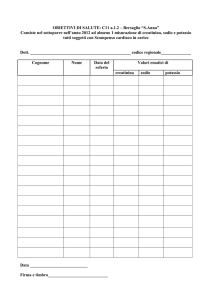
Saggio di Lassaigne Fusione, pirolisi e carbonizzazione riduttiva con sodio metallico. Saggio qualitativo di carattere generale per la ricerca di zolfo, azoto, alogeni nei composti organici. Procedura operativa: Indossare occhiali da laboratorio (flessibili) e guanti (ma non del tipo sottile da chirurgia, nel qual caso meglio nulla, in quanto un eventuale lapillo fonderebbe la gomma/plastica causando un’ustione anche peggiore, in caso di incidente). Si mescolano una piccola aliquota (circa 20‐30 mg) di campione solido finemente polverizzato in un tubicino da via secca insieme a due scagliette di Na metallico, tagliato sul momento (con i guanti!) e ben asciugato dall’etere di petrolio in cui è immerso, di circa la stessa massa o in lieve ulteriore eccesso, l’una posta sopra la sostanza, l’altra a metà tubicino. I campioni liquidi, specialmente quando basso bollenti e molto volatili, non sono adatti a essere usati tal quali per le perdite elevate che subirebbero al riscaldamento. In questi casi conviene farli adsorbire su solidi inerti, come il lattosio, o allumina, zeolite, kieselguhr o altre polveri minerali purificate, e solo dopo mescolarli alle scaglie di sodio. Successivamente si intiepidisce su fiamma, regolata a temperatura media (fiamma incolore silenziosa). È molto importante che nelle fasi iniziali il riscaldamento sia lento, graduale ed intermittente, per dare modo al sodio di fondere e al campione di completare progressivamente le reazioni di demolizione riduttiva senza che le stesse procedano in modo esplosivo ed incompleto (cosa che tendono spesso a fare ugualmente!), con perdite e spruzzi di sostanze fuse. Si noterà quasi sempre emissione di fumi e vapori (non foss’altro che per l’etere di petrolio che bagna il sodio stesso). Dopo il riscaldamento preliminare a calore moderato (di durata proporzionale alla durata dei processi reattivi evidenti), si prosegue la calcinazione al calor rosso. Si regola la fiamma del Bunsen alla massima temperatura (apertura massima dell’aria, con fiamma corta, bicolore azzurra–blu e ruggente) e si arroventa il tubo da saggio sul cono ossidante. Nel frattempo si dispone un becker piuttosto alto e stretto con circa 5 ml di acqua distillata e, non appena pronti, ci si lascia cadere dentro la provettina ancora incandescente (appena tolta dalla fiamma) orientata orizzontalmente. È importante che lo shock termico frantumi il più possibile il tubo da saggio. Malgrado gli occhiali gettare la provetta a braccio teso, senza guardare verso l’imboccatura del becker (residui di sodio metallico si incendieranno e potrebbero lanciare spruzzi di soluzione basica calda e/o minuscoli frammenti di metallo fuso o vetro). In ogni caso la procedura è pericolosa soltanto quando eseguita distrattamente o senza le dovute precauzioni. Finito lo sfrigolio, si frantumano per bene con pinze di metallo i “resti” della provettina, stemperando il residuo solido insolubile, per far dissolvere meglio le componenti saline utili ai saggi specifici successivi. Soluzioni molto colorate di colori vivaci non sono segnali rassicuranti sulla corretta esecuzione (anche se vi sono eccezioni). Quindi si filtra su carta l’intera soluzione, detta soluzione madre di Lassaigne. Questa soluzione deve bastare per tutti i saggi paralleli seguenti, per cui deve essere prelevata in piccole aliquote senza sprecarla (essendo laboriosa da ripreparare). Ricerca degli elementi 1) Ricerca dello zolfo Far cadere una goccia della soluzione alcalina sopra della carta da filtro imbevuta con (CH3COO)2Pb: la formazione di una macchia scura dovuta alla formazione di solfuro di piombo nero insolubile. 2) Ricerca dello azoto Si aggiunge una spatolina di FeSO4 (solfato ferroso) finemente macinato. Si formerà subito un precipitato gelatinoso e denso di colore dal grigio al verdastro, che col tempo si scurisce in superficie diventando giallastro, poi ruggine, sino a marrone dovuto alla precipitazione di idrossido ferroso come gel insolubile che, al contatto con aria, si ossida facilmente ad idrossido ferrico ed ossido ferrico idrato. Contemporaneamente, anche se ciò non è visibile ad occhio, qualora sia presente azoto in forma di ione cianuro, si forma anche un complesso solubile e incolore, molto stabile, il ferrocianuro di sodio: Na4[Fe(CN)6]. Si riscalda insufflando aria per alcuni minuti, sin quando si è formata una discreta quantità di ferro (III) per ossidazione con aria del ferro (II) in ambiente basico. Il ferro (III) complessa pure esso con il cianuro, per dare ferricianuro sodico, ma in maniera molto meno stabile del ferro (II). Si acidifica poi la soluzione con H2SO4 2 N fino a pH nettamente acido, ma non in grande eccesso. Se il cianuro era assente, a questo punto il gel bruno verdastro si ridissolve tutto e la soluzione diventa sostanzialmente incolore, o al limite giallina pallida e limpida. Viceversa, se c’era cianuro e quindi azoto, il complesso ferrico (ferricianuro) viene decomposto dall’acido forte a ione ferrico libero (Fe3+), mentre lo ione ferrocianuro rimane integro [Fe(CN)6]4–. Quando si liberano in reciproca presenza, questi due ioni formano un composto insolubile detto Blu di Prussia, di formula grossomodo: Fe4[Fe(CN)6]3 ferrocianuro o esacianoferrito ferrico Il Blu di Prussia a volte colora la soluzione in azzurro tenue quando disperso finemente e/o l’azoto era stato fissato poco; in questo caso si può filtrare la soluzione su carta da filtro: la formazione di parti azzurro chiaro non rimuovibili anche dopo lavaggio indica saggio positivo per la ricerca dell’azoto. 3) Ricerca degli alogeni : (Cl, Br, I) (a) In presenza accertata di zolfo e azoto, che interferiscono per la capacità di precipitare il reattivo di indagine, bisogna eliminare tutti gli ioni solfuro e cianuro aggiungendo HNO3 2 N in eguale quantità alla soluzione alcalina. Si riscalda la soluzione su fiamma bassa agitando in continuazione riducendone il volume della metà: fare attenzione a non riscaldare troppo per evitare di perdere elementi. Una volta ridotto il volume si reintegra aggiungendo un po’ di acqua. (b) In assenza di zolfo e azoto, si acidifica l’aliquota di soluzione madre con HNO3 sino a pH acido (cartina indicatore rossa) ma non in largo eccesso. Poi si aggiungono poche gocce di AgNO3 (argento nitrato) circa 0.1 M. In presenza di alogeni si formano precipitati densi e “caseosi” di alogenuri di argento, dai colori simili ma non uguali. AgCl: bianco latteo, AgBr: color crema, AgI: giallo. È facile distinguere il cloruro dagli altri due, in quando esso non è giallo, ma una prova più sicura consiste nel valutare la resistenza di questi precipitati insolubili alla ridissoluzione con reattivi ammoniacali. Per saggiarla ed identificare con precisione l’alogeno in questione, si centrifuga la sospensione quindi si decantano i precipitati solidi, eliminando la soluzione madre acida. Si aggiunge al solido (NH4)2CO3 (carbonato di ammonio) che solubilizza bene AgCl e poco AgBr e non solubilizza AgI. Quindi si agita bene in maniera da solubilizzare completamente il cloruro di argento, si centrifuga di nuovo e si decanta il liquido in una nuova provetta e a questo si aggiunge KI (ioduro di potassio): la formazione di un precipitato giallo di argento ioduro indica la positività del saggio per il cloro. (c) Conferma presenza cloro e bromo con acqua di cloro: alla soluzione alcalina, acidificata con HNO3 2N, viene addizionato 1 mL di CHCl3 e goccia a goccia acqua di cloro. Si dibatte vigorosamente in modo di far entrare in contatto le due fasi e facilitare l’estrazione dell’alogeno elementare (Br2 o I2 o entrambi) da parte dello strato organico che si colorerà di rosa intenso/viola se è presente lo I2, o lo I2 e il Br2 o di giallo intenso se è presente Br2. Continuando ad aggiungere acqua di cloro I2 viene ulteriormente ossidato a IO3‐ che è incolore: la fase cloroformica sarà incolore se è presente solo I2 oppure assumerà una colorazione giallo intenso se è presente anche il Br2. Aggiungendo un eccesso di acqua di cloro (specialmente quando è preparata da poco e quindi molto ricca di Cl2) può essere ulteriormente ossidato anche il bromo a BrCl che è anch’esso incolore. Particolare attenzione deve essere posta quando si aggiunge acqua di cloro perché si potrebbe passare direttamente alla massima ossidazione senza vedere le colorazioni intermedie di I2 e Br2 e quindi avere dei falsi negativi. Cenni sui processi e reazioni Innanzi tutto occorre sottolineare che molti dettagli di ciò che avviene durante la prima fase del saggio, la mineralizzazione degli eteroatomi, non sono neppure ben noti. Il campione viene fuso in presenza di sodio (un forte agente riducente, attivo prevalentemente come agente S.E.T., o “single electron transfer”, cioè riducente radicalico) a temperature anche superiori a 400°. Le temperature effettive variano notevolmente durante gli stadi del saggio. Inizialmente, quando sono presenti frazioni e prodotti volatili di pirolisi, l’aumento di temperatura è frenato dalla loro vaporizzazione, ma sul finale, quando rimangono soltanto frammenti minerali, salini e residui carboniosi, la temperatura è quella del vetro arroventato (500°‐
600°). L’unico evento ragionevolmente sicuro è l’addizione ossidativa al metallo da parte dei legami carbonio alogeno, radical–anionica (questa reazione è parente di un processo chiamato accoppiamento di Wurtz), subita da rottura e formazione di ioni alogenuro e radicali al carbonio, che reagiscono ulteriormente condensando o venendo ridotti ancora dal sodio. Il residuo nerastro è SEMPRE costituito da : 1) CGraph ‐ carbone (carbonio amorfo), originatosi per pirolisi, disidratazione e deidrogenazione termiche. Si forma in prevalenza nelle fasi finali dell’arroventamento. Può contenere “intercalati” atomi di sodio (come il catodo delle pile al litio), idrossido di sodio ed altro. In effetti uno dei rari frammenti metallorganici inerti a queste condizioni estreme che si può formare in tracce è l’acetiluro di sodio NaC≡CNa, salino e resistente alla riduzione, che però non viene cercato nei saggi dato che non è diagnostico di alcun gruppo funzionale particolare. Inoltre, nell’istante stesso dell’idrolisi della miscela rovente con acqua si libera e si incendia insieme all’idrogeno. 2) Na2CO3 ‐ carbonato di sodio (originato dall’azione combinata dell’umidità atmosferica e/o dell’ossigeno estratto riduttivamente dal campione, quando presente, e poi dalla carbonatazione con CO2 atmosferica). 3) Na ‐ tracce di sodio metallico residuo (possono abbondare se l’arroventamento è stato troppo breve e/o il dosaggio di sodio metallico iniziale troppo generoso). Questo residuo causa spesso un’esplosione abbastanza vivace nella rottura della provettina da via secca rovente gettata nell’acqua fredda, dato che il sodio, e l’idrogeno che produce, si incendiano. 4) Na2O / NaOH ‐ tracce di idrossido/ossido di sodio. A seconda del tipo di campione, il residuo può poi contenere anche : 5) NaCN ‐ cianuro di sodio : se e solo se il campione contiene azoto, ma non sempre. Esistono infatti alcuni falsi negativi, ossia campioni azotati che fissano molto male l’azoto in queste condizioni. Varie possono essere le ragioni, ma principalmente appartenenti a due categorie: a. Composti contenenti azoto ammoniacale inorganico o in forma di ammine (anche le solfonammidi primarie possono a volte dare il problema). Entrambi i casi perdono azoto troppo facilmente ma in una forma molto volatile, che non reagisce bene col sodio, non si fissa come cianuro e viene persa nei vapori. Talvolta un uso oculato della cartina indicatrice avvicinata al tubicino da via secca durante l'emissione di fumi consente di intercettare questo particolare tipo di “azoto volatile” (non elemento, ovviamente!). Bisogna però saper distinguere il viraggio al blu tipico di eventuali schizzi di idrossido di sodio fuso, non significativo, dal viraggio omogeneo e diffuso dovuto alle ammine/ammoniaca. In ogni caso di norma questi casi possono comunque essere identificati mediante la pirolisi per via secca con KOH e la cartina indicatore universale, che denuncerà meglio la presenza di vapori basici ammoniacali o di ammine. b. Alcuni composti contengono azoto in una forma troppo labile e che si frammenta subito, ma in vapori non basici. Ad esempio alcuni nitro e nitroso‐
composti oppure azo e diazo‐composti possono perdere rispettivamente vapori nitrosi o persino azoto molecolare (NO, NO2, N2). Nessuno di questi vapori è basico, e non sempre il secondo, il biossido di azoto, che invece è acido, riesce a causare un viraggio acido nella cartina. È possibile però identificare questi composti con saggi appositi complementari (come quello con ioduro di sodio/HCl/salda d’amido per i nitro e nitroso composti). Bisogna non interpretare mai il Lassaigne con il “paraocchi”, come ogni altro saggio. In ogni caso il falso negativo al Lassaigne non è una regola, ma dipende dalla struttura (ad esempio è più comune nei derivati sostituiti a un carbonio alifatico privo di idrogeni). c. Alcuni composti, molto rari, contengono azoto in una forma chimicamente assai stabile (come gli eterocicli aromatici più stabili, tipo la piridina, e gli “acidi” all’azoto, come le immidi) e che richiede molto sodio e prolungato arroventamento prima di essere fissato come cianuro (se invece si forma cianato, cianammidi o urea, il saggio da un falso negativo). In questi casi un’esecuzione poco accurata può fornire falsi negativi facilmente. 6) Na2S ‐ solfuro di sodio: se e solo se il campione contiene zolfo. Esistono esempi di falsi negativi, molto rari, come accade in alcuni solfoni ciclici o che comunque frammentano molto facilmente per riscaldamento “estrudendo” anidride solforosa gassosa, che non ha quindi il tempo di essere fissata dal sodio metallico. Nuovamente, l’indagine con cartina indicatore, che in tali casi si arrossa per l’acidità della SO2, o anche con la cartina impregnata di dicromato di potassio ed acido acetico (che subisce un viraggio riduttivo dall’arancio pallido al verdazzurro) può far sospettare qualcosa, anche se non si tratta di indizi sicuri e univoci. Anche certi acidi solfonici e loro sali e gli alchil/aril solfati e loro sali, possono “fare cilecca” ed essere convertiti a solfiti e/o solfati minerali, che si riducono a solfuri molto lentamente e parzialmente. 7) Alogenuri di sodio : NaCl, NaBr, NaI ‐ Sono presenti quando il campione contiene i rispettivi alogeni. I falsi negativi sono sostanzialmente assenti per gli alogeni citati. Per contro il fluoro, che forma col carbonio un legame fortissimo, non viene mai fissato come fluoruro di sodio (e se anche è già contenuto nel campione come fluoruro ione, non viene evidenziato dal reattivo precipitante, il nitrato di argento in acido).