Renato Borgatti
Neuropsichiatra infanzia e adolescenza
IRCCS E. Medea.
Bosisio Parini - Lecco
I.R.C.C.S. “Eugenio Medea”
Bosisio Parini (Lecco)
2
Indice
• Cervelletto
• Corpo Calloso
Note introduttive anatomia e fisiologia
Clinica neuropsichiatrica
Diagnosi prenatale e outcome neuro-evolutivo
3
Anatomia della fossa cranica posteriore
Fossa Cranica Posteriore =
(Brainstem)
Mesencefalo (Midbrain)
+
25 giorno
37 giorno
Rombencefalo (Hindbrain):
•Cervelletto (rombomero1)
•Ponte (metencefalo)
•Bulbo (mielencefalo)
4
Sviluppo embriologico complesso
Inizia alla 3° settimana di gestazione dopo la chiusura del tubo neurale quando le
vescicole primarie (prosencefalo, mesencefalo e romboencefalo) si formano lungo
l’asse antero-posteriore del cervello in via di sviluppo. Prosegue fino al 20° mese
di vita postnatale.
5
6
Tra le strutture accolte nella fossa cranica posteriore il cervelletto è
quello che si sviluppa più tardivamente e molto lentamente secondo
un processo che va dal 3-4 mese di gestazione (il verme) fino ai 20
mesi di vita post natale
A ciò consegue una elevata
VULNERABILITA’ perché
diversi agenti, su base genetica
e/o acquisita possono
comprometterne lo sviluppo.
Ne deriva che PATOLOGIE CEREBELLARI (malformative o
non) possono essere il risultato di una precoce distruzione o di
un’alterata programmazione dello sviluppo.
7
Il cervelletto è organizzato in strati e circuiti che gli consentono un
continuo controllo delle diverse funzioni a cui è deputato.
Le connessioni cervelletto si attuano
mediante fibre in entrata ed in uscita
che passano per le coppie di peduncoli
e decorrono con varia direzione nel
tronco encefalico. Le fibre afferenti al
cervelletto hanno origine da elementi
funicolari del midollo spinale o da
nuclei del tronco dell'encefalo od infine
da nuclei di nervi cranici.
Le fibre efferenti provengono dai nuclei profondi che a loro volta
ricevono fibre da vari territori della corteccia cerebellare. Sono
dirette alla periferia (vie cerebello-spinali) ai nuclei del
troncoencefalo e alla corteccia cerebrale.
Organizzazione Funzionale
Le funzioni attribuite al cervelletto si esplicano in rete con
aree corticali (associative) e con strutture sottocorticali
(talamo, nuclei della base, ponte)
Questo sistema di rete controlla le sequenze temporali con
cui le diverse strutture entrano in gioco (dysmetria of
thought) e la velocità di esecuzione (quindi l’efficacia).
Si tratta quindi di una funzione di controllo di competenze
allocate in altre sedi
Funzioni Cerebellari
Motorie
Non motorie
Postura e Tono muscolare
Funzioni esecutive
Equilibrio
Apprendimento
Diadococinesi
Memoria
Coordinazione motoria e metria
Attenzione
Motilità oculare
Competenze visuo-spaziali
Fonazione e deglutizione
Linguaggio
Affettività e comportamento
The cerebellar cognitive affective
syndrome.
Schmahmann JD, Sherman JC
Brain 1998; 121: 561–79.
Vengono descritti adulti andati incontro ad una
lesione acquisita del cervelletto che presentano
un complesso pattern comportamentale
denominato
“Cerebellar Cognitive Affective Syndrome”
(CCAS)
caratterizzato da una generale riduzione delle
funzioni cognitive associato a specifici deficit
neuropsicologici.
In particolare la CCAS si caratterizza per:
1. Compromissione delle funzioni esecutive:
deficit di pianificazione, “set-shifting”, ragionamento
astratto, fluenza verbale, memoria di lavoro;
2. Compromissione delle competenze spaziali
associata a perseverazione, inattenzione, povertà
nell’organizzazione e nella memoria visuo-spaziale;
3. Disordini del linguaggio e della comunicazione
come disprosodia, agrammatismi e anomie;
4. Modificazioni della personalità
con fluttuazioni dello stato affettivo, comportamenti
inappropriati e disinibiti
Dan Doherty, Kathleen J Millen, A James Barkovich Lancet Neurol 2013;
Malformazioni cerebellari e
della fossa cranica posteriore
Tasso stimato di incidenza:
> 1/5000 nati vivi
Tasso stimato di interruzione di gravidanza:
fino all’80%
Tasso stimato di mortalità precoce:
10‐66%
Tasso stimato di disabilità tra i sopravvissuti:
45‐100%
Valente E. et al
2013
Quadri clinici associati a
malformazioni cerebellari isolate:
la nostra esperienza.
La casistica
27 soggetti (17 maschi)
n.1 Agenesia cerebellare
completa
n.5 Agenesia isolata del
verme
n. 17 Ipoplasia cerebellare
diffusa
n.4 Agenesia/displasia
emisferica (uno o due)
Ritardo Mentale:
20/27 (74%) presentano ritardo mentale
10 profondo
4 medio
6 lieve
5/27 (19%) presentano livello cognitivo borderline
2/27 (7%) hanno QI nella norma
Agenesia del verme: ritardo mentale profondo e DGS
Ipoplasia Verme ed Emisferi: evoluzione molto varia (ritardo da
profondo a lieve)
Lesioni solo emisferiche: ritardo lieve o border o QI normale ma
deficit nps specifici (deficit visuo-spaziali e/o delle funzioni esecutive)
Linguaggio:
Tutti i soggetti studiati presentano una compromissione del
linguaggio (seppure variabile per entità e caratteristiche)
In 2 casi il linguaggio è assente in 5 casi limitato a poche
singole parole (agenesie del verme)
Nei rimanenti interessamento meno grave (più deficitarie le
performance rispetto al verbale)
I deficit del linguaggio riguardano sia la comprensione che
l’espressione
Competenze affettive e relazionali
10/27 (37%) presentano un DGS (tutti i soggetti con agenesia del
verme; 1/3 di quelli con ipoplasia del verme e degli emisferi)
2 soggetti (un soggetto con iplopasia del verme e degli emisferi
;uno con agenesia dell’emisfero destro) presentano un quadro di
ADHD
In un caso (con iplopasia del verme e degli emisferi) quadro clinico
compatibile con diagnosi di disturbo ossessivo compulsivo
In tutti i casi rimanenti, pur non evidenziandosi un franco quadro
psicopatologico è possibile osservare fragilità emotiva, rigidità del
comportamento, note d’ansia e tono dell’umore depresso tra loro
variamente associati.
Sviluppo ed evoluzione nel tempo
In tutti i casi esaminati è presente un ritardo dello sviluppo.
In generale l’area motoria è quella che presenta il miglior
andamento evolutivo.
Nel tempo si osserva sempre una lenta ma continua evoluzione
con acquisizione di competenze anche in età più avanzata
Unica eccezione rispetto ad una buona evoluzione è rappresentata
dai soggetti con DGS i quali si presentano con un quadro di
estrema gravità destinato a rimanere invariato nel tempo
Congenital cerebellar cognitive
affective syndrome (1)
1. Malformazioni a carico del cervelletto non determinano solo deficit
motori ma anche un interessamento delle funzioni cognitive e del
comportamento confermando il quadro di CCAS.
2. Una grave ritardo nelle prime fasi dello sviluppo non preclude ad
un successivo miglioramento anche in età più avanzata.
3.
Il quadro di CCAS osservabile nei soggetti con malformazioni è
più severo e meno specifico di quanto osservabile negli adulti e
nei bambini andati incontro a lesione acquisita.
Congenital cerebellar cognitive
affective syndrome (2)
4.
La compromissione della comunicazione e del comportamento
è tanto più grave tanto più sono interessate le strutture
filogeneticamente più antiche
5.
Deficit neuropsicologici specifici (competenze visuo-spaziali;
compiti di pianificazione; abilità di problem-solving) si
riscontrano nelle lesioni emisferiche senza chiara distinzione di
lato
6.
La compromissione dell’eloquio (disartria) e del linguaggio
(fluenza verbale, agrammatismi) è sempre presente anche se di
entità minore rispetto ai deficit visuospaziali
MRI fetali : 20 ipoplasia isolata
MRI post-natale
12 confermata
2 malformazioni extra
6 normali (1 autistico)
Strutture Commessurali
Nei mammiferi placentati si
distinguono 5 commessure
• Corpo Calloso
• Commessura Anteriore
• Commessura Ippocampale
• Commessura Abenulare
• Commessura Posteriore
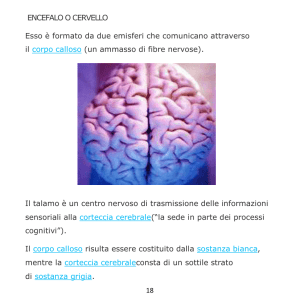
Il Corpo Calloso rappresenta il maggiore insieme di fibre
nervose nell’encefalo dei mammiferi. Nell’uomo esso è costituito da
circa 2 milioni di assoni paria a circa il 2-3% di tutte le
fibre nervose della corteccia.
Anatomia
genu
body
rostrum
splenium
Il Corpo Calloso è topograficamente organizzato: la porzione anteriore connette le regioni più anteriori
della corteccia (aree associative prefrontali, area premotoria e motoria supplementare, parietale inferiore ed anteriore)
mentre la porzione più posteriore connette le aree associative più posteriori dei lobi temporali e parietali e il lobo
occipitale.
Fisiologia
La principale funzione del CC è la
coordinazione ed il trasferimento di
informazioni tra i due emisferi.
Le connessioni sono sia di tipo
inibitorio (consentendo ai due
emisferi di inibirsi l’un l’atro e
funzionare così in modo indipendente)
che di tipo eccitatorio (consentendo
di integrare le informazioni tra i due
emisferi). Queste ultime sono
prevalenti
I due emisferi cerebrali pur essendo organizzati in aree simili
presentano una diversa organizzazione intrinseca cui consegue una
specifica specializzazione emisferica .
Questa si realizza appieno solo in presenza del corpo calloso. Una
sua assenza congenita determina una ridondanza funzionale tra gli
emisferi
Il CC consente il continuo scambio di informazioni tra i due
emisferi indispensabile per un corretto funzionamento del nostro
sistema nervoso centrale
Una sezione del corpo calloso determina una particolare condizione
neuropsicologica definita : sindrome da disconnessione
Soggetti nati senza tutto il corpo calloso (agenesia totale)
o senza una sua parte (agenesia parziale) NON presentano
il classico quadro da disconnessione ma possono
presentare diversi gradi di ritardo mentale o specifici
deficit neuropsicologici.
In generale è stato dimostrato che in tutti i soggetti con l’assenza
dalla nascita del cc la comunicazione tra i due emisferi viene
conservata attraverso vie vicarianti comunque meno efficaci del
corpo calloso
32
2006
2014
63 pazienti
162 pazienti
AGENESIE DEL CORPO CALLOSO
No lesioni acquisite
No CC dismorfici
•
anamnesi positiva per sofferenza,
prematurità
•
RM indicativa di lesioni acquisite
asfittiche
•
Diagnosi di malattie metaboliche,
degenerative, …
SI
agenesie complete o parziali
Agenesia totale: cc completamente assente
Agenesia parziale: almeno una regione,
ma non tutto il cc, mancante.
NO
dismorfismi
CC dismorfico: normale estensione antero-posteriore ma forma
irregolare: più sottile (ipoplasico, nastiforme) o più spesso (ipertrofico)
35
Agenesie del Corpo Calloso (ACC)
162 soggetti
2/3
1/3
Sindromici
112 (69%)
Isolati
57(51 %)
Complete
31 (54%)
Parziali
26 (46%)
Non Sindromici
50 (31%)
Plus
55 (49%)
Complete
25 (46%)
Parziali
30 (54%)
Isolati
27 (54 %)
Complete
19 (70%)
Plus
23 (46 %)
Parziali
8 (30%)
Complete
9 (39 %)
Parziali
14 (61%)
ACC sindromici: malformazioni extra SNC
Anomalies involving:
number
%
Cranium
72/112
64
Ocular region
98/112
88
External ear anomalies
56112
50
Oral region and palate
44/112
40
Heart
39/112
34
Genitalia
21/112
19
Urinary apparatus
9/112
8
Minor skeletal
47/112
42
Skin and adnexa
3/112
3
Specific syndromes associated to Agenesis of Corpus Callosum
Diagnosis
Number of cases
type of ACC
ACC frequency
Sotos syndrome
3
partial and complete
frequent
Tuberous sclerosis
2
complete
occasional
Acrocallosal syndrome
2
complete
mandatory
Facio-auriculo-vertebral syndrome
2
partial and complete
occasional
Prader-Willi syndrome
1
complete
not previously described
Aicardì syndrome
9
complete
mandatory/ very frequent
Kabuki syndrome
1
partial
occasional
Malpuech syndrome
1
partial
occasional
Winter-Baraitser syndrome
1
partial
occasional
FG syndrome
2
partial
frequent
ARX syndrome
1
partial
frequent
Wolf-hirsprung syndrome
1
partial
occasional
Megalocornea-MR syndrome
1
partial
not previously described
Syndromic craniostenosis
1
partial
occasional
Neurofibromatosis 1
1
partial
occasional
De Morsier syndrome
2
partial
frequent
Seckel syndrome
1
partial
occasional
Joubert syndrome
1
partial
occasional
Opitz trigonecephaly syndrome
1
partial
occasional
Holoprosencephaly-ectrodactyly-cleft-lip association
1
partial
occasional
Ito hypomelanosis
1
complete
occasional
L1 Syndrome
1
partial
mandatory
ACC SINDROMICI
DATI CLINICI
Quadri sindromici clinicamente definiti si sono riscontrati nel
33% (37/112 pazienti sindromici) diagnosi confermata da test
genetico quando possibile.
La sindrome di Aicardì rappresenta la condizione più
frequentemente riscontrata (9 pazienti)
DATI GENETICI
L’esame del cariotipo ha evidenziato riarrangiamenti
cromosomici in 17 casi (10% -17/162)
Il cariotipo molecolare (CGH-array) ha evidenziato microriarrangiamenti in 19 casi (17% -19/108) in 11 diagnostici, in 8 di
significato incerto.
ACC PLUS 39
Agenesie del Corpo Calloso (ACC)
162 soggetti
Sindromici
112 (69%)
Isolati
57(51 %)
Complete
31 (54%)
Parziali
26 (46%)
Non Sindromici
50 (31%)
Plus
55 (49%)
Complete
25 (46%)
Parziali
30 (54%)
Isolati
27 (54 %)
Complete
19 (70%)
Plus
23 (46 %)
Parziali
8 (30%)
Complete
9 (39 %)
Parziali
14 (61%)
Malformazioni cerebrali associate sono più frequenti in ACC parziale (57%) rispetto ad
ACC completa (43%) mentre non differiscono tra sindromici e non sindromici
ACC PLUS
Nei soggetti con ACC completa si osserva una
prevalenza di malformazioni sovratentoriali (65%), in
particolare malformazioni dello sviluppo corticale.
Dato, questo, confermato dalla più recente letteratura.
Al contrario nei soggetti con ACC parziale le
malformazioni del brainstem (cervelletto e tronco)
sono maggiormente rappresentate (56% ).
RITARDO MENTALE
Ritardo dello sviluppo psicomotorio e Ritardo mentale si
riscontrano nel 90%
Tutti i pazienti sindromici presentano ritardo mentale
Ritardo mentale è presente anche soggetti non sindromici (64 %)
con diversa distribuzione a seconda della presenza di malformazioni
cerebrali associate.
(93%) con ACC plus vs (43%) con ACC isolata
I soggetti non sindromici con ACC isolata presentano il miglior
profilo di funzionamento e tra questi quelli con ACC completa
appaiono essere meno compromessi
RITARDO MENTALE
12/27 (44%) soggetti non sindromici con ACC isolata presentano
un QI normale (8 di 12) o borderline (4 di 12).
10/12 (83%) presentano ACC completa
Il profilo cognitivo è sempre disarmonico.
Una discrepanza tra il punteggio di performance e verbale >= 10
punti è stato osservato in 9 casi (tutti soggetti con QI borderline) a
causa di un disturbo del linguaggio.
Meno frequentemente si osservano deficit di memoria (uguale
compromissione) tra la memoria verbale e visuospaziale.
Difficoltà negli apprendimenti scolastici sono presenti in tutti i
soggetti
Patient
Global
intellectual
functioning
FIQ VIQ PIQ
age
(years)
Attention
Visuospatial
memory
Verbal
memory
Visual perceptual skills,
motor and visuospatial
and graphic skills
Language
Academic
Performances
reading writing
calculation
ACC type
handed
9
C
6
P
R
110
113
104
n
+/++
n
b
n
n
n
n
109
114
103
n
+/b
n
b
n
nt
nt
nt
R
10
C
R
102
94
108
n
n
+
n
b
+
+
b
10
C
R
92
89
98
n
n
n
b
b
++
++
b
16
P
L
86
96
78
n
+/b
n
+
b
+
+
b
5
C
R
102
95
109
b
nt
+
n
b
nt
nt
nt
12
C
L 95
90
100
n
n
n
n
b
n
b
n
20 C
R
72
52
119
n
n
+/++
n
++
n
b
n
10 C
R
75
72
83
+
++
+
b
++
+
+
+
11 C
L
70
60
70
+
+
+/++
+
+
++
++
++
13 C
R
71
61
87
n
+
b
++
++
+/++
+/++
++
DISTURBI MOTORI
Disturbi motori si riscontrano più frequentemente nel gruppo dei soggetti
sindromici
Tra i soggetti non sindromici quelli con ACC isolata completa sono i meno
compromessi (esame neurologico normale nel 53.8% dei casi ). La
compromissione neurologica può essere rappresentata da segni cerebellari
maggiori (dismetria, atassia, adiadococinesia), segni piramidali
(ipereccitabilità dei riflessi tendinei e segno di Babinski) oppure un
impaccio motorio globale
Nel gruppo dei soggetti non sindromici con malformazioni cerebrali
associate i disturbi motori appaiono di entità medio-grave con profilo
peggiore nei pazienti con ACC parziale (tra questi nessun soggetto presenta
esame neurologico normale; prevalentemente sono presenti ipotonia e
impaccio motorio)
Grave compromissione neurologica come tetraparesi spastica si osserva nel
55.5 % dei soggetti con ACC parziale plus e nel 28% dei soggetti con ACC
completa plus (in particolare si associano malformazioni sovratentoriali)
EPILESSIA
Epilessia presente nel 41% dei casi
Quadri severi di epilessia anche farmacoresistente si osserva più
frequentemente in pazienti sindromici con malformazioni
cerebrali associate (plus ACC) in particolare sovratentoriali
(malformazioni sviluppo corticale)
Nei pazienti non sindromici epilessia è presente nel 32%
soggetti. L’incidenza è maggiore (56.2%) in quadri con
malformazioni cerebrali associate,
Ben il 15% dei soggetti con ACC isolata presenta epilessia
Le crisi sono focali nel 46% dei pazienti e generalizzate nel 38% .
Presentano più tipologie di crisi 16% dei pazienti
EPILESSIA
• Le crisi focali sono prevalentemente di tipo motorio con
successiva generalizzazione
• I tracciati EEG presentano anomalie epilettiformi focali con
tendenza alla diffusione
• Le crisi generalizzate più frequenti sono rappresentate da spasmi
(sindrome di West), assenze con mioclonie palpebrali, crisi TC
• Le anomalie EEG comprendono ipsaritmia, ed anomalie
epilettiformi diffuse talvolta con componente focale
• Sette pazienti con ACC plus hanno presentato uno stato di male
convulsivo (5/7) e non covulsivo (2/7) almeno una volta
In sintesi:
1.
2.
3.
4.
5.
Dati riferiti a popolazione patologica; non valore
epidemiologico
Lieve prevalenza di sesso maschile (55% vs 45%), dato in
linea con la letteratura; non differenze significative di
incidenza tra ACC completa e parziale (52 e 48%
rispettivamente)
Elevata percentuale (69%) di ACC inserita in quadri
sindromici
Nei casi con malformazioni cerebrali associate (ACC plus) le
MCD sono le più frequenti
Il ritardo mentale (90%), i disturbi motori (75%) e
l’epilessia (41%) rappresentano patologie frequentemente
associate
6.
7.
8.
9.
Outcome più sfavorevole (per deficit motori, ritardo di
sviluppo ed epilessia) nei quadri sindromici e con
malformazioni cerebrali associate.
La prognosi è migliore nell’ACC completa rispetto alla
parziale
I soggetti con ACC Isolata hanno una prognosi migliore
anche se l’elevata percentuale di ritardo neuropsicomotorio
(70 %), ritardo mentale (63 %), ed epilessia (34%) osservato
nei nostri pazienti con ACC isolata consente di ipotizzare che
tale condizione rappresenti la “punta di un iceberg” associata
ad anomalie non individuabili con le tecniche di neuroimaging
al momento disponibili.
Anche i soggetti senza ritardo presentano profilo
neuropsicologico peculiare e andrebbero seguiti per favorire
un armonico sviluppo
Diagnosi prenatale di ACC: diversa prognosi?
ACC fetale isolata (Ultra Sound)
71% outcome normale; (74.3% complete; 65.5% parziali)
14% outcome borderline (14.3% complete; 6.9% parziali)
15% outcome severo ritardo (11.4% complete; 27.6 % parziali)
ACC MRI
22.5% non isolate
Prognosi nettamente peggiore
(12.5% sviluppo normale; 75% grave ritardo)
però ….
• solo 6 studi hanno valutato lo sviluppo cognitivo e
neuropsicologico con test appropriati (e non solo dati
anamnestici)
• Età del follow-up in genere molto bassa (in genere < 4
anni) e difficilmente consente di rilevare problemi di
linguaggio, di apprendimento, di comportamento che si
rendono più evidenti tardivamente. (per es. nella serie di
Ghi et al. l’epilessia, in un caso poi classificato come
grave, compare a 10 anni)
Nella serie descritta da Moutard alcune difficoltà appaiono
con gli anni: 2 di16 (12%) a 2 anni; 5 di 9 (55%) a 4 anni e 5
di 7 (71%) a 6 anni, testimoniando così l’importanza del
tempo di follow-up per poter sciogliere una prognosi.
1. Malformazioni del SNC si associano quasi
sempre a ritardi di sviluppo, disturbi neuromotori,
del linguaggio, intellettivi e ad epilessia
2. Malformazioni associate extra SNC peggiorano
largamente la prognosi
3. In presenza di lesioni del SNC nessun individuo
presenta uno sviluppo tipico
REGOLA D’ORO DELLA
NEURORIABILITAZIONE
Un intervento riabilitativo è tanto più efficace
quanto più è:
Precoce
Specifico
Intensivo
considerando la plasticità neuronale spiccatissima
nei primi mesi di vita …
Lo sviluppo del cervello umano
nei primi due anni di vita
2-4 settimane di
età: circa il 36% di
quello di un adulto
12 mesi: circa il 72%
di quello di un adulto
24 mesi: circa l’83% di
quello di un adulto
(Knickmeyer et al., 2008)
REGOLA D’ORO DELLA
NEURORIABILITAZIONE
Un intervento riabilitativo è tanto più efficace
quanto più è:
Precoce
Specifico
Intensivo
considerando la plasticità neuronale è spiccatissima
nei primi mesi di vita … la diagnostica fetale è una
importante risorsa da non sprecare
60
grazie per l’attenzione