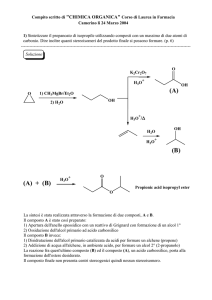
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
UNIVERSITA’ DEGLI STUDI DELLA TUSCIA
DIPARTIMENTO DI SCIENZE DEI BENI CULTURALI
CORSO TRIENNALE IN CONSERVAZIONE DEI BENI
CULTURALI (CLASSI L1-L43)
DISPENSE PER CHIMICA ANALITICA
EQUILIBRI CHIMICI
ELETTROCHIMICA
DOTT.SSA CLAUDIA PELOSI
1
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
CAPITOLO 1
1.1 L’equilibrio chimico
L’equilibrio chimico è un equilibrio dinamico. L’equilibrio si raggiunge quando la
velocità della reazione diretta diventa uguale alla velocità della reazione inversa.
All’equilibrio le concentrazioni dei reagenti e dei prodotti non variano ulteriormente nel
tempo. Lo stesso stato di equilibrio può essere raggiunto partendo sia dal lato dei
reagenti che quello dei prodotti dell’equazione chimica.
Lo stato di equilibrio è caratterizzato dalla costante di equilibrio della reazione che
lega le concentrazioni dei reagenti e dei prodotti che partecipano al processo chimico.
L’espressione della costante di equilibrio di una reazione chimica è uguale al rapporto
tra il prodotto delle concentrazioni dei prodotti ed il prodotto delle concentrazioni dei
reagenti, con ciascuna concentrazione elevata ad una potenza pari al coefficiente
stechiometrico di quella specie nell’equazione bilanciata.
La legge di azione di massa definisce definisce l’espressione della costante di
equilibrio di una reazione chimica. Per una generica reazione:
aA + bB
cC + dD
la costante di equilibrio, in base alla legge di azione di massa risulta:
KC =
[C]ceq [D]deq
[A]aeq [B]beq
Il pedice C indica che la reazione si svolge in soluzione. Nel caso di reazioni in fase
gassosa si utilizza la costane KP e al posto delle concentrazioni si utilizzano le pressioni.
Da un punto di vista termodinamico, una reazione chimica può essere descritta
prendendo in considerazione la sua variazione di energia libera di Gibbs. In una
reazione spontanea ΔG < 0 mentre in condizione di equilibrio ΔG = 0. La costante di
equilibrio e l’energia libera standard sono grandezze correlate e si può dimostrare che è
valida la seguente realzione:
2
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
ΔG° = - RT lnK
Quando una reazione si trova in una condizione diversa da quella presente all’equilibrio
il rapporto delle concentrazioni dei reagenti e dei prodotti non sarà più uguale alla
costante di equilibrio ma verrà denominato quoziente di reazione Q ovvero una
grandezza che ha la stessa espressione della costante di equilibrio ma a valori di
concentrazione delle specie chimiche arbitrari, non necessariamente all’equilibrio. K è
la costante di equilibrio. L’espressione dell’energia libera sarà:
ΔG = ΔG° + RT lnQ
Ovvero: ΔG = - RT lnK + RT lnQ
ΔG = RT ln(Q/K)
All’equilibrio Q/K = 1, ovvero Q = K e ΔG = 0. Quando Q/K < 1, ovvero Q < K, ΔG < 0
e la reazione procede spontaneamente da sinistra a destra; Q/K > 1, ovvero Q > K, ΔG >
0 e la reazione procede spontaneamente da destra a sinistra, ovvero in senso opposto a
come è scritta.
1.2 Il principio di Le Châtelier
Henri Le Châtelier (1850-1936) formulò, nel 1884, il principio che porta il suo nome:
Se una reazione chimica viene sottoposta ad un cambiamento delle condizioni di
reazione che la spostano dal suo stato di equilibrio allora la reazione procede verso
una nuova condizione di equilibrio nella direzione in cui il cambiamento delle
condizioni viene, almeno in parte, annullato. Le condizioni che possono influenzare
una reazione all’equilibrio chimico sono: 1 – concentrazione di un reagente o di un
prodotto; 2 – volume di reazione o pressione applicata 3 – temperatura.
In generale, una diminuzione del volume di reazione provoca uno spostamento
dell’equilibrio di reazione verso il lato con il minor numero di moli di specie gassose.
In una reazione endotermica un aumento della temperatura provoca uno spostamento
della reazione all’equilibrio verso destra, ovvero verso i prodotti.
3
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Per una reazione esotermica, ovvero una reazione che avviene con liberazione di
energia, quando la temperatura aumenta l’equilibrio di reazione si sposta verso sinistra.
Da un punto di vista termodinamico è possibile esprimere matematicamente la
dipendenza della costante di equilibrio dalla temperatura, infatti, tenendo conto della
relazione – RTlnK = ΔG° = ΔH° - TΔS, si ottiene:
ln K = - ΔG°/RT = -ΔH°/RT + ΔS°/R
Riportando in grafico lnK in funzione di 1/T si ottiene una retta con pendenza –ΔH°/R e
intercetta ΔS°/R. Oltre al metodo grafico si possono collegare con un’espressione
matematica i valori delle costanti di equilibrio a diverse temperature. Si considerino K1
e K2 le costanti di equilibrio alle temperature T1 e T2. Esprimendo le due costanti di
equilibrio con l’equazione su riportata e sottraendo le due equazioni si ottiene una
relazione nota come nequazione di van’t Hoff:
ln (K2/K1) = -ΔH°/R [1/T2 – 1/T1]
Queste equazioni presuppongono che ΔH e ΔS non varino con la temperatura, almeno in
un limitato intervallo.
4
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
CAPITOLO 2
2.1 Equilibri acido-base
Le reazioni acido-base sono molto importanti nella chimica perché si riscontrano in
tantissimi processi della vita di tutti i giorni. La maggior parte dei processi chimici sono
reagolati dall’acidità presente nel sistema; nel settore dei beni culturali l’acidità e la
basicità sono fattori importanti per le possibili alterazioni dei materiali e quindi per la
loro conservazione (si pensi come esempio alle famose piogge acide che determinano
l’alterazione chimica dei monumenti esposti all’aperto, soprattutto, come già visto,
quelli costituiti da carbonato di calcio). Le reazioni acido-base avvengono sia in fase
gassosa che in fase solida che in soluzione (acquosa e non), tuttavia per semplicità e
importanza verranno trattiti solo gli equilibri in soluzione acquosa.
La prima definizione di acidi e basi fu data da Arrhenius nel XIX secolo. Secondo
Arrhenius un acido è una sostanza che in soluzione acquosa produce ioni H+(aq) e
una base è una sostanza che in soluzione acquosa produce ioni OH-(aq).
Una teoria più moderna, e oggi comunemente accettata e utilizzata, è quella formulata
indipendentemente da Johannes Brønsted e Thomas Lowry nel 1923 e che non
considera gli acidi e le basi come separati, ma introduce il concetto di equilibri acidobase. Secondo la teoria di Brønsted-Lowry: un acido è definito come un donatore di
protoni, una base come un accettore di protoni. Quindi, secondo la teoria di
Brønsted-Lowry, si parla in termini di reazioni acido-base. Si consideri ad esempio la
reazione tra acqua e acido acetico:
CH3COOH(aq) + H2O(l) = H3O+(aq) + CH3COO-(aq)
acido1
base2
acido2
base1
Gli acidi e le basi si presentano come coppie coniugate acido-base.
Gli acidi e le basi forti sono completamente dissociati in soluzione acquosa. Questa
affermazione è stata verificata sperimentalmente mediante misure di conducibilità. Si
dice anche che l’acqua ha un effetto livellante su un certo gruppo di acidi e basi nel
senso che questi si comportano da acidi forti e basi forti quando il solvente è l’acqua.
5
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Gli acidi e le basi che in soluzione acquosa non sono completamente dissociati sono
detti acidi e basi deboli. Gli acidi organici sono quasi sempre acidi deboli; quelli più
comuni sono gli acidi carbossilici tra i quali il più importante è l’acido acetico
(CH3COOH).
___________________________________________________________________
ACIDI FORTI
BASI FORTI
HClO4
acido perclorico
LiOH
idrossido di litio
HNO3
acido nitrico
NaOH
idrossido di sodio
H2SO4
acido solforico
KOH
idrossido di potassio
HCl
acido cloridrico
RbOH
idrossido di rubidio
HBr
acido bromidrico
CsOH
idrossido di cesio
HI
acido iodidrico
TlOH
idrossido di tallio
Ca(OH)2
idrossido di calcio
Sr(OH)2
idrossido di stronzio
Ba(OH)2
idrossido di bario
___________________________________________________________________
Alcune sostanze si comportano come acidi o come basi a seconda delle condizioni di
reazione e sono dette anfotere. L’esempio più importante è quello dell’acqua poiché a
seconda che essa si trovi a reagire con un acido o con una base si comporterà da base o
da acido rispettivamente.
Oltre le teorie di Arrhenius e Brønsted-Lowry, esiste la teoria acido-base di Lewis in
base alla quale gli acidi vengono definiti come accettori di doppietti elettronici
mentre le basi vengono definite come donatori di doppietti elettronici.
La definizione di Lewis permette di spiegare e descrivere il comportamento di molti
ossidi binari che possono essere considerati come anidridi di acidi o basi. Ad esempio la
6
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
maggior parte degli ossidi dei non metalli si comportano come anidridi acide ovvero
reagiscono con l’acqua formando soluzioni acide:
N2O5(s) + H2O(l) → 2H+(aq) + 2NO3-(aq)
SO3(g) + H2O(l) → H+(aq) + HSO4-(aq)
Un altro esempio importante di reazione acido-base di Lewis è quella tra un’anidride
acida e una basica:
CaO(s) + CO2(g) → CaCO3 (s)
In questa reazione la base di Lewis CaO cede un doppietto elettronico all’acido CO2 per
formare un legame covalente dativo nello ione CO32-. Questa reazione ricorda il
processo di carbonatazione, ovvero la reazione che avviene in fase acquosa negli
affreschi dove la calce viva, applicata sotto forma di malta in miscela con altri materiali
(sabbia, pozzolana, coccio pesto, fibre vegetali e animali, ecc) reagisce con l’anidride
carbonica presente nell’aria formando il carbonato di calcio che provoca il processo di
presa e indurimento e fissa i pigmenti dello strato pittorico.
2.2 Il concetto di pH
In acqua esiste un equilibrio:
H2O(l) + H2O(l) = H3O+(aq) + OH-(aq)
Ovvero: 2H2O(l) = H3O+(aq) + OH-(aq)
La costante per questo equilibrio è: Kw = [H3O+][OH-]
La concentrazione dell’H2O non compare in quanto in soluzioni acquose [H2O] è
costante. La costante Kw si chiama prodotto ionico dell’acqua. A 25°C il valore della
Kw é: Kw = 1,00x10-14 M2. Dalla stechiometria della reazione si può notare che in acqua
pura gli ioni H3O+ e gli ioni OH- sono prodotti in un rapporto 1:1, quindi la loro
concentrazione è uguale: [H3O+] = [OH-] = 1,00x10-7 M. Una soluzione acquosa
7
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
neutra viene definita una soluzione in cui: [H3O+] = [OH-]. Una soluzione acquosa
acida viene definita una soluzione in cui: [H3O+] > [OH-]. Una soluzione acquosa
basica viene definita una soluzione in cui: [H3O+] < [OH-]
Il pH è semplicemente una misura convenzionale dell’acidità di una soluzione
adottata perché numericamente comoda. Per esprimere l’acidità in termini di pH si
utilizza una scala logaritmica:
pH = -log10[H3O+]
analogamente: pOH = -log10[OH-]
In acqua pura a 25 °C [H3O+] = 1,0x10-7 M, quindi: pH = -log10 (1,0x10-7) = 7,00.
Sfruttando le proprietà dei logaritmi, si può scrivere:
-log10[H3O+][OH-] = -log10[H3O+] - log10[OH-] = 7,00 + 7,00 = 14,00 = -log10 Kw
pertanto: pH + pOH = pKw = 14,00
Ricapitolando, in soluzione acquosa, a 25 °C:
pH < 7
soluzione acida
pH = 7
soluzione neutra
pH > 7
soluzione basica
2.3 Tipi di acidi e basi
Nel caso di acidi e basi deboli la dissociazione in soluzione acquosa non è completa e
viene descritta da un equilibrio chimico. Ad esempio per un acido generico HA si ha:
HA(aq) + H2O(l) = H3O+(aq) + A-(aq)
Questo equilibrio è descritto dalla costante di ionizzazione acida Ka scritta in accordo
con il principio di azione di massa:
Ka = [H3O+] [A-]/[HA]
8
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Il valore della costante di ionizzazzione acida da una misura quantitativa della forza di
un acido in un dato solvente. Come per il pH spesso è conveniente esprimere la Ka in
forma logaritmica: pKa = -logKa.
Nel caso di basi deboli si parla di costante di ionizzazione basica o costante di
protonazione Kb. Ad esempio nel caso di una base generica B:
B(aq) + H2O(l) = BH+(aq) + OH-(aq)
Kb = [BH+] [OH-]/[B]
In genere la base coniugata di un acido debole è una base debole e quindi la reazione di
dissociazione può essere scritta come un equilibrio. Se HB è un acido debole:
HB(aq) + H2O(l) = H3O+(aq) + B-(aq)
L’acqua si comporta da base accettando un protone. Nella reazione inversa lo ione
idronio trasferisce un protone allo ione B- comportandosi quindi come acido mentre Bsi comporta come base. La base B- è detta base coniugata dell’acido HB e la coppia
HB/B- è detta coppia coniugata acido-base. Per questa reazione si può scrivere:
Ka=[H3O+][B-]/[HB]
Analogamente per la base coniugata:
B-(aq) + H2O(l) = HB(aq) + OH-(aq)
si può scrivere:
Kb=[HB][OH-]/[B-]
Moltiplicando le due costanti si ottiene: Ka Kb = [H3O+][OH-] = Kw, ovvero, passando
ai logaritmi: pKa + pKb = 14,00.
2.4 Indicatori acido-base
Esistono numerosi acidi organici deboli che cambiano colore per perdita di un protone:
composti con queste caratteristiche vengono chiamati indicatori, perché sono in grado
di indicare, mediante il loro colore, il pH di una soluzione. Gli indicatori cambiano
colore in uno stretto intervallo di pH e possiedono colori così intensi che possono essere
utilizzati a concentrazioni bassissime, in modo che, aggiunti ad una soluzione, non ne
influenzino praticamente il pH (fig. 67).
9
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Fig. 67 Variazioni di colore di diversi indicatori a diversi valori di pH, (da Donald A. McQuarrie, Peter A.
Rock, Chimica generale, Zanichelli, 1991)
Indicando con HIn la forma acida dell’indicatore e con In- la forma della base
coniugata, in soluzione acquosa si ha il seguente equilibrio:
HIn(aq) + H2O(l) = H3O+(aq) + In-(aq)
Questo equilibrio è descritto dalla costante di dissociazione acida dell’indicatore:
Kai = [H3O+][In-]/[HIn]. Questa relazione può essere anche scritta nella forma:
[H3O+]/Kai = [HIn]/ [In-]
10
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Se la concentrazione dello ione idronio è grande rispetto a Kai, allora il rapporto
[H3O+]/Kai è grande e la concentrazione di indicatore nella forma indissociata è molto
maggiore di quelle dell’indicatore nella forma dissociata: la soluzione avrà quindi la
colorazione della forma acida dell’indicatore. Se nella soluzione sono presenti le due
forme dell’indicatore in quantità simile, cioè se [HIn] ≈ [In-] allora la soluzione appare
color arancio. In questo caso [H3O+] ≈ Kai, ovvero pH ≈ pKai.
2.5 Idrolisi
Non sempre gli acidi e le basi sono specie elettricamente neutre, ci sono anche dei sali
che, disciolti in soluzione, liberano cationi o anioni acidi o basici. Ad esempio il cloruro
di ammonio, NH4Cl, disciolto in acqua libera ioni NH4+ che reagisce con l’acqua
secondo una reazione acido-base:
NH4+(aq) + H2O(l) = H3O+(aq) + NH3(aq)
La costante dell’equilibrio è: Ka=[H3O+][NH3]/[NH4+] = 5,6 x 10-10.
Idrolisi è un termine generale che viene utilizzato per indicare la reazione di una
sostanza con l’acqua ed è applicato in particolare ad una reazione nella quale il pH
cambia dal suo valore neutro, 7, quando si scioglie in acqua un sale che si comporta da
acido o da base. Si tratta semplicemente di reazioni acido-base che avvengono però solo
per quei sali contenenti ioni che sono acidi coniugati di basi deboli o basi coniugate di
acidi deboli. In generale si può dire che:
- le basi coniugate di acidi forti monoprotici sono anioni neutri che non reagiscono
quindi con l’acqua per formare ioni OH-. Le basi coniugate di acidi deboli, come lo ione
acetato, sono anioni basici che reagiscono con l’acqua per formare OH-, dando quindi
idrolisi basica. Lo ione idrogenosolfato, HSO4-, è un anione acido a causa della sua
reazione di seconda dissociazione acida, in acqua può quindi dare luogo ad idrolisi
acida: HSO4 -(aq) + H2O(l) = H3O+(aq) + SO42-(aq);
– non vi sono cationi basici, ma solamente cationi acidi e cationi neutri. Gli ioni dei
metalli alcalini ed alcalino terrosi (tranne Be2+) sono tutti neutri. Gli acidi coniugati di
basi deboli, come ad esempio NH4+, sono acidi e danno quindi idrolisi acida;
11
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
– molti ioni metallici sono acidi in soluzione acquosa, dove esistono legati ad un certo
numero di molecole d’acqua. Essi vengono quindi detti ioni solvatati. Un esempio di
catione acido solvatato è Fe(H2O)63+(aq).
2.6 Soluzioni tampone
Si dice soluzione tampone, una soluzione che mantiene il pH pressochè costante anche
quando si aggiungono piccole quantità di un acido o dia una base. Una soluzione
normalmente contiene un acido debole e la sua base coniugata in concentrazione circa
uguali. Le soluzioni tampone sono importantissime nel controllo della solubilità di molti
ioni e nel mantenere costante il pH in processi biochimici e fisiologici. Le proteine e in
particolare gli enzimi sono estremamente sendibili al pH e le loro funzioni come
molecole biologiche sono fortemente legate al valore del pH. Si consideri un acido HA
e la sua base coniugata A-, l’espressione dell’equilibrio può essere scritta nella forma:
[H3O+] = Ka[HA]/[A-]. La concetrazione degli ioni idronio e quindi il pH dipende dal
rapporto tra la concentrazione dell’acido debole e della sua base coniugata. Se queste
concentrazioni sono circa uguali e abbastanza grandi, una loro piccola variazione per
aggiunta di un acido o di una base non varierà in maniera significativa il loro rapporto e
quindi il pH si manterrà pressochè costante. Un tampone viene dunque creato
scegliendo un acido debole disciolto in soluzione acquosa insieme ad una il più
possibile simile concentrazione della sua base coniugata. Poiché si tratta di acidi e basi
deboli, la dissociazione si può considerare minima e quindi si può scrivere con buona
approssimazione che la concentrazione dell’acido all’equilibrio è uguale a quella
dell’acido iniziale, [HA] = [HA]0, e che la concentrazione della base coniugata
all’equilibrio è uguale a quella della base iniziale, [A-] = [A-]0. Quindi la relazione
scritta sopra diventa: [H3O+] = Ka[HA]0/[A-]0. Trasformando tutte le grandezze in
termini logaritmici si ottiene la seguente importante relazione, nota come equazione di
Henderson-Hasselbalch:
pH ≈ pKa – log10 ([HA]0/[A-]0)
Questa equazione, seppur approssimata, può essere utilizzata per progettare dei tamponi
a valori prefissati di pH, come già detto un tampone ottimale è quello nel quale l’acido e
12
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
la sua base coniugata hanno concentrazioni il più possibile simili. Chiaramente un
tampone non possiede una capacità infinita di annullare le variazioni di pH: se si
aggiunge un acido forte o una base forte in quantità tale da consumare tutta la base o
tutto l’acido è chiaro che il tampone perde la sua funzione.
2.7 Titolazioni acido-base
Le reazioni acido – base sono reazioni di neutralizzazione e risultano complete quando
il numero delle moli della base che ha reagito è uguale al numero delle moli di acido
che ha reagito, ovvero quando: MacidoVacido = MbaseVbase.
Si supponga di aggiungere lentamente una soluzione acquosa di una base forte, come
NaOH(aq), ad una soluzione acquosa di un acido forte, come HCl(aq): questa
operazione viene detta titolazione ed il grafico del pH della soluzione risultante in
funzione del volume della soluzione aggiunta, cioè il titolante, è detta curva di
titolazione. La forma della curva di titolazione dipende dal valore della costante Ka (o
Kb, a seconda del tipo di titolazione che si sta eseguendo) e dalle concentrazioni
dell’acido e della base reagenti. Il punto di equivalenza di una titolazione è il punto in
cui sono presenti quantità stechiometricamente equivalenti di acido e base. Nelle
titolazioni gli indicatori vengono utilizzati per evidenziare il completamento della
reazione acido base: il punto in cui l’indicatore cambia il colore è detto punto finale o
punto di viraggio. Nel caso della titolazione di un acido forte (HCl) con una base forte
(NaOH), la curva di titolazione avrà la forma di fig. 68.
Chiaramente in una titolazione la concentrazione del titolante è esattamente nota e viene
utilizzata per determinare la concentrazione incognita di titolato. Quello che occorre
determinare in una titolazione è il punto di fine e quindi il volume esatto di titolante
impiegato per completare la reazione di neutralizzazione. Il punto di fine può essere
valutato o con un indicatore, come già detto, o in modo molto più preciso con una
misura strumentale del pH, come si vedrà in seguito.
Nel caso di una titolazione di un acido debole con una base forte la curva avrà una
forma diversa da quella vista per la titolazione di un acido forte con una base forte (fig.
69). Il punto di mezzo corrisponde alla formazione di una soluzione tampone poiché le
concentrazioni di acido e base coniugata sono uguali. In questo punto, infatti, piccole
13
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
aggiunte di base forte non portano ad una significativa variazione del pH, inoltre si
verifica la condizione per cui pH = pKa.
Fig. 68 Illustrazione schematica dell’apparecchitura utilizzata in una titolazione acido-base e curva di
titolazione di HCl con NaOH, (da Donald A. McQuarrie, Peter A. Rock, Chimica generale, Zanichelli,
1991)
Fig. 69 Curva di titolazione dell’acido acetico con NaOH, (da Donald A. McQuarrie, Peter A. Rock,
Chimica generale, Zanichelli, 1991)
14
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Nel caso di una titolazione di una base con un acido, la curva di titolazione avrà un
andamento analogo ma si potrebbe dire “opposto” a quello visto per la titolazione di un
acido con una base forte (fig. 70).
Fig. 70 Curva di titolazione di NH3 con HCl, (da Donald A. McQuarrie, Peter A. Rock, Chimica generale,
Zanichelli, 1991)
2.8 Acidi poliprotici
Gli acidi che hanno più di un protone dissociabile sono detti acidi poliprotici. Alcuni
esempi sono costituiti dall’acido solfidrico H2S e solforico H2SO4 (acidi diprotici),
dall’acido fosforico H3PO4 (acido triprotico). Chiaramente questi acidi danno origine a
diversi equilibri di dissociazione acida anche se la costante dell’equilibrio, dopo la
prima dissociazione, diventa molto piccola. Questo si verifica perché, la carica negativa
che si forma per la perdita di uno ione idrogeno nella prima ionizzazione fa si che il
secondo idrogeno sia più fortemente legato e quindi più difficilmente cedibile. L’acido
solforico è un acido forte relativamente alla prima dissociazione mentre diventa debole
nella seconda ionizzazione. Un esempio importante di acido diprotico debole sia in
prima che seconda ionizzazione è l’acido carbonico formato per solvatazione della CO2
(acqua carbonatata).
H2CO3(aq) + H2O(l) = H3O+(aq) + HCO3-(aq)
Per questo equilibrio Ka1 = 4,3x10-7
15
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
HCO3-(aq) + H2O(l) = H3O+(aq) + CO32-(aq)
Per questo equilibrio Ka2 = 4,8x10-11
In realtà gli equilibri di dissociazione dell’acido carbonico sono complicati dal fatto che
la maggior parte della CO2 disciolta rimane come tale, CO2(aq), e solo una piccola
frazione si trasforma in acido carbonico. Approssimativamente 0,034 moli di CO2 si
sciolgono in 1 litro d’acqua a 25 °C e a pressione atmosferica.
16
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
CAPITOLO 3
3.1 Equilibri eterogenei, concetto di attività
Nel caso di gas ideali o soluzioni ideali, è possibile descrivere l’equilibrio attraverso la
legge di azione di massa, utilizzando le pressioni parziali o le concentrazioni. Nel caso
però siano presenti solidi o liquidi allo stato puro che partecipano all’equilibrio, la
concentrazione non descrive più bene l’equilibrio e occorre introdurre il concetto di
attività. Questo parametro descrive lo stato termodinamico di una sostanza
confrontandolo con uno stato di riferimento; è legato alla pressione e alla
concentrazione di una specie attraverso il coefficiente di attività. L’attività dello stato
di riferimento ha sempre valore unitario e quindi la variazione di energia libera di Gibbs
per portare un sistema dallo stato di riferimento ad uno stato termodinamico generico è
determinata solo dall’attività dello stato generico. Il coefficiente di attività γi di una
specie gassosa non ideale è legato alla pressione dalla relazione:
ai = γi Pi/Prif
Lo stato di riferimento per un gas ideale è quello ad 1 atm di pressione. Per i gas ideali
γi = 1 e dunque l’attività coincide con la pressione. Nel caso di una soluzione non
ideale, l’attività di un soluto i sarà data dalla relazione:
ai = γi ci/crif
Anche in questo caso γi = 1 per una soluzione ideale nello stato di riferimento, crif = 1M,
e quindi l’attività coincide con la concentrazione. Per le sostanze pure (solidi e liquidi)
gli stati di riferimento sono quelli stabili ad 1 atm e le attività in questi stati sono pari a
1. La costante di equilibrio può a questo punto essere espressa in modo più generale
attraverso le attività, ovvero:
K = aCc aDd/aAa aBb
Questa equazione rappresenta la legge di azione di massa per casi più generali.
17
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
3.2 Equilibri di solubilità
Le reazioni di dissoluzione e precipitazione dei solidi nelle soluzioni costituiscono una
classe di reazioni chimiche estremamente importanti in molti settori, non solo della
chimica, ma anche della medicina, dell’ingegneria, dell’ecologia e dei beni culturali (si
ricordi il problema già accennato dei sali solubili).
Una soluzione nella quale è stato disciolto tanto soluto da stabilire un equilibrio tra la
sua forma disciolta e quella solida, presente come corpo di fondo, è detta soluzione
satura. Se si aggiunge tanto solvente da portare in soluzione tutto il solido disciolto (in
accordo con il principio di le Châtelier) si dice che la soluzione è insatura. La
precipitazione controllata, che sfrutta le diverse solubilità dei solidi, viene utilizzata
ampiamente in chimica per la purificazione dei prodotti di reazione. Durante questo
processo però possono separarsi insieme alla sostanza desiderata anche delle impurezze
che debbono in qualche modo essere eliminate per ottenere dei prodotti di reazione
altamente purificati. Si ricorre allora al processo di ricristallizzazione per cui un
prodotto impuro viene sciolto e riprecipitato più volte, controllando opportunamente le
condizioni di reazione, sfruttando le diverse solubilità tra il prodotto desiderato e le
impurezze. Le reazioni di dissoluzione e precipitazione raggiungono spesso molto
lentamente la condizione di equilibrio e possono servire anche settimane o più per
ottenere una soluzione satura. Inoltre, spesso si formano delle soluzioni soprasature
ovvero soluzioni nelle quali la concentrazione del soluto supera il suo valore di
equilibrio e può occorrere anche moltissimo tempo perché si raggiunga la condizione di
soluzione satura.
La solubilità di una sostanza in un certo solvente, come già visto, è definita come la
quantità massima di quella sostanza che si scioglie all’equilibrio in un volume
definito del solvente ad una data temperatura. La maggior parte delle reazioni di
solubilizzazione di composti ionici è endotermica pertanto, in base al principio di Le
Châtelier, la solubilità aumenta all’aumentare della temperatura. Non tutti i composti
ionici presentanto lo stesso grado di solubilità e si possono riassumere dei criteri
empirici per individuare velocemente se un composto ionico è più o meno solubile.
1 – Tutti i sali di sodio, potassio e ammonio sono solubili , solubilità maggiore di 0,1
mol l-1.
2 – Tutti i nitrati, gli acetati e i perclorati sono solubili.
18
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
3 – Tutti i sali d’argento, di piombo e di mercurio (I) sono insolubili.
4 – Tutti i cloruri, i bromuri e gli ioduri sono solubili.
5 – Tutti i carbonati, i solfuri, gli ossidi e gli idrossidi sono insolubili.
6 – Tutti i solfati, tranne quelli di calcio e di bario, sono solubili.
I criteri di solubilità devono essere applicati nell’ordine in cui sono elencati: in caso
di discordanza ha la prevalenza il criterio che viene enunciato prima. Ad esempio il
solfuro di sodio Na2S è solubile perché il criterio 1 afferma che tutti i sali di sodio sono
solubili. Il solfato di piombo, PbSO4, è insolubile perché, anche se il criterio 6 afferma
che i solfati sono solubili, il criterio 3 afferma che tutti i sali di piombo sono insolubili.
L’equilibrio tra un solido ionico ed i suoi ioni costituenti in soluzione è regolato dalla
legge di azione di massa.
AgBrO3(s) = Ag+(aq) + BrO3-(aq)
La costante di equilibrio per questa reazione è:
Kps = [Ag+][BrO3-]
Il pedice ps sta per prodotto di solubilità e Kps viene detta prodotto di solubilità.
Il bromato d’argento solido non compare nella costante in quanto è un solido puro e
quindi la sua concentrazione è costante. Il valore sperimentale per questa costante è
5,8x10-5 M2. La solubilità si può ottenere dalla Kps. Infatti, sempre considerando il
bromato d’argento in acqua a 25 °C, all’equilibrio si ottiene: [Ag+][BrO3-] = 5,8x10-5
M2. Dalla stechiometria della reazione si ha che [Ag+] = [BrO3-] = solubilità = s.
Dall’espressione della Kps si ottiene: Kps = 5,8x10-5M2 = [Ag+][BrO3-] = s2 ; perciò s =
(5,8x10-5M2)1/2 = 7,6x10-3 M. Per un sale generico AxBy la solubilità è data da:
s = [A]/x = [B]/y
3.3 Effetto dello ione a comune
La solubilità di un solido ionico diminuisce se nella soluzione è presente uno ione a
comune. Infatti, la presenza di un eccesso di ioni (positivi o negativi), che erano già in
19
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
soluzione, riduce la concentrazione consentita dal prodotto di solubilità di quegli ioni e
quindi la solubilità diminuisce. Questo fenomeno è noto come effetto dello ione
comune: se la soluzione ed il sale che in essa deve essere disciolto hanno uno ione in
comune, allora la solubilità del sale diminuisce. Per comprendere meglio questo
concetto consideriamo l’esempio del bromato d’argento a 25 °C in una soluzione
acquosa 0,10 M di bromato di sodio. Il bromato di sodio è un sale solubile e quindi è
completamente dissociato in acqua; lo ione sodio Na+ è uno ione spettatore che non
rientra nei calcoli di solubilità. La solubilità del bromato d’argento è data da: s = [Ag+].
Gli ioni bromato provengono sia dalla dissociazione del bromato d’argento che da
quella del bromato di sodio, pertanto la loro concentrazione sarà: [BrO3-] = s + 0,10 M.
Quindi, sostituendo nell’espressione della Ksp si avrà: s(s+0,10 M) = 5,8x10-5 M2.
Poichè s è molto più piccolo di 0,10 M si può trascurare e quindi l’equazione diviene:
s(0,10 M) ≈ 5,8x10-5 M2 da cui s ≈ 5,8x10-4 M. Questo valore di solubilità è
decisamente inferiore a quello del bromato d’argento puro che si è visto essere uguale a
7,6x10-3 M.
3.4 Effetto del pH sulla solubilità
Molti solidi, poco solubili in acqua, aumentanto moltissimo la loro solubilità in
presenza di acidi. Ad esempio solfuri di rame e nichel, presenti nei minerali, possono
essere portati in soluzione con acidi forti per poter poi isolare i metalli che sono molto
ricercati ed impiegati in vari settori dell’industria. Come già visto, il carbonato di calcio
presenta scarsissima solubilità in acqua, ma in presenza di acidi la solubilità aumenta e
può essere portato facilmente in soluzione: è il caso delle piogge acide che provocano la
dissoluzione e quindi il degrado dei manufatti esposti all’aperto e costituiti da carbonato
di calcio (marmo, travertino e pietre calcaree in generale). Nel caso degli idrossidi
metallici il pH ha un effetto diretto sulla solubilità. Infatti, gli ioni OH- partecipano
direttamente all’equilibrio di solubilità e quindi un aggiunta di ioni H3O+, reagendo con
gli OH-, determina uno spostamento dell’equilibrio di dissoluzione verso destra. È il
caso, ad esempio, dell’idrossido di zinco:
Zn(OH)2(s) = Zn2+(aq) + 2OH-(aq)
20
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
3.5 Effetto della formazione di complessi
La formazione di un complesso di coordinazione di uno ione metallico con dei leganti,
porta ad un aumento della solubilità di un solido. Ad esempio gli ioni Ag+(aq)
reagiscono con NH3(aq) formando un complesso di coordinazione secondo la reazione:
Ag+(aq) + 2NH3(aq) = Ag(NH3)2+(aq)
La costante per questo equilibrio è nota come costante di formazione e in questo caso ha
il valore di: Kf = 2,0x107M-2. Un valore così elevato della costante indica che la
formazione del complesso è favorita e quindi in presenza di ammoniaca gli ioni argento
reagiranno con essa formando il complesso di coordinazione. Per il principio di Le
Châtelier, l’equilibrio di dissociazione di un sale d’argento, ad esempio:
AgBr(s) = Ag+(aq) + Br-(aq)
Ksp = 7,7x10-13
si sposterà verso destra poiché gli ioni Ag+ vengono sottratti all’equilibrio: questo
risulterà pertanto in una maggiore dissociazione e quindi in una maggiore solubilità del
sale.
3.6 Reazioni di ossido-riduzione
Le reazioni che avvengono con un trasferimento di elettroni da un reagente ad un altro
sono dette reazioni di ossidoriduzione o reazioni a trasferimento di elettroni (più
comunemente sono indicate come reazioni redox). Per capire le reazioni di
ossidoriduzione occorre definire il numero di ossidazione degli elementi nelle specie
chimiche e definire delle semplice regole per determinarlo. Il numero di ossidazione
esprime la carica (positiva o negativa) che l’atomo possiede nella reazione redox e che
non coincide necessariamente con la carica effettiva. In pratica i numeri di ossidazione
servono
per
bilanciare
stechiometricamente
le
equazioni
delle
reazioni
di
ossidoriduzione. Anche in questo caso esistono delle regole generali per poter assegnare
i numeri di ossidazione ai vari elementi:
1 – ad atomi allo stato elementare è assegnato numero di ossidazione 0;
21
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
2 – la somma algebrica dei numeri di ossidazione di tutti gli atomi di una specie chimica
deve essere uguale alla carica della specie;
3 – i metalli alcalini nei loro composti hanno sempre numero di ossidazione +1;
4 – il fluoro nei suoi composti ha sempre numero di ossidazione -1;
5 - i metalli alcalino-terrosi lo zinco e il cadmio nei loro composti hanno sempre
numero di ossidazione +2;
6 – alluminio e gallio nei loro composti hanno sempre numero di ossidazione +3;
7 – l’idrogeno nei suoi composti ha numero di ossidazione +1;
8 – l’ossigeno nei suoi composti ha numero di ossidazione -2;
Nei casi in cui possono sorgere dubbi nell’applicazione di queste regole si deve tenere
conto del fatto che la priorità delle regole è decrescente dalla n. 1 alla n. 8.
Si consideri la semplice reazione di ossido-riduzione:
Zn(s) + Cu2+(aq) = Cu(s) + Zn2+(aq)
Si dice che lo ione rame 2+ è ridotto a rame metallico perché il processo evolve
attraverso un decremento (riduzione) del numero di ossidazione del rame (da +2 a 0):
Cu2+(aq) + 2e- = Cu(s)
riduzione
Si dice che lo zinco metallico è ossidato a zinco 2+ perché la reazione implica un
aumento del numero di ossidazione dello zinco (da 0 a 2+) :
Zn(s) = Zn2+(aq) + 2e-
ossidazione
Il reagente che contiene l’elemento che è ridotto è detto agente ossidante
(elettronaccettore) o più semplicemente ossidante, mentre quello che contiene l’atomo
che si ossida è detto agente riducente (elettrondonatore) o più semplicemente
riducente.
Le reazioni di ossido-riduzione possono essere scomposte in due semireazioni: la
semireazione di ossidazione e la semireazione di riduzione e possono essere più
agevolmente bilanciate procedendo separatamente al bilanciamento delle due
semireazioni.
Si consideri ad esempio la semplice reazione di ossido-riduzione:
Fe(s) + Cl2(aq) = Fe3+(aq) + Cl-(aq)
22
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
L’equazione così come è scritta non è bilanciata. Il metodo più semplice e sistematico
per bilanciare la reazione è il metodo delle semireazioni, che risulta utile soprattutto
per reazioni di ossidoriduzione particolarmente complesse.
Si procede per passaggi successivi seguendo l’ordine riportato di seguito.
1 – Dividere l’equazione in due equazioni distinte per la semireazione di ossidazione e
la semireazione di riduzione:
Fe(s) = Fe3+(aq)
Cl2(aq) =
Cl-(aq)
ossidazione
riduzione
2 – Bilanciare l’equazione di ciascuna semireazione rispetto a tutti gli elementi tranne
ossigeno e idrogeno:
Fe(s) = Fe3+(aq)
Cl2(aq) = Cl-(aq)
3 – Bilanciare l’equazione di ciascuna semireazione rispetto all’ossigeno aggiungendo
l’appropriato numero di molecole d’acqua al membro in difetto di ossigeno:
Fe(s) = Fe3+(aq)
Cl2(aq) = Cl-(aq)
4 – Bilanciare l’equazione di ciascuna semireazione rispetto all’idrogeno aggiungendo
l’appropriato numero di ioni H+ al membro in difetto di idrogeno:
Fe(s) = Fe3+(aq)
Cl2(aq) = Cl-(aq)
5 - Bilanciare l’equazione di ciascuna semireazione rispetto alla carica aggiungendo
l’appropriato numero di elettroni al membro con eccesso di carica positiva:
Fe(s) = Fe3+(aq) + 3eCl2(aq) + 2e- = 2Cl-(aq)
6 – Moltiplicare membro a membro ciascuna equazione delle semireazioni per numeri
interi in modo tale che il numero totale degli elettroni ceduti dalla semireazione di
ossidazione sia uguale al numero di quelli acquistati nella semireazione di riduzione:
2Fe(s) = 2Fe3+(aq) + 6e3Cl2(aq) + 6e- = 6Cl-(aq)
7 – Scrivere l’equazione completa bilanciata sommando membro a membro le due
semireazioni bilanciate e semplificando i termini simili.
23
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
2Fe(s) + 3Cl2(aq) = 2Fe3+(aq) + 6Cl-(aq)
Si consideri adesso una reazione di ossido-riduzione un po’ più complessa, nella quale
entrano in gioco anche i bilanciamenti degli ioni H+ e degli atomi di ossigeno, quindi in
soluzione acida:
Fe2+(aq) + Cr2O72-(aq) = Fe3+(aq) + Cr3+(aq)
Seguendo lo stesso procedimento visto in precedenza, si ottiene:
1 – Dividere l’equazione in due equazioni distinte per la semireazione di ossidazione e
la semireazione di riduzione:
Fe2+(aq) → Fe3+(aq)
ossidazione
Cr2O72-(aq) → Cr3+(aq)
riduzione
2 – Bilanciare l’equazione di ciascuna semireazione rispetto a tutti gli elementi tranne
ossigeno e idrogeno:
Fe2+(aq) → Fe3+(aq)
Cr2O72-(aq) → 2Cr3+(aq)
3 – Bilanciare l’equazione di ciascuna semireazione rispetto all’ossigeno aggiungendo
l’appropriato numero di molecole d’acqua al membro in difetto di ossigeno:
Fe2+(aq) → Fe3+(aq)
Cr2O72-(aq) → 2Cr3+(aq) + 7H2O(l)
4 – Bilanciare l’equazione di ciascuna semireazione rispetto all’idrogeno aggiungendo
l’appropriato numero di ioni H+ al membro in difetto di idrogeno:
Fe2+(aq) → Fe3+(aq)
14H+(aq) + Cr2O72-(aq) → 2Cr3+(aq) + 7H2O(l)
5 - Bilanciare l’equazione di ciascuna semireazione rispetto alla carica aggiungendo
l’appropriato numero di elettroni al membro con eccesso di carica positiva:
Fe2+(aq) → Fe3+(aq) + e14H+(aq) + Cr2O72-(aq) + 6e- → 2Cr3+(aq) + 7H2O(l)
6 – Moltiplicare membro a membro ciascuna equazione delle semireazioni per numeri
interi in modo tale che il numero totale degli elettroni ceduti dalla semireazione di
ossidazione sia uguale al numero di quelli acquistati nella semireazione di riduzione:
6Fe2+(aq) → 6Fe3+(aq) + 6e14H+(aq) + Cr2O72-(aq) + 6e- → 2Cr3+(aq) + 7H2O(l)
24
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
7 – Scrivere l’equazione completa bilanciata sommando membro a membro le due
semireazioni bilanciate e semplificando i termini simili.
6Fe2+(aq) + 14H+(aq) + Cr2O7-2(aq) → 6Fe3+(aq) + 2Cr3+(aq) + 7H2O(l)
Le reazioni di ossidoriduzione che avvengono in ambiente basico si bilanciano
aggiungendo ioni OH-, anziché H+, e, di conseguenza, sarà necessario aggiungere
molecole d’acqua nell’altro membro dell’equazione chimica per ottenere il
bilanciamento degli atomi di idrogeno e di quelli di ossigeno.
Si consideri, ad esempio, la reazione di dissoluzione del cloro in soluzione basica:
Cl2(g) → ClO3-(aq) + Cl-(aq)
Questa reazione di ossido riduzione viene detta disproporzione poiché la stessa
sostanza, ovvero il cloro, viene sia ossidata che ridotta nella stessa reazione.
1 – Dividere l’equazione in due equazioni distinte per la semireazione di ossidazione e
la semireazione di riduzione:
Cl2(g) → ClO3 -(aq)
ossidazione
Cl2(g) → Cl-(aq)
riduzione
2 – Bilanciare l’equazione di ciascuna semireazione rispetto a tutti gli elementi tranne
ossigeno e idrogeno:
Cl2(g) → 2ClO3 -(aq)
Cl2(g) → 2Cl-(aq)
3 – Bilanciare l’equazione di ciascuna semireazione rispetto all’ossigeno aggiungendo
l’appropriato numero di ioni OH- al membro in difetto di ossigeno:
Cl2(g) + 6OH-(aq) → 2ClO3 -(aq)
Cl2(g) → 2Cl-(aq)
4 – Bilanciare l’equazione di ciascuna semireazione rispetto all’idrogeno aggiungendo
l’appropriato numero di molecole d’acqua al membro in difetto di idrogeno e
bilanciando la reazione:
Cl2(g) + 12OH-(aq) → 2ClO3 -(aq) + 6H2O(l)
Cl2(g) → 2Cl-(aq)
5 - Bilanciare l’equazione di ciascuna semireazione rispetto alla carica aggiungendo
l’appropriato numero di elettroni al membro con eccesso di carica positiva:
25
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Cl2(g) + 12OH-(aq) → 2ClO3 -(aq) + 6H2O(l) + 10eCl2(g) + 2e-→ 2Cl-(aq)
6 – Moltiplicare membro a membro ciascuna equazione delle semireazioni per numeri
interi in modo tale che il numero totale degli elettroni ceduti dalla semireazione di
ossidazione sia uguale al numero di quelli acquistati nella semireazione di riduzione:
Cl2(g) + 12OH-(aq) → 2ClO3 -(aq) + 6H2O(l) + 10e5Cl2(g) + 10e-→ 10Cl-(aq)
7 – Scrivere l’equazione completa bilanciata sommando membro a membro le due
semireazioni bilanciate e semplificando i termini simili.
6Cl2(g) + 12OH-(aq) → 2ClO3-(aq) + 10Cl-(aq) + 6H2O(l)
L’equazione ottenuta può anche essere divisa per due, pertanto la reazione finale sarà:
3Cl2(g) + 6OH-(aq) → ClO3-(aq) + 5Cl-(aq) + 3H2O(l)
3.7 Cenni di elettrochimica
L’elettrochimica è lo studio dei processi chimici che si verificano al passaggio di una
corrente elettrica attraverso un materiale. Il passaggio di corrente elettrica attraverso un
elettrolita viene detto elettrolisi. Le modificazioni chimiche durante l’elettrolisi
avvengono in corrispondenza degli elettrodi. Da una reazione di ossidoriduzione si può
ricavare una corrente elettrica mediante un apparecchio chiamato cella elettrochimica.
Una cella elettrochimica è caratterizzata dal suo voltaggio, che è una misura della forza
con cui una corrente elettrica viene spinta attraverso un conduttore. Il voltaggio della
cella dipende dalla concentrazione delle varie specie presenti nella cella di reazione.
Lo studio dell’elettrochimica ebbe inizio nel 1791, quando lo scienziato italiano Luigi
Galvani dimostrò che una corrente elettrica era in grado di provocare la contrazione di
una zampa di rana. Partendo dagli studi di Galvani, Alessandro Volta costruì un
apparecchio, detto pila voltaica, costituito da parecchi dischi di metalli diversi, ad
esempio zinco e rame, alternati e separati tra loro da tamponi di stoffa impregnati di una
soluzione salina. Michael Faraday impiegò le pile voltaiche per studiare l’effetto del
passaggio di una corrente elettrica attraverso soluzioni di vari elettroliti e scoprì che in
26
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
certe condizioni il passaggio di corrente provoca reazioni chimiche che non potrebbero
verificarsi altrimenti. Una reazione chimica provocata dal passaggio di corrente è detta
elettrolisi (fig. 71).
Fig. 71 Elettrolisi dell’acqua contenente solfato di sodio. Il gas che si svolge sull’elettrodo di platino
collegato con il disco di zinco superiore della serie di celle è idrogeno mentre il gas che si svolge
sull’elettrodo di platino collegato con il disco di rame sul fondo della pila è ossigeno. La corrente è
trasportata attraverso la soluzione da ioni Na+(aq) e ioni SO42-(aq), (da Donald A. McQuarrie, Peter A. Rock,
Chimica generale, Zanichelli, 1991)
Faraday scoprì che al passaggio di una corrente elettrica attraverso le soluzioni di molti
sali si formano depositi dei metalli corrispondenti. Controllando la corrente elettrica che
attraversa la soluzione si può controllare il numero di elettroni forniti alla reazione
elettrochimica. L’intensità di corrente elettrica viene misurata in ampere (A): un ampere
è definito come il flusso di un coulomb di carica al secondo.
corrente = carica/tempo
I = Q/t
carica totale = corrente x tempo
Q=I·t
(coulomb = ampere x secondi)
Le sperimentazioni e le osservazioni di Faraday sono raccolte nelle leggi di Faraday che
dicono:
Prima legge:
L’entità di una reazione elettrochimica dipende esclusivamente dalla quantità di
elettricità che attraversa la soluzione. Ovvero la massa di una sostanza, prodotta o
consumata ad un elettrodo, è proporzionale alla quantità di carica elettrica che è passata
attraverso la cella.
27
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Seconda legge:
La quantità in peso di una sostanza depositata come metallo o liberata come gas dal
passaggio di una determinata quantità di elettricità è direttamente proporzionale al peso
molare della sostanza diviso il numero degli elettroni scambiati per unità formula nel
corso della reazione.
Faraday introdusse una costante, che si ritroverà in tutte le relazioni principali
dell’elettrochimica, e che viene detta costante di Faraday, indicata con il simbolo F.
F = 96485,31 coulomb per mole = 9,648531 x104 Cmol-1.
Le leggi di Faraday furono scoperte nel 1833, quindi mezzo secolo prima che fosse
scoperto l’elettrone e che fosse compresa la base atomica dell’elettricità.
Il valore di F può essere ricavato considerando che la carica dell’elettrone è pari a
1,6021773x10-19 C e che il numero di Avogadro è pari a 6,022137x1023mol-1. Quindi,
una mole di elettroni possiede una carica pari a:
Q = (6,022137x1023mol-1)(1,6021773x10-19 C) = 96.485,31 C mol-1, che rappresenta
appunto la costante di Faraday.
Nell’apparecchio per l’elettrolisi visto prima la corrente viene trasportata da cationi e
anioni che si muovono in direzioni opposte verso due barrette di platino che vengono
dette elettrodi. Un elettrodo è costituito da una fase solida sulla cui superficie
avvengono reazioni ossidoriduzione:
l’elettrodo su cui avviene la semireazione di riduzione è detto catodo;
l’elettrodo su cui avviene la semireazione di ossidazione è detto anodo.
Molti prodotti chimici vengono preparati industrialmente mediante elettrolisi. Ad
esempio i metalli alcalini ed alcuni metalli alcalino-terrosi vengono preparati
industrialmente mediante elettrolisi. Tutto l’idrossido di sodio e gran parte del cloro
prodotto negli Stati Uniti vengono preparati mediante il processo cloro-alcali, basato
sull’elettrolisi di una soluzione acquosa concentrata di NaCl.
La reazione complessiva è:
2NaCl(aq) + 2H2O(l) → 2NaOH(aq) + Cl2(g) + H2(g)
Le due semireazioni sono:
2Na+(aq) + 2H2O(l) + 2e- → 2NaOH(aq) + H2(g)
2Cl-(aq) → Cl2(g) + 2e-
al catodo
all’anodo
28
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
3.8 Celle elettrochimiche
Una cella elettrochimica produce elettricità direttamente da una reazione chimica. Si
consideri la reazione spontanea:
Zn(s) + CuSO4(aq) → Cu(s) + ZnSO4(aq)
Una cella elettrochimica che sfrutta questa reazione è formata da due diversi metalli, Zn
e Cu, immersi in soluzioni elettrolitiche contenenti rispettivamente gli ioni metallici
Zn2+ e Cu2+ e mantenute in contatto elettrico mediante un ponte salino. Il ponte salino è
costituito da una soluzione satura di KCl miscelata con agar, una sostanza gelatinosa
aggiunta allo scopo di trattenere nel tubo la soluzione salina e di evitare la miscelazione
delle soluzioni. Il ponte salino costituisce un percorso per la corrente ionica tra la
soluzione di solfato di zinco e quella di solfato di rame impedendone al contempo la
miscelazione (fig. 72).
Fig. 72, Esempio di cella elettrochimica, (da Donald A. McQuarrie, Peter A. Rock, Chimica generale,
Zanichelli, 1991)
Gli elettrodi sono collegati a cavi elettrici che permettono alla cella elettrochimica di
fornire corrente elettrica ad un circuito esterno.
Le semireazioni di cella sono:
Zn(s) → Zn2+(aq) +2e-
ossidazione dello zinco
Cu2+(aq) + 2e- → Cu(s)
riduzione del rame
Gli elettroni prodotti per ossidazione dello zinco all’elettrodo di zinco si spostano
attraverso il circuito esterno verso l’elettrodo di rame, dove vengono consumati nella
riduzione di Cu2+(aq) a Cu(s). Poiché ioni Zn2+(aq) vengono prodotti nella soluzione che
29
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
contiene l’elettrodo di zinco, degli ioni negativi, in questo caso Cl-(aq), devono fluire
attraverso il ponte salino verso la soluzione di solfato di zinco per mantenere la
neutralità elettrica della soluzione. D’altra parte, poiché dalla soluzione contenente
l’elettrodo di rame vengono rimossi ioni Cu2+(aq), degli ioni positivi, in questo caso
K+(aq), devono fluire attraverso il ponte salino verso la soluzione di solfato di rame per
mantenere la neutralità elettrica.
Un diagramma di cella è una rappresentazione grafica sintetica di una cella
elettrochimica:
Zn(s) | ZnSO4(aq) || CuSO4(aq) | Cu(s)
Le barre verticali semplici indicano la separazione tra fasi distinte in contatto tra loro, la
doppia barra indica il ponte salino. La convenzione adottata per scrivere le equazioni
che descrivono le reazioni di cella è quella di scrivere la semireazione dell’elettrodo di
sinistra come equazione della semireazione di ossidazione e la semireazione
dell’elettrodo di destra come equazione della semireazione di riduzione.
Le celle elettrochimiche che funzionano spontaneamente vengono dette anche celle
galvaniche o celle voltaiche. Collegando gli elettrodi ad un circuito esterno, in
particolare ad un voltmetro, è possibile misurare la differenza di potenziale che si
produce in seguito alla reazione di ossido-riduzione che avviene spontanemente nelle
soluzioni della celle voltaica.
Un elettrodo che coinvolge specie chimiche gassose è detto elettrodo a gas.
L’elettrodo a gas più importante è l’elettrodo ad idrogeno. Questo elettrodo è costituito
da una spirale di platino inserita nel compartimento della cella elettrochimica che
contiene le specie H+(aq) e H2(g). Il platino è un metallo relativamente non reattivo, che
fornisce semplicemente la superficie su cui avviene la reazione di riduzione senza
parteciparvi. Accoppiando un elettrodo ad idrogeno con un elettrodo di zinco si ottiene
il seguente diagramma di cella (fig. 73):
Zn(s) | Zn2+(aq) || H+(aq) | H2(g) | Pt(s)
30
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Fig. 73 Esempio di cella elettrochimica con elettrodo a gas, (da Donald A. McQuarrie, Peter A. Rock,
Chimica generale, Zanichelli, 1991)
Facendo riferimento alla termodinamica, occorre introdurre un tipo di lavoro
fondamentale in elettrochimica, ovvero il lavoro elettrico welettr che sarà dato dalla
relazione welettr = -QΔE. Il segno negativo è dovuto al fatto che si tratta di lavoro
eseguito dal sistema (in questo caso la cella elettrochimica), in base alla convenzione
stabilita in termodinamica. ΔE è la differenza di potenziale che si genera per
spostamento attraverso il circuito della carica Q. A temperatura e pressione costante,
dalla termodinamica sia ha che il lavoro massimo che può eseguire il sistema sarà dato
dalla variazione di energia libera: welettr,max = ΔG. Pertanto ΔG = -QΔE = -nFΔE (per un
processo reversibile). Per reazioni che avvengono in una cella elettrochimica, la
variazione di energia libera standard, ΔG°, sarà in relazione al voltaggio standard di
cella, ΔE°: ΔG° = -nFΔE°. Il voltaggio standard della cella elettrochimica è quello
calcolato quando i reagenti e i prodotti sono nei loro stati standard.
È importante sottolineare il fatto che non è possibile misurare potenziali elettrici di un
singolo elettrodo, mentre è possibile misurare solamente differenze di potenziale. Se
però si attribuisce un valore arbitrario al potenziale standard di una semicella di
riferimento, è possibile poi, per differenza, determinare i potenziali standard di tutte le
altre semicelle. Per convenzione si pone uguale a zero il potenziale standard di
riduzione dell’elettrodo a idrogeno, cioè si pone E° = 0 per la semireazione di elettrodo:
2H3O+(aq, 1M) + 2e- → H2(g, 1 atm) + 2H2O(l)
31
E° = 0 per convenzione
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Quando due semicelle vengono combinate per formare una cella galvanica, avviene la
riduzione nella semicella con il potenziale maggiore (catodo) e l’ossidazione nella
semicella con il potenziale minore (anodo). Quindi per la cella elettrochimica completa
si può scrivere:
ΔE°cella = E°(catodo) - E°(anodo)
Queste equazioni possono essere utilizzate per compilare la tabella dei potenziali
standard di riduzione. Infatti, i potenziali standard delle singole semireazioni vengono,
per convenzione, riportati come potenziali di riduzione partendo da quelli più positivi
via via fino a quelle più negativi. Un forte agente ossidante è una specie chimica che si
riduce facilmente e sarà quindi caratterizzata da un potenziale di riduzione molto
positivo. Ossidanti forti sono il fluoro, F2, l’acqua ossigenata, H2O2, l’ozono O3, il
cloro, Cl2, l’ossigeno, O2, ecc. Un forte agente riducente è una specie chimica che si
ossida facilmente e sarà quindi caratterizzata da un potenziale molto negativo. Riducenti
forti sono generalmente i metalli alcalini e alcalino-terrosi, l’idrogeno, H2, ecc.
3.9 L’equazione di Nernst
Walther Nernst fu uno dei pionieri dell’elettrochimica e ricevette il premio Nobel per
la chimica per i suoi studi sulla dipendenza dei potenziali di cella dalla concentrazione
degli elettroliti, dalle dimensioni degli elettrodi e da altri fattori. Il potenziale di cella è
una misura della forza motrice della reazione e quindi l’effetto della variazione di
concentrazione di un reagente o di un prodotto su di esso può essere spiegato in termini
qualitativi applicando il principio di Le Châtelier alla reazione di cella.
Da un punto di vista termodinamico si ha che ΔG = ΔG° + RT lnQ, dove si ricordi che
Q è il quoziente di reazione. Considerando che ΔG = -nFΔE e che ΔG° = -nFΔE°, si
ottiene la seguente relazione che rappresenta appunto l’equazione di Nernst:
ΔE = ΔE° - RT/nF lnQ
Considerando che lnQ = 2,303 log10Q, e sostituendo i valori delle costanti R e F a
temperatura di 298,15 K, si ottiene l’equazione di Nernst nella forma:
32
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
ΔE = ΔE° - 0,0592/n log10Q
L’elettrochimica fornisce un modo conveniente ed accurato per determinare le costanti
di equilibrio di molte reazioni chimiche in soluzioni. Infatti, poiché la variazione di
energia libera standard è legata alla costante di equilibrio dalla relazione: ΔG° = -RTlnK
e poiché: ΔG° = -nFΔE°, combinando le due espressioni si ha: RTlnK = nFΔE°, lnK =
nF/RT ΔE° e quindi:
log10 K = n/0,0592 ΔE°
(a 25 °C)
Lo stesso risultato può essere ottenuto a partire dall’equazione di Nernst che a 25° C ha
la seguente espressione:
ΔE = ΔE° - 0,0592/n log10Q
Quando si raggiunge la condizione di equilibrio non si ha più variazione del potenziale
e dunque ΔE = 0 e Q = K, pertanto vale la relazione ΔE° = 0,0592/n log10K.
3.10 Alcuni tipi di elettrodi
Una branca molto importante della chimica analitica è costituita dalla potenziometria
che sfrutta la misura del voltaggio delle celle per ottenere informazioni chimiche. La
cella per la misura potenziometrica è costituita da un elettrodo indicatore (ad esempio
platino), che risponde alla concentrazione dell’analita, e da un elettrodo di riferimento
che mantiene un potenziale costante e definito.
Un comune elettrodo di riferimento quello ad argento-cloruro d’argento (fig. 74a),
basato sulla reazione:
AgCl(s) + e- → Ag(s) + Cl-
E (KCl saturo) = + 0,197 V
Un altro elettrodo di riferimento molto utilizzato in elettrochimica è l’elettrodo a
calomelano saturo (E.C.S, fig. 74b), basato sulla reazione:
33
Corso di Chimica analitica applicata ai beni culturali
Hg2Cl2(s) + 2e- → 2Hg(l) + 2Cl-
Dott.ssa Claudia Pelosi
E (KCl saturo) = + 0,197 V
Il cloruro mercuroso, Hg2Cl2, è detto calomelano.
(a)
(b)
Fig. 74 Elettrodi di riferimento ad argento-cloruro d’argento (a) e a calomelano saturo (b), (da D. C.
Harris, Chimica analitica quantitativa, Zanichelli, 1991)
In potenziometria sono particolarmente importanti gli elettrodi iono-selettivi ovvero
queli elettrodi che rispondono selettivamente ad una sola specie in soluzione. Questi
elettrodi sono costituiti da una membrana che separa il campione incognito dall’interno
dell’elettrodo dove è presente lo ione da determinare a concentrazione nota e costante.
La differenza di concentrazione dello ione da misurare, nella soluzione interna
all’elettrodo e nel campione, genera attraverso la membrana una differenza di potenziale
che, una volta misurata, permetterà di determinare la concentrazione dello ione nel
campione, essendo quella interna all’elettrodo nota e costante. L’elettrodo iono-selettivo
più importante e utilizzato è l’elettrodo a vetro per la misura del pH. Gli elettrodi a
vetro utilizzati oggi sono elettrodi combinati (fig. 75), ovvero elettrodi che
comprendono in un unico corpo sia l’elettrodo a vetro vero e proprio che l’elettrodo di
riferimento. Questo elettrodo può essere schematizzato come:
34
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
Ag(s) | AgCl(s) | Cl-(aq) || H+(aq, esterno) ¦ H+(aq, interno), Cl-(aq) | AgCl(s) | Ag(s)
La linea tratteggiata verticale indica la membrana di vetro. È stato dimostrato da studi
sperimentali che nell’elettrodo a vetro, la membrana si porta in equilibrio con gli ioni
H+ su ciascuna superficie e che sono gli ioni Na+ che trasportano la carica attraverso la
membrana coordinandosi con gli atomi di ossigeno carichi negativamente e presenti nel
reticolo del vetro della membrana. Poiché la concentrazione di Cl- è costante in ciascun
compartimento dell’elettrodo e quella di H+ è costante all’interno della membrana di
vetro, l’unico fattore che può produrre una differenza di potenziale è una variazione di
pH nella soluzione al di fuori della membrana di vetro.
Fig. 75 Elettrodo combinato per la misura del pH, avente un elettrodo di riferimento ad argento-cloruro
d’argento, (da D. C. Harris, Chimica analitica quantitativa, Zanichelli, 1991)
L’elettrodo a vetro viene utilizzato normalmente nei pH-metri, strumenti di laboratorio
o portatili, che vengono appunto impiegati per la misura del pH. Gli elettrodi a vetro,
prima di essere utilizzati, devono essere tarati per mezzo di soluzioni tampone a pH
noto e definito. In genere, per la maggior parte dei pH metri, la taratura viene effettuata
immergendo l’elettrodo a vetro in una soluzione a pH = 7 e aspettando che si equilibri.
Poi, l’elettrodo viene immerso nella seconda soluzione tampone il cui pH viene scelto in
funzione dell’intervallo di misura che sperimentalmente si pensa di utilizzare, in genere
35
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
pH = 4 o pH = 9. Ogni volta, prima di immergere l’elettrodo in una soluzione, esso deve
essere accuratamente sciacquato e asciugato. Infine, l’elettrodo a vetro deve sempre
essere conservato in una soluzione acquosa di KCl per evitare che lo strato di gel
idratato della membrana si asciughi e quindi occorrano poi tempi molto lunghi per
rigenerarlo.
3.11 La corrosione dei metalli
La corrosione dei metalli è uno dei problemi più gravi per le società industriali con costi
elevatissimi per le nazioni più industrializzate. La corrosione coinvolge chiaramente il
settore dei beni culturali e della conservazione dei manufatti metallici soprattutto quelli
esposti all’aperto (statue e portali) nelle aree urbane ad alta densità abitativa. Gli effetti
della corrosione sono sia visibili (formazione di strati superficiali ad esempio di ruggine
nel caso del ferro) che invisibili (rotture e indebolimenti del metallo sotto la superficie).
La corrosione può essere vista come una cella galvanica “corto-circuitata” nella quale
alcune zone della superficie del metallo funzionano da catodo e altre da anodo e il
circuito elettrico si chiude con il flusso degli elettroni attraverso il metallo stesso.
Queste celle elettrochimiche si formano nelle parti del metallo dove sono presenti
impurezze oppure stress meccanici del materiale. Nel caso del ferro, uno dei metalli
notoriamente più facilmente soggetti a corrosione, la reazione anodica è:
Fe(s) → Fe2+(aq) + 2eSono possibili diverse reazioni catodiche a seconda che sia presente ossigeno oppure
questo sia carente (ad esempio manufatti immersi o seppelliti). Nel secondo caso le
reazioni di corrosione sono:
Fe(s) → Fe2+(aq) + 2e2H2O(l) + 2e- → 2OH- + H2(g)
(anodo)
(catodo)
Fe(s) + 2H2O(l) → Fe2+(aq) + 2OH- + H2(g)
La reazione di corrosione del ferro è molto più rilevante in presenza di ossigeno e
acqua, in questo caso la reazione catodica è:
36
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
½ O2(g) + 2H3O+(aq) + 2e- → 3H2O(l)
Gli ioni Fe2+ che si formano nello stesso tempo all’anodo, migrano al catodo dove
vengono ulteriormente ossidati per azione di O2 e passono allo stato di ossidazione +3
formando la ruggine (Fe2O3 · xH2O), un ossido di ferro (III) idrato:
2Fe2+(aq) + ½ O2(g) + (6+x)H2O(l) → Fe2O3 · xH2O + 4H3O+(aq)
Sul catodo si forma la ruggine mentre all’anodo il metallo si consuma per perdita di ioni
che, una volta formatisi per ossidazione, migrano verso il catodo. I sali disciolti
favoriscono la corrosione poiché forniscono un elettrolita che migliora il flusso della
carica attraverso la soluzione. Anche l’acidità aumenta la corrosione, infatti gli ioni
H3O+ partecipano attivamente alla reazione vera e propria di corrosione; la CO2 (che
produce ioni H3O+ e HCO3-) aumenta la corrosione; l’inquinamento da ossidi di zolfo,
che formano acido solforico disciolto nelle piogge acide, favorisce la corrosione. Un
metodo per proteggere i metalli dalla corrosione è quello di verniciarli, tuttavia la
vernice può deteriorarsi o graffiarsi. Una protezione molto più efficace è quella della
passivazione, mediante la quale si forma sulla superficie del metallo un sottile strato di
ossido che previene ulteriori reazioni elettrochimiche. Alcuni metalli si passivano
spontaneamente per esposizione all’aria, ad esempio l’alluminio forma uno strato di
Al2O3. Un altro metodo, infine, è quello di usare un anodo sacrificale, ovvero un altro
metallo che si ossida molto più facilmente di quello che deve essere protetto e quindi si
consuma per primo. Chiaramente, l’anodo sacrificale deve essere periodicamente
sostituito prima che si consumi completamente.
37
Corso di Chimica analitica applicata ai beni culturali
Dott.ssa Claudia Pelosi
INDICE
CAPITOLO 1
1.1 L’equilibrio chimico
p. 2
1.2 Il principio di Le Châtelier
p. 3
CAPITOLO 2
2.1 Equilibri acido-base
p. 5
2.2 Il concetto di pH
p. 7
2.3 Tipi di acidi e basi
p. 8
2.4 Indicatori acido-base
p. 9
2.5 Idrolisi
p. 11
2.6 Soluzioni tampone
p. 12
2.7 Titolazioni acido-base
p. 13
2.8 Acidi poliprotici
p. 15
CAPITOLO 3
3.1 Equilibri eterogenei, concetto di attività
p. 17
3.2 Equilibri di solubilità
p. 18
3.3 Effetto dello ione a comune
p. 19
3.4 Effetto del pH sulla solubilità
p. 20
3.5 Effetto della formazione di complessi
p. 21
3.6 Reazioni di ossido-riduzione
p. 21
3.7 Cenni di elettrochimica
p. 26
3.8 Celle elettrochimiche
p. 29
3.9 L’equazione di Nernst
p. 32
3.10 Alcuni esempi di elettrodi
p. 33
3.11 La corrosione dei metalli
p. 36
INDICE
p. 38
38