EMOGLOBINA E MIOGLOBINA
Emoglobina e Mioglobina sono proteine globulari in grado di legare l’ossigeno molecolare. Queste
due proteine si differenziano tra loro per le funzioni e per le caratteristiche chimico-fisiche che
mostrano nei confronti dell’O2 che le differenziano significativamente.
L’emoglobina (Hb) è una proteina in grado di legare molecole di O2 (quattro in tutto) a livello
polmonare e di trasportarla nel sangue fino ai tessuti ed alle cellule che si trovano ad immediato
contatto del letto capillare del sistema vascolare. Al contrario è in grado di trasportare dai tessuti
ed organi periferici fino ai polmoni, CO2 e protoni (H+)
La mioglobina (Mb) è una proteina capace di legare o liberare O2 a seconda della sua
concentrazione nel citoplasma delle cellule muscolari; essa presenta una catena polipeptidica ed
un sito di legame per l’ossigeno, al contrario dell’emoglobina, costituita da quattro catene
globiniche, contenente ognuna un sito di legame per l’O2 ed a due a due uguali.
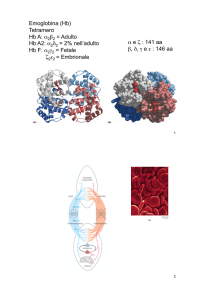
L’emoglobina HbA1 è la forma maggiormente rappresentata nell’uomo (circa il 98%) ed è
costituita da due catene globiniche α, di 141 residui aminoacidici ciascuno e due β costituite da
146 residui.
E’ anche presente l’HbA2 (circa il 3%) in cui le due catene β sono sostituite da due catene δ.
Emoglobine umane e loro subunità
Fonte principale
Simbolo
Subunità
Adulto
Hb A1
α2β 2
Adulto
HbA2
α2δ2
Fetale
HbF
α2γ2
Embrionale
HbGower-2
α2ε2
(stato fetale precoce)
Sia le quattro catene globiniche dell’emoglobina che quelle della mioglobina contengono un
gruppo prostetico in grado di legare una molecola di O2 (gruppo Eme).
L’eme ha una struttura porfirinica contenente un atomo di ferro centrale. Questa porfirina
(protoporfirina IX) contiene otto catene laterali e più precisamente.
O - due residui di acido propionico
X - quattro gruppi metilici legati agli anelli pirrolici
+ - due gruppi vinilici
L’atomo di ferro presente nell’eme presenta un numero di ossidazione 2+ ed è in grado di formare
5 o 6 legami di coordinazione.
Quattro legami di coordinazione sono formati con i quattro atomi di azoto degli anelli pirrolici
della protoporfirina e giacciono sullo stesso piano degli anelli pirrolici quando l’Hb è ossigenata,
mentre quando l’O2 è assente il ferro si trova ad una distanza dal piano di 0.6 Å.
Il quinto legame di coordinazione lo ione Fe2+ lo forma con un atomo di azoto presente nell’anello
imidazolico di un residuo di istidina (istidina distale F8) della catena polipeptidica.
Il sesto legame può essere libero o impegnare il ferro con la molecola di ossigeno. Questi due
ultimi legami sono perpendicolari al piano dell’anello tetrapirrolico dell’eme.
Quando l’ossigeno è legato allo ione Fe2+ si verrà a trovare in prossimità dell’anello imidazolico di
un secondo residuo di istidina della catena polipeptidica (istidina distale E7).
Quando l’eme non è legato a proteine, la reazione dell’ossigeno con uno dei due siti di
coordinazione del ferro (perpendicolari al piano dell’anello porfirinico) genera l’ossidazione
irreversibile del Fe2+ a Fe3+. Quando l’eme è inserito in una proteina, questa reazione non avviene
in quanto il gruppo eme è immerso in profondità nella struttura proteica è l’accessibilità ai siti di
coordinazione è limitata.
L’importanza dell’impedimento sterico presentato dall’ambiente in cui si trova il sito di legame
dell’eme, sia nell’emoglobina che nella mioglobina, è dimostrata dall’affinità di queste due
proteine per il monossido di carbonio, che compete con l’ossigeno per tale sito. In soluzione
acquosa l’affinità del monossido di carbonio per l’eme libero è circa 25.000 volte maggiore di
quella dell’ossigeno, mentre l’affinità del CO nei confronti dell’eme legato alla proteina è solo 250
volte superiore a quella dell’ossigeno. Questo a causa dell’impedimento sterico determinato
dall’istidina E7 (Figura (b), che causa un’inclinazione nella conformazione del complesso globina-
CO con conseguente riduzione dell’affinità di legame. Anche il monossido di azoto (nitric oxide)
(NO), si può coordinare al ferro dell’eme con una affinità anche in questo caso superiore a quella
dell’ossigeno.
Quando si lega l’ossigeno, le proprietà elettroniche del ferro si modificano; ciò spiega il diverso
colore che ha il sangue venoso povero di ossigeno (rosso scuro) rispetto al sangue arterioso ricco
di ossigeno (rosso brillante).
Il gruppo eme si trova alloggiato all’interno di una tasca idrofobica formata dal ripiegamento di
ciascuna catena polipeptidica all’interno del quale le catene laterali di circa 18 aminoiacidi
stabiliscono all’incirca ottanta interazioni non covalenti con gli atomi di carbonio dell’eme. Le
due catene di acido propionico sono invece stabilizzate dalla interazione con la catena laterale
priva di carica di un residuo di istidina e con molecole d’acqua, entrambe presenti sulla superficie
esterna dellamolecola. La diffrazione ai raggi X ha mostrato che le catene globiniche
dell’emoglobina cosi come quelle della mioglobina si presentano costituite da numerose regioni ad
α-elica interrotte per la presenza di residui di prolina, che permettono il ripiegamento della
catena a cui si accompagna spesso una inversione della direzione della stessa catena. Questo tipo
di andamento fa assumere a queste catene globiniche una struttura grossolanamente sferoidale.
Le catene a sono caratterizzate da sette zone ad α-elica, mentre le β da otto. Lo studio delle
sequenze aminoacidiche delle catene globiniche di diverse specie animali ha evidenziato una
notevole variabilità anche se la variazione nella composizione aminoacidici è tale da permettere
sempre le interazioni apolari ed a idrogeno con i residui che si trovano sulla superficie delle altre
unità globiniche.
Si assiste, invece, alla costante presenza dei due residui di istidina prossimale e distale e degli altri
residui presenti nella tasca idrofobica, dove si posizione l’eme, necessari per la stabilizzazione del
gruppo prostetico con la proteina. Questo permette alle catene globiniche di conservare una
struttura secondaria e terziaria simile, anche se piccole variazioni in punti specifici della loro
struttura comportano delle differenze significative nelle proprietà delle catene come ad esempio
avviene tra le catene α, β, γ, δ.
Esiste una notevole somiglianza tra la struttura della catena globinica della mioglobina (A) e
la subunità β dell’emoglobina (B). Queste, infatti, si differenziano soltanto per la disposizione
del residuo carbossilico e amminico terminale. Il posizionamento verso l’esterno di tali gruppi,
nella catena β dell’emoglobina permetterà l’interazione tra le due catene beta.
La funzione fisiologica della mioglobina e della emoglobina dipende dalla reversibilità del legame
fra ossigeno e gruppo eme, processo il cui andamento è descritto dalle curve di saturazione con
l’O2.
Figura: Rappresentazione in grafico del legame di un ligando. La frazione di siti di legame occupata dal ligando, θ, è
messa in grafico in funzione della concentrazione di ligando libero. Entrambe le curve sono iperboli rettangolari.
(a) Una curva di legame ipotetica del ligando L. La [L] a cui metà dei siti totali sono occupati corrisponde a 1/Ka,
oppure a Kd,. La curva ha un asintoto orizzontale quando θ = 1 e un asintoto verticale (non mostrato) quando [L] = l/Ka.
(b) Curva di legame dell'ossigeno alla mioglobina: la pressione parziale di ossigeno presente nell'aria in contatto
con la soluzione è espressa in kilopascal (kPa). L'ossigeno si lega saldamente alla mioglobina con una P50 di soli
2,6 kPa.
In genere, il legame reversibile di un ligando (L) a una proteina (P) può essere descritto
dall'espressione all'equilibrio:
P+L → PL
La costante di equilibrio, Ka, di questa reazione è
[PL]
Ka = ———
[P] [L]
(1)
II termine Ka è la costante di associazione (da non confondere con la Ka che indica la costante di
dissociazione di un acido). La costante di associazione è una misura dell'affinità del ligando L per
la proteina P. Ka ha come unità M-l; un valore di Ka elevato corrisponde a un'elevata affinità del
ligando per la proteina. Un riarrangiamento dell'equazione 1 mostra che il rapporto tra proteina
con il ligando legato e proteina libera è direttamente proporzionale alla concentrazione del
ligando libero:
[PL]
Ka [L] = ———
[P]
(2)
Quando la concentrazione del ligando è molto più alta della concentrazione dei suoi siti di legame
sulla proteina, il legame del ligando alla proteina non modifica in modo apprezzabile la
concentrazione del ligando libero (non legato), cioè [L] resta costante. Questa condizione è
applicabile a molti ligandi che si associano a proteine nelle cellule e semplifica la nostra
descrizione dell'equilibrio di legame.
Consideriamo l'equilibrio di legame mettendo in rapporto i siti di legame occupati con i siti di
legame presenti nella proteina, θ (theta):
siti di legame occupati
θ = ———————————
totale dei siti di legame
[PL]
= ————— (3)
[PL] + [P]
Sostituendo il termine Ka [L][P] a [PL] (Equaz. 2) e riarrangiando i termini, si ha
Ka [L][P]
θ = ——————— =
Ka [L][P] + [P]
Ka [L] [PL]
[L]
—————— = ————— (4)
Ka [L] + l
[L] + 1/Ka
II termine Ka può essere determinato dal grafico di θ in funzione della concentrazione del ligando
libero [L] (Figura). Qualsiasi equazione nella forma di x = y (y + z) descrive un'iperbole e quindi
θ è una funzione iperbolica di [L].
La frazione di siti che legano il ligando occupata tende ad arrivare asintoticamente a saturazione
quando [L] aumenta. Il valore di [L] a cui metà dei siti di legame sono occupati dalligando (a θ =
0,5) corrisponde a l/Ka.
Qualche volta è intuitivamente più semplice considerare la costante di dissociazione, Kd, cioè il
reciproco di Ka (Kd = 1/Ka) , espressa in unità di concentrazione molare (M). Kd è la costante di
equilibrio della reazione di rilascio del ligando.
[L] [PL]
Kd = ———— (5)
[PL]
[P] [L]
[PL] = ——— (6)
Kd
[L]
θ = ———— (7)
[L] + Kd
Quando [L] è uguale a kd metà dei siti di legame sono occupati dal ligando. Quando [L] è minore
di Kd, la quantità di ligando legata alla proteina è piccola. Perché il 90% dei siti di legame siano
occupati, il valore di [L] deve essere nove volte più alto di quello di Kd. In pratica, il termine Kd
viene usato molto più spesso di Ka per esprimere l'affinità di una proteina per il suo ligando.
Notate che un valore molto basso di Kd corrisponde a un'elevata affinità della proteina per il
ligando.
La matematica può ridurre questo concetto a una semplice asserzione:
Kd corrisponde alla concentrazione di ligando a cui metà dei siti di legame disponibili sono occupati
dal ligando. A questo punto la proteina ha raggiunto metà della saturazione rispetto ai siti di
legame. Quanto più saldamente la proteina lega il ligando, tanto più bassa è la concentrazione di
ligando necessaria a occupare metà dei siti di legame e minore è il valore di Kd.
Il legame dell'ossigeno alla mioglobina ha un andamento simile a quello discusso prima; essendo
l'ossigeno un gas, bisogna effettuare alcune variazioni nell'equazione. Dobbiamo semplicemente
sostituire la concentrazione di ossigeno disciolta a [L] nell'Equazione 7:
[O2]
θ = ———— (8)
[O2] + Kd
Come accade per ogni ligando, Kd corrisponde alla [O2] a cui metà dei siti disponibili sono
occupati e quindi è uguale a [O2]0,5. L'Equazione 8 diventa:
[O2]
θ = —————— (9)
[O2] + [O2]0,5
La concentrazione di una sostanza volatile in una soluzione è sempre proporzionale alla sua
pressione parziale nella fase gassosa in equilibrio con la soluzione. Negli esperimenti in cui viene
usato ossigeno come ligando, è la pressione parziale di questo gas, pO2, che viene variata, in
quanto è molto più facile misurare quest'ultima che non la concentrazione del gas disciolta nella
soluzione. Se definiamo la pressione parziale di ossigeno [O2]0,5 come P50, sostituendo
nell'Equazione 9 otteniamo
pO2
θ = —————— (9)
pO2 + P50
Nel caso in cui una proteina leghi più di una molecola di ligando come nel caso dell’emoglobina
L’equazione (9) diventerà:
[pO2]n
θ = —————— (10)
P50 + [pO2]n
Questa equazione rappresenta la forma non linearizzata della equazione di Hill ed n rappresenta
il coefficiente di Hill.
Se il valore di n risulta diverso da 1, la proteina legante o l’enzima presenterà fenomeni
cooperativi.. In particolare per n<1 saranno presenti fenomeni cooperativi negativi, mentre per
n>1 la proteina presenterà fenomeni cooperativi positivi.
Non essendo possibile calcolare dall’equazione (10), se non con l’ausilio di un calcolatore, il valore
di “n” viene utilizzato la sua forma linearizzata.
Forma linearizzata dell’equazione di “Hill”
Keq
[Hb]+nO2 Hb(O2)n
vf= K1[Hb(O2)n] (1); vr=K-1[Hb]*[O2]n (2)
[Hb] = [Hbt]-[Hb(O2)n] (3)
quando [Hbt] = [Hb(O2)n] tutta l’emoglobina sarà saturata ed avremo che:
vf= K1[Hb(O2)n] sarà uguale a Vmax = K1[Hbt]
ponendo Vmax (100% di saturazione) uguale ad 1 avremo: 1 = K1[Hbt] (4)
se moltiplichiamo tutti i membri dell’eq. (3) per K1 otteremo:
K1 [Hb] = K1 [Hbt] - K1 [Hb(O2)n] (5)
e sostituendo i termini comuni con la (1) e al (2) otterremo:
K1 [Hb] = Vmax-v
ovvero:
K1 [Hb] = 1-y
dividiamo ambo i membri per l’eq. (1)
K1 [Hb]
1-y
—————— = ——— (10)
K1[Hb(O2)n]
v
Se applichiamo la legge di azione di massa all’equilibrio di saturazione dell’Hb avremo:
[Hb] [O2]n
Keq = ——————
[Hb(O2)n]
riarrangiando l’eq. precedente questa diventerà:
[Hb]
Keq
————— = ———
[Hb(O2)n]
[O2]n
sostituendo nella (10) e facendo il reciproco otterremo:
y
[pO2]n
v
[O2]n
—— = ——— ovvero —— = ———
1-y
Keq
1-y
P50
questa espressa in forma logaritmica diventerà:
y
Log —— = nLog [pO2]n – Log P50
1-y
La presenza nell’emoglobina di quattro subunità non differenzia questa proteina dalla
mioglobina soltanto per il numero di molecole di O2 legate ma anche per la presenza di fenomeni
cooperativi positivi. Tutto ciò comporterà che il legarsi della prima molecola di O2 alla
emoglobina renderà più semplice il legarsi delle successive alle altre subunità dell’Hb, cosi come il
distacco della prima molecola di O2 comporterà un aumento nella velocità di rilascio delle
successive. La presenza di fenomeni cooperativi ha un preciso significato e svolge un ruolo
significativo nel regolare l’attività
nh
Rx
0,5
0,6
0,7
0,8
0,9
6560
1520
533
243
132
1,0
81,0
Osservazioni
metabolica della proteina. Infatti,
come mostrato in tabella, dalla
Cooperatività
negativa da
substrato
valutazione
dell’indice
di
cooperatività (Rx) si evince come una
variazione di 4,8 volte la pO2 sia in
Assenza di
cooperatività
grado di far passare lo stato di
saturazione dell’Hb dal 10% al 90%
mentre per la Mb (coeff. di Hill 1) sia
1,5
2,0
18,7
9,0
2,8
4,8
Cooperatività
3,5
3,5
positiva da
6,0
2,1
substrato
10,0
1,6
presenza di fenomeni di cooperatività
20,0
1,3
positiva. Al contrario, fenomeni di
necessario un incremento di 81 volte.
Da quanto detto si evince come la
velocità
di
saturazione
e
di
desaturazione sia accelerata dalla
cooperatività negativa decrementano la velocità di saturazione all’aumentare della
concentrazione del ligando.
Il legarsi dell’ossigeno all’emoglobina è regolato non solo dalla cooperatività, ma anche da
interazioni allosteriche (dal greco allos, altro; steros, spazio). Si tratta di interazioni che hanno
luogo quando una particolare molecola, di piccole dimenzioni, si unisce a una proteina,
modulandone l’attività. Tali molecole sono chiamate effettori allosterici e possono essere
classificate come attivatori o come inibitori, a seconda della loro influenza sulla funzionalità della
proteina a cui si legano. La proteina la cui attività è modulata da un effettore allosterico viene
definita proteina allosterica. L’effettore allosterico non deve essere considerato come un
interruttore che accende o spegne l’attività della proteina ma piuttosto come un potenziometro in
grado di regolarne l’attività. Verranno ora presi in esame due modelli per le interazioni
allosteriche: un modello concertato (Monod-Wyman-Changeux) e uno sequenziale (Koshland,
Nèmethy e Filmer). In seguito si cercherà di chiarire la base molecolare delle interazioni
allosteriche che regolano il legarsi dell’ossigeno alle quattro subunità dell’emoglobina.
Un modello concertato dell’interazione allosterica: il modello di Monod-WymanChangeux
L'emoglobina è un tipico esempio di proteina allosterici; pertanto, ogni teoria proposta per
spiegare il comportamento delle proteine allosteriche deve essere applicabile anche ad essa. Negli
anni sessanta, J. Monod, J. Wyman e J.P. Changeux hanno presentato un modello di cinetica
enzimatica che riusciva a spiegare la curva di saturazione con l'ossigeno dell'emoglobina e di altre
proteine oligomere con caratteristiche analoghe per quanta riguarda il legame del ligando. Questo
modello si basa sulle seguenti ipotesi:
1. Le proteine allosteriche sono oligomeri Ie cui subunita occupano posizioni equivalenti nella
struttura.
2. Ogni subunità possiede uno specifico sito di legame per ciascuno dei possibili ligandi. Poichè le
subunità occupano posizioni equivalenti, i ligandi occupano anch'essi posizioni equivalenti.
3. Ciascuna subunità e limitata nella sua conformazione dal fatto di essere associata alle altre
subunità dell'oligomero.
4. L'oligomero puo esistere in due diverse conformazioni: uno stato teso, o rigido, T, che ha
un'affinità minore per il substrato, e uno stato rilassato, R. I due stati T e R sono in una
situazione di equilibrio, regolata dalla costante allosterica L:
L
R T; L = T/R
La figura mostra l'effetto di differenti valori di L sulla frazione di saturazione (Y) di una proteina
allosterica con il suo ligando primario.
Un inibitore allosterico sposta l'equilibrio tra le conformazioni R e T verso la forma T, facendo
cosi aumentare il valore di L. Un attivatore allosterico, al contrario, sposta l'equilibrio verso la
conformazione R, facendo diminuire L e aumentare la frazione di saturazione della proteina per
una data concentrazione di ligando.
5. Quando un ligando si unisce allo stato T, ha luogo un cambiamento conformazionale
concertato che provoca il passaggio allo stato R. Questo cambiamento conformazionale, a volte
detto anche transizione allosterica, coinvolge tutte le subunità della proteina. Cioè, il modello di
Monod-Wyman-Changeux ipotizza che non siano possibili conformazioni intermedie in cui
alcune subunità siano R e altre T (figura), ed è per questo che esso viene anche chiamato modello
concertato per le proteine allosteriche.
Il ligando primario di una proteina allosterica (per l'emoglobina, l'ossigeno) può unirsi
all'una o all'altra delle due conformazioni T e R. Il modello di Monod-Wyman-Changeux
ipotizza che l'affinità di una subunità per un certo ligando dipenda solo dalla particolare
conformazione di quella subunità e non dall'eventuale occupazione dei siti di legame nelle
altre subunità. Di conseguenza, sono necessarie solo due costanti di dissociazione per
specificare l'interazione tra un ligando e una proteina allosterica: una costante, KT relativa
alla dissociazione dei ligandi (S) dallo state T, e una costante, KR, relativa alla dissociazione
dei ligandi S dallo stato R:
Nel caso dell'emoglobina le due costanti di dissociazione sono molto diverse: l'ossigeno si lega,
infatti, in modo preferenziale alla conformazione R. Analogamente, in quasi tutte le proteine
allosteriche, una delle due conformazioni ha un’affinità per il ligando primario superiore a
quella dell'altra conformazione.
Il modello sequenziale dell'interazione allosterica ipotizza l'esistenza di
conformazioni intermedie
II modello concertato è piuttosto restrittivo poiché consente solo due stati conformazionali per
ogni proteina. Questa limitazione non esiste in un modello alternativo, proposto per la prima
volta nel 1925 da G. S. Adair e successivamente sviluppato da D. E. Koshland, G. Nemethy e D.
Filmer.
Il modello sequenziale dell'interazione allosterica prevede una serie di cambiamenti nelle
conformazioni delle subunità. Cioè, nel corso della transizione dallo stato tutto-T a quello tutto-R
possono formarsi conformazioni intermedie, in cui alcune subunità sono nello stato T e altre sono
nello stato R (figura).
Il modello sequenziale si fonda su tre ipotesi:
1. In assenza di ligandi la proteina esiste in una sola conformazione.
2. L'unione di un ligando a una subunità induce in questa un cambiamento conformazionale.
3. II cambiamento conformazionale influisce sull'affinità delle subunità adiacenti per i ligandi.
Si può fare un'analogia tra la transizione allosterica di un oligomero e una fila di birilli che
cadono: secondo il modello concertato essi cadono tutti in una volta; secondo il modello
sequenziale cadono in rapida successione. II modello sequenziale, che ipotizza l'esistenza di molti
più stati conformazionali di quelli previsti dal modello concertato, simula meglio la realtà, ma è
concettualmente più complesso. Per entrambi i modelli di transizione allosterica, la curva della
frazione di saturazione in funzione della concentrazione del ligando è una sigmoide. In alcuni casi
è però possibile fare una scelta tra i due modelli grazie ad una ben precisa differenza. Nel modello
di Monod-Wyman-Changeux un substrato sposta sempre l'equilibrio allosterico verso la
conformazione a cui il substrato stesso si lega preferenzialmente. Di conseguenza, secondo questo
modello, tutti gli effetti cooperativi sono positivi e non c'e posto per un meccanismo grazie al
quale l'unione del primo ligando inibisce quella del ligando successivo, un fenomeno noto come
cooperatività negativa. II modello sequenziale, invece, permette anche questo comportamento,
perchè il cambiamento conformazionale, provocato dall'interazione di un ligando con una
subunità, può indurre nelle subunità adiacenti modificazioni che sfavoriscono l'unione del
ligando con tali subunità.
La funzione dell'emoglobina comporta modificazioni conformazionali.
Il legame con l'ossigeno produce un cambiamento conformazionali nell'emoglobina.
Quando cristalli di emoglobina conservati sotto azoto vengono a contatto con l'ossigeno, si
sbriciolano perché il legame con l'ossigeno determina un rilevante cambiamento conformazionale,
sufficiente a vincere le forze che tengono assieme le molecole di emoglobina nel reticolo cristallino.
Come conseguenza, infatti, del legarsi dell'ossigeno una coppia di subunità aβ ruota di circa 15°
rispetto all'altra coppia (vedi fig.).
Si tratta di un cambiamento nella struttura quaternaria che, in parte, è di natura elettronica e, in
parte, è di natura sterica. Gli effetti elettronici sono determinati dalla struttura elettronica dello
ione Fe2+. Nella deossiemoglobina, infatti, dei sei elettroni dell'orbitale d dello ione Fe2+ quattro
sono spaiati e due formano un doppietto; sono perciò possibili legami con cinque ligandi (i
quattro atomi di azoto porfirinici e l'istidina F8). In questa situazione, l'atomo di ferro è
paramagnetico e si trova nello stato ad alto spin, che si trasforma in stato a basso spin quando
l'ossigeno si lega al gruppo eme. In questa nuova situazione gli elettroni del fèrro sono disposti in
tre doppietti e l'atomo di ferro è diamagnetico. Il passaggio dalla forma ad alto spin a quella a
basso spin, provocato dall'ossigenazione, è dovuto in parte alla repulsione fra gli elettroni
dell'ossigeno e gli elettroni spaiati dello ione Fe2+ ad alto spin. Come effetto del cambiamento dello
stato di spin, si riduce la distanza di legame tra lo ione Fe2+ e l'istidina F8. Inoltre, l'ossigenazione
rende più forti i legami fra l'atomo di ferro e gli atomi di azoto porfirinici, in quanto ne accresce il
carattere covalente.
L'ossigenazione produce anche effetti sterici che alterano la struttura dell'emoglobina. Lo ione
Fe2+, quando è nello stato ad alto spin, ha un volume atomico maggiore di quando è nello stato a
basso spin, perché i suoi quattro elettroni spaiati sono distribuiti in quattro orbitali anziché in
due. Nella deossiemoglobina, a causa della repulsione sterica fra gli atomi di azoto porfirinici e
l'istidina F8 e fra gli elettroni π della porfirina e gli orbitali dello ione Fe2+, l'atomo di ferro viene
a trovarsi al di fuori del piano della porfirina di circa 0,06 nm. In seguito all'ossigenazione, la
configurazione elettronica dello ione Fe2+ cambia ed esso si sposta verso il piano della porfirina di
circa 0,039 nm fino a una posizione che è 0,021 nm al di fuori del piano stesso. Sembra
inverosimile che un cambiamento conformazionale così piccolo possa influire da un punto di vista
biologico; eppure, questo leggero spostamento dell'atomo di ferro innesca una complessa serie di
modificazioni conformazionali, di vitale importanza sotto l'aspetto fisiologico. Nello spostarsi, lo
ione Fe2+ trascina con sé l'intera catena polipeptidica dell'emoglobina.
Cambiamenti conformazionali indotti dall'ossigenazione.
La deossiemoglobina è rappresentata in verde, l'ossiemoglobina in blu.
(Disegno di Mike Webb.) Baldwin, J.; Chothla, C. (1979) “Hemoglobin: the structural changes related to ligand binding and
its allosteric mechanism” J. Mol. Biol. 129:175-179
Al momento dell'ossigenazione, l'elica F della subunità β si avvicina all'elica H spingendo la
tirosina HC2 (vicina all'estremità C-terminale) al di fuori di una tasca in cui normalmente, viene
mantenuta tramite un legame idrogeno con il gruppo carbonilico della catena peptidica al livello
della valina FG5. Lo spostamento della tirosina HC2 costringe la valina FG5 a spostarsi,
rompendo le coppie ioniche che formano legami trasversali fra le catene polipeptidiche della
deossiemoglobina. Quando l'ossigeno si lega allo ione Fe2+ di una subunità, il conseguente
cambiamento conformazionale di quella subunità si trasmette alle altre subunità. L'emoglobina
resiste inizialmente all'ossigenazione perché le coppie ioniche conferiscono stabilità alla
deossiemoglobina. Tuttavia, il legarsi dell'ossigeno è cooperativo, perché, quando le interazioni
elettrostatiche tra le coppie ioniche in una subunità si interrompono, le altre subunità risentono
meno delle modificazioni conformazionali indotte dal formarsi del legame con l'ossigeno.
L'acido 2,3-bifosfoglicerico fa diminuire l'affinità dell'emoglobina per l’ossigeno
La curva di saturazione dell'emoglobina con l'ossigeno nel sangue intero e la curva
corrispondente in soluzioni acquose tampone non contenenti tutte le sostanze normalmente
disciolte nel sangue presentano molte differenze. Anche se le loro forme sono simili, l'emoglobina
nel sangue intero ha un'affinità per l'ossigeno minore di quella che presenta l'emoglobina in vitro,
in soluzione acquosa.
Il componente del sangue, responsabile della minore affinità dell'emoglobina per l'ossigeno, è un
effettore allosterico, l'acido 2,3-bifosfoglicerico [2,3-BPG (2,3-bisphosphoglycerate)].
Negli eritrociti la concentrazione di 2,3BPG e di emoglobina sono all'incirca uguali (4,5 mM).
La cavità centrale dell'emoglobina è tappezzata dalle catene laterali, dotate di carica positiva,
della lisina 82, dell'istidina 2, dell'istidina 143 e del residuo N-terminale di ciascuna delle due
subunità β . La disposizione di questi residui con carica positiva nella cavità centrale della
deossiemoglobina è complementare alla conformazione e alla carica del 2,3BPG.
Le due catene β sono unite al 2,3BPG mediante legami ionici che conferiscono stabilità alla
conformazione della deossi-emoglobina e diminuiscono l'affinità di questa per l'ossigeno
I cambiamenti conformazionali che hanno luogo, con l'ossigenazione rompono il sito di legame
con il 2,3BPG, sicché questo composto non può più legarsi all'ossiemoglobina. I siti di legame per
l'ossigeno e quello per il 2,3BPG sono diversi. Questo non impedisce che i legami allosterici, che
queste due sostanze stabiliscono con la proteina, si escludono a vicenda.
L'effetto Bohr influenza il legarsi dell'ossigeno con l'emoglobina
Agli inizi del secolo, Christian Bohr, fisiologo danese, padre del celebre fisico Niels Bohr, osservò
che l'anidride carbonica prodotta dal metabolismo dei tessuti fa diminuire l'affinità
dell'emoglobina per l'ossigeno (vedi fig.).
20
40
60
80
100
Figura: Effetto dell’anidride carbonica sul legame tra emoglobina e ossigeno. L’anidride carbonica prodotta dal
metabolismo dei tessuti fa diminuire la frazione di saturazione (Y) dell’emoglobina con l’ossigeno, per qualunque
valore della pressione parziale dell’ossigeno. (Disegno di Patricia Johnson.)
Nei globuli rossi del sangue l'anidride carbonica catalizza la reazione in cui, a partire da anidride
carbonica e acqua, si formano uno ione bicarbonato e un protone. La formazione di H+ determina
una diminuzione del pH all'interno dei globuli rossi.
CO2 + H2O H2CO3 HCO3- + H+
La diminuzione del pH, ancor più che la stessa anidride carbonica, fa diminuire l'affinità
dell'emoglobina per l'ossigeno. Questo fenomeno è noto come effetto Bohr. Anche se, al diminuire
del pH, la curva di saturazione con l'ossigeno si sposta verso destra, il meccanismo di legame con
l'ossigeno rimane cooperativo.
Figura: Effetto Bohr. La diminuzione del pH da 7,8 a 7,2 fa diminuire l’affinità dell'emoglobina per l'ossigeno,
spostando a destra la curva cl saturazione con l'ossigeno.
In realtà, i protoni che causano l'effetto Bohr non sono effettori allosterici dell'emoglobina, dato
che non interagiscono con un particolare sito della molecola proteica. L'effetto del pH sull’affinità
dell'emoglobina per l'ossigeno é legato al grado di protonazione di vari gruppi coinvolti in coppie
ioniche esistenti fra le subunità. Uno di questi é lo ione imidazolio del residuo di istidina Cterminale (Istidina 146) di ciascuna delle due catene β che, nella deossiemoglobina, forma una
coppia ionica con il gruppo carbossilico dell'acido aspartico 94. Il cambiamento conformazionale
che avviene in seguito all’ossigenazione spezza queste coppie ioniche e gli ioni imidazolio liberano
protoni.
Coppia ionica fra ione imidazolio e ione carbossilato nell’Hb. In ciascuna delle due catene β della
deossi Hb lo ione imidazolio del residuo di istidina 146 (l’a.a. C-terminale) è reso più stabile dal
gruppo carbossilato adiacente della catena laterale dell’acido aspartico 94, con cui forma un
legame ionico. L’ossigenazione rompe tale legame: lo ione carbossilato si allontana e si libera un
protone. Il pKa dello ione imidazolio é quindi minore nell’ossiemoglobina che nella
deossiemoglobina. Una diminuzione del pH, favorisce la formazione di coppie ioniche. Pertanto,
la diminuzione del pH favorisce la formazione della deossiemoglobina, la cui conformazione è
resa più stabile da tali coppie ioniche.
Anche i gruppi amminici N-terminali della deossiemoglobina sono coinvolti nelle coppie ioniche
la cui formazione è favorita da minori valori di pH. Esiste un equilibrio tra forma protonata e
forma neutra dell'emoglobina. Il pKa della prima è 8,0. L'equazione di Henderson-Hasselbalch
permette di calcolare il rapporto, a pH 7,4, tra i gruppi amminici N-terminali neutri (Hb-NH2) e
protonati (Hb-NH3+):
pH = pK'+log[Hb-NH2]/[Hb-NH3+] (1)
7,4 = 8,0 + log[Hb-NH2]/[Hb-NH3+] (2)
[Hb-NH2]/[Hb-NH3+] = 0,25
(3)
Una diminuzione del pH fa diminuire il rapporto tra la forma neutra e quella protonata,
favorendo così la conformazione deossi- con coppie ioniche.
Nei tessuti, dove avviene la respirazione, la concentrazione di anidride carbonica è
relativamente elevata e, pertanto, il pH è relativamente basso. Per l'effetto Bohr, che favorisce la
formazione della deossi-emoglobina, l’ossigeno è ceduto efficientemente dall’emoglobina ai
tessuti. Nei polmoni, si ha una situazione esattamente opposta: il pH più elevato favorisce la
formazione dell'ossiemoglobina.
A differenza del trasporto dell'ossigeno, il trasporto dell'anidride carbonica dai tessuti ai
polmoni è fondamentalmente un fenomeno passivo. Lo ione bicarbonato in soluzione è
trasportato mediante il sangue ai polmoni dove si ricombina con i protoni liberati
dall'emoglobina ed è eliminato con l'espirazione sotto forma di anidride carbonica. Una parte
dell'anidride carbonica è trasportata dalla stessa emoglobina. Dall'eq. (3) risulta infatti che a pH
7.4, circa il 25% dei residui N-terminali dell'emoglobina non è protonato. Questi gruppi amminici
neutri possono reagire con l'anidride carbonica formando derivati dell'acido carbamminico
(Vedi Figura)
La reazione di formazione di tali derivati è facilmente reversibile. Nei tessuti, sede di attività
metaboliche, la pressione parziale dell'anidride carbonica è relativamente elevata e, pertanto,
l'equilibrio della suddetta reazione è spostato nel senso della formazione di tali derivati. A livello
dei polmoni, dove la pressione dell'anidride carbonica è molto più bassa, l'equilibrio della
reazione si sposta in senso inverso e si libera l'anidride carbonica.
Meccanismo di trasporto della CO2 da parte dell’Hb
Il meccanismo del trasporto indiretto isoidrico della CO2, da parte dell'emoglobina è
schematizzato nella figura seguente.
A livello dei tessuti la CO2 che entra nel sangue è idratata, per azione dell’anidrasi carbonica
tissutale, a H2C03 che si dissocia in HCO3- e H+. L'aumento di H+ facilita il rilascio di O2 dalla
ossiemoglobina (HbO2-). La deossiemoglobina che si forma lega i protoni ([H+]↓
↓) sottraendoli al
mezzo.
Trasportati dal sangue venoso, HbH e HCO3- (questi ultimi salificati con Na+ e K+) arrivano ai
polmoni dove la HbH, combinandosi con l'O2, diventa HbO2- che dissocia H+. Questi si associano
con l'HCO3- formando H2CO3 mentre la HbO2- si mette in equilibrio con Na+ e K+. Il ricostituito
H2CO3, per azione dell'anidrasi carbonica polmonare, si dissocia in CO2 che viene eliminata e
H2O. Si noti che la “riserva alcalina", cioè l'insieme dei K+ e Na+, nel sangue venoso è associata
agli HCO3-, in quello arterioso alla HbO2-. Durante l'intero processo i protoni non sono mai liberi,
in quanto vengono catturati dalla HbO2- in corrispondenza dei tessuti, o dagli anioni HCO3- in
corrispondenza dei polmoni. Per questo in condizioni fisiologiche non vi è mai nel sangue
variazione dell’acidità attuale, ma soltanto una leggerissima diminuizione del pH nel sangue
venoso rispetto a quello arterioso. Processi di questo tipo che avvengono senza variazioni attuali
della concentrazione idrogenionica (pH) si chiamano isoidrici. A livello dei tessuti una buona
parte dello ione HCO3- formatosi entro gli eritrociti esce nel plasma, con contemporaneo
passaggio di ioni Cl- negli eritrociti. A livello polmonare avviene il processo inverso:
l'eliminazione di CO2 provoca una diminuzione di HCO3- negli eritrociti, con richiamo di HCO3plasmatico e fuoriuscita di Cl- dagli eritrociti. E’ questo lo "scambio dei cloruri" tra eritrociti e
plasma che accompagna il trasporto isoidrico della CO2 nel sangue.
VARIANTI FISIOLOGICHE DELL'EMOGLOBINA
Nei globuli rossi degli individui normali sono presenti diverse emoglobine a seconda delle varie
fasi dello sviluppo, e precisamente (vedi anche la Fig. 5.13):
1) la Hb-E (α2ε2) o emoglobina embrionale (HbGower-2), che è la forma prevalente di Hb nel
sangue dell'embrione nelle prime fasi dello sviluppo (4°-10° settimana);
2) la Hb-F (α2γ2) o emoglobina fetale, caratteristica del feto (3°-9° mese). La Hb-F, non legando
2,3BPG ha una affinità per 1'02 più elevata della emoglobina adulta: ciò consente il
trasferimento dell'O2 dal sangue materno a quello fetale. Nel contempo, essendo la pO2 del
sangue periferico fetale più bassa che nel sangue p1acentare (10-12 mm Hg contro 25-40),
l'Hb-F rilascia efficacemente O2 ai tessuti fetali (Fig. 5.14);
3) la Hb-A1 (α2β 2), che costituisce più del 90% dell'emoglobina totale durante la vita
postnatale (sopra i 7 mesi di vita), insieme alla Hb-A2 (α2δ2), presente nella percentuale del
3%.
Tutte le emoglobine posseggono le due catene α, mentre diversificano per le due catene non α
(β,
β, γ, ε, δ).
δ Le catene "non α" hanno tutte la stessa lunghezza (146 residui di amino acidi), ma si
differenziano per la natura di alcuni residui nella sequenza. La Figura mostra la variazione delle
catene polipeptidiche costituenti le emoglobine umane nelle varie fasi di sviluppo.
Il significato delle varianti di emoglobina va probabilmente ricercato in un adattamento alle
condizioni ambientali che si succedono nel corso della vita intra- ed extrauterina.
DERIVATI DELL’EMOGLOBINA
Metaemoglobina. La metaemoglobina, o ferriemoglobina, è il prodotto di ossidazione (non di
ossigenazione) della emoglobina. Nella metaemoglobina il ferro dell'eme è allo stato ossidato
(Fe3+) e non è capace di legare 1'O2. La metaemoglobina è quindi inefficiente nel trasporto
dell'O2. La formazione di metaemoglobina è accompagnata da quella dell'anione superossido O2•-,
specie radicalica dell'O2.
Nell'uomo adulto normale la metaemoglobina è presente nella percentuale del 1,7%
dell'emoglobina totale. Una più elevata quantità di metaemoglobina (metaemoglobinemia) può
conseguire o ad aumentata formazione di metaemoglobina (come ad esempio dopo
somministrazione di certi farmaci: antipirina, fenacetina, ecc.), o a sua diminuita riduzione da
parte dei sistemi enzimatici, presenti negli eritrociti, normalmente addetti alla riconversione della
metaemoglobina in emoglobina desossigenata (vedi figura).
Questi sono imperniati sull'azione della metaemoglobina riduttasi che provvede alla riduzione
della metaemoglobina a spese del NADH(H+), e della superossido dismutasi ed enzimi ancillari,
che rimuovono l'anione superossido, con generazione di O2 molecolare, a spese del NADPH(H+).
La metaemoglobina riduttasi è deficitaria in una malattia ereditaria, la metaemoglobinemia
familiare, nella quale la percentuale di metaemoglobina raggiunge il 30-40%, il che comporta,
episodicamente, insufficienza respiratoria e cianosi.
Carbossiemoglobina o carbonilemoglobina (Hb-CO). L'emoglobina lega l'ossido di carbonio (CO)
con una affinità 250 volte superiore a quella per l'O2, Legandosi con l'emoglobina in
corrispondenza del Fe2+ dell'eme, il CO impedisce all'O2 di legarsi all'emoglobina, di qui la sua
elevata tossicità. Anche legando si ad uno solo dei 4 gruppi eme dell'emoglobina, il CO diminuisce
notevolmente l'affinità per l'O2 dei rimanenti 3. Per questa ragione la tossicità del CO è molto più
elevata di quanto sia prevedibile in base alla sua concentrazione nel sangue. Si spiega così perché
la semisaturazione di CO nel sangue è mortale, mentre la disponibilità del 50% di emoglobina,
come si può avere nelle anemie, non compromette affatto la vita.
Cianometaemoglobina. L'azione tossica micidiale del cianuro (CN-) è dovuta alla sua capacità di
legarsi alla citocromo ossidasi bloccando così la respirazione mitocondriale. Il cianuro non
reagisce con l'emoglobina ma si combina con grande facilità con la metaemoglobina per formare
la cianometaemoglobina. Data la elevata affinità del cianuro per la metaemoglobina e la scarsa
tossicità della cianometaemoglobina, nella intossicazione da cianuro si cerca di trasferire questo
tossico dalla citocromo ossidasi alla metaemoglobina, favorendone la formazione dalla
emoglobina per somministrazione endovenosa di sodio nitrato. Contemporaneamente si
somministra anche sodio tiosolfato che reagisce con il cianuro per formare tiocianato, prodotto
non tossico e facilmente eliminabile attraverso i reni.
EMOGLOBINE ATIPICHE
Si tratta di emoglobine anomale nella loro struttura primaria e soprapprimaria (malattie
molecolari), a causa di mutazioni dei geni che le codificano. Le più tipiche sono:
1. Emoglobina S
2. Emoglobina M
3. Emoglobina Riversale-Bronx
4. Talassemie
Emoglobina S (Hb-S). Questa denominazione deriva dalla forma a falce (falce = sickle, donde la
sigla S) che assumono gli eritrociti degli individui che ne sono affetti quando siano esposti a basse
tensioni di O2. La emoglobina S è costituita da due catene α normali e da due catene β modificate
nella struttura primaria per un errore genetico. Nella posizione 6 a partire dall'amino acido Nterminale contengono infatti un residuo di valina al posto di un residuo di acido glutammico:
α2β 2val6. La sostituzione di un residuo apolare (valina) ad un residuo polare (acido glutammico),
localizzato sulla superficie dell'emoglobina, impartisce all’emoglobina S proprietà elettroforetiche
diverse da quelle della emoglobina normale. Possedendo infatti due cariche negative in meno la
Hb-S migra più lentamente verso il polo positivo (vedi figura).
Anche la solubilità della Hb-S allo stato deossigenato è notevolmente diminuita (25 volte meno
solubile della forma deossigenata della Hb-A) a causa di una interazione idrofobica fra valina
terminale di una delle due catene β e la valina-6 dell'altra. La polimerizzazione che ne deriva da
luogo ad un precipitato fibroso (si parla dell'Hb-S come di emoglobina “appiccicosa”) che induce
la deformazione a falce del globulo rosso.
I globuli rossi falciformi hanno una vita media più breve dei normali, sono più facilmente
emolizzabili e agglutinabili nei piccoli vasi che ne possono essere ostruiti, creando piccole zone
ischemicbe. Gli eritrociti falciformi sono facilmente fagocitati dai reticoli endoteliali e rimossi dal
circolo, donde lo stato anemico (anemia).
Il decorso cronico della malattia é costellato da crisi accessuali cianotiche, che si manifestano
allorché i pazienti si trovino esposti a relativamente basse tensioni di O2, per esempio dopo sforzo
prolungato.
L'anemia falciforme è una malattia ereditaria, che si esprime in forma conclamata solo negli
omozigoti, vale a dire nei figli che ricevono il gene anomalo da entrambi i genitori. Negli omozigoti
la Hb-S é la sola forma di emoglobina presente negli eritrociti. Gli omozigoti non vivono
generalmente oltre l'età di 25 anni. Gli eterozigoti, cioè i figli che ricevono il gene anomalo da uno
solo dei genitori, posseggono il 50% di Hb-S ed il 50% di Hb-A. In essi l'anemia falciforme è
asintomatica, salvo crisi sporadiche che possono verificarsi in condizioni di bassa tensione di O2:
maschio falciforme (sickle cell trait).
L'anemia falciforme è molto comune nelle popolazioni africane. Tale elevata percentuale viene
raggiunta nelle zone malariche. Si ritiene infatti che l'anemia falciforme costituisca un
adattamento difensivo del globulo rosso contro il Plasmodium falciparum. In effetti una fase del
ciclo vitale di questo parassita si svolge nei globuli rossi: la emolisi immediata dei globuli rossi
falciformi, allorché vengono invasi dal parassita. ne interrompe il ciclo vitale.
Emoglobina M. L’istidina prossimale all'eme, in entrambe le catene α e β è sostituita da tirosina,
che si lega con legame coordinativo al ferro dell’eme stabilizzandolo nella forma ferrica (Fe3+). Da
ciò la sigla M, iniziale di Metaemoglobina. In tale condizione l’eme non può legare l'O2 e la
malattia è infatti letale per gli omozigoti. L’anemia da emoglobina M si manifesta con cianosi, più
o meno intensa ed estesa, dovuta a carenza di O2 nei piccoli vasi dei tessuti periferici.
Emoglobina Riverdale-Bronx. In questa emoglobina un residuo di glicina delle catene β , viene
sostituito da uno di arginina, molto più ingombrante. La struttura terziaria del protomero ne
viene deformata in modo tale da non essere più in grado di tener legato l'eme. Così si spiega come
l’emoglobina Riversale-Bronx possegga 2 o 3, ma non 4 gruppi eme.
Talassemie. Sono emoglobinopatie causate da errori genetici che si traducono nella difettosa
sintesi non di un solo aminoacido -vedi emoglobine atipiche- ma di una o più catene
polipeptidiche (subunità) costitutive. Si distinguono in talassemie α e β a seconda che il difetto sia
a carico della subunità α o β .
Le talassemie α sono dovute a delezione di uno o più geni codificanti le subunità α. I due geni
codificanti le subunità α sono localizzati, uno adiacente all'altro, nel cromosoma 16, per cui un
soggetto normale, diploide, ne possiede 4 copie. Se di queste ne manca una si ha la talassemia α-l,
se ne mancano due la talassemia α-2. Entrambi i casi comportano una modesta anemia. Quando
invece mancano 3 dei 4 geni, e conseguentemente la subunità α è pressoché assente, si ha
produzione di un tetramero β 4. Se tutti i geni sono deleti, la malattia è letale.
Le talassemie β sono prodotte da delezione dei geni che codificano le subunità β o alla incapacità
di essere espressi. Nella variante omozigote, cioè nella talassemia β , nella quale tutte le subunità
sono assenti si ha la produzione compensatoria di subunità γ che continuano ad essere
sintetizzate anche dopo il parto. Si ha così emoglobina fetale (α
α2γ2), la cui produzione continua
fino alla fanciullezza, seguita sempre dalla morte. Le emoglobine delle talassemie β analogamente
alla emoglobina fetale possono trasportare l'ossigeno, ma non sono sede dell'effetto Bohr e quindi
insensibili all'effetto allosterico degli H+. Pertanto l'affinità per l'O2 è aumentata ed il rilascio
dell'O2 ai tessuti è insufficiente.
Oltre alle numerose emoglobinopatie associate ad uno stato patologico più o meno grave, sono
possibili alterazioni della molecola dell'emoglobina che, non comportando alcuna disfunzione
della molecola. si definiscono "silenti". Ciò si verifica quando l'anomalia, genetica implica la
sostituzione di amino acidi non essenziali per la funzionalità dell'emoglobina.
BIOSINTESI DELL’EME
Sia nei batteri che negli animali i gruppi prostetici dell’eme dell’emoglobina, della mioglobina,
del citocromo c, delle per ossidasi, della catalasi e del sistema policiclico corrinico della vitamina
B12, derivano tutti dalla glicina e dal succinil-CoA.
Nella biosintesi dell’eme, la reazione che determina la velocità dell’intero processo è la
condensazione della glicina con il succinil-CoA,
che avviene con la perdita di anidride carbonica. Questa reazione di condensazione è
catalizzata dalla δ-aminolevulinico sintetasi, PLP dipendente, presente nei mitocondri. Questo
enzima è controllato dall’eme; l’eme, infatti, è in grado sia di inibire allostericamente
quest’attività enzimatica sia di reprimerne la sintesi.
La successiva tappa biosintetica prevede la condensazione di due molecole di acido δaminolevulinico con sintesi di porfobilinogeno (Vedi schema seguente). Tale reazione è catalizzata
dalla δ-aminolevulinico deidratasi.
Questa reazione è sensibile al piombo e in caso di avvelenamento da Pb si verificherà la
eliminazione, con le urine, di grandi quantità di acido δ-aminolevulinico.
Dalla successiva condensazione di quattro molecole di porfobilinogeno, si produrrà il
tetrapirrilmetano. La reazione è catalizzata dalla Uroporfobilinogeno I-sintetasi.
Quando è presente soltanto questo enzima (Uroporfobilinogeno I-sintetasi) il tetrapirrilmetano
ciclizza dando l’uroporfobilinogeno I che possiede doppi legami simmetrici rispetto ai piani di
simmetria della molecola e gruppi acetato e proprionato al centro della molecola (vedi schema
successivo).
Affinché si sintetizzi l’uroporfobilinogeno III, che è il vero precursore dell’eme,
l’Uroporfobilinogeno I-sintetasi deve agire in presenza dell’Uroporfobilinogeno III-cosintetasi
(Vedi schema seguente).
La cosintetasi tautomerizza i doppi legami dell’anello D del tetrapirrilmetano e la successiva
ciclizzazione da origine ad un composto asimmetrico, le cui catene di acetato e propionato non
sono disposte simmetricamente rispetto al centro della molecola.
L’Uroporfibilinogeno III verrà trasformato in protoporfirina IX, il diretto precursore
dell’eme. Nella prima reazione, per decarbossilazione dei gruppi acetato in gruppi metinici, si
origina il coproporfobilinogeno III. La reazione è catalizzata dall’Uroporfobilinogeno
decarbossilasi.
Successivamente, i gruppi proponici degli anelli A e B verranno convertiti in gruppi vinilici ad
opera della Coproporfobilinogeno ossidasi, con formazione del Protoporfirinogeno IX. Infine i
quattro legami metilenici, che uniscono gli anelli pirrolici, verranno ossidati in gruppi metenici
dalla Protoporfirinogeno ossidasi con produzione della Protoporfirina IX.
A tal punto una Ferrochelatasi inserirà lo ione ferroso (Fe2+) nella Protoporfirina IX formando
l’eme.
Gli errori congeniti del metabolismo dell'eme sono noti complessivamente come porfirie. Ne
esistono tre classi. Nella porfiria congenita eritropoietica, la concentrazione dell'uroporfirinogeno
III-cosintetasi è pari a circa un terzo di quella normale. Di conseguenza, vengono sintetizzate
notevoli quantità di uroporfirina I, che sono accumulate nei tessuti e, alla fine, eliminate con le
urine e con le feci. Gli individui affetti emettono un'urina rossa e i loro denti sono fluorescenti alla
luce ultravioletta. Inoltre, soffrono di una grave fotosensibilità cutanea, spesso debilitante.
Questa malattia ereditaria è trasmessa come carattere autosomico recessivo.
La seconda classe di porfiria ereditaria comprende la protoporfiria eritropoietica, che ha sintomi
analoghi, ma generalmente più lievi, della porfiria congenita eritropoietica. Pare sia dovuta a una
parziale deficienza della ferrochelatasi ed è trasmessa come carattere autosomico dominante.
La porfiria intermittente acuta, che rappresenta la terza classe, è una malattia autosomica
dominante, che può provocare forti dolori addominali e disfunzioni neurologiche. Dal punta di
vista biochimico, è caratterizzata da un incremento nell'escrezione urinaria dell'acido δamminolevulinico e di porfobilinogeno. Un tempo si riteneva che fosse dovuta a un difetto
primario nella regolazione della δ-amminolevulinico-sintetasi, ma studi più recenti hanno
dimostrato che il difetto primario e rappresentato, invece, dalla parziale (50%) deficienza della
uroporfirinogeno I-sintetasi. Ne risulta uno stato di relativa carenza di eme e una mancanza di
inibizione retrograda della δ-amminolevulinico-sintetasi da parte dell'eme, il che conduce a un
incremento della sintesi di acido δ-amminolevulinico e di porfobilinogeno per compensare il
blocco parziale della reazione catalizzata dall'uroporfirinogeno I-sintetasi nella biosintesi dell'eme.
La maggior parte degli individui affetti da porfiria intermittente acuta sono asintomatici fino a
quando non assumono farmaci come estrogeni, sulfamidici, barbiturici e altre sostanze che
influiscono sulla sintesi dell'acido δ-amminolevulinico.