il più comune di tutti gli ossidi, e le sue modificazioni polimorfe, non sono
trattati in questa classe, ma in quella dei silicati, in quanto le loro strutture sono più vicine a
quelle degli altri composti Si-O.
Corindone (Al2O3, trigonale) impiccamento compatto degli ossigeni e Al in coordinazione
ottaedrico: 2/3 degli ottaedri sono occupati e 1/3 vuoti.
Ematite (Fe2O3, trigonale). Isostrutturale con il corindone. E’ il più abbondante e diffuso (si
trova in quasi tutte le rocce) minerale di ferro (contiene il 70% di ferro).
Ilmenite (FeTiO3, trigonale) (H=5.5-6; G=4.7). è un comune minerale accessorio.
Serie degli spinelli.
Gli spinelli, che hanno notevoli soluzioni solide, mostrano un impaccamento compatto di
anioni ossigeno nelle cui cavità trovano posto i cationi . La formula generale di questa serie di
minerali è XY2O4, per gli spinelli “normali” e Y(XY)O4 per gli”inversi”: nei primi gli X occupano
le cavità tetraedriche e gli Y quelle ottaedriche; nei secondi metà degli Y sono nelle cavità
tetraedriche, l’altra metà, insieme agli X, occupano posizioni ottaedriche.
Spinello (MgAl2O4, cubico). E’ un comune minerale di calcari e rocce argillose, povere in
silice, che hanno subito metamorfismo di contatto. Si trova, per la sua resistenza all’attacco
fisico e chimico, anche come minerale residuale (ciottoli di fiue,ecc.).
Magnetite (Fe3O4, cubica). Di solito granulare o massiva, ma anche in ottaedri (raramente in
rombododecaedri), ha lucentezza metallica, colore e polvere neri; è opaco e ferromagnetico.
E’ un minerale accessorio di rocce ignee dove talvolta può accumularsi in grande quantità
dando luogo a importanti depositi, sfruttati per il ferro.
L’ossidazione di Fe2+ a Fe3+ può portare a formule non stechiometriche, Fe3-xO4.
Cromite (FeCr2O4, cubico). Normalmente massivo o granulare, raramente in piccoli ottaedri:
E’ costituente comune di peridotiti e di altre rocce ultrabasiche o desse derivate come le
serpentine. Si trova anche come minerale residuale (sabbie a cromite).
Crisoberillo (BeAl2O4, rombico
Idrossidi
Tutte le strutture degli idrossidi sono caratterizzate dalla presenza di ioni (OH)-.
La brucite, Mg(OH)2 è trigonale con una struttura a strati paralleli con Mg e Al in
coordinazione ottaedrica. Si trova in cristalli tabulari, facilmente sfaldabili, in lamine e
massiva. Ha lucentezza perlacea sulla rottura fresca, colore chiaro, è trasparente o
traslucida, ed è flessibile, ma non elastica. Si distingue dal talco, perché è
più dura ((5-6) e dalle miche, perché non è elastica.
La gibbsite, Al(OH)3 è simile alla brucite, ma con 1/3 dei cationi in coordinazione ottaedrico
mancanti.
I miscugli degli idrossidi costituiscono importanti giacimenti: bauxite (idrossidi di Al); limonite
(idrossidi di Fe); lateriti (idrossidi di Al e Fe con silice);