Sindromi mielodisplastiche
Premessa degli autori
Stimato lettore, stimata lettrice,
Contrariamente ad un tumore situato, ad esempio, nello stomaco o nei polmoni, la sindrome mielodisplastica è una malattia difficilmente rappresentabile per le persone che ne
sono affette. Infatti si diffonde in tutto l’organismo attraverso il sangue ed è perciò impossibile da visualizzare per mezzo di ecografie o radiografie. Che cosa si deve allora
intendere per questa malattia dal nome difficile?
La sindrome mielodisplastica è una malattia delle cellule staminali che producono il sangue e. Insorge principalmente a causa di una maturazione difettosa delle cellule ematiche nel
midollo osseo. Nel sangue si verifica quindi una carenza di globuli rossi, bianchi o piastrine o piastrine completamente maturi. Ciò può comportare stanchezza, debolezza,
maggiore predisposizione a infezioni o a sanguinamenti spontanei. La sindrome mielodisplastica può presentarsi in diverse sottoforme e alcune di queste possono trasformarsi
in una leucemia acuta.
Negli ultimi anni sono stati compiuti significativi progressi nel trattamento delle sindromi mielodisplastiche. I nuovi medicinali sono in grado di neutralizzare parzialmente il blocco
della maturazione, con conseguente miglioramento dei valori ematici e della qualità di vita possono anche migliorare l’aspettativa di vita e ridurre il rischio di trasformazione
in leucemia acuta. Per i pazienti più giovani il trapianto di cellule staminali può essere un’opzione promettente. Infine, oggi è possibile curare con farmaci anche il sovraccarico di
ferro, spesso pericoloso, causato dalle trasfusioni di sangue.
Il seguente opuscolo, grazie ad illustrazioni adatte, intende aiutarvi a comprendere meglio questa malattia, così come il meccanismo d’azione dei medicinali.
Vi incoraggiamo pertanto a leggere questa pubblicazione e a rivolgervi al vostro oncologo o ematologo, qualora insorgessero domande.
Dtt. R. Benz
Dtt. U. Hess
L. D. Dtt. G. Stüssi
Dtt. J. Voegeli
Dirigente medico
Primario
Servizio di Ematologia
Ematologo FMH/FAMH
Oncologia medica
Oncologia ed ematologia medica
Istituto Oncologico della Svizzera Italiana
La Chaux-de-Fonds
Ospedale cantonale di Münsterlingen
Ospedale cantonale di San Gallo
Bellinzona
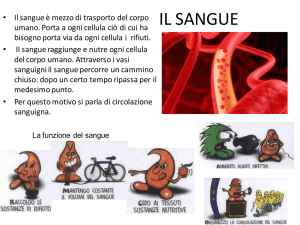
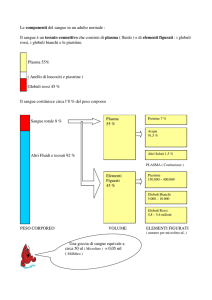
I diversi tipi di cellule ematiche e le loro funzioni
Piastrine (trombociti)
Globuli rossi (eritrociti)
Funzione: coagulazione del sangue
Le piastrine sono importanti per
Funzione: trasporto dell’ossigeno
la coagulazione del sangue in caso
I globuli rossi contengono l’emoglobina.
Questa lega l’ossigeno che viene
assorbito nei polmoni. Grazie a questo
legame l’ossigeno può essere
trasportato nei vari tessuti e organi del
corpo e lì essere rilasciato.
Globuli bianchi (leucociti)
Sottogruppi:
• Granulociti (per es. neutrofili)
•Monociti
• Linfociti B e T
Funzione: difesa contro le infezioni
I globuli bianchi fanno parte del sistema
immunitario. Aiutano a prevenire e
combattere le infezioni.
I rapporti dimensionali delle cellule raffigurate non corrispondono alle proporzioni reali.
di lesioni. Arrestano l’emorragia
«incollandosi» tra loro in modo da
chiudere la ferita con un coagulo
(trombo).
Il midollo osseo: la fabbrica delle cellule ematiche
Sviluppo normale delle cellule ematiche
Cellule progenitrici
linfatiche
Linfocita B
Linfocita T
Cellula staminale
Cellula progenitrice
mieloide (blasto)
Granulocita
(per es. neutrofilo)
Piastrine
Globuli rossi
In caso di sindrome mielodisplastica (SMD)
Cellule progenitrici
linfatiche
Linfocita B
Linfocita T
Dettaglio del midollo osseo
Nel midollo osseo si trovano le cellule
staminali che maturano fino a diventare
cellule ematiche.
Negli adulti l’emopoiesi avviene esclu­
sivamente nelle ossa del tronco (per es.
bacino, costole, sterno, vertebre).
Cellula staminale
Cellule progenitrici mieloidi
modificate (blasti)
Cellule ematiche con difetti di
maturazione (displastiche),
per es.:
Pseudo-Pelger
Piastrine giganti
Macrocita con
presenza di basofili
Nella sindrome mielodisplastica
(SMD) la maturazione nel
midollo osseo non può più
avvenire normalmente a causa
delle cellule progenitrici mieloidi
alterate. Le cellule finali mature
che arrivano nel sangue sono
perciò molto poche.
Sintomi
Quadro ematologico
normale
Carenza di globuli rossi
Y Anemia
Conseguenze:
• Stanchezza
• Vertigini
• Pallore
• Dispnea
• Tachicardia
• Riduzione della capacità fisica
Anemia
Carenza di globuli bianchi
Principalmente granulociti neutrofili (neutropenia)
Y Predisposizione alle infezioni
Conseguenze:
• Febbre
• Brividi
• Tosse
• Insufficienza respiratoria
• Diarrea
Quadro ematologico anomalo
dovuto a SMD
Neutropenia
Carenza di piastrine
Y Riduzione della coagulazione del sangue (trombocitopenia)
Conseguenze:
• Emorragie puntiformi (petecchie):
Per «petecchie» si intende un grande numero
di emorragie delle dimensioni della
capocchia di uno spillo sulla pelle o sulle mucose
• Ecchimosi
• Epistassi
• Sanguinamento gengivale
Trombocitopenia
Rappresentazione schematica
Blasto
Granulocita
Piastrine
Piastrine giganti
Globuli rossi
Linfocita
I cromosomi come portatori di informazioni genetiche
Ogni cellula umana contiene 23 coppie di cromosomi di forma
e dimensioni differenti, nei quali il DNA è compresso in strutture
spiraliformi. I cromosomi sono anche i portatori dei geni.
Un gene è un segmento determinato del DNA che contiene le
istruzioni per la produzione di una specifica proteina o enzima.
Nel nucleo di ogni singola cellula del nostro corpo si trovano
memorizzate tutte le informazioni genetiche dell’organismo,
paragonabili di costruzione di un edificio molto complesso.
Questo progetto è immagazzinato sotto forma di acido
desossiribonucleico o, più brevemente, DNA. Il DNA è il
composto chimico dal quale si sviluppa il patrimonio genetico.
Il DNA presente nelle cellule di tutte le forme di vita superiori
ha la forma di lunghe strutture filiformi, i cromosomi.
Le proteine sono molecole biologiche composte da molti
aminoacidi e sono essenziali per la vita di ogni singola cellula:
regolano importanti funzioni dell’espressione genica, servono
come trasportatori, agiscono come recettori, prendono parte
Nucleo cellulare
Cromosoma
Gene
Il cariotipo rappresenta il corredo cromosomico di un individuo, che è definito dal numero e dalla forma dei cromosomi.
Il numero e la forma sono identici sia per gli uomini (22 coppie
di cromosomi e una coppia XY) sia per le donne (22 coppie di
cromosomi e una coppia XX). Ogni cromosoma ha una sua
forma specifica.
Corredo cromosomico normale (cariotipo) di un uomo
Cellula
DNA
allo spostamento/movimento delle cellule e, come proteine
strutturali, costituiscono l’impalcatura delle cellule, ecc.
Mutazioni genetiche nella SMD
Tipiche alterazioni cromosomiche nella SMD (uomo)
Solo nella metà dei casi di SMD il cariotipo subisce un’alterazione. In questi casi è possibile confermare
la diagnosi di SMD (al contrario, nel caso di un cariotipo normale non è possibile rilevare una SMD).
La determinazione del cariotipo è un elemento fondamentale della diagnostica della SMD, in quanto le
alterazioni delle informazioni genetiche possono fornire importanti indizi sulla classificazione, la prognosi
e il trattamento. Ad esempio, in caso di una SMD con assenza del ramo lungo del cromosoma 5 è possibile
prendere in considerazione la terapia con Revlimid® – diversamente da tutti gli altri tipi di SMD.
Cellula
Nucleo cellulare
Cromosoma
DNA
Gene
Nelle persone con SMD può verificarsi un’alterazione dei cromosomi (portatori del patrimonio genetico).
Queste alterazioni cromosomiche si ritrovano solo nelle cellule ematopoietiche malate, quindi non
possono essere trasmesse alla discendenza. Il più delle volte è possibile osservare alterazioni dei
cromosomi 5, 7 e 8.
La conoscenza di tali alterazioni del patrimonio genetico è importante per la prognosi e per la scelta
della terapia.
La SMD può insorgere senza alcuna causa apparente (SMD de novo) oppure può essere dovuta ad una
radioterapia o chemioterapia (SMD secondaria). Molto spesso la malattia insorge con l’avanzare dell’età.
7
8
5
Monosomia
Trisomia
Delezione (del)
Si parla di monosomia
quando si verifica la perdita
completa di uno dei due
cromosomi che compongono la coppia cromosomica.
Si parla di trisomia quando
un cromosoma si presenta
tre volte invece di due.
La delezione indica la perdita
di una parte di un cromosoma. Qui è illustrata la delezione del cromosoma 5
(del5q–).
Gli esami più importanti
Prelievo di sangue
Biopsia del midollo osseo
Il sangue viene prelevato da una vena del braccio. Il sangue prelevato serve per la
determinazione del numero di cellule ematiche e altri esami di laboratorio.
Il medico preleva un campione di midollo osseo con un ago (in anestesia locale).
Generalmente si tratta di un piccolo cilindro di midollo prelevato dal bacino per eseguire
degli esami microscopici. Con il sangue così ottenuto dal midollo osseo è possibile
identificare alterazioni formali, stadi iniziali (per es. i blasti), ma anche alterazioni
genetiche e cromosomiche. La biopsia del midollo osseo non è una biopsia del midollo
spinale, non comporta quindi alcun rischio di paraplegia.
Determinazione del numero delle varie
cellule ematiche:
Altri test ematologici:
Esami microscopici:
Determinazione del cariotipo:
Questi test permettono di escludere altre
malattie con sintomi simili (per es. la carenza
di vitamina B12 o di acido folico).
• Numero di cellule ematiche nel
midollo osseo
• Forma delle cellule ematiche nel
midollo osseo
• Stadio di maturazione delle cellule
ematiche nel midollo osseo
• Numero di blasti (cellule progenitrici
immature)
Il cariotipo contiene le alterazioni cromosomiche e geniche delle cellule ematiche nel
midollo osseo.
• Globuli rossi
• Globuli bianchi
• Piastrine
Tutti questi esami, nel loro insieme, permettono al medico
di classificare meglio la malattia: di tratta di uno stadio iniziale
oppure la malattia è già avanzata? Si sviluppa lentamente
o rapidamente?
I seguenti criteri sono rilevanti per la valutazione:
• Numero di cellule ematiche
• Alterazioni cromosomiche
• Numero di blasti
• Necessità di trasfusioni di sangue
Grazie a questi criteri è possibile calcolare un indice di rischio
(IPSS, IPSS-R, WPSS: v. appendice) che è determinante per la
scelta della terapia individuale.
Se la quantità di blasti nel midollo osseo non supera il 20 % si
tratta di una SMD – ma se tale quantità supera il 20 % è possibile
che si tratti di una leucemia acuta. Durante il decorso della SMD
circa un terzo dei pazienti va incontro ad una leucemia acuta.
Possibilità di trattamento della SMD
Trattamenti della SMD allo stadio precoce
Terapie di supporto
Trasfusioni di globuli rossi
Trasfusioni di piastrine
Chelazione del ferro: Exjade® (Déférasirox) per l’eliminazione del ferro in eccesso nell’organismo e per stimolare l’ematopoiesi
Sostegno alla maturazione dei globuli bianchi (G-CSF)
Sostegno alla maturazione dei globuli rossi (eritropoietina, EPO)
Le terapie immunosoppressive
Durante la SMD le normali cellule progenitrici nel midollo osseo sono attaccate dallo stesso sistema immunitario.
Le seguenti terapie riducono questa reazione eccessiva del sistema immunitario contro le proprie cellule:
Globulina anti-timociti (ATG)
Sandimmun® (ciclosporina A)
Trattamento specifico
Revlimid® (lenalidomide) per il trattamento della sindrome mielodisplastica con una delezione del cromosoma 5 (del5q)
Trattamenti della SMD allo stadio avanzato
Altre terapie farmacologiche
Vidaza® (azacitidina)
Chemioterapia intensiva
Chemioterapia a basso dosaggio
Trapianto allogenico di cellule staminali
Nel caso del trapianto allogenico di cellule staminali, queste sono prelevate da un donatore – perciò in questo caso il donatore
e il ricevente non sono la stessa persona. Il donatore può essere un familiare o una persona estranea che possiede un midollo
osseo con determinate caratteristiche il più possibile simili a quelle del ricevente.
Il trapianto è l’unica forma di terapia in grado di offrire una possibilità di guarigione. Tuttavia il presupposto per il successo della
terapia non è solo la corrispondenza delle caratteristiche genetiche dei globuli bianchi, ma anche la presenza del migliore stato
generale di salute possibile nel paziente.
Trasfusioni di sangue: azione dell’eritropoietina
(EPO) sui globuli rossi
Trattamento con Exjade® (deferasirox)
per le periodiche trasfusioni di sangue
I pazienti con SMD sono soggetti a frequenti trasfusioni di sangue a causa del loro ridotto
numero di globuli rossi. Attraverso queste trasfusioni l’organismo riceve un eccesso di ferro.
Ad ogni trasfusione l’organismo riceve 200–250 mg di ferro. Questo ferro in eccesso non può
essere eliminato spontaneamente dall’organismo e tende ad accumularsi in diversi tessuti.
Poiché il sovraccarico di ferro è nocivo per il cuore, il fegato e per le ghiandole, il livello può
essere tenuto sotto controllo per mezzo di una terapia chelante.
Produzione di globuli rossi nel midollo
osseo e rilascio nel sangue
Trasfusione di globuli rossi
++
EPO
In caso di riduzione del contenuto
di ossigeno del sangue (anemia),
tale deficienza viene rilevata dai
sensori di ossigeno contenuti nei
reni che, di conseguenza, stimolano la produzione di EPO.
Rappresentazione schematica
Livello del ferro
Fe++
Fe++
Esc r ez
ione d
e l fe r r o
++
Fe
im
As sor b
e n to d
i fe r r o
Fe++ = ferro contenuto nel siero
A lungo andare la quantità di ferro somministrata all’organismo è superiore a quella
che può essere eliminata. La conseguenza è un grave sovraccarico di ferro.
Fe++
Equilibrio del ferro
L’eritropoietina (EPO) è uno degli
ormoni naturali prodotti dai reni,
ha la funzione di stimolare la produzione di globuli rossi nel midollo
osseo. In medicina l’EPO è utilizzata
per il trattamento dell’insufficienza
renale cronica e dell’anemia. L’EPO
aumenta il numero di globuli rossi
anche nelle persone sane ed è utilizzata in modo illecito come doping
negli sport di resistenza.
Rene
Quanti più globuli rossi sono
presenti nel sangue, tanto
maggiore è la quantità di
ossigeno ad essi legato che
può essere trasportata agli
organi/tessuti.
Terapia chelante
Globuli rossi
Sovraccarico di ferro
Fe
Midollo osseo
Chelatore
Ferro
Fe++
Chelato
Escrezione da parte dell’organismo
Il sovraccarico di ferro deve essere trattato per evitare gravi danni al fegato, al cuore e
alle ghiandole. La terapia chelante del ferro ha il compito di ridurre il contenuto di ferro
nell’organismo.
Fe++
Fe++
Fe++
Assorbimento di ferro
Fe++
Escrezione del ferro
Il legame del ferro con il chelatore conduce all’escrezione della quantità in eccesso,
rendendo nuovamente possibile l’equilibrio nell’organismo.
Trattamento con Revlimid® (lenalidomide) nella SMD del5q
Terapia
con Revlimid®
Midollo osseo
Cromosoma difettoso
nella cellula alterata
5
Eliminazione delle cellule
recanti la delezione del
cromosoma 5
Molte cellule nel midollo osseo contengono
cromosomi con una delezione del
cromosoma 5 (del5q–).
Cromosoma 5 normale in
una cellula sana
5
Nel midollo osseo sono presenti ancora poche
cellule con delezione del cromosoma 5 (del5q–).
La maturazione delle cellule
ematiche sane è difettosa
perché le cellule con una delezione del cromosoma 5
non sono in grado di produrre
cellule ematiche mature.
Maturazione
delle cellule mature
La modalità d’azione del Revlimid® non è ancora
del tutto chiara. Dopo trattamenti di durata diversa
le cellule recanti la delezione del cromosoma 5
(del5q–) scompaiono dal midollo osseo e inizia di
nuovo la produzione di un numero sufficiente di
cellule progenitrici sane.
Quadro ematico
Anemia
Rappresentazione esemplificativa
Blasto con delezione del cromosoma 5
Quadro ematologico
normale
Granulocita
Piastrine
Piastrine giganti
Globuli rossi
Linfocita
Trattamento con Vidaza® (azacitidina)
Situazione
nella
SMD
Situation
Situation
en
encas
casde
de
SMD
SMD
® ®®
Situazione
terapia con
Vidaza
Situation
Situation
avec
avecnella
un
untraitement
traitement
avec
avec
Vidaza
Vidaza
Le cellule alterate a causa della SMD producono una quantità eccessiva di una specifica
molecola (gruppo metilico) che si lega al
DNA bloccando la produzione di proteine
essenziali. Questo blocco comporta
l’interruzione della corretta maturazione
delle cellule ematiche.
DNA
Il Vidaza® è una molecola dalla struttura
simile al DNA. Grazie alla penetrazione di
Vidaza® nel DNA delle cellule geneticamente
alterate, é possibile ostacolare l’inserzione
del gruppo metilico. Potendo proseguire
regolarmente la produzione delle proteine
essenziali, la maturazione completa delle
cellule ematiche é garantita.
Il principio attivo del Vidaza® penetra
nel DNA, rendendo impossibile il
legame del gruppo metilico (CH3) allo
stesso DNA.
Inserzione di un
gruppo metilico (CH3)
Ingrossamento della struttura del DNA
DA
OFF
OFF
Cellule
ematiche
immature
(displastiche)
Cellules
Cellules
sanguines
sanguines
anormales
anormales
Rappresentazione esemplificativa
A
ON
ON
Normalizzazione
del
quadro
ematico
Normalisation
Normalisationdes
des
cellules
cellules
sanguines
sanguines
Il DNA cellulare deve essere diviso e moltiplicato affinché Vidaza® possa penetrarvi per
svolgere la propria azione. È necessaria una
terapia continua con Vidaza® per almeno
6 mesi, affinché Vidaza® possa ridurre
progressivamente, ad ogni divisione
cellulare, il numero delle cellule alterate.
Esempio per la valutazione del rischio
Il trattamento della SMD viene adattato al profilo di rischio individuale del paziente. I principi di riferimento
sono l’IPSS (International Prognostic Scoring System), l’IPSS-R (Revised International Prognostic Scoring
System) e il WPSS (WHO classification – based Prognostic Scoring System). Grazie a queste scale è
possibile determinare se la malattia si trova in uno stadio iniziale o avanzato.
IPSS (International Prognostic Scoring System)
Punteggio
0
0.5
1
1.5
2
Percentuale di blasti nel midollo osseo (%)
0–4
5–10
–
11–20
21–29
Numero di serie cellulari con valore ridotto1
0–1
2–3
–
–
–
Gruppo di rischio citogenetico2
Basso
Medio
Alto
–
–
Piastrine <100.000/μl, emoglobina <10 g/l, neutrofili <1.500/μl
Gruppi di rischio citogenetico:
– Rischio ridotto: corredo cromosomico normale, 5q meno, 20q meno, meno Y
– Rischio elevato: alterazioni cromosomiche complesse (> 3 anomalie), difetti del cromosoma 7
– Rischio intermedio: tutte le altre anomalie
1
2
Inoltre l’età del paziente e le patologie concomitanti possono avere un’influenza rilevante sul decorso della malattia.
Gruppi di rischio
Punteggio aggiuntivo
Rischio basso
0
Rischio intermedio 1
0.5–1
Rischio intermedio 2
1.5–2
Rischio elevato
> 2.5
IPSS-R (Revised International Prognostic Scoring System)
Punteggio
0
Gruppo di rischio citogenetico
Molto basso
Percentuale di blasti nel midollo
osseo (%)
2
3
4
Basso
Medio
Alto
Molto alto
≤2
Da > 2
a<5
5 a 10
> 10
Emoglobina (g/dl)
≥ 10
8 à < 10
Piastrine (x 10 9/l)
≥ 100
50 a < 100
Neutrofili (ANC, x 10 9/l)
≥ 0.8
< 0.8
Gruppi di rischio citogenetico
Alterazioni citogenetiche
Molto basso
Semplice:
–Y, del (11q)
Basso
0.5
Cariotipo normale
1
1.5
<8
< 50
Semplice:
del (5q), del (12p), del (20q)
Doppia:
Inclusa del (5q)
Medio
Semplice:
del (7q), +8, +19, i (17q), tutti i cloni indipendenti successivi
Doppia:
Tutti i successivi
Alto
Semplice:
–7, inv (3) / t (3q) / del (3q)
Complessa:
3 alterazioni
Molto alto
Complexe:
> 3 modifications
Gruppo di rischio IPSS-R
Punteggio aggiuntivo
Rischio molto basso
≤ 1.5
Rischio basso
> 1.5–3
Rischio medio
> 3–4.5
Rischio elevato
> 4.5–6
Rischio molto elevato
>6
Doppia:
Inclusa–7 / del (7q)
del = delezione
inv = inversione
t = traslocazione
WPSS (WHO classification-based Prognostic Scoring System)
Punteggio
0
1
2
3
Categoria OMS
RA/RARS/5q
RCMD/RCMD-RS
RAEB I
RAEB II
Gruppo di rischio citogenetico1
Bassa
Media
Alta
–
Necessità di trasfusione2
No
Sì
–
–
Gruppi di rischio citogenetico:
– Rischio ridotto: corredo cromosomico normale, 5q meno, 20q meno, meno Y
– Rischio elevato: alterazioni cromosomiche complesse (≥ 3 anomalie), difetti del cromosoma 7
– Rischio intermedio: tutte le altre anomalie
2
La necessità di trasfusione è definita da una frequenza di almeno 1 trasfusione di globuli rossi ogni 8 settimane per 4 mesi
1
Categorie OMS:
5q
= delezione isolata sul cromosoma 5q (sindrome 5q)
RA
= anemia refrattaria
RARS
= anemia siderobalstica refrattaria
RCMD
= citopenia refrattaria con displasia di più linee cellulari
RCMD-RS= citopenia refrattaria con displasia di più linee cellulari e sideroblasti
RAEB I
= anemia refrattaria con eccesso di blasti (5–10 %)
RAEB II = anemia refrattaria con eccesso di blasti (11–20 %)
Gruppi di rischio
Punteggio aggiuntivo
Rischio molto basso
0
Rischio basso
1
Rischio intermedio
2
Rischio elevato
3 – 4
Rischio molto elevato
5 – 6
Prima pagina: cellule ematiche in caso di sindrome mielodisplastica | Retro: quadro ematologico normale
Questo opuscolo sulla descrizione della sindrome mielodisplastica
può anche essere scaricato in formato PDF:
www.pomcanys.ch/zeigebuch_mds
Questa brochure non ha ricevuto alcun tipo di sostegno vincolante da parte delle aziende Celgene GmbH e Novartis (25733/10.2013). © 2013 Celgene and Novartis Corporation. All rights reserved.
26244