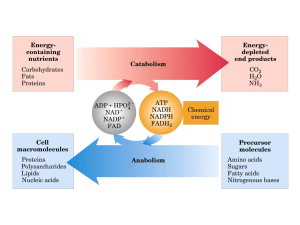
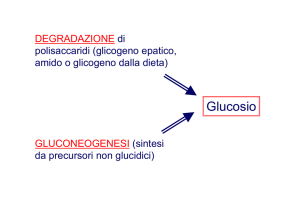
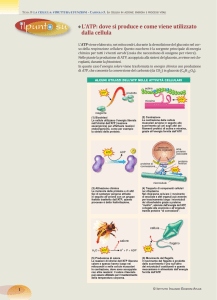
caricato da
common.user13271
Biochimica Umana - 1° Anno Scienze Motorie

BIOCHIMICA UMANA - 1°ANNO SCIENZE MOTORIE TESTO: INTRODUZIONE ALLA BIOCHIMICA DI LEHNINGER (NELSON DL – EDIZIONI ZANICHELLI) PROF: ARCONE-MASULLO CAPITOLO 0: BIOCHIMICA = CHIMICA DELLA VITA (NOTE INTRODUTTIVE) La biochimica ha come programma base: Struttura e funzione delle molecole di interesse biologico Metabolismo delle biomolecole, cioè sintesi, degradazione e regolazione Evoluzione molecolare Nonostante le diversità tra tutti gli organismi viventi, dal punto di vista biologico, tutti presentano delle similitudini, tipo: Molte importanti vie metaboliche sono conservate (tipo la glicolisi è uguale in tutti gli organismi viventi) Informazione genetica e la sua trasmissione CAPITOLO 1: CARATTERISTICHE UNIVERSALI DELLE CELLULE E BIOMOLECOLE Tutte le cellule posseggono: Una membrana plasmatica formata da molecole lipidiche e proteiche Un nucleo sede del DNA e della sua duplicazione, circondato da una doppia membrana detta membrana nucleare Citoplasma, formato da citosol e organelli Il materiale di scarto di una cellula che sta per morire può essere utilizzato per fare altre funzioni (tipo le cellule proteiche della membrana). In condizioni particolari, tipo sotto sforzo fisico, il corpo prende questi materiali anche da cellule che non stanno per morire (tipo durante uno sforzo atletico, il corpo prenderà questi materiali dalle cellule del muscolo, se lo sforzo è continuo, si preleva sempre più materiale e si rischia lo strappo del muscolo dato dall’indebolimento delle sue cellule). Le biomolecole possono essere: Semplici, cioè composti organici e inorganici formati da un numero limitato di atomi Complesse, composti formati da un numero più grande di atomi (lo sono le proteine, i polisaccaridi, gli acidi nucleici e i lipidi [i primi 3 sono detti biopolimeri]). 1 Le proteine sono formate da 20/22 diversi tipi di amminoacidi. Le basi azotate sono 5. Gli acidi grassi, glicerolo e colina sono i principali costituenti dei lipidi. Il D-glucosio (6 atomi di C) è il precursore di molti zuccheri. Essendoci un numero pressocchè infinito di combinazioni di basi, amminoacidi ecc, si può creare una molecola specifica di ogni cosa. CAPITOLO 1.1: COMPOSIZIONE CHIMICA DEGLI ORGANISMI VIVENTI La materia vivente è costituita da un numero relativamente ridotto di elementi chimici; se eliminiamo l’acqua da essi infatti (peso secco), il 98% è costituito da C, N, O, H, Ca, P, K ed S; il restante 2% è rappresentato da elementi traccia ed è comunque fondamentale per la vita anche se è formato da sostanze presenti in quantità nettamente minore (tipo lo iodio). Il composto più abbondante è ovviamente l’acqua (70% di un organismo) e quindi gli elementi più abbondanti sono H e O. Tranne ossigeno e calcio, il resto degli elementi sopra citati è poco presente nella crosta terrestre (cioè è più presente nell’insieme di tutti gli esseri viventi che sulla crosta). CAPITOLO 1.2: LA TAVOLA PERIODICA (IN SEGUITO INDICATA COME “TP”) Sulla tavola periodica sono localizzati tutti gli elementi chimici, quelli molto rappresentati e quelli di traccia sono tutti nella parte alta e hanno un basso numero di protoni e di conseguenza elettroni. Tutti gli elementi più complessi infatti, hanno una minore variabilità ed ecco perché, nell’evoluzione, sono stati “scelti” elementi più semplici. Meno di 30 elementi sono essenziali alla vita e quasi tutti hanno un numero atomico basso. Numero atomico e simbolo identificano univocamente un elemento. Per indicare un composto, è ovviamente più diffuso e usato il simbolo rispetto al numero, in quanto questo permette di indicare più precisamente una sostanza (esempio: l’acqua con l’uso dei simboli è H2O, se usassimo i numeri sarebbe indicata con 128, numero nettamente più “anonimo”). Gli elementi più abbondanti in natura sono H, C, O ed N. Numero atomico: riguarda la struttura sub-atomica, considera il numero di protoni presenti nel nucleo dell’atomo Le sostanze, sono nel gruppo che indica il numero di elettroni presenti nel loro guscio di valenza (esempio: gruppo 3 = 3 elettroni nell’orbita più esterna) CAPITOLO 1.3: L’ATOMO L’atomo è la parte più piccola della materia a mantenere le sue caratteristiche, è formato da tre particelle sub-atomiche: 2 Protoni, carichi positivamente Neutroni, privi di carica (insieme ai protoni forma il nucleo dell’atomo), si mettono tra i vari protoni evitando che questi si respingano reciprocamente data la stessa polarità Elettroni, carichi negativamente, sono quelli più mobili e variabili nella struttura, pertanto non servono a identificare un elemento, sono impiegati principalmente nella formazione dei legami; si muovono alla velocità della luce sulle orbite intorno al nucleo A distinguere gli elementi, è la composizione quantitativa di queste tre particelle, infatti i protoni, gli elettroni e i neutroni di tutti gli elementi sono materialmente uguali, è la loro quantità a definire le differenze degli elementi. Ogni atomo di una determinata sostanza, possiederà sempre lo stesso numero di sub-unità. La struttura elettrica degli atomi è fata dal numero di protoni e dal numero di elettroni; se varia il numero di protoni si avrà un cambio totale di identità dell’atomo, se varia invece il numero di elettroni si avrà un cambio di polarità. Per la legge dell’elettroneutralità, se il numero di protoni e di elettroni è uguale, un elemento sarà elettricamente neutro. Ogni particella occupa un determinato spazio (un elettrone per esempio, non si troverà mai nello spazio di un protone), gli elettroni occupano uno spazio nettamente superiore rispetto a quello occupato, nel nucleo, da protoni + neutroni, e la maggior parte di questo spazio occupato dagli elettroni è uno spazio vuoto. Un elettrone se è molto lontano dal nucleo perde il suo senso di appartenenza ad esso e non subirà più nessuna forza di attrazione da questo. La struttura elettrica degli atomi si riassume con il modello atomico ad orbitali. Un orbitale atomico è definito da tre numeri quantistici (n, l, m). L’elettrone è caratterizzato da un quarto numero quantico (s) associato al moto di spin dell’elettrone. Se gli elementi hanno numero atomico basso, come sappiamo, hanno meno elettroni e quindi anche gli orbitali intorno al nucleo saranno di meno. CAPITOLO 1.4: LEGAMI CHIMICI Gli atomi tendono a combinarsi tra loro formando legami, nella formazione degli elementi conta la quantità di atomi presenti in quel momento. Per legame chimico si intende: l’insieme delle forze che tengono uniti due o più atomi, forze elettrostatiche ma soprattutto covalenti. Il legame si forma perché due atomi legati insieme, sono più stabili rispetto a quando sono isolati e corrispondono meno energia (in breve: fisicamente più stabili e con meno energia quando sono legati). Un legame quando si crea, libera energia, quando unisce due o più atomi abbassa l’energia totale del composto. Un legame si forma per permettere alle sostanze di completare i loro gusci di valenza e quindi “rispettare” la regola dell’ottetto. 3 CAPITOLO 1.5: REGOLA DELL’OTTETTO Durante la formazione di un legame, l’atomo tende a cedere, acquistare o condividere elettroni, in modo da avere nel livello (orbita) più esterno (sarebbe nel guscio di valenza) 8 elettroni (non sono sempre 8 comunque, basta che il livello più esterno sia completo per quella sostanza). I gas nobili non si combinano con nessun elemento e sono già completi. CAPITOLO 1.6: ELETTRONEGATIVITA’ L’elettronegatività è un parametro che fa capire se un atomo si lega cedendo/acquistando o mettendo in condivisione i suoi elettroni. E’ una misura relativa delle capacità di un atomo di attrarre elettroni quando prende parte ad un legame chimico. E’ un valore ricavato mettendo in contatto ogni elemento con l’ossigeno che è l’atomo più reattivo che esiste in natura, questo infatti non lo si trova mai isolato (cioè sotto forma di un singolo atomo). CAPITOLO 1.7: LEGAME IONICO Un legame ionico si forma quando c’è una grande differenza di elettronegatività tra gli atomi interessati, si instaura tra due ioni che hanno carica opposta (un anione e un catione), è una forza di natura elettrostatica. Questo tipo di legame prevede il trasferimento di elettroni al fine di equilibrare i gusci di valenza degli atomi (uno cederà elettroni e l’altro li acquisterà, determinando un catione ed un anione appunto, ricordando che gli elettroni sono uguali in tutte le sostanze e che, in natura, per la legge dell’elettroneutralità, tutte le sostanze sono neutre). Il suffisso –uro indica una sostanza ionizzata, cioè che ha perso/acquistato elettroni e non è più pura. Se un atomo ha elettronegatività maggiore rispetto all’atomo dell’altra sostanza che si vuole legare al primo, questo acquisterà l’elettrone che quello con elettronegatività minore cederà. I composti ionici sono sostanze solide a struttura cristallina e con alti punti di fusione. Se sono solide non conducono la corrente mentre sono ottimi conduttori allo stato liquido. Quando passano allo stato liquido, vengono rotti i legami che tenevano fermi gli ioni nel reticolo cristallino, conferendo mobilità a questi ultimi. Nel loro movimento, verso il catodo andranno i cationi (+), mentre verso l’anodo andranno gli anioni (-) (cioè gli ioni vanno verso i poli opposti una volta liberi dal reticolo, cioè allo stato liquido). 4 CAPITOLO 1.8: LEGAME COVALENTE Un legame covalente si ha quando l’elettronegatività tra i due atomi interessati non è tanto grande da permettere il trasferimento di elettroni (cioè il delta/differenza tra i due valori è minima) da un atomo all’altro. In questo caso, si ha la condivisione degli elettroni ottenendo un legame covalente, che poi è anche il legame più diffuso. Si forma per esempio, tra atomi della stessa sostanza (quindi differenza di elettronegatività nulla) e si indica con una “linea/stecchetta” tra i simboli dei due atomi, tipo H---H (1 linea [---] se legame singolo, 2 [=] se doppio legame, 3 linee se legame triplo) Il legame covalente tra atomi uguali, quindi della stessa sostanza, è detto puro e forma, quasi sempre, molecole apolari, perché avviene tra atomi di sostanze gassose, è bene precisare però che i legami covalenti possono avvenire in sostante in tutti e tre gli stadi della materia (solido, liquido e appunto gassoso; ricorda che se una sostanza non è solida, allora presenterà sicuramente un legame covalente). In caso di legame covalente simmetrico, quindi nel caso di atomi di sostanza uguale, gli elettroni condivisi sono esattamente al centro tra i due atomi. Si può anche avere un legame covalente eteropolare (sarebbe quello asimmetrico). Questo tipo di legame covalente, si ha tra atomi che hanno elettronegatività differente tra loro (ovviamente differenza non elevata come quella del legame ionico, ma più grande rispetto a quella del legame covalente simmetrico). Di conseguenza, gli elettroni saranno più vicini a un atomo che all’altro e non più al centro tra i due. Questo tipo di legame è un legame polarizzato e rendono polari le molecole che creano (a meno di geometrie molecolari simmetriche). Il legame covalente può essere (in base al numero di coppie di elettroni condivise): Semplice/singolo Doppio Triplo Tutti gli atomi coinvolti nel legame raggiungono la configurazione elettronica esterna stabile (regola dell’ottetto). CAPITOLO 1.9: LEGAME INTERMOLECOLARI: INTERAZIONI DEBOLI Non riguardano i composti ionici, riguardano infatti, solo le sostanze che formano molecole. Sono legami che non seguono la Legge di Coulomb ma quella di van der Waals. Sono forze più piccole rispetto a quelle di altri legami, cioè di minor intensità. Nel dettaglio, questi legami sono: Legami a idrogeno (avvengono SOLO in presenza di idrogeno (H) Interazioni tra gruppi carichi (cioè tra poli opposti, sono forze di orientazione perché pongono il polo positivo “contro” quello negativo, orientando appunto le molecole) 5 Forze di van der Walls Interazioni idrofobiche (cioè la combinazione di 2 legami detti in precedenza) Singolarmente, questi legami hanno importanza relativa (anche se dal punto di vista biologico può essere importante anche un singolo legame, vedi legami a idrogeno del DNA), assumono però importanza, sia chimica che biologica, quando li consideriamo collettivamente. Questi tipi di legami possono anche essere transitori/temporanei e conferiscono flessibilità e stabilità alle biomolecole, fattori importanti soprattutto dal punto di vista biologico. Il legame a idrogeno si può formare ogni volta che un atomo di idrogeno legato covalentemente ad un atomo fortemente elettronegativo e di piccole dimensioni (atomo di F, O [caso dell’acqua], N) si trova ad una certa distanza da un altro atomo di questo di tipo di elementi. E’ un legame che non segue, ovviamente, la legge di Coulomb ed è un legame più debole e modellabile rispetto agli altri. L’intensità del legame a idrogeno dipende anche dalla disposizione dei tre atomi considerati (cioè se sono allineati oppure no, il legame è più forte se sono orientati sullo stesso asse, questo fattore determina la solidità del legame). Questo tipo di legame, date le sue caratteristiche, è anche detto legame a ponte di idrogeno dove un atomo è accettore di questo ponte e l’altro è un donatore. CAPITOLO 1.10: INTERAZIONI DIPOLO-DIPOLO Sono interazioni deboli che coinvolgono molecole neutre, la forza di ogni dipolo è indicata dallo spessore della freccia che “collega” i due dipoli (simbologia grafica). E’ un altro tipo di legame debole tra diverse molecole, può essere un interazione tra dipoli permanenti o un interazione dipolo-dipolo indotto. CAPITOLO 2: MOLECOLE DI ACQUA Nella molecola dell’acqua, l’ossigeno è legato a due atomi di idrogeno mediante legami covalenti polarizzati, l’angolo di legame è di circa 105°. 6 A sinistra la molecola dell’acqua è rappresentata con il metodo “balls and sticks” (sfere e bastoncini), a destra invece è rappresentata con la rappresentazione di van der Walls, cioè rappresentate come spazio realmente occupato. La formula di questa molecola è H2O ma può anche essere scritta, secondo la simbologia dei legami covalenti, come H---O---H. Due molecole di acqua, si legano tra loro, con legami a idrogeno. CAPITOLO 3: LE PRINCIPALI SOSTANZE E I LORO TIPI DI LEGAMI Gli atomi delle sostanze che ci interessano maggiormente sono: Idrogeno (H), è una sostanza del 1° gruppo della TP quindi ha un solo elettrone sul livello esterno (questo dato è importante perché sono proprio questi elettroni che vanno a formare i legami) Carbonio (C), è una sostanza del 4° gruppo della TP quindi ha quattro elettroni sul livello esterno Ossigeno (O), è una sostanza del 6° gruppo della TP quindi ha sei elettroni sul livello esterno Azoto (N), è una sostanza del 5° gruppo della TP quindi ha cinque elettroni sul livello più esterno I tipi di legami che intercorrono tra queste sostanze (variano a seconda di come si dispongono gli elettroni esterni [ogni “linea” indica come sono disposti gli elettroni, ogni linea = 1 elettrone]) sono: H---, l’idrogeno infatti può formare un solo legami covalenti semplici causa il singolo elettrone presente sul livello più esterno ---O---, l’ossigeno può formare, nel caso in cui abbia questa disposizione degli elettroni, due legami covalenti singoli O=, in questo caso invece, l’ossigeno può formare un legame covalente doppio il carbonio con questa disposizione può formare quattro legami covalenti semplici; se la disposizione è =C= allora forma due legami covalenti doppi; se la disposizione invece è C≡C allora forma un singolo legame covalente triplo l’azoto con questa disposizione forma tre legami covalenti singoli; con la disposizione ---N= forma un legame doppio e uno singolo; con la disposizione N≡ forma un singolo legame covalente triplo 7 CAPITOLO 3.1: IL CARBONIO Solo con il carbonio si può fare una catena illimitata di atomi, collegandoli uno vicino all’altro anche con vari tipi di legami. Il carbonio infatti, può avere anche la seguente struttura allotropica: Il carbonio ha struttura tetraedrica (4 elettroni, legami singoli e forma classica “a diamante”). La sua configurazione elettronica è 2sp22p2. Quando non c’è ramificazione nella catena, cioè gli atomi di carbonio hanno struttura lineare, il composto è identificato dalla desinenza “-ano” (i primi 4 composti sono metano (CH4), etano (CH3CH3), propano (CH3-CH2-CH3) e butano (CH3-CH2-CH2-CH3); varia il numero di idrogeni perché per legarsi l’uno con l’altro perdono un atomo di idrogeno)). Al carbonio si lega l’idrogeno per formare gli idrocarburi. Sono definiti composi organici solo se presentano carbonio. CAPITOLO 3.2: IBRIDAZIONE Per ibridazione si intende: nello stabilire legami, gli orbitali più esterni di un atomo si combinano tra loro, questo perché sono simili energeticamente ma diversi per disposizione spaziale. Questa combinazione, che vale per tutti i tipi di atomo e avviene proprio nel momento in cui i singoli atomi si stanno combinando tra loro (un atomo singolo nello spazio infatti non presenterà mai questo fenomeno), forma degli orbitali ibridi identici energeticamente e identici come forma/disposizione nello spazio. E’ un processo che riguarda solo gli orbitali dello stesso livello energetico. Gli ibridi si dispongono nello spazio tridimensionale secondo un criterio che varia a seconda di come si legano gli atomi, e stabilisce che: Ibridazione sp3 se sono uniti con legami singoli, gli orbitali tra loro formano angoli di 109,5° Ibridazione sp2 se sono uniti con legami doppi, gli orbitali tra loro formano angoli di 120° Ibridazione sp se sono uniti con un unico legame triplo, gli orbitali tra loro formano angoli di 180° CAPITOLO 3.3: INTERAZIONE TRA CARBONIO (C) E IDROGENO (H) = IDROCARBURI Questi composti, come da titolo, sono formati solamente da carbonio e idrogeno e si identificano con la descrizione della struttura con la quale si organizzano gli atomi e con che legami ci sono tra loro. Se tutti gli atomi di “C” sono legati con legami singoli (sp3) allora il composto sarà detto alcano; se invece sono legati con legami doppi (sp2) allora sarà un alchene; se invece saranno legati con un singolo legame triplo (sp) allora il composto sarà un alchino. Siccome l’idrogeno forma sempre e 8 solo un legame singolo, causa singolo elettrone di valenza, i legami doppi e tripli si avranno per forza tra i vari atomi di carbonio. CAPITOLO 3.4: INTERAZIONE (GRUPPI FUNZIONALI) TRA CARBONIO (C) E OSSIGENO (O) Nota bene: a ogni “C” (tranne quello centrale) sono legati 3 atomi di idrogeno (uno per lato, con legami singoli). Visto che i gruppi funzionali si possono combinare tra loro, se condividono lo stesso atomo di “C” allora variano le loro proprietà, se invece sono legati a atomi di “C” diversi, mantengono le loro proprietà (cioè quelle che avrebbero da singoli). I gruppi funzionali servono per scrivere tutte le molecole. C—O—H Gruppo alcolico (presenta anche un atomo di idrogeno, se attorno a “C” mancano 1,2 o 3 idrogeni allora il gruppo sarà alcolico primario, secondario e terziario) C—O—C Gruppo etereo __________________ Gruppo carbonilico (versione generica/base, per “X” si intende una parte variabile) __________________ Gruppo aldeidico (è un carbonilico base + un “H” al posto di una delle “X”) __________________ Gruppo chetonico (è un carbonilico base + due “C” al posto delle “X”) __________________ Gruppo carbossilico (è un gruppo alcolico base [--O—H] legato al “C” centrale + un gruppo carbonilico legati allo stesso “C”) __________________ Acido carbossilico (è il comune aceto, formato da un carbossilico + un atomo di “C” alla “X”) 9 __________________ Gruppo esteri (presenti nei trigliceridi) __________________ Cheto Acido – Acido Piruvico (formato da un gruppo carbossilico + un gruppo carbonilico chetonico) __________________ Piruvato (sarebbe l’acido piruvico che si ossida e perde l’idrogeno del gruppo carbossilico [parte superiore]) __________________ Acido Lattico __________________ Lattato 10 CAPITOLO 3.5: INTERAZIONI (GRUPPI FUNZIONALI) TRA AZOTO (N) E CARBONIO (C) Gruppo amminico terziario (perché presenta “3C”) __________________ C—NH2 Gruppo amminico primario (perché presenta un solo atomo di “C”) __________________ Gruppo amminico secondario (perché presenta “2C”) __________________ X—C=N—X Gruppo imminico __________________ Gruppo ammidico CAPITOLO 3.6: INTERAZIONI (GRUPPI FUNZIONALI) CON ZOLFO (S) Nota bene: nella “X” ci deve essere un alchino, quindi sarà “CHx”. X—S—H Tialcoli X—S—X Tioeteri o solfuri (la “X” avrà basi alchiniche) __________________ Gruppo tioestereo 11 CAPITOLO INTRO-4: NOTE GENERALI DI BIOLOGIA (APPROFONDIRE NEL CASO CON IL FILE DI BIOLOGIA, SOPRATTUTTO CELLULA E SUA MEMBRANA) Dalla rottura dei legami si libera energia, questa energia verrà trasportata dall’ATP. Il glucosio è il principale carburante delle cellule ed entra in esse grazie all’insulina prodotta dal pancreas. Trigliceridi: forma di deposito degli acidi grassi Le differenze tra le varie proteine sono date dalla successione/sequenza dei 20 tipi diversi di amminoacidi che le compongono. Per attivare le funzioni della cellula serve un segnale ormonale dato appunto dagli ormoni. Questi ormoni possono essere anabolici o catabolici e arrivano alle cellule nei suoi recettori che poi leggono il segnale e lo trasmettono fino al nucleo con la trasduzione del segnale e attiva i geni necessari alla funzione richiesta (la funzione può essere la sintesi di una proteina, la replicazione del DNA ecc.). CAPITOLO INTRO-4.0.1: NOTE BASILARI DELLE PROTEINE Le differenze tra le varie proteine sono date dalla successione/sequenza dei 20 tipi diversi di amminoacidi che le compongono. Le principali sono mioglobina (la sua funzione è il trasporto intracellulare di ossigeno in cellule specializzate che hanno bisogno di quest’ultimo per fare la loro funzione al meglio) ed emoglobina (responsabile del trasporto di ossigeno nei tessuti periferici, è un tetramero con 4 sub-unità uguali a due a due). La mioglobina presenta struttura terziaria dove presenta un gruppo prostetico che lega l’ossigeno. Una proteina funziona grazie alla sua struttura tridimensionale (cioè la funzione è dettata dalla disposizione nello spazio delle varie catene polipeptidiche che la compongono). Gruppo Prostetico: parte non amminoacida della proteina Il gene che codifica una determinata proteina è situato su un locus genico preciso. Le proteine possono essere di forma: Globulare, cioè a forma di gomitolo Fibrosa, cioè a forma distesa Una proteina ha tre tipi di strutture: Primaria, singola catena di amminoacidi, da sola non ha alcuna funzione Secondaria, cioè il ripiegamento periodico di amminoacidi adiacenti, può essere ad alfa-elica o beta-foglietto Terziaria, sarebbe la disposizione finale tridimensionale delle catene che “attiva la proteina” 12 Se varia la struttura di una proteina, questa vedrà variare anche la sua funzione a causa di questo mutamento, una variazione di funzione può anche arrecare danni all’organismo (tipo anemia falciforme dovuta a mutazione della forma dell’emoglobina S). Ogni proteina assolve uno specifico compito. [APPROFONDIMENTO SULLE PROTEINE NEL CAPITOLO 5] CAPITOLO 4: GLI AMMINOACIDI – I MONOMERI DELLE PROTEINE Questa a lato, è la struttura base di un amminoacido in forma ionizzata (ionizzata infatti presenta le polarità è l’idrogeno del gruppo carbossilico viene acquisito dal gruppo amminico). Come si può vedere, i suoi costituenti/sostituenti sono: Un gruppo carbossilico COOH (nella forma ionizzata diventa COO-, è un gruppo acido perché può cedere un protone che verrà assorbito dal gruppo amminico, reazione intramolecolare) Un gruppo amminico H2N (nella forma ionizzata diventa H3N perché acquista il protone del gruppo carbossilico, reazione intramolecolare), è un gruppo basico Un atomo di idrogeno (H) legato con legame covalente singolo (una sola coppia di elettroni condivisa) al carbonio centrale Una variabile R (detta anche residuo amminoacido), questa variabile può essere sia basica che acida; può essere un anche un altro gruppo funzionale Ogni amminoacido ha almeno due gruppi funzionali. Gli amminoacidi sono molto abbondanti in natura e, oltre a comporre le proteine, possono anche svolgere altre funzioni, in questo caso si parla di amminoacidi non proteinogenesi, cioè amminoacidi che non partecipano alla costruzione delle proteine ma svolgono altre funzioni. Tutti e 20 diversi tipi di amminoacidi che compongono le proteine sono detti alfa-amminoacidi e hanno sempre (parte costante): un gruppo carbossilico (va scritto sempre nella parte alta, vedi dopo per dettagli sulla scrittura) e un gruppo amminico (va scritto sempre in posizione alfa, cioè adiacente al carbonio centrale [C2 o carbonio alfa, il primo carbonio sarà quello del gruppo carbossilico]), legati allo stesso atomo di carbonio (quello alfa appunto). La figura a lato mostra proprio la struttura di un amminoacido alfa generico (si definisce anche chirale in quanto i suoi costituenti sono tutti e quattro diversi tra loro [si suppone ovviamente che la variabile R sia diversa dagli altri tre], in questo caso, il carbonio centrale alfa è detto anche chirale (se sono uguali anche solo due costituenti ovviamente non lo è più) e si indica con un *). Tutti gli alfa-amminoacidi, esclusa la glicina, presentano questo centro chirale. Gli amminoacidi si distinguono tra loro grazie alle varie composizioni che la variabile R può assumere. 13 CAPITOLO 4.1: ENANTIOMERI Quando il carbonio centrale è chirale, la struttura tridimensionale di un amminoacido può essere scritta in due modi speculari (a specchio, non sovrapponibili) che differiscono per la sola inversione di posizione dei due costituenti “laterali”. Questi due modi vengono detti enantiomeri. Due enantiomeri presentano le stesse proprietà chimiche ma differiscono per un’unica proprietà fisica: fanno variare il piano di vibrazione della luce polarizzata in maniera perfettamente opposta. E’ bene comunque precisare che, due enantiomeri rappresentano comunque lo stesso amminoacido, varia solamente il comportamento di questi. CAPITOLO 4.2: PROIEZIONI DI FISCHER Le proiezioni di Fischer sono il modo che si usa per rappresentare gli amminoacidi (sia la struttura con lettere e linee classica, sia quella con “balls and stickes”). In queste proiezioni, l’atomo centrale di carbonio giace sul piano del foglio (piatto), i legami che si allontanano dall’osservatore vengono indicati con linee verticali (sarebbero quelli dietro nella struttura 3D, W e Z in figura), i legami che invece si dirigono verso l’osservatore sono indicati con linee orizzontali (X e Y in figura). CAPITOLO 4.3: SERIE STEREOCHIMICA Una serie stereochimica è la base per capire se gli amminoacidi sono “L” o “D”, in base a dove è collocato il secondo gruppo funzionale con più rilevanza. In una proteina infatti, è raro se non impossibile trovare degli enantiomeri, questi infatti, presentando uno spostamento di due dei due costituenti, andrebbero ad alterare la funzione della proteina oltre che a richiedere altri enzimi per essere riconosciuti. Nelle proteine infatti, si trovano solamente gli “L-alfa-amminoacidi”, cioè quelli che hanno il secondo gruppo funzionale a sinistra del carbonio alfa (al posto della X nella figura sopra tanto per capirci). Per stabilire la nomenclatura infatti, si è deciso che se il secondo gruppo principale di un amminoacido si trova a sinistra allora questo sarà un “l-amminoacido”, cioè un amminoacido con “Configurazione L”; viceversa se questo gruppo è situato a destra del carbonio alfa, in questo caso sarà un “D-amminoacido” e avrà quindi una “Configurazione D”. 14 CAPITOLO 4.3.1: COME SI SCRIVE UN AMMINOACIDO ALFA Il primo gruppo funzionale per importanza/priorità/forza ossidativa in un amminoacido è quello carbossilico o aldeidico (CHO o COOH) e va scritto sempre sopra al carbonio alfa (sarebbe il gruppo con priorità maggiore e che contiene anche il C1) Il secondo gruppo funzionale per importanza/priorità è quello amminico (NH2) e va sempre scritto alla sinistra del carbonio alfa nella “Configurazione L” (ci interessa quasi solo questo tipo di configurazione in quanto è quella presente negli amminoacidi delle proteine), o a destra in caso di “Configurazione D” (quindi in caso di un amminoacido che non fa parte delle proteine in quanto non è riconosciuto dagli enzimi che le formano) (in pratica si mette a sinistra il gruppo con priorità più alta tra quelli rimasti ed è proprio quello amminico che ha priorità più alta rispetto agli altri, anche rispetto al gruppo ossidrilico) Tutti gli altri gruppi funzionali rimasti hanno priorità minore rispetto a quello amminico e vengono scritti nel lato libero (destro in caso di “Configurazione L”, sinistro in caso di “Configurazione D”) La parte variabile R va scritta sempre in basso rispetto al carbonio centrale. Nelle proteine, questa variabile è sempre uno “L-stereosimoero”. [Esempio in figura: il secondo gruppo principale che detta la configurazione (L in questo caso) è la “X”, quello primario è la “W”, l’altro gruppo con priorità inferiore è la “Y”. Nel caso la “Y” andasse a destra e la “X” a sinistra (in caso di enantiomeri appunto) si avrebbe una configurazione D] CAPITOLO 4.4: CLASSIFICAZIONE DEGLI ALFA-AMMINOACIDI Gli alfa-amminoacidi che si trovano nelle proteine si differenziano per le diverse catene laterali (variabile R) legate all’atomo di carbonio alfa. Questa distinzione rappresenta un criterio per la loro classificazione, la variabile R infatti può essere e dare all’amminoacido la caratteristica (cioè può essere un amminoacido): Non polare (idrofobica) Polare non carico (con carica parziale, delta carica positiva o negativa) Polare carico (con carica netta) Un’altra classificazione distingue gli amminoacidi in essenziali (questi non sono sintetizzabili attraverso il metabolismo e quindi devono essere introdotti con l’alimentazione, cioè come avviene per gli acidi grassi) e non essenziali. 15 CAPITOLO 4.4.1: AMMINOACIDI NON POLARI Gli amminoacidi non polari sono (ricorda, TUTTI i seguenti amminoacidi [e quelli dei capitoli successivi, sono i 20 diversi tipi e si indicano con dei simboli] variano solo per la composizione della variabile R che sarà composta come indicato accanto al nome, il resto è uguale per tutti, cioè hanno tutti un gruppo carbossilico COOH, un gruppo amminico H2N e un atomo di idrogeno (H) legati al carbonio alfa centrale con legami covalenti singoli): Glicina, -H Alanina, -CH3 Valina, -CH(CH3)2 Leucina, -CH2CH(CH3)2 Isoleucina, -CH(CH3)CH2CH3 Metionina, -(CH3)2SCH3 Prolina CAPITOLO 4.4.2: AMMINOACIDI POLARI NON CARICHI Serina, -CH2OH Treonina, -CH(OH)CH3 Cisteina, -CH2SH Asparagina, -CH2CONH2 Glutammina, -CH2CH2CONH2 CAPITOLO 4.4.3: AMMINOACIDI POLARI CARICHI Acido Aspartico (aspartato), -CH2COOAcido Glutammico (glutammato), -CH2CH2COO- Questi due presentano una variabile R ionizzata (infatti hanno la polarità negativa sul gruppo carbossilico). Lisina, -(CH2)4NH3+ (positivo quindi basico) Istidina (basico) Arginina (presente nel pompaggio degli spermatozoi) (basico) CAPITOLO 4.4.4: AMMINOACIDI AROMATICI Fenilalanina 16 Tirosina Triptofano Questi tre amminoacidi sono anche dei marcatori che servono per quantizzare le proteine. CAPITOLO 4.5: GLI AMMINOACIDI POSSONO COMPORTARSI DA ACIDI O DA BASI Allo stato puro, gli amminoacidi sono sostanze ioniche cristalline (non ionizzate e neutre): in soluzione acquosa, i gruppi –COOH e –NH2 negli alfa-amminoacidi si trovano in forma ionizzata, cioè si ionizzano e possono comportarsi sia come una base che come un acido. Il comportamento come acido o base (quindi la carica che acquisisce il singolo gruppo) è determinato dal pH (sarebbe la concentrazione dei protoni in una soluzione; da 1 a 7 è acido, da 7 a 14 è basico, 7 è neutro) della soluzione acquosa nel quale si trovano. Nelle soluzioni acquose quindi, l’amminoacido è sempre ionizzato e diventa uno ione dipolare/zwitterione. Per forma non ionica si intende l’amminoacido non ionizzato, senza polarità ed è assente nelle soluzioni acquose. Per ogni amminoacido esiste un pH a cui predomina la forma zwitterionica (cioè carica totale=0, neutro). Uno zwitterione può comportarsi come un acido o come una base; i composti con questa doppia natura acido-base sono detti anfoliti. CAPITOLO 4.5.1: EQUILIBRIO ACIDO-BASE NEGLI ALFA-AMMINOACIDI Partendo dalla forma completamente protonata (forma cationica, presenta un atomo di idrogeno in più nel gruppo amminico dato dalla ionizzazione) un amminoacido si comporta come un acido diprotico. Se questa forma protonata/cationica perde un atomo di idrogeno (quello del gruppo COOH) diventa uno ione dipolare/anfoione e si presenta in forma zwitterionica. Se lo ione dipolare perde un altro atomo di idrogeno (uno dei 3 del gruppo amminico) allora diventa un amminoacido con forma anionica. Questo processo di trasformazione è reversibile, basta semplicemente cambiare il pH. CAPITOLO 4.5.2: PUNTO ISOELETTRICO (pI): VALORE DI pH AL QUALE LA FORMA ZWITTERIONICA E’ QUELLA PREDOMINANTE Il punto isoelettrico (pI) è il valore di pH al quale la carica netta dell’amminoacido è pari a 0 e il suo comportamento non è nè acido nè basico (quindi neutro). E’ un valore specifico di ogni sostanza. In 17 questa situazione l’amminoacido è comunque ionizzato ma la somma delle sue cariche è pari a 0 quindi è neutra. E’ il valore con il quale l’amminoacido si presenta in forma zwitterionica. CAPITOLO 4.6: UN AMMINOACIDO CON CATENA LATERALE MOLTO REATTIVA – LA CISTEINA Questo amminoacido prevede la formazione di un ponte disolfuro tra due sue molecole poste a una certa distanza tramite ossidazione. Questo ponte però, che serve a stabilizzare la struttura, può essere rotto usando un agente riducente, e far tornare quindi separate le due molecole di cisteina. Il ponte disolfuro, si forma solo in una determinata posizione delle molecole. CAPITOLO 4.7: AMMINOACIDI NON COMUNI – NON PROTEINOGENESI Ornitina Citrullina Sono amminoacidi intermedi fondamentali nella biosintesi dell’arginina e nel ciclo dell’urea. Fanno cioè altre funzioni e NON partecipano alla formazione delle proteine. CAPITOLO 5: LE PROTEINE Le proteine, sono generalmente grandi, e svolgono numerose funzioni fondamentali all’interno della cellula. Le più ricorrenti sono: Funzione strutturale (cheratine, collageno ecc.) Funzione di trasporto (emoglobina, albumina ecc.) 18 Funzione catalitica (enzimi) Funzione specializzate (immunoglobuline) Le proteine sono biopolimeri costituiti da “L-alfa-amminoacidi” legati tra loro mediante legami petpidici. In base alla loro struttura, si possono classificare in proteine fibrose e proteine globulari. CAPITOLO 5.1: STRUTTURA DELLE PROTEINE Nella struttura tridimensionale delle proteine si possono riconoscere quattro livelli di organizzazione strutturale, caratterizzati da tipi di legami diversi alla base della loro stabilizzazione. Vediamo nel dettaglio i quattro livelli di struttura: Struttura primaria, catena di peptidi in sequenza legati da legami peptidici/amminici (amminici perchè si forma tra i gruppi amminici dei vari amminoacidi), questo tipo di legame è covalente e molto forte Struttura secondaria, ripiegamento periodico (cioè ogni struttura secondaria ha sempre le stesse caratteristiche in tutte le proteine) e spaziale di amminoacidi adiacenti della struttura primaria, presenta legami a idrogeno tra gruppi peptidici (è un legame più debole rispetto a quello della struttura primaria) Struttura terziaria, ripiegamento tridimensionale di tutta la catena peptidica (parte dalla conformazione nativa), è specifica per ogni proteina e imprevedibile, presenta legami intermolecolari tra le catene laterali (sarebbero le variabili R), questa struttura determina la funzione della proteina, può presentare anche ponti salini (legami tra acidi e basi), sono tutti legami deboli Struttura quaternaria, presenta legami intermolecolari tra le catene laterali di catene polipeptidiche diverse (cioè tra R di catene diverse) CAPITOLO 5.1.1: STRUTTURA PRIMARIA – IL LEGAME PEPTIDICO I gruppi –COOH e –NH2 di due diversi alta-amminoacidi possono reagire e attraverso l’eliminazione di una molecola di acqua, formano il legame peptidico. Nella foto a lato si vede chiaramente che, per ottenere questo legame peptidico, si forma una molecola di acqua unendo il gruppo OH del primo amminoacido (quello che parte del gruppo funzionale –COOH) e un atomo di idrogeno (H) del gruppo amminico dell’altro amminoacido (quello –NH2). Questa molecola di acqua viene poi eliminata e il legame peptidico si forma tra 19 l’atomo di azoto (N) e l’ossigeno (O) dei due amminoacidi (sarebbe la parte nell’ellisse in basso a destra in foto). Un legame peptidico è molto forte perché viene stabilizzato per risonanza. Questo spostamento di elettroni modifica quindi la geometria della struttura creando legami parziali (sono doppi legami ibridi, quelli col tratteggio in foto), questo porta tutta la struttura sullo stesso piano in quanto gli atomi di C, N e O impegnati nel legame sono tutti ibridati sp 2. Il secondo amminoacido legato è ribaltato strutturalmente rispetto al primo (cioè è sul piano trans, se fosse stato sullo stesso piano sarebbe stato allora cis). Per far avvenire i ripiegamenti nella struttura primaria, c’è bisogno di punti di flessibilità, cioè punti precisi nella geometria della struttura. Si considerano quindi gli angoli “phi e psi” che presentano rotazione data dalla natura del gruppo R. Se il gruppo R è più grande e ingombrante infatti, la catena avrà maggior rigidità e viceversa. A causa di questo limite, due atomi non possono assumere tutte le posizioni, ma solo quelle che il gruppo R permette (è anche possibile che, quando R è molto ingombrante, non si verifica nessun ripiegamento). Nella prolina per esempio, il legame N-C(alfa) non può ruotare. Come detto, la sequenza degli amminoacidi in una catena polipeptidica rappresenta la struttura primaria di una proteina e costituisce il primo livello di organizzazione strutturale. I vari amminoacidi sono legati con legami che si formano tra N e C-alfa e C-alfa e C1. In ogni catena, si identifica un’estremità C-terminale (C = carbossilico) e una estremità N-terminale (N = amminico), queste estremità definiscono anche la polarità della catena. La parte R è impiegata in reazioni di legame. La catena primaria, quando presenta la cisteina, vede la formazione di un ponte disolfuro tra i vari atomi di questa, o ponti disolfuro intercatena, cioè tra atomi di cisteina appartenenti a due catene (in questo caso vengono considerate come una singola catena). 20 CAPITOLO 5.1.2: STRUTTURA SECONDARIA DELLE PROTEINE I legami che stabilizzano questa struttura sono i legami a idrogeno tra i gruppi–NH e –C=O di gruppi peptidici diversi (gruppi peptidico diversi = amminoacidi diversi), intorno a uno stesso asse longitudinale. Questi legami, provocano un ripiegamento nello spazio della catena polipeptidica, questi ripiegamenti possono essere: Alfa-elica, è la più comune struttura secondaria, è dovuta a legami a idrogeno tra il –C=O di un gruppo peptidico e l’idrogeno di un –NH di un altro gruppo peptidico distante 3 residui/R (per esempio si lega quello dell’amminoacido 1 con quello dell’amminoacido 4, cioè con 3 amminoacido di distanza tra loro). In questo tipo di struttura, la catena laterale (cioè la variabile R) è sempre all’esterno dell’elica che si forma. Il diametro dell’elica è uguale in tutte le proteine. Sono coinvolti tutti i gruppi peptidici. La geometria di questa struttura prevede una distanza costante di 5,4Å (Angstrom) tra due punti dell’elica (sarebbe il passo dell’elica, cioè la distanza tra un “giro di elica” e l’altro) e ogni passo contiene 3,6 residui/R. La prolina, a causa della sua struttura, non può formare questo tipo di struttura, stesso discorso vale per la glicina che raramente la forma causa mancanza di catena laterale (cioè dipende dalle caratteristiche di R se l’elica si può avere oppure no) Beta-foglietto, è dovuta alla formazione di legami a idrogeno tra il –C=O di un gruppo peptidico e l’idrogeno di un –NH di un altro gruppo peptidico molto distante nella sequenza. Si ottiene una organizzazione strutturale in cui le catene polipeptidiche sono allineate (struttura a fisarmonica regolare). Ogni ripiegamento presenta alle sue estremità i gruppi R, i C-alfa sono situati nei vertici dei ripiegamenti (nelle “cerniere” che uniscono i vari ripiegamenti). Il foglietto presenta più filamenti (tratti della stessa catena polipeptidica che si ripiega, arriva a un’estremità e torna indietro [a modi serpente]), i vari filamenti sono tra loro antiparalleli in quanto presentano estremità diverse (si alternano C-terminale e Nterminale), in questa forma antiparallela quindi, le catene sono orientate in direzioni opposte (stessa direzione invece nella forma parallela, di conseguenza le estremità saranno tutte uguali ai due lati, cioè tutte C-terminale da una parte e N-terminale dall’altra). La struttura a foglietto è mantenuta da legami a idrogeno tra tutti i gruppi peptidici, anche molto lontani che entrano in contatto grazie al ripiegamento. Le varie catene laterali/R sono in trans tra loro. Ripiegamenti inversi (anse o angoli beta), si ritrovano molto spesso nelle proteine perché provano un brusco cambiamento della direzione della catena polipeptidica (formano quindi un’ansa/gomito). Sono di due tipi (1 e 2) e sono costituiti da 4 residui amminoacidi. Sono stabilizzati da un legame a idrogeno tra i gruppi peptidici dei residui 1-4 e 2-3. Il 2 è quasi sempre Pro, il 3 è quasi sempre Gly. 21 CAPITOLO 5.1.3: STRUTTURA TERZIARIA DELLE PROTEINE Questa struttura descrive la posizione nello spazio di tutti gli atomi di una proteina. E’ caratteristica delle proteine globulari. Non presenta legami a idrogeno perché questi sono impegnati a stabilizzare le strutture secondarie. Questa struttura definisce anche la funzione della proteina, se si altera cambia la funzione. Questa struttura terziaria viene definita/studiata mediante tecniche spettroscopiche sofisticate come: Diffrazione dei raggi X (strutture cristalline e senza acqua) Risonanza magnetica nucleare (in soluzione, vale solo per proteine piccole) Le coordinate atomiche di tutti gli atomi, che servono per definire la struttura terziaria, sono registrate in banche dati e possono essere visualizzate con appositi programmi di visualizzazione biochimica. Questa struttura non è prevedibile (cosa che invece è quella secondaria), in quanto il ripiegamento funzionale è specifico per ogni proteina e omologo solo per proteine uguali. Questa struttura è stabilizzata dalle interazioni che si instaurano tra le catene laterali/R dei residui amminoacidici, anche quelli già organizzati in struttura secondaria. Queste interazioni sono (in ordine di forza): Ponti disolfuro (unico legame forte, si forma tra i residui di cisteina) Interazioni Ioniche (ponti salini, legami cioè tra un acido e una base) Legami ad idrogeno (solo se è presente una parte R con un atomo polare) Interazioni dipolo-dipolo Interazioni idrofobiche Interazioni di van der Waals (con R apolare) La struttura terziaria delle proteine porta al ripiegamento delle strutture secondarie (se presenti) nello spazio. I ripiegamenti vengono spesso utilizzati per collegare diversi segmenti di struttura secondaria. Tali organizzazioni sono definiti motivi strutturali. Nella foto a destra, si osservano i vari motivi strutturali dati dalla combinazione di più strutture secondarie (quelle in rosso sono le beta-foglietto, le verdi sono le alfa-elica). Un barile si ottiene quando una struttura beta- 22 foglietto si chiude su stessa, nel fare questa operazione può anche presentare pezzi di struttura secondaria ad alfa-elica (barile alfa-beta). Le proteine di grandi dimensioni (oltre i 200 amminoacidi costituenti) sono organizzate in unità strutturalmente indipendenti denominate domini. Per dominio si intende una “zona/parte” di una proteina che svolge una funzione specifica e diversa da quelle degli altri domini che compongono la stessa proteina. Ogni dominio presenta la struttura di una piccola proteina globulare. Un dominio può avere funzione strutturale, funzionale, catalitica, regolatoria. Se si produce solamente un dominio di una proteina, questo sarà funzionante e svolgerà la sua funzione nonostante non sia collegato agli altri domini della proteina. E’ bene precisare però che, una cellula, necessità di una proteina completa, quindi di una proteina con tutti i suoi domini. CAPITOLO 5.1.4: STRUTTURA QUATERNARIA DELLE PROTEINE Questa struttura quaternaria si ha quando diverse catene polipeptidiche con strutture terziarie indipendenti si associano. Viene stabilizzata dagli stessi legami che stabilizzano quella terziaria (tranne i ponti disolfuro, vedi sotto). Le diverse catene che la compongono vengono definite subunità e possono essere identiche o diverse (l’emoglobina per esempio ne presenta 4, uguali a due a due per esempio), generalmente queste si dispongono in maniera simmetrica. Hanno questa struttura quaternaria: Gliceraldeide 3-P deidrogenasi Emoglobina Proteine G Chaperoni Molecolari Le forze che stabilizzano la struttura proteica sono: Effetto idrofobico, cioè la tendenza delle sostanze non polari a minimizzare i loro contatti con il solvente acquoso (se la proteina è in acqua quindi, si ripiega su se stessa al fine di mettere a contatto con il liquido solo la sua parte idrofila, un po come fanno i lipidi anfipatici della membrana biologica) Interazioni elettrostatiche, ponti salini e forze di van der Waals Legami chimici trasversali Per denaturazione della struttura della proteina si intende un processo spesso reversibile che, spezza i vari legami (ponti disolfuro) per far tornare la proteina alla sua struttura primaria e vedere se questa torna in modo spontaneo alla forma nativa. Questo procedimento si ottiene con un agente denaturante che prima viene avvicinato per rompere i legami e poi allontanato. Si osserva quindi che, una volta allontanando l’agente denaturante, la proteina torna nella sua forma nativa in modo spontaneo. Nel farlo però, se si fa questo esperimento in vitro, può succedere che si formino, erroneamente, ponti disolfuro tra molecole diverse che quindi alterano la forma della proteina. Ciò non accade invece nelle cellule, in quanto queste hanno particolari enzimi che evitano che si verifichino tali errori. 23 CAPITOLO 5.2: LE PROTEINE FIBROSE Le proteine fibrose sono ricche di elementi con struttura secondaria. Questa classe di proteine svolge un ruolo di supporto fisico (strutturale) o di protezione. Queste sono: Cheratine (alfa e beta), sono i principali componenti degli strati epidermici esterni e delle appendici da essi derivati (pelle, capelli, unghie ecc.) Collageno, è il principale costituente dei tessuti connettivi (ossa, denti, tendini, cartilagini ecc.) L’alfa-cheratina è una proteina molto resistente allo stress fisico, è costituita da una coppia di alfaeliche avvolte una intorno all’altra generando un superavvolgimento (coiled coil). Presenta una sequenza di 7 residui/R di cui “a” e “d” sono idrofobici. Le due eliche-alfa sono tenute insieme da interazioni idrofobiche che si formano tra i residui “a” e “d” di un elica e “a1” e “d1” dell’altra elica. Il passo dell’elica in questo caso è ridotto a 5,1Å. La struttura fibrosa dell’alfa-creatina è data da un protofilamento in cui si instaurano interazioni deboli tra le estremità C-terminale e N-terminale di ogni dimero avvolto (il dimero è l’unità base del protofilamento, questi si uniscono nelle estremità e formano il protofilamento appunto). Più protofilamenti insieme formano le protofibrille. L’associazione di quattro protofibrille forma le microfibrille di questa struttura. Questa struttura può presentare anche numerosi ponti disolfuro, cioè un legame debole reversibile. La beta-cheratina è una proteina molto ricca di residui di piccole dimensioni (variabili R molto piccole, sono Gly, Ala e Ser) e non contiene cisteina (quindi non può fare ponti disolfuro). Questa assenza di ponti disolfuro da a questa proteina maggior rigidità rispetto a quella alfa. E’ costituita prevalentemente da strutture a beta-foglietto. Le catene laterali di glicina e alanina/serina sono disposte in maniera alternata. I gruppi di una catena laterale si adattano perfettamente a quelli della catena adiacente (ci chiudono cioè come i pezzi di un puzzle o i denti di una cerniera). Tra le catene laterali di residui più ingombranti si instaurano interazioni idrofobiche. Il collageno è una delle proteine più abbondanti nei mammiferi, è costituita da fibre insolubili che le danno un’elevata resistenza alla tensione. La sua composizione in amminoacidi è: Un residuo su tre (33%) è glicina 15% di prolina Presenza di amminoacidi modificati che costituiscono il restante 52%, questi amminoacidi sono la 4-idrossiprolina, la 3-idrossiprolina e la 4-idrossilina La presenza abbondante di prolina e glicina impedisce la formazione di strutture ad alfa-elica. Questa proteina però assume comunque una conformazione elicoidale dovuta alla configurazione dei residui di prolina: genera un’elica più distesa e con passo più largo, un’elica che si forma quando tre catene si avvolgono una sull’altra generando così una tripla elica. Questa tripla elica viene stabilizzata da legami a idrogeno tra gruppi peptidici di residui/R appartenenti ad eliche diverse. Non forma ponti disolfuro data la mancanza di cisteina. Presenta legami covalenti trasversali dati dai residui di lisina, idrossilisina e istidina. 24 CAPITOLO 6: LE PROTEINE CHE LEGANO CON L’OSSIGENO – MIOGLOBINA ED EMOGLOGINA Mioglobina (Mb): mio perché riguarda tutto ciò che a che fare con i muscoli, è nei tessuti periferici ed è il deposito di O2 (ossigeno) nei tessuti Emoglobina (Hb): si trova nel sangue (emo), precisamente è contenuta nei globuli rossi, trasporta O2 (ossigeno) dai polmoni/alveoli polmonari ai tessuti periferici Mioglobina ed emoglobina sviluppano una nuova capacità grazie alla loro struttura quaternaria (eme), cioè quella di legarsi e trasportare ossigeno che si lega al gruppo prostetico (all’atomo di ferro dell’eme appunto). Sono due proteine globulari e presentano 8 alfa-eliche (tutte indicate con una lettera maiuscola dell’alfabeto) nella loro struttura terziaria. L’emoglobina è formata da 4 monomeri uguali a due a due (a destra in figura), ogni unità è uguale strutturalmente a quella della mioglobina (a sinistra in figura) che invece è formata da una sola struttura di questo tipo. Ogni monomero dell’emoglobina possiede un eme, quindi a ogni molecola di emoglobina si possono legare 4 molecole di ossigeno. Pur avendo la struttura terziaria praticamente uguale, mioglobina ed emoglobina hanno una struttura primaria diversa tra loro, hanno uguale solo alcune porzioni che permettono loro di avere i ripiegamenti uguali (questi ripiegamenti si chiamano ripiegamenti globinici e sono comuni ad altre proteine che si legano con l’ossigeno). Operando in zone del corpo diverse, mioglobina ed emoglobina hanno caratteristiche diverse (vedi dopo per dettagli). L’ossigeno dell’emoglobina, deve passare alla mioglobina che poi lo farà entrare nella cellula (passaggio che avviene secondo gradiente, la mioglobina si trova nei pressi dell’esterno della cellula) e nei mitocondri di questa. Queste due proteine quindi, devono essere in grado di legare e di rilasciare ossigeno a seconda della condizione nella quale si trovano. L’emoglobina che ha ceduto l’ossigeno alla mioglobina (deossiemoglobina), quindi scarica di ossigeno, acquista CO2 e protoni e torna nei polmoni (sottoforma di sangue venoso) dove verrà riossigenato e ricomincerà la sua circolazione (come sangue arterioso e quindi carico di ossigeno). L’emoglobina lascerà l’anidride carbonica (CO2) nei polmoni che la espelleranno con l’espirazione. L’ossigeno trasportato da queste due proteine, è fondamentale per la fosforilazione ossidativa (cioè l’ultima fase della glicolisi, quella con l’ATP sintetasi per capirci), più precisamente serve ossigeno perché gli elettroni liberati da NADH e FADH2 vengono accettati dall’acqua che sarebbe ossigeno ridotto. In questa fase, si consuma quasi il 95% di tutto l’ossigeno acquisito tramite la respirazione. L’eme è localizzato in una tasca idrofobica e ad esso si lega la molecola di ossigeno assorbita. 25 Quindi per ricapitolare: le proteine che legano ossigeno sono proteine specializzate all’interazione con questo gas, sono essenziali per la vita degli organismi aerobi, queste proteine accelerano la diffusione di ossigeno nel corpo, l’ossigeno si lega in modo reversibile a queste proteine (cioè queste proteine lo possono accettare e rilasciare quando serve), nessuna catena laterale R di queste proteine interagisce/si lega con l’ossigeno, l’ossigeno infatti si lega grazie ad alcuni ioni di metalli (ferro nell’uomo, rame in alcuni animali) situati nella struttura quaternaria della proteina (eme), senza eme queste proteine non funzionano, la parte peptidica di queste proteine serve a regolare la reversibilità (cioè l’eme da solo non può rilasciare ossigeno ma solo assorbirlo, questo arrecherebbe danni all’organismo creando una specie radicalica, la parte proteica quindi conferisce proprio questa capacità di rilasciare l’ossigeno che si è legato ad esso). CAPITOLO 6.1: LA STRUTTURA DELL’EME L’eme è formato da: Tetrapirrolo ciclico (cioè 4 gruppi pirrolici situati attorno al ferro centrale e legati ad esso con 4 dei 6 legami di coordinazione), [parte rossa in figura]) 4 Gruppi metilici [parte viola in figura] 2 Gruppi vinilici [parte gialla in figura] 2 Gruppi Propionato [parte azzurra in figura], rappresentano l’unica parte polare dell’eme 1 Atomo di ferro (Fe centrale), presenta legami di coordinazione (sono 6 e partono dal ferro centrale, 4 sono impegnati con i gruppi pirrolici, 2 sono liberi e sono perpendicolari al piano, vedi dopo per dettagli) L’elevato numero di doppi legami coniugati e la presenza dello ione ferroso, conferiscono alla mioglobina e all’emoglobina il classico colore rosso. CAPITOLO 6.1.1: LA STRUTTURA DELL’EME L’atomo di ferro è situato al centro dell’eme e forma una struttura bipiramidale insieme ai suoi legami. Le due punte delle due piramidi, rappresentano i 2 legami di coordinazione liberi, ai quali si attacca l’ossigeno (ad un legame si lega sempre ossigeno [sesto legame, quello in E7], l’altro è vuoto ma può ospitare ossigeno). Questi due legami sono in posizione F8 ed E7 (coordinate nella struttura terziaria 3D della proteina, cioè sull’elica F in posizione 8 perché un elica si divide in più zone in base a quanti R contiene al suo interno ecc.), coordinate che invece diventano His93 e His64 se si fa riferimento alla struttura primaria della proteina (cioè alla singola catena, per His = istidina, sarebbero la 93sima e la 64esima istidina della catena). 26 Come detto, l’ossigeno si lega all’atomo di ferro con il sesto legame di coordinazione (quello in E7), quindi l’istidina distale (quella His64) fornisce la geometria giusta di legame, questa istidina schiaccia il legame, impedendo che si leghi ad esso un monossido di carbonio (un veleno). CAPITOLO 6.2: LA MIOGLOBINA (Mb) La mioglobina è una proteina abbastanza piccola (è composta infatti da 153 amminoacidi), ha una sola struttura monomerica (quindi un solo eme contenuto in una tasca idrofobica della struttura globulare, in prossimità della superficie), comprende 8 alfa-eliche indicate tutte con una lettera maiuscola dell’alfabeto (AB, CD sono i gomiti, cioè le curve). La maggior parte dei residui (gruppi R della proteina) polari e quindi idrofili sono situati sulla superficie esterna della molecola (idrofobici all’interno), tranne due residui di istidina (His93 e His64 appunto, dette anche prossimale e distale) coinvolte nel legame con l’ossigeno. Trasporta ossigeno a livello locale e lo immagazzina. CAPITOLO 6.3: EMOGLOBINA (Hb) L’emoglobina è più complessa e grande (è formata da 574 amminoacidi), presenta una struttura quaternaria tetramerica (4 sub-unità, uguali a due a due tra loro, 2 alfa e 2 beta), 4 gruppi eme (uno per ogni sub-unità) situati in cavità vicine alla superficie esterna. Ogni sub-unità alfa e in contatto con le due beta, mentre esistono poche interazioni tra le due alfa e tra le due beta. Trasporta ossigeno a lungo raggio. CAPITOLO 6.4: CURVA SATURAZIONE DELLA MIOGLOBINA Nel grafico a lato, sull’asse delle X sono indicate le varie pressioni parziali di ossigeno, mentre sull’asse delle Y è indicata la percentuale delle molecole di mioglobina che si sono legate all’ossigeno a quella data pressione. Come si può vedere, la curva cresce in modo netto nella prima parte, per poi veder diminuita sempre più la sua crescita fino al punto di saturazione quando non si registra più nessuna crescita (questo perché tutte le molecole si sono legate all’ossigeno e, pur aumentando la pressione, non succede niente). La curva disegnata è un iperbole rettangolare. La formula indicata nel grafico, presenta una doppia freccia che indica la reversibilità del processo, la doppia freccia inoltre, 27 rappresenta il momento nel quale la reazione è in equilibrio, cioè quando il 50% delle molecole è legata all’ossigeno e il 50% non ancora, quindi se si legge verso destra si avrà anche l’altro 50% gradualmente ossigenato e viceversa. La massima affinità della mioglobina a legarsi all’ossigeno si ha quando la pressione è pari a 2,8 torr cioè a p50. Sopra questo valore oltre il 50% delle molecole sarà ossigenato, al di sotto invece saranno scariche di ossigeno. Questo valore a p50 di 2,8 torr definisce la massima affinità di legarsi all’ossigeno come detto, di conseguenza a tutte le pressioni superiori, la mioglobina sarà sempre più carica di ossigeno e satura, la mioglobina infatti è quasi sempre satura ed ecco perché questa è un ottimo deposito di ossigeno (ossigeno che poi rilascia in condizioni di scarso apporto di questo ai tessuti). La pressione dell’ossigeno nei muscoli a riposo è di 40 torr, mentre in quelli in attività è di 20 torr (inutile dire quindi, che in entrambi i muscoli, la mioglobina è satura di ossigeno, infatti oltre il 90% delle molecole sono ossigenate nei muscoli a riposo [funzione di riserva di ossigeno], mentre nei muscoli sotto sforzo circa il 60% è ossigenato [questo ossigeno serve per produrre ATP necessario alla contrazione muscolare]). CAPITOLO 6.5: CURVA DI SATURAZIONE DELL’EMOGLOBINA Come si vede dall’immagine a lato, la curva di saturazione dell’emoglobina (cioè quando l’emoglobina si satura di ossigeno) ha un andamento sigmoidale, presenta un punto di flesso (cioè dove “gira la curva”, a p50 = 26 torr) dal quale poi la curva diventa esponenziale fino al valore massimo di saturazione. Sull’asse X abbiamo sempre i valori della pressione parziale di ossigeno, mentre sull’asse Y è presente la percentuale di molecole ossigenate. Come detto, il punto di equilibrio (p50) è situato a 26 torr, quindi è quasi 10 volte superiore a quello della mioglobina, ciò significa che l’emoglobina necessita di una pressione più alta per legarsi ossigeno (ecco perché, nei tessuti periferici, dove la pressione è sotto i 20 torr, l’emoglobina cede ossigeno, causa sua bassa affinità di legarsi ad esso con la pressione dei muscoli, alla mioglobina che lo accetta data la sua grande affinità a legarsi con esso anche con pressioni bassissime (2,8 torr). Al punto p50 si ha l’effetto operativo positivo, cioè la curva cambia verso e cresce esponenzialmente perchè i legami delle molecole che hanno già accettato la molecola di ossigeno, “aiutano” le altre non ancora ossigenate a ricevere la molecola di 28 O2 (ecco perché dopo questo punto, l’ossigenazione è più rapida). L’emoglobina si ossigena negli alveoli polmonari perché, in quel punto, la pressione dell’ossigeno è molto alta. Alla pressione arteriosa, sia mioglobina che emoglobina sono sature (è una pressione molto alta ed ecco perché il sangue di questa pressione è ricco di ossigeno). Alla pressione venosa invece, la mioglobina è già satura, mentre l’emoglobina no. Le curve di saturazione delle due proteine hanno andamento diverso, nonostante abbiamo praticamente la stessa struttura terziaria, perché nell’emoglobina ha l’effetto allosterico, cioè arrivate a un certo punto, le molecole già cariche di ossigeno, aiutano le altre a ossigenarsi, velocizzando il processo (la cooperazione appunto). CAPITOLO 6.5.1: EFFETTI SFERICI CAUSATI DAL LEGAME DI OSSIGENO NELLA MIOGLOBINA Quando il primo ossigeno si lega all’eme della mioglobina, questo modifica la struttura e la forma dell’elica e quindi dell’eme stesso (praticamente la struttura si appiattisce e “stringe” la molecola di ossigeno legata) CAPITOLO 6.6: STRUTTURA QUATERNARIA DELL’EMOGLOBINA Quando si lega il primo ossigeno, cosi come avviene nella mioglobina, anche i monomeri dell’emoglobina variano la loro forma: in questo caso, i monomeri si avvicinano e danno vita alla struttura R State (struttura rilassata, più compatta e che “stringe” la molecola di ossigeno all’interno di essi), quando invece non c’è ossigeno (desossiemoglobina), i monomeri formano una struttura T State (struttura tesa); questa mutazione serve a favorire l’entrata di ossigeno (infatti la cavità centrale è più larga nella struttura T) e bloccarne l’uscita (la cavità si stringe nella struttura R quando si avvicinano i monomeri). Questa trasformazione avviene perché l’ossigeno sposta la His e quindi modifica l’elica di 0,6 Angstrom di un monomero, lo spostamento poi si riflette poi gradualmente anche sulle altre eliche. Questa mutazione serve a stabilizzare i legami della struttura R. Come detto, il passaggio da struttura T a R avviene in modo sequenziale e graduale: si modifica infatti, prima un monomero, la modifica di questo favorisce la modifica degli altri tre partendo da quella più vicina (ecco perché dopo il punto di equilibrio, le restanti molecole di emoglobina si ossigenano più in fretta, effetto allosterico). A modulare questo processo è l’ossigeno, quindi il ligando stesso che aumenta l’affinità di legame dell’emoglobina per l’ossigeno (questo prende il nome di modulatore omotropico) che induce una cooperatività positiva tra le molecole di ossigeno. 29 CAPITOLO 6.7: FATTORI CHE REGOLANO IL LEGAME DI OSSIGENO ALL’EMOGLOBINA – EFFETTO BOHR In condizioni normali, il legame dell’ossigeno all’emoglobina è regolato da diversi fattori: Gli effettori che stabilizzano la forma T provocando il rilascio dell’ossigeno e viceversa: il 2,3 bisfosfoglicerato (2,3-BPG) La diminuzione del pH Aumento della concentrazione di CO2 sul legame Questi tre fattori sono modulatori negativi del legame tra ossigeno e Hb quindi favoriscono il rilascio di ossigeno stabilizzando la struttura T. Per effetto Bohr si intendono appunto questi fattori che favoriscono il rilascio di ossigeno. All’aumento della concentrazione di CO2 scende il pH (diventa acido), abbassandosi il pH aumenta la capacità di rilascio di ossigeno da parte dell’Hb (avviene anche nei muscoli, infatti l’acido lattico abbassa il pH). Nei polmoni avviene l’opposto, cioè aumenta l’affinità di legame con l’ossigeno anche perché il pH è basico. CAPITOLO 6.7.1: IL 2,3-BPG E’un intermedio anche di altre vie metaboliche Presenta 5 cariche negative che formano 5 coppie ioniche con l’emoglobina Sono coinvolti residui basici positivamente che formano una tasca polare Si lega nello spazio inter-subunità dell’Hb stabilizzando la forma T e dando bassa affinità nel legarsi con l’ossigeno all’Hb Nella forma R non è presente in quanto favorisce il rilascio di ossigeno, non l’assorbimento (manca anche per questioni di spazio nella struttura R) CAPITOLO 7: PROTEINE SPECIALIZZATE – GLI ENZIMI Gli enzimi sono macromolecole, di forma globulare deputate alla catalisi/catalizzazione delle reazioni biologiche (più reazioni consecutive formano le vie metaboliche). Rimangono inalterati alla fine della reazione e non modificano il risultato finale di questa, accelerano/diminuiscono (raramente, solo in caso di enzimi negativi, noi studieremo solo enzimi positivi che aumentano) la velocità con la quale la reazione tra due reagenti avviene. Si differenziano dai catalizzatori chimici per le seguenti proprietà: Sono più efficienti, gli enzimi infatti, aumentano (quelli positivi) la velocità di una reazione di alcuni ordini di grandezza in più rispetto alla catalisi chimica 30 Hanno condizioni di reazione molto più moderate, essendo noi degli organismi omeotermi infatti, la nostra temperatura è relativamente bassa ma gli enzimi lavorano lo stesso anche a questa temperatura, anzi una temperatura più alta potrebbe denaturarli e renderli inattivi. In generale, la temperatura deve essere inferiore ai 100°C e il pH simile a neutro (tranne che in casi particolari). Anche la pressione atmosferica può incidere sulla funzionalità degli enzimi Sono specifici di una reazione, ogni enzima infatti catalizza una sola reazione, cioè è specifico solo per quella reazione (più enzimi possono catalizzare una stessa reazione ma non viceversa) Hanno una possibilità di regolazione, cioè si può regolare la velocità con la quale questi catalizzano le reazioni, ci sono due tipi di regolazioni: si/no e più veloce/meno veloce CAPITOLO 7.1: LA CLASSIFICAZIONE DEGLI ENZIMI Nella nomenclatura comune, per indicare un enzima, si aggiunge il suffisso “-asi” alla reazione catalizzata o al composto su cui questo agisce (esempio: lipasi, proteasi ecc.). Visto il numero elevato di enzimi conosciuti, il loro nome viene identificato in tre modi diversi: 1. Nome tradizionale, breve e per l’uso comune (ambiguo) 2. Nome sistematico, identifica la reazione (più specifico, esempio: piruvato carbossilasi) 3. Numero di identificazione/classificazione (numero EC), identificazione univoca e che non lascia spazio ad ambiguità Facciamo un esempio con la seguente reazione (si ha in caso di attività fisiche intense ma di breve durata e conseguente riposo): ATP + creatina ADP + fosfocreatina L’enzima di questa reazione, prende i seguenti nomi (i numeri indicano il tipo di nomenclatura tra i tre indicati qui sopra): 1. Creatina chinasi (da questo nome capiamo che fosforila la creatina ma sappiamo che forma anche ATP se viene letta al contrario) 2. ATP : creatina fosfotransferasi (si capisce che c’è il trasferimento del gruppo fosfato ed è una reazione reversibile) 3. EC 2.7.3.2 (i numeri indicano classi, sotto-classi e sotto-sottoclassi, vedi dopo per dettagli) CAPITOLO 7.1.1: DETTAGLI DELLA CLASSIFICAZIONE CON EC Nel numero di classificazione il termine di riferimento è il meccanismo della reazione catalizzata. Sappiamo che è l’unica nomenclatura che indica con estrema precisione l’enzima che viene usato, e per fare ciò, sono state identificate sei classi di reazioni principali, indicate con numeri arabi (sarebbe il primo numero, nell’esempio è il primo 2). Se il primo numero è: 31 1 – si tratta allora di una ossidoriduttasi/reazione redox 2 – si tratta allora di una transferasi, cioè un trasferimento di gruppi funzionali (come nell’esempio di sopra) 3 – si tratta allora di una idrolasi, cioè di una reazione di idrolisi 4 – si tratta allora di una liasi, cioè un’eliminazione di gruppi (forma doppi legami, si indica con il prefisso “de-“) 5 – si tratta allora di una isomerasi, cioè di isomerizzazioni 6 – si tratta allora di una ligasi, cioè la formazione di legami con idrolisi di ATP In questo tipo di classificazione inoltre, vengono identificate delle sottoclassi e delle sottosottoclassi con lo stesso criterio (cioè in base a ciò che fanno) (si indicano con il secondo e con il terzo numero dell’EC, numeri 7 e 3 nell’esempio). Infine si aggiunge un numero che identifica lo specifico enzima (sarebbe il quarto è ultimo numero, nell’esempio sarebbe il secondo 2). Quindi, rianalizzando nel dettaglio, il nome dell’enzima della reazione ATP + creatina ADP + fosfocreatina Si osserva che il suo EC 2.7.3.2 indica: 2 = transferasi 7 (sottoclasse) = fosfotranferasi 3 (sotto-sottoclasse) = accettore gruppo azotato 2 (numero specifico) = creatina chinasi CAPITOLO 7.2: SPECIFICITA’ DEGLI ENZIMI Il composto (il reagente) su cui agisce un enzima viene definito substrato, termine che sottolinea la sua specificità, cioè un enzima è capace di interagire solo e solamente con un tipo di molecola o con una classe molto ristretta di molecole (cioè con molecole correlate e che hanno una parte uguale tra loro, tipo le glucochinasi che hanno tutte glucosio). La parte dell’enzima che interagisce con il substrato (sito attivo o sito catalitico) può presentare già la forma adatta all’accomodamento di quest’ultimo (questo secondo l’ipotesi di Fischer), cioè l’enzima ha già pronta la cavità nella quale si inserirà senza problemi il substrato. Questa rigidità intrinseca di un sito attivo cosi fatto però non giustifica i cambiamenti dinamici alla base della catalisi enzimatica, infatti un enzima, durante la reazione può variare. Si introduce quindi l’ipotesi di Koshland, che prevede che in seguito all’interazione, l’enzima può subire un cambio conformazionale (adattamento indotto) che stabilizza il legame del substrato (cioè l’enzima adatta il suo sito quando entra in contatto con il substrato, cioè il sito attivo non è già pronto ma si adatta solo quando entra la proteina giusta, rivelando il suo sito catalitico ed evidenziando la specificità di un enzima, questo si attiva infatti, solo quando entra in contatto con il substrato “giusto”). L’interazione tra enzima e substrato dalla reversibilità dei legami deboli che si instaurano tra i gruppi di atomi presenti nel sito attivo dell’enzima e il substrato (cioè si stabiliscono dei legami deboli 32 reversibili tra le due componenti, infatti i substrati entrato, si trasformano in prodotti ed escono dal sito dell’enzima, permettendo il riutilizzo di quest’ultimo che, come sappiamo, ne esce senza alcuna variazione). CAPITOLO 7.3: ENZIMI - COFATTORI Molto spesso gli enzimi vengono aiutati nella loro funzione da molecole relativamente piccole che partecipano al meccanismo della reazione enzimatica: i cofattori. Questi possono essere di diverso tipo, sono indispensabili alla funzione degli enzimi e vengono denominati in maniera diversa in base a come si legano con gli enzimi. Si distinguono quindi in: Cofattori, quando sono molto piccoli (ioni metallici, molecole inorganiche) Coenzimi, se sono più complessi e si legano all’enzima con legami deboli (lo sono il NADH e il FADH2) Gruppi prostetici, se sono legati covalentemente all’enzima (sono detti appunto sia cofattori sia coenzimi) (l’eme è erroneamente detto gruppo prostetico, nonostante sia legato con legami deboli) La presenza del cofattore è essenziale per l’attività dell’enzima. Il complesso enzima-cofattore viene definito oloenzima (ed è attivo). L’enzima senza il cofattore viene definito apoenzima (ed è inattivo proprio perché non ha i cofattori). CAPITOLO 7.4: COME LAVORANO GLI ENZIMI Gli enzimi, come detto, modificano la velocità della reazione ma non gli stati di equilibrio, ne quindi la resa finale della reazione. E + S ↔ ES ↔ EP ↔ E + P Con: E = enzima S = substrato ES = composto enzima-substrato EP = composto enzima-prodotto P = substrato che si trasforma in prodotto ed esce dall’enzima La trasformazione del substrato a prodotto avverrebbe anche senza enzima, ma molto più lentamente. 33 CAPITOLO 7.5: ENERGETICA DEGLI ENZIMI Gli enzimi accelerano la velocità di una reazione abbassando l’energia di attivazione. Lo stato intermedio della reazione è diverso di quello che si avrebbe senza gli enzimi, proprio questa variazione fa aumentare la velocità nella seconda parte della reazione. Gli enzimi, fanno raggiungere la condizione di equilibrio più in fretta. CAPITOLO 7.6: MECCANISMI DI CATALISI ENZIMATICA (CIOE’ COME INTERAGISCONO ENZIMI E SUBSTRATO) I meccanismi di catalisi enzimatica sono gli stessi dei catalizzatori chimici ma più specifici. Si distinguono i seguenti meccanismi: Catalisi acido-base, per esempio: si lega un substrato acido con un enzima basico Catalisi covalente, genera un legame covalente tra sito attivo e substrato Catalisi favorita da ioni metallici, cioè tra polarità, gli ioni metallici possono funzionare da catalizzatori elettrofili o generare un nucleofilo Catalisi elettrostatica, genera legami deboli come le forze di van der Waals Catalisi di prossimità e di orientamento, cioè vengono orientati nella giusta posizione i substrati che interagiscono Catalisi favorita dal legame preferenziale del complesso di transizione, cioè si lega lo stadio intermedio della reazione, accelerando il proseguimento di essa CAPITOLO 7.7: CINETICA ENZIMATICA Nelle reazioni, la velocità dipende dalla concentrazione di substrato in maniera non univoca. Inoltre la concentrazione di substrato è nella realtà, molto maggiore di quella dell’enzima. Per misurare questa velocità, si considera quanto substrato si trasforma in prodotto in un dato intervallo di tempo, o quanto prodotto si forma nello stesso intervallo di tempo. Se osserviamo il grafico che viene fuori (figura a lato) mettendo sull’asse delle X il tempo, e sull’asse delle Y la quantità di prodotto che si forma, si osserva che, unendo i vari valori sperimentali, viene fuori un’iperbole che ha come asintoto (quello in blu) il valore massimo di prodotto che si può formare (non si tocca mai questo valore). Si osserva anche che, all’aumentare del tempo, aumenta la 34 percentuale di prodotto che si è formato, ciò succede fino a quando tutto il substrato non è stato trasformato, una volta “finito” il substrato non succede più niente. Più aumenta la quantità di substrato più aumenta la quantità di prodotti. Nella prima parte della reazione, precisamente tra i valori 0 e 3 dell’asse delle X, si osserva che c’è linearità tra i valori, cioè c’è una diretta proporzionalità tra il tempo passato e i prodotti fatti, in questo intervallo è possibile misurare la velocità istantanea (V0). Più scende la quantità di substrato non ancora trasformato (dopo il valore 3 delle X nel grafico), più diminuisce la quantità di prodotti in un intervallo di tempo. La velocità aumenta se aumenta la quantità di enzima usato (questo ovviamente fa variare il valore della V0). Anche se la quantità di substrato sarà di molto superiore alla quantità di enzima, ci sarà sempre una minima parte di enzima che non si legherà e che causerà la reversibilità del processo (cioè che i substrati possono entrare e uscire dal sito attivo). Dal grafico a lato, si osserva che: La velocità è proporzionale alla quantità di substrato Anche questa curva è un’iperbole asintotica perché anche se aumentiamo all’estremo la quantità del substrato, la velocità si stabilizza dopo un certo periodo, se calcoliamo il valore dell’asintoto (facendo il limite che tende all’infinito) troviamo il valore della velocita massima (Vmax) Nella prima parte (quella evidenziata da Km) la velocità è sensibile alla reazione E + S ↔ ES (cioè la velocità dipende dalla quantità di substrato presente e varia al suo diretto variare), condizione che invece non vale nella seconda parte della curva/reazione (reazione di ordine 0) Dai due grafici, si evince che c’è un fenomeno di saturazione (assente invece nei catalizzatori chimici) della velocità massima. Il valore di V0 sarà dato dall’ equazione di Michaelis-Menten: Con: Vmax = asintoto, non ricavabile con dati sperimentali perché non si raggiunge mai come valore, rappresenta la massima efficacia degli enzimi V0 = velocità inziale, quando c’è proporzionalità tra t e P Km = concentrazione di substrato alla quale la velocità di reazione è meta di quella massima, è uguale a [S] 35 CAPITOLO 7.8: EFFICIENZA ENZIMATICA Questo parametro è un indice del modo di funzionamento di un enzima, cioè misura efficacia ed efficienza di quest’ultimo. Tiene conto sia di Km che di Vmax. L’efficienza catalitica è uguale a: Kcat / Km Con: Kcat = costante catalitica = Vmax / [Etot] [Etot] = quantità totale di enzima coinvolto nella reazione Questa costante catalitica viene anche definita come numero di turnover e rappresenta il numero di volte che la reazione viene catalizzata a ogni sito attivo dell’enzima. I parametri Km e Vmax sono parametri cinetici, e si possono ricavare dalla determinazione della velocità di una reazione catalizzata da un enzima (equazione di Michaelis-Menten) facendo il reciproco di entrambi i membri di questa equazione (cioè girando l’equazione). Questa prende il nome di equazione di Lineweaver-Burk. CAPITOLO 7.9: REAZIONI A DUE SUBSTRATI Sono le reazioni più comuni catalizzate da enzimi e il modello/equazione di Michaelis-Menten non è di semplice applicazione con esse (non vale). Queste sono: Reazioni a spostamento singolo, cioè quando entrambi i substrati si devono legare all’enzima per poter generare il prodotto; si legano cioè, in maniera sequenziale e ordinata o casuale i due substrati insieme Reazioni a spostamento doppio, cioè i due substrati non saranno mai legati all’enzima nello stesso momento Vediamo nel dettaglio le fasi delle reazioni a spostamento doppio: 1. Entra il primo substrato 2. Si trasforma il primo substrato ed esce, la sua trasformazione in prodotto modifica anche la struttura dell’enzima 3. Entra il secondo substrato (ingresso permesso esclusivamente dal prodotto del primo substrato che ha modificato l’enzima) 4. Il secondo substrato si trasforma in prodotto e riporta l’enzima nel suo stato originale e rendendolo quindi disponibile a una nuova catalizzazione (questo avviene per esempio, nelle trasferasi, dove il gruppo funzionale del primo substrato “rimane” nell’enzima modificandolo, in attesa di legarsi al secondo substrato) 36 CAPITOLO 7.10: REGOLAZIONE DELL’ATTIVITA’ ENZIMATICA Un enzima deve funzionare solamente quando la sua funzione è necessaria (al fine di evitare sprechi energetici e soprattutto arrecare danni all’organismo), cioè solo quando il substrato deve essere catalizzato per compiere una reazione. Gli enzimi possono funzionare più o meno bene, a seconda dei seguenti fattori che ne influenzano l’attività (questi fattori infatti devono rientrare in parametri ottimali, specifici di ogni enzima a seconda di dove si trovano nell’organismo, che permettono a questo di lavorare al meglio): Temperatura, ogni enzima ha una sua temperatura ottimale alla quale esso lavora al 100%, nell’uomo deve essere di circa 38/40°, se è più alta l’enzima si denatura/sfalda e non lavora più o lavora male e a regime ridotto. Fanno eccezione gli enzimi che “abitano” negli organismi estremofili che vivono in luoghi molto caldi, questi enzimi infatti sono adattati alla situazione grazie a delle strutture termostatiche che permettono loro di lavorare ugualmente nonostante la temperatura molto elevata Forza ionica, ogni enzima opera con una salinità precisa (fanno sempre eccezione quelli degli estremofili che si adattano) pH, un enzima lavora bene solo nella zona in cui si trova perché questa ha un pH preciso e specifico, se mettiamo l’enzima in un’altra parte del corpo, con conseguente variazione del pH, questo non funzionerà Modifiche covalenti, modifiche ai gruppi R degli enzimi, modifiche reversibili e fatte da altri enzimi (enzima che modifica un altro enzima) detti chinasi, sono processi regolati dagli ormoni (vedi dopo per dettagli) Presenza di inibitori o di attivatori, sono piccole molecole, quando sono in grande quantità influenzano l’attività enzimatica aumentando o abbassando la sua velocità (vedi dopo per dettagli) In una via metabolica, possono esserci più meccanismi di controllo che agiscono in varie fasi. CAPITOLO 7.10.1: EFFETTO DELLA TEMPERATURA (CIOE’ COME LA TEMPERATURA INFLUISCE SULL’ATTIVITA’ ENZIMATICA) Come detto, un enzima lavora a pieno regime solo a una determinata temperatura, specifica e che varia da enzima a enzima. Un aumento della temperatura, cosi come in tutte le reazioni, provoca un aumento dell’attività enzimatica ma solo se il valore della temperatura rientra in un determinato intervallo (vedi grafico a campana a lato), se va oltre l’enzima perde efficacia, si denatura e non è più utilizzabile. Quelli umani, lavorano al top quando la temperatura va dai 38° ai 40° gradi, è un valore che varia a seconda della specie. 37 CAPITOLO 7.10.2: EFFETTO DEL pH (CIOE’ COME IL pH INFLUISCE SULL’ATTIVITA’ ENZIMATICA) Come sappiamo, ogni enzima ha un valore di pH caratteristico al quale la loro attività è massima. La variazione del pH influenza direttamente l’efficacia dell’enzima, a valori molto più bassi/alti infatti, l’enzima non funziona. Questa variazione di pH avviene quando si sposta l’enzima dalla sua zona naturale a un’altra zona del corpo. CAPITOLO 7.11: INIBIZIONE ENZIMATICA Anche la quantità di substrato influisce sull’attività enzimatica (tipo se il substrato è poco l’attività sarà più lenta). Generalmente però, esistono degli inibitori che riducono l’attività di un enzima legandosi reversibilmente a esso (cioè la molecola inibitrice si lega ma può anche staccarsi dall’enzima e farlo tornare normale e facente attività a pieno regime). Questi inibitori influenzano quindi l’affinità per il substrato e/o il numero di turnover. Esistono vari tipi di inibizione, differenziati in base al loro meccanismo (questo è solo un elenco, vedi dopo per i dettagli): Inibizione competitiva, è reversibile Inibizione incompetitiva, è reversibile Inibizione mista, è reversibile Inattivazione irreversibile, se viene inibito in questo modo, l’enzima non è più utilizzabile e deve essere degradato per poi essere risintetizzato da zero Il tipo di inibizione si identifica mediante l’analisi dei grafici dei doppi reciproci (cioè si guarda la cinetica enzimatica e come variano i valori di Vmax e Km. CAPITOLO 7.11.1: INIBIZIONE COMPETITIVA In questo tipo di inibizione, l’inibitore è molto simile al substrato che si lega con l’enzima. L’inibitore si lega al sito attivo, impedendo l’accesso al substrato. E’ un processo reversibile (se l’inibitore esce dal sito infatti, l’enzima può riaccogliere regolarmente il substrato e fare la sua funzione). Varia a seconda della quantità di inibitore o di substrato, se c’è più substrato infatti, tenderà a legarsi proprio quest’ultimo, rendendo vano l’inibitore o viceversa (cioè entra chi è più numeroso e quindi “più forte”). Si definisce “competitiva” proprio perché, essendo simili, inibitore e substrato “fanno a gara” per entrare nel sito attivo dell’enzima. In questo tipo di inibizione, a causa proprio della competizione, varia solamente il valore di Km all’aumentare della concentrazione di inibitore; Vmax non varia. 38 Questo tipo di inibizione, sul grafico dei doppi reciproci (grafico cioè che rappresenta l’attività in base alla quantità di inibitore presente, vedi a lato), si presenta con tre rette che hanno un punto in comune, punto rappresentato proprio dal valore di Vmax. CAPITOLO 7.11.2: INIBIZIONE INCOMPETITIVA In questo tipo di inibizione, l’inibitore si lega in un altro sito dell’enzima (non in quello attivo, in questo continua a legarsi il substrato, ecco perché è incompetitiva perché non c’è “gara” tra inibitore e substrato) e proprio a causa di questo legame, si influenza l’attività enzimatica, causando una distorsione del sito attivo che diventa meno efficace. E’ un processo reversibile. In questo tipo di inibizione diminuisce l’affinità a legare il substrato (aumenta quindi Km) e aumenta anche Vmax. Entrambi i valori aumentano in maniera proporzionale e infatti, il loro grafico dei doppi reciproci presenta tre rette parallele. CAPITOLO 7.11.3: INIBIZIONE MISTA In questo tipo di inibizione, l’inibitore si lega sia al complesso enzima-substrato sia all’enzima libero nel sito attivo (cioè o sta insieme al substrato o da solo). E’ un processo reversibile. Variano sia l’affinità nel legare il substrato (aumenta quindi Km), sia la Vmax ma in modo diverso (non più proporzionale quindi come nell’inibizione incompetitiva). Il grafico dei reciproci evidenzia che le tre rette si intersecano nel quadrante negativo dell’asse delle ascisse e con ordinata positiva. Esistono alcuni inibitori misti che non fanno variare Km, in questo caso si parla di inibizione non competitiva, e varia solamente la Vmax. CAPITOLO 7.11.4: INATTIVAZIONE IRREVERSIBILE Questo tipo di inibizione vede la modifica dei gruppi R dell’enzima (cioè l’inibitore si lega covalentemente all’enzima e ne modifica il gruppo funzionale essenziale per la sua attività), questo modifica l’affinità con il substrato in modo irreversibile. Le molecole di enzima non inattive presentano la stessa affinità per il substrato. Il grafico dei doppi reciproci è uguale a quello dell’inibizione non competitiva solo che questo processo non è reversibile e l’enzima, dopo questo, è da buttare. 39 CAPITOLO 7.12: MECCANISMI DI REGOLAZIONE Esistono altri meccanismi di regolazione dell’attività enzimatica che le cellule di un organismo mettono in atto per poter coordinare tutti i numerosi processi metabolici. Quelli che riguardano il controllo dell’attività enzimatica sono (vedi dopo per dettagli): Meccanismi di inibizione retroattiva (a feedback), cioè quando una molecola è presente in grande quantità, non c’è più bisogno che si sintetizzi, quindi la stessa molecola va a bloccare il processo enzimatico andando a inibire il primo enzima della reazione (prodotto della reazione che inibisce la reazione stessa, si inibisce sempre il primo enzima o quello che richiede più energia al fine di evitare sprechi e la formazione di prodotti parziali in caso di interruzione a metà della reazione) Utilizzo di regolatori allosterici (cooperatività), molecole che vanno in altri siti dell’enzima regolando l’attività con il loro legame, aumentano o diminuiscono l’attività enzimatica Modificazioni covalenti reversibili, fatte da altri enzimi (chinasi, enzima che modifica enzima) Modificazioni per maturazione proteolitica da un precursore inattivo, cioè sono enzimi che si attivano solo in particolari situazioni, sono già presenti nel corpo, inattivi, sottoforma di zimogeni che vengono maturati all’occorrenza e diventano enzimi attivi, la loro maturazione è regolata da ormoni Un altro meccanismo è quello del controllo della disponibilità di un enzima (cioè viene regolato il processo di sintesi dell’enzima stesso che viene prodotto o no a seconda della situazione, oppure viene regolata la sua post-traduzione, cioè l’enzima non subisce le modifiche post-traduzionali fondamentali per la sua funzione) Nel metabolismo cellulare, gli enzimi spesso agiscono in sequenza in una via definita metabolica (cioè nelle reazioni a catena, il prodotto di un enzima e il substrato di quello dopo e cosi via). Si inibisce quasi sempre il primo enzima in quanto è quello regolatore di tutto il processo, infatti esiste una gerarchia tra le varie reazioni di una via metabolica. CAPITOLO 7.12.1: INIBIZIONE RETROATTICA (A FEEDBACK) Lo stesso prodotto della reazione blocca la reazione stessa, avviene quando il prodotto è già presente in grandi quantità nella cellula e sarebbe superfluo continuare a produrlo. Il prodotto si lega al primo enzima della reazione inibendolo e quindi comportandosi come modulatore allosterico (cioè si lega in un altro sito dell’enzima, non nel sito attivo, ma il suo legare influenza l’attività enzimatica diminuendo l’affinità di legame con il substrato). 40 CAPITOLO 7.12.2: REGOLAZIONE ALLOSTERICA E’ un meccanismo di controllo dell’attività enzimatica eseguito da un modulatore. Il modulatore può essere omotropico se è uguale al substrato (tipo quando è presente molto substrato) o può essere eterotropico se il modulatore è diverso dal substrato (si parla infatti di enzimi omotropici o eterotropici). Questi modulatori modificano l’affinità di legame con il substrato in modo negativo (inibitore allosterico) o positivo (attivatore allosterico), legandosi in un altro sito dell’enzima e per effetto di questo legame, varia l’affinità del sito attivo. Negli enzimi con struttura quaternaria, il legame tra questi modulatori e l’enzima, all’inizio è generalmente più difficile ma, una volta avviato il processo, essi cooperano e quindi diventa sempre più facile il legame tra queste due componenti causa cooperatività di legame. A causa di questa cooperazione, il grafico di questa regolazione è sigmoidale (un grafico del genere identifica sempre una proteina allosterica). Sono attivatori o inibitori allosterici l’ATP (attivatore) e il CTP (inibitore, citidina trifosfato che sarebbe il prodotto finale della reazione che vedremo qui di seguito). In questo tipo di regolazione, si evidenziano negli enzimi, sub-unità catalitiche e sub-unità regolatorie. Un esempio di regolazione allosterica si ha nell’aspartato transcarbamilasi (ATCasi). Questo enzima, è coinvolto nella prima tappa della biosintesi delle basi pirimidiniche, il cui prodotto finale è il CTP. L’ATCasi è un enzima costituito da 12 sub-unità (6 cataliticamente attive e 6 cataliticamente inattive, in questi ultimi si legano i modulatori e creano l’effetto allosterico). Ogni sub-unità cataliticamente attiva, può assumere due forme: forma T o forma R (tesa o rilassata come l’emoglobina, forme presenti in tutti gli enzimi allosterici, i modulatori quindi stabilizzano una delle due forme, accelerando o rallentando l’attività enzimatica (cioè il modulatore stabilizza la forma che fa quella funzione, cosi regola l’attività) CAPITOLO 7.12.3: MODIFICHE COVALENTI Sono modifiche covalenti reversibili dei gruppi R di uno o più residui amminoacidici che compongono l’enzima. Si modificano solo gruppi particolari (gruppi fosforici, acetilici, adenilici, uridilici, metilici, ammidici ecc.) e la modifica comporta una variazione dell’attività dell’enzima che può sfavorire o favorire la reazione catalizzata. Vengono modificati solo gruppi R di particolari sequenze. 41 Questa modifica è fatta da enzimi particolari detti chinasi, un tipo di modifica fatto da queste chinasi è la fosforilazione fatta dalle fosfatasi che rimuove gruppi fosforici (è la modifica più importante che le chinasi fanno). CAPITOLO 7.12.4: ATTIVAZIONE PROTEOLITICA DEGLI ZIMOGENI (ATTIVAZIONE/MATURAZIONE ZIMOGENI) E’ la conversione irreversibile di un enzima dallo stato inattivo (zimogeno o proenzima) alla forma attiva mediante idrolisi di pochi o anche di un solo legame peptidico (cioè lo zimogeno viene tagliato di una parte grazie all’idrolisi e diventa l’enzima corrispondente attivo). E’ un processo che si attiva solo quando ci sono particolari condizioni e quindi solo quando c’è bisogno di quell’enzima che deve catalizzare una particolare reazione. Questi enzimi sono sempre presenti nel corpo sottoforma di zimogeni e quindi sono inattivi (i composti che finiscono sempre per “-ogeno” sono sempre inattivi). La maturazione di questi zimogeni viene fatta da un altro enzima. Questo meccanismo di regolazione si verifica: Negli enzimi digestivi (chimotripsina, tripsina, pepsina) Negli enzimi della coagulazione del sangue, hanno una riposta rapida in seguito a un trauma o un danno (la coagulazione si serve di questo meccanismo in quanto non deve sempre produrre trombina, che è un coagulante, in quanto questa proteina creerebbe trombosi di sangue e quindi ci farebbe morire, deve quindi attivarsi solo quando serve per fermare l’emorragia). Questa avviene con la cascata della coagulazione del sangue che vede tanti zimogeni diventare attivi, è quindi un processo a catena Negli enzimi coinvolti nella morte cellulare programmata o apoptosi L’antitrombina, sempre presente nel sangue, si lega o alla trombina inibendola, o si lega all’eparina che fa aumentare di oltre 50 volte la sua affinità con la trombina, favorendo quindi ulteriormente l’anti-coagulazione del sangue. CAPITOLO 7.12.5: ISOENZIMI Gli isoenzimi/isozimi sono forme multiple di uno stesso enzima, che quindi è presente in varie forme nel nostro organismo (sono cioè enzimi che fanno e catalizzano la stessa reazione, variano solamente come struttura primaria e possiedono valori di Km e Vmax diversi e proprietà regolatrici diverse). Gli isoenzimi sono espressi in forma tessuto specifica, cioè hanno una forma particolare a seconda di dove si trovano (in un tessuto specifico, in un organello o in uno stato embrionale preciso, tipo l’emoglobina che si evolve dopo la nascita). In pratica, sono enzimi che fanno la stessa cosa ma hanno forma e caratteristiche diverse a seconda di dove si trovano o in che stato embrionale sono presenti, giustificano il fatto che diversi enzimi catalizzano la stessa reazione. 42 CAPITOLO 8: BIOENERGETICA E’ lo studio dei fenomeni termodinamici applicati alle reazioni biochimiche, è l’energetica dei sistemi biologici, studio del metabolismo con le leggi della termodinamica applicate alle reazioni biochimiche. Essendo i sistemi biologici, soprattutto quello umano, omeotermico (o isotermico, cioè con temperatura costante), questi sistemi utilizzano solo energia chimica per compiere i processi vitali. I sistemi biologici quindi, seguono le leggi generali della termodinamica. La spontaneità di un fenomeno/reazione (cioè la velocità con la quale questa avviene naturalmente) tiene conto sia della variazione totale di energia (entalpia, ∆H) sia della variazione dello stato totale di disordine (entropia, ∆S). Questi due parametri si bilanciano tra loro (cioè se uno sale, l’altro scende e viceversa, questo non fa variare la temperatura) e concorrono alla definizione di variazione di energia libera di Gibbs (∆G): ∆G = ∆H - T∆S Con: ∆H = calore in gioco T = temperatura costante, valore sempre positivo e preciso (sarebbero 37° gradi celsius espressi in Kelvin, quindi 310,15K) ∆S = grado di disordine totale Se il ∆G < 0 allora il fenomeno sarà spontaneo quanto più il valore è basso (tipo ∆G = -100 allora reazione spontanea). Questi fenomeno avvengono con una diminuzione di energia libera e vengono definiti esoergonici. Viceversa, se ∆G > 0 allora l’energia libera tenderà ad aumentare, questo avviene nei fenomeni NON spontanei e sono definiti endoergonici. Se ∆G = 0 allora il sistema è in equilibrio chimico e non evolve, cioè non succede niente. Per una reazione chimica, il ∆G è correlato alla costante di equilibrio (Keq) secondo la seguente relazione: ∆G = ∆G° + RT ln Keq Con: Keq = valore costante, mai negativo, o è molto alto o è molto basso ∆G° = variazione di energia libera nello stato standard (cioè in condizioni ottimali, cioè allo stato fermo, con concentrazione di sostanze unitaria e temperatura di 25° gradi) Queste condizioni standard però, nei sistemi biologici sono leggermente modificate, date le caratteristiche di questi. Deve esserci infatti, presente obbligatoriamente un pH = 7 (cosa non possibile con concentrazione unitaria), una concentrazione di acqua costante e una temperatura che va dai 37° ai 40 gradi°. Pertanto, la variazione di energia libera standard nei sistemi biologici si indica con ∆G°’ (l’equazione di sopra quindi diventa ∆G = ∆G°’ + RT ln Keq) (cioè il ∆G si adatta al sistema). 43 Quando una relazione raggiunge l’equilibrio, la variazione di energia libera (∆G) sarà nulla, per cui: ∆G = ∆G°’ + RT ln Keq = 0 ∆G°’ = -RT ln Keq Per molte delle reazioni metaboliche, questo ∆G = 0 è la condizione normale, per cui le variazioni delle concentrazioni dei componenti regolano il decorso della reazione, cioè nel caso di una reazione biochimica, un aumento della concentrazione del substrato determina la sua trasformazione in prodotto e viceversa (cioè all’aumentare del substrato aumenta il prodotto e viceversa). L’azione di un enzima in questo caso, è quella di accelerare il raggiungimento dello stato di equilibrio. Per altre reazioni biochimiche invece, le concentrazioni hanno valori molto diversi da quelli previsti per l’equilibrio, è il caso delle reazioni irreversibili che hanno un ∆G << 0 (cioè molto basso o molto maggiore di 0). Non si ragiona quindi più sul sistema di equilibrio e, in questo caso, anche aumentando le concentrazioni di substrato non varia l’effetto della catalisi che non sarà sufficiente, e quindi porterà ad un accumulo di prodotti (se ∆G molto maggiore di 0, anche aumentando il substrato la reazione non avverrà in quanto non è spontanea e farà accumulare substrato). Sia il substrato accumulato che il prodotto accumulato fungeranno quindi da inibitori della reazione. Il decorso di queste reazioni, può avvenire solo con una variazione di attività dell’enzima (mediante modulazione allosterica, rimozione di eventuali inibitori o aggiunta attivatori). Questo tipo di reazioni sono circa 50 e si trovano tutte in punti cruciali delle vie metaboliche in quanto conferiscono alle vie una direzione precisa. In generale, i processi vitali che richiedono energia (sintesi, trasporto, contrazione ecc.) prendono l’energia necessaria attraverso l’accoppiamento con reazioni esoergoniche ed endoergoniche (cioè accoppiano reazioni collegate energeticamente, quindi con un punto in comune, al fine di conferire l’energia necessaria al processo vitale). Si accoppiano un processo favorevole e un processo sfavorevole (cioè uno con ∆G positivo quindi non spontanea e una con ∆G negativo che però deve essere superiore come valore, a quello positivo [esempio: ∆G = +25 e ∆G = - 49, ci deve stare un refuso negativo]), questi processi, come detto, devono avere un punto della reazione in comune. I meccanismi di accoppiamento tra reazioni esoergoniche ed endoergoniche sono due: In un primo caso abbiamo la presenza di un intermedio comune (punto in comune della reazione) che partecipa ad entrambe le reazioni ed è specifico per ogni coppia di reazioni, quindi: A + C I B + D (con I che può essere uno stato di transizione tra le due fasi, quindi invisibile, o un prodotto reale) Nell’altro caso abbiamo invece la sintesi di un intermedio ad alto contenuto energetico durante la reazione esoergonica che trasferisce successivamente questo potenziale alla reazione endoergonica (cioè potenziale che passa dalla prima alla seconda reazione grazie alla sintesi dell’intermedio). In questo caso c’è il vantaggio che il composto intermedio piò esistere indipendentemente dalle due reazioni accoppiate (cioè non è il prodotto di una delle due per esempio, già è presente nell’organismo). Questo intermedio ad alto contenuto energetico può quindi essere utilizzato da più reazioni endoergoniche. Il principale composto ad alta energia (intermedio) nei sistemi biologici è l’adenina trifosfato (ATP) 44 Vediamo, nella figura che segue, degli esempi di reazioni accoppiate (quindi reazioni con un punto in comune, l’intermedio appunto che però può anche essere indipendente): Nel primo esempio, l’intermedio è il gruppo fosfato (Pi) che nella prima riga si accoppia al glucosio mentre nella seconda viene separato dall’ATP, quindi le due reazioni si possono accoppiare perché c’è una reazione che ne ha bisogno e l’altra che lo cede (prodotto di una che diventa substrato dell’altra). Il ∆G°’ rappresenta l’energia che si libera nell’idrolisi e quando si aggiunge acqua. CAPITOLO 8.1: FORMULA DI STRUTTURA DELL’ATP L’ATP, come si vede dal grafico qui sopra, è formato da adenina (si distingue grazie all’NH2 in alto), da uno zucchero (ribosio, per la precisione beta di ribofuranosio) e da 3 gruppi fosfato che si legano con un legame fosfoestereo (cioè legame che si instaura tra due acidi) al C5 dello zucchero, e tra loro sono legati con legami fosfoanidridici. Il legame fosfoestereo è molto più stabile di quelli fosfoanidridici, questi ultimi infatti, sono definiti ad alto contenuto energetico in quanto la loro idrolisi (cioè la loro rottura), è altamente esoergonica. 45 Le reazioni che si possono avere con l’idrolisi di questi legami, a seconda di quale legame fosfoanidridico si rompe, sono: ATP ADP + Pi con ∆G°’ = - 30,5 kJ/mol ATP AMP + PPi con ∆G°’ = - 32,2 kJ/mol PPi 2Pi con ∆G°’ = - 33,5 kJ/mol Esistono anche altri composti fosforilati, vediamoli di seguito in ordine di energia che posseggono (in rosso le parti che li contraddistinguono): Fosfoenolpiruvato 1,3-Bisfosfoglicerato come formula totale) Fosfocreatina (parte specifica OPO3-2) (parte specifica OPO3-2 ma più lunga Questi erano i composti fosforici ad alta energia. Dopo questi, sempre in ordine di energia, c’è l’ATP, e a seguire i composti fosforici a bassa energia che sono (sempre in ordine di energia): Glucosio-6-fosfato Glicerolo-3-fosfato CAPITOLO 8.2: FOSFOCREATINA Nei vertebrati, la fosfocreatina viene sintetizzata a partire dall’ATP secondo la seguente reazione, catalizzata dall’enzima creatina chinasi: Creatina + ATP fosfocreatina + ADP con ∆G°’ = +12,6 kJ/mol Come si vede dal valore di ∆G°’, questa NON è una reazione spontanea (perché ∆G°’ maggiore di 0). Questo però avviene solo in condizioni standard, cioè non è spontanea solo con le condizioni standard non adattate. Nella realtà infatti, causa concentrazioni intracellulari dei reagenti e dei prodotti, questa reazione si trova in condizioni di equilibrio e ha un ∆G simile a 0, quindi basta regolare le concentrazioni dei reagenti e dei prodotti per farla avvenire (cioè le loro concentrazioni relative regolano il decorso della reazione. Quindi si osserva che: Se la quantità di ATP nella cellula è alta, allora viene sintetizzata la fosfocreatina Se la quantità di ATP nella cellula è bassa la fosfocreatina genera ATP 46 La fosfocreatina infatti, come detto, può anche essere usata per la biosintesi dell’ATP, trasformandosi in creatina è diventando ATP unendosi all’ADP (cioè se c’è poco ATP si fa la reazione “ADP + fosfocreatina” che genera creatina e ATP appunto). Se invece l’ATP è già presente in grande quantità si sintetizza normalmente fosfocreatina con la formula detta ad inizio capitolo. La fosfocreatina quindi, mantiene costante la concentrazione di ATP nei tessuti che contengono creatina chinasi (muscoli e nervi). Se c’è troppa fosfocreatina, questa viene espulsa dal nostro corpo sotto forma di creatinina, questa espulsione porta ad un accumulo di liquidi. CAPITOLO 8.3: ALTRI COMPOSTI AD ALTO CONTENUTO ENERGETICO: I TIOESTERI DERIVATI DAL COENZIMA A (CoA) Il cuore di questo capitolo è di questi composti, come si evince dal titolo, è l’acetil-coenzima A (CoA). Come si vede dalla figura qua in alto, l’acetil-coenzima A è composto da: Adenina, ribosio e tre gruppi fosfati (è la parte in nero a destra; il fosfato in più è quello legato al C3 che permette già di individuare una caratteristica chiave di questo coenzima) Un gruppo acetile collegato a un atomo di zolfo (S) del residuo di beta-mercaptoetilammina con legame tioestereo (è la parte rossa in alto, altra caratteristica chiave per identificarlo) Residuo di acido pantotenico o vitamina B3 Questo coenzima ha energia in quanto, la rottura del legame tioestero tra il gruppo acetile e l’atomo di zolfo, libera la stessa energia che libera l’idrogenazione/idrolisi di un legame dell’ATP (- 31,5 kJ/mol). Dalla figura, precisamente in basso a destra, osserviamo anche i derivati dell’acetilcoenzima A (succinil-CoA, propionil-CoA e acil-CoA). 47 CAPITOLO 8.3.1: SINTESI DEI COMPOSTI AD ALTO CONTENUTO Negli organismi aerobi, si ottengono attraverso l’ossidazione dei carburanti metabolici (carboidrati, grassi, amminoacidi ecc.) con consumo di O2 e produzione di CO2. Negli organismi anaerobi, si ottengono attraverso l’ossidazione diretta di specifiche molecole. In ogni caso, sono le reazioni di ossido-riduzione/redox che forniscono la maggior parte dell’energia libera necessaria alle reazioni biochimiche e quindi ridurre questi composti. Gli equivalenti riducenti (gli elettroni e l’idrogeno che si liberano grazie all’ossidazione), vengono trasferiti da una molecola all’altra, fino all’accettore finale attraverso alcuni trasportatori di elettroni (per noi è l’ossigeno). CAPITOLO 8.3.2: I TRASPORTATORI DI ELETTRONI I trasportatori di elettroni più comuni sono quattro coenzimi nucleotidici: FAD, FMN, NAD+ e NADP+. Vediamoli nel dettaglio: Per riconoscere il FAD si vede la parte a sinistra (quella in nero) cioè il residuo isoallossazinico e il ribitolo che insieme formano la riboflavina o più comunemente nota come Vitamina B2 (ecco perché importante assumere questa vitamina in quanto è proprio lei a far sintetizzare i FAD). L’FMN ha la stessa struttura del FAD solo che non ha l’adenosina-fosfato (parte rossa in figura) In questi due coenzimi flavinici (FAD e FMN, flavinici perché hanno la riboflavina), gli elettroni e i protoni si legano al residuo isoallossazinico per essere trasportati, quindi è proprio in questa parte che avviene la funzione energetica. Questo residuo piò accettare 1 o 2 elettroni in maniera reversibile. Entra un elettrone per volta e ne cambia la forma. La forma più ridotta di questi (idrochinonica) è il FADH2 o il FMNH2 (l’ossidazione/riduzione avviene gradualmente e, come si vede 48 dalle formule, oltre ai 2 elettroni, questi coenzimi possono accettare anche fino a 2 idrogeni [H]). La forma intermedia è quella semichinonica (FADH e FMNH, quindi con un solo idrogeno accettato, quando accetta il secondo diventa la forma ridotta idrochinonica detta prima). Il NAD (nicotinammide adenin-dinucleotide), ha la seguente struttura: Come si vede dalla figura, presenta nella parte alta (quella in nero), la nicotinammide che accetta gli elettroni e che è una vitamina derivata, mentre nella parte bassa (quella rossa) ha l’adenosina. Nel NADP+ l’unica differenza rispetto al NAD è la presenza di un fosfato al posto dell’idrogeno (è quello cerchiato in basso a sinistra della parte rossa). Questi coenzimi pirinidinici (NAD e NADP+) è, come detto, la porzione nicotinammidica che può accettare fino a 2 elettroni, legando in maniera reversibile uno ione idruro. Questi coenzimi hanno una sola forma ridotta (NADH) e possono accettare quindi, oltre ai 2 elettroni, anche un solo idrogeno (H). Negli organismi aerobi (come l’uomo), l’ossigeno può accettare solo elettroni non accoppiati (cioè uno alla volta), per cui c’è bisogno di altre molecole che possono trasportare un elettrone per volta, per esempio il FAD o l’FMN, non bastano quindi solo NAD e NADP +. Come detto, gli equivalenti riducenti (elettroni e idrogeni) trasportati dai coenzimi ridotti, vengono utilizzati dalle cellule in diversi processi metabolici. I coenzimi flavinici ridotti e il NADH vengono riossidati a FAD, FMN e NAD+ a spese dell’ossigeno nel processo di fosforilazione ossidativa in seguito a processi catabolici. In questo processo, che è l’ultimo della respirazione cellulare, vengono prodotte circa 3 molecole di ATP per ognuna di NADH ossidata e 2 da ogni FADH2 (cioè i coenzimi ridotti tornano nella loro forma originale e facendolo producono ATP). Il NADPH (NADP ridotto) viene utilizzato nei processi anabolici come fornitore di equivalenti riducenti nelle reazioni redox di sintesi di molecole e/o precursori di macromolecole. 49 I due gruppi di coenzimi ridotti vengono prodotti attraverso processi metabolici diversi (cioè NADH, NADPH e FADH2 si ottengono in modo diverso in base alla loro destinazione): FADH2 e NADH, destinati alla fosforilazione ossidativa, si ottengono con il catabolismo di carboidrati, lipidi e amminoacidi, cioè con la degradazione di questi NADPH si ottiene con la “via dei pentosi-fosfato” PER RICAPITOLARE: Nella parte alta (in rosso) si osserva come, composti ricchi di energia vengono degradati per creare i composti finali privi di energia, questo passaggio fa ridurre i coenzimi (catabolismo, da molecole a composte a più semplici). Nella parte bassa (in blu) invece avviene l’opposto, con le molecole precursori vengono usate per formare macromolecole cellulari (anabolismo, da più semplici a molecole composte), in questa fase i coenzimi tornano alla loro forma originale riossidandosi. CAPITOLO 9: I CARBOIDRATI I carboidrati sono i composti organici più comuni in natura. Sono noti anche come zuccheri, saccaridi, glucidi o idrati di carbonio. Tale nome deriva dal fatto che hanno formula generale: Cn (H2O)m Con: “n” ed “m” sempre uguali (minimo 3, massimo 7; lo zucchero più semplice infatti ha formula C3 (H2O)3 C3 H6 O3) I carboidrati sono composti bifunzionali in quanto presentano nella stessa molecola sia un gruppo carbonilico (aldeidico o chetonico), sia almeno due gruppi alcolici (OH) che coinvolgono/si legano, ad atomi di carbonio diversi. 50 Si classificano in: Monosaccaridi Disaccaridi, cioè 2 monosaccaridi legati Oligosaccaridi, cioè da 2 fino a 10 monosaccaridi legati Polisaccaridi, diverse decine/centinaia di monosaccaridi legati insieme I vari monosaccaridi si legano tra loro con legami glicosilici: avviene la disidratazione (cioè viene tolta una molecola di H2O) tra il C4 di uno e il C1 dell’altro (il C4 perde solo H, il C1 perde OH, resta quindi solo una molecola di ossigeno dove appunto avviene il legame tra i due monosaccaridi), questo tipo di legame è detto legame 1-4 glicosilico. Questo però non è l’unico modo in cui più zuccheri uguali si possono legare tra loro: questi infatti, si possono “ramificare” grazie ai legami 1-6 glicosilici (come si può capire, il legame si forma tra il C1 del primo zucchero e il C6 dell’altro). Solitamente, questa ramificazione avviene quando si forma il glicogeno (insieme di più glucosi) e può essere una ramificazione organizzata se avviene ogni 6 residui (cioè ogni 6 unità, in questo caso si otterrà sempre e solo glicogeno), oppure se la ramificazione ha organizzazione diversa (cioè si trova ogni “X” residui/unità) allora si formerà amido. Quando si ha il legame 1-6 nel glicogeno animale, al C4 che sarà libero si legherà una proteina. CAPITOLO 9.1: I MONOSACCARIDI I monosaccaridi possono essere aldosi o chetosi (a seconda se hanno un gruppo carbonilico aldeidico o chetonico [C=O al C2] legato ad essi). I principali monosaccaridi sono: La D-gliceraldeide è la forma aldeidica (gli aldeidi finiscono tutti con il suffisso “-ale”, i chetoni invece con il suffisso “-one”), mentre il Didrossiacetone è la forma chetonica. Il C2 della gliceraldedide, come si può vedere, è chirale (perché ha quattro composti diversi legati ad esso, il carbonio chirale sarà sempre quello più lontano dal C con il gruppo carbonilico, cioè sarà sempre quello più in basso) ed è quello che quindi detta la serie stereochimica (questo vale per tutti gli zuccheri, è sempre il carbonio chirale a definire se è uno zucchero “L” o “D”, il carbonio chirale avrà sempre legato ad esso H, CH2OH e OH), per fare ciò si vede su che lato sta il gruppo OH (se a 51 destra allora “D”, se a sinistra “L” [l’insieme della forma “D” e della forma “L” di uno zucchero forma gli enantiomeri, cioè zuccheri uguali come formula ma diversa posizione dell’OH del carbonio chirale], due zuccheri che variano solo la serie stereochimica sono detti isomeri, cioè stessa formula ma struttura diversa). Tutti gli zuccheri utili sono quelli “D” cioè quelli naturali (quelli quindi con l’OH indicato dalla freccia, sempre a destra). CAPITOLO 9.1.1: I MONOSACCARIDI ALDOTETROSI (PERCHE’ HANNO IL GRUPPO COH ALDEIDICO IN ALTO E QUATTRO ATOMI DI CARBONIO) C4H8O4 La parte nel riquadro, identifica il carbonio chirale che si usa per definire la serie stereochimica. CAPITOLO 9.1.2: I MONOSACCARIDI ALDOPENTOSI (PERCHE’ HANNO IL GRUPPO COH ALDEIDICO IN ALTO E CINQUE ATOMI DI CARBONIO) C5H10O5 Il carbonio chirale è quello evidenziato in rosa. Sono possibili 8 isomeri con questa formula ma noi ne interessa solo uno: è il D-aldopentoso o più comunemente noto come D-Ribosio (vedi capitolo 9.2 per dettagli). 52 CAPITOLO 9.1.3: I MONOSACCARIDI CHETOPENTOSI (PERCHE’ HANNO IL GRUPPO C=O CHETONICO AL C2 E CINQUE ATOMI DI CARBONIO) C5H10O5 In questo tipo di zucchero, cioè quello chetonico (in azzurro in figura), il gruppo carbonile chetonico va sempre al C2, alle due estremità ci sarà sempre il gruppo CH2OH e il carbonio chirale sarà il C4 (quello in rosa). Questo che interessa a noi è il D-Ribulosio (uno zucchero che finisce per “-osio” sarà sempre chetoso). CAPITOLO 9.1.4: I MONOSACCARIDI ALDOESOSI (PERCHE’ HANNO IL GRUPPO COH ALDEIDICO IN ALTO E SEI ATOMI DI CARBONIO) C6H12O6 Il gruppo aldeidico è sempre in alto (in azzurro), il C6 sarà sempre CH2OH, il carbonio chirale è il C5 (in rosa), essendo della serie stereochimica “D”, il C5 dell’OH sarà a destra. Il D-galattosio, come si può vedere anche dalla figura, è uguale come struttura al D-glucosio, l’unica differenza è che il gruppo OH del C4 è a sinistra e non a destra (resta comunque della serie stereochimica D perché si considera solo l’OH del carbonio chirale per questa). 53 CAPITOLO 9.1.5: I MONOSACCARIDI CHETOESOSI (PERCHE’ HANNO IL GRUPPO C=O CHETONICO AL C2 E SEI ATOMI DI CARBONIO) C6H12O6 Come tutti gli zuccheri chetosi, ha il gruppo chetonico al C2, il C6 sarà, come in tutti gli altri zuccheri, sempre CH2OH e il carbonio chirale sarà il C5 (OH a destra perché serie stereochimica “D”). 54 CAPITOLO 9.2: IL D-RIBOSIO E LE SUE STRUTTURE CICLICHE C5H10O5 Il D-Ribosio, come detto, è uno zucchero aldopentoso. Nella figura, è scritto in forma aperta. I suoi gruppi possono interagire tra loro (per esempio, il gruppo del C1 può interagire con il gruppo OH C4 di un altro ribosio): quando succede, avviene la reazione di addizione, cioè da due molecole si passa a una sola molecola (rifacendoci al nostro esempio, ciò avviene con la rottura del doppio legame di CHO del C1 e la rottura del legame tra O e H del C4). Tale interazione può avvenire anche tra gruppi della stessa molecola, questo crea due epimeri, cioè è sempre ribosio ma variano i gruppi legati al C1 e al C4. Se l’O del C1 si troverà a destra allora si avrà l’alfa-D-ribosio, se invece è a sinistra allora si parla di beta-D-ribosio (vedi figura sotto per capire meglio). 55 CAPITOLO 9.3: IL D-GLUCOSIO E LE SUE STRUTTURE CICLICHE Stesso procedimento del D-ribosio, interagiscono gruppi della stessa molecola tra loro. Nel caso del D-glucosio, l’interazione avverrà tra il C1 e il C5 (perché il C5 è quello chirale) poi a seconda della posizione che assumerà l’O trasferito (ricorda, si rompe sempre il doppio legame del C1 e il legame tra O e H del C5) si parla di beta-D-glucosio (se va a sinistra) o di alfa-D-glucosio (se va a destra). CAPITOLO 9.4: COME SI SCRIVE UNO ZUCCHERO E IL PASSAGGIO DALLA FORMA APERTA ALLA RAPPRESENTAZIONE DI HAWORT (FORMA CHIUSA) Per scrivere qualsiasi zucchero in forma aperta, si seguono le seguenti istruzioni: 1. 2. 3. 4. Catena di carbonio uno sotto l’altro Si scrive il C6 che sarà sempre CH2OH Si piazza il gruppo carbonilico che può essere aldeidico (allora va al C1) o chetonico (va al C2) Poi a seconda della serie stereochimica, “D” o “L”, si mettono gli OH e gli H per completare la formula (in caso di serie “D” gli OH vanno a destra e gli H a sinistra) Per passare alla forma chiusa di Hawort: 1. 2. 3. 4. Si parte dall’ossigeno Per ogni carbonio che compone lo zucchero si fa un lato fino a formare il poligono In ogni angolo, che rappresenta l’atomo di C, si mette una linea verticale I gruppi OH e H andranno sopra la linea verticale, se nella forma aperta sono a sinistra, o sotto la linea verticale se si trovano a destra nella forma aperta 56 CAPITOLO 10: LE PRINCIPALI VIE DI UTILIZZAZIONE DEL GLUCOSIO NELLE CELLULE DELLE PIANTE SUPERIORI E DEGLI ANIMALI Il glucosio è il carboidrato principale dal quale si trae energia (in quanto è ricco di energia potenziale), sia con processi catabolici che con processi anabolici. Il glucosio si trova nella matrice extracellulare e come polisaccaride nella parete cellulare, oppure si trova come glicogeno (forma di deposito del glucosio). Dalla sua glicolisi vengono fuori: 2 molecole di piruvato (molecole a tre atomi di carbonio) 4 molecole di ATP 2 NADH Il glucosio si può anche ossidare e al posto della glicolisi, fare la via dei pentosio-fosfati che da come prodotti [VEDI DOPO PER DETTAGLI]: Ribosio-5-fosfato che viene usato durante la duplicazione del DNA, RNA o nel FAD in quanto è costituente di questi CAPITOLO 10.1: IL CATABOLISMO DEL GLUCOSIO – LA GLICOLISI La glicolisi è un processo che si verifica nel citoplasma delle cellule e produce piruvato, ATP e NADH. E’ un processo che è composto da due fasi, ciascuna fase comprende 5 diverse reazioni per un totale di 10 reazioni ognuna catalizzata da un enzima diverso. E’ un processo che avviene anche senza ossigeno. (è l’unica via metabolica che avviene anche senza ossigeno). Il risultato finale, espresso come formula è il seguente: GLUCOSIO + 2ADP + 2P + 2NAD+ 2 PIRUVATO + 2 ATP + 2 NADH + 2H+ + 2 H2O La prima fase è quella preparatoria ed è formata, come detto, da 5 tappe, questa fase consuma 2 molecole di ATP, in questa il glucosio viene scisso in due molecole di griceraldeide-3-fosfato (ovviamente risultato che si ha al termine della quinta tappa). La seconda fase invece, è quella di recupero energetico, anch’essa è composta da 5 tappe. Le due molecole di gliceraldeide-3-fosfato sono convertite in piruvato con produzione di 4 molecole di ATP (2 per ogni molecola di gliceraldeide-3-fosfato) e 2 molecole di NADH. L’ATP prodotto può essere utilizzato per ricavare energia libera mentre piruvato e NADH prenderanno diversi destini a seconda della presenza o assenza di ossigeno (senza ossigeno è fondamentale riportare il NADH a NAD+ al fine di far ripartire la glicolisi e quindi la produzione di ATP) 57 CAPITOLO 10.2: LE 10 TAPPE/REAZIONI DELLA GLICOLISI 1) ESOCHINASI (INIZIO FASE PREPARATORIA) a. Il glucosio viene fosforilato in posizione 6 da parte dell’enzima esochinasi (enzima attivato da ioni di magnesio) con consumo di 1 molecola di ATP che serve a formare il legame fosfoestereo (cioè viene usato 1ATP per fare il legame tra il C6 del glucosio e un gruppo fosfato strappato all’ATP utilizzato). Questo processo trasforma il glucosio in glucosio-6-fosfato (G-6P). E’ una reazione irreversibile. Nelle cellule epatiche, questa reazione viene catalizzata anche dalla glucochinasi (isoenzima della esochinasi, Km basso in quanto è poco affine al glucosio, ecco perché si attiva solo quando il glucosio è presente in grandi quantità). Questo enzima non è un enzima della glicolisi, la sua funzione infatti è mantenere costante il livello di glucosio nel sangue e si attiva solo quando c’è molto glucosio nel sangue (questo enzima prende il glucosio e lo porta nel fegato dove si trasforma in glicogeno e viene immagazzinato) b. Differenze tra esochinasi e glucochinasi: l’esochinasi presenta una più bassa specificità, questo infatti fosforila anche il fruttosio e il mannosio oltre al glucosio; la glucochinasi invece fosforila solo il glucosio (quindi varia la specificità). Un’altra differenza è la cinetica: l’esochinasi infatti ha alta affinità per il glucosio (Km = 0,1 mM), presenta una curva di saturazione come quella di “Michaelis-Menten” e può essere inibito in modo allosterico con il glucosio-6P; la glucochinasi invece, ha un affinità più bassa per il glucosio (Km = 5mM), ha una curva di saturazione sigmoidale (quindi più glucosio c’è più si attiva questo enzima), non viene inibito dal glucosio-6P ed è una proteina monomerica 2) FOSFOGLUCOSIO ISOMERASI a. Il glucosio-6-fosfato viene isomerizzato a fruttosio-6-fosfato (da aldoso a chetoso, da struttura esagonale a pentagonale ma sempre con 6C) da parte dell’enzima fosfoglucosio isomerasi (cioè si passa dal glucosio-6-fosfato al fruttosio-6-fosfato semplicemente modificando la disposizione dei costituenti nello spazio, questi due zuccheri infatti sono isomeri quindi composti dalla stessa cosa ma cambia la disposizione, cioè si apre l’anello del glucosio e si richiude come pentangono). E’ una reazione reversibile e non usa energia. 3) FOSFOFRUTTOCHINASI a. E’ la fase nella quale viene usata la seconda molecola di ATP. Si passa infatti dal fruttosio-6-fosfato al fruttosio-1,6-bifosfato (FBP), aggiungendo, mediante fosforilazione fatta dalla fosfofruttochinasi-1 (PFK-1), un gruppo fosfato strappato all’ATP al C1 dello zucchero. Anche il PFK-1 è attivato da ioni di magnesio. Esiste anche la fosfofruttochinasi-2 (PFK-2) che fa la stessa funzione di quella 1, solo che mette il gruppo fosfato al C2 invece che al C1 dello zucchero creando quindi il fruttosio-2,6-fosfato. La PFK-1 regola la glicolisi (cioè la fa avvenire quando c’è molto glucosio a disposizione, questa regolazione è di natura ormonale, cioè è un ormone che regola l’attivazione/inibizione di questo enzima), la PFK-2 invece fa esattamente la funzione opposta, cioè sintetizza glucosio. E’ un processo irreversibile 58 4) ALDOLASI a. Il fruttosio-1,6-bifosfato viene scisso in gliceraldeide-3-fosfato (GAP, aldosio) e diidrossiacetone-fosfato (DHAP, chetosio) grazie all’enzima aldolasi (si passa dal fruttosio-1,6-bifosfato a GAP e DHAP spezzando il fruttosio con l’aldolasi, cioè si dividono gli atomi di carbonio del fruttosio, si fa quindi la scissione aldolica che divide quindi in due monosaccaridi GAP e DHAP appunto, questi zuccheri sono isomeri). Successivamente, il DHAP diventerà anch’esso GAP nella fase 5 della glicolisi, mentre il GAP passa direttamente alla fase 6. E’ una reazione reversibile 5) TRIOSO FOSFATO ISOMERASI a. E’ la fase nella quale il DHAP si trasforma in GAP. E’ una reazione reversibile e richiede energia blanda. Il passaggio da una forma all’altra avviene riordinando nello spazio i costituenti dei due zuccheri che, come detto, sono isomeri quindi formati dalle stesse cose, questo riordino è fatto appunto dall’enzima trioso fosfato isomerasi. In questo modo si ottengono 2 molecole di GAP (1 avuta nella fase 4 + questa della fase 5) che proseguiranno la glicolisi. Questa è l’ultima reazione della fase preparatoria che si chiude quindi con l’uso di 2 molecole di ATP e la produzione di 2 molecole di GAP (gliceraldeide-3-fosfato), tutta questa fase avviene nel citoplasma. 6) GLICERALDEIDE-3-P DEIDROGENASI (INIZIO FASE DI RECUPERO ENERGETICO) a. Il GAP viene ossidato e fosforilato a causa del NAD+ e di un gruppo fosfato P, entrambe le reazioni sono catalizzate dall’enzima GAP-deidrogenasi (GAPDH). L’ossigenazione toglie due H al GAP, questi due H vengono assorbiti dal NAD+ che diventa cosi NADH, mentre la fosforilazione avviene aggiungendo un gruppo fosfato inorganico P al C1 del GAP che così diventa 1,3-bifosfoglicerato (1,3-BPG, forma ossidata di GAP+P). Questa tappa ospita due reazione reversibili e non usa ATP in quanto il fosfato aggiunto al GAP è inorganico e quindi non prelevato da nessun ATP. 7) FOSFOGLICERATO CHINASI a. L’1,3-BPG si trasforma in 3-fosfoglicerato con produzione di ATP a causa dell’enzima fosfoglicerato chinasi (PGK, anche questo enzima si attiva con gli ioni di magnesio) (questo passaggio avviene perché il fosfato legato al C1 passa a un ADP libero formando quindi la prima molecola di ATP). Questa tappa avviene sempre in conseguenza alla 6 in quanto le due tappe sono reazioni accoppiate (cioè la prima è endoergonica mentre questa è esoergonica, si accoppiano per renderle spontanee, cioè ∆G°’<0). E’ una reazione reversibile anche se il passaggio da 3-fosfoglicerato a 1,3-BPG è più difficile dell’opposto 8) FOSFOGLICERATO MUTASI a. Il 3-fosfoglicerato viene isomerizzato (cioè cambia la disposizione dei suoi costituenti) a 2-fosfoglicerato ad opera dell’enzima fosfoglicerato mutasi (in breve, questo enzima sposta il gruppo fosfato dal C3 al C2, questo passaggio si definisce mutasi). Il passaggio da una all’altra forma è graduale, infatti un intermedio di questa tappa è il 2,3-bifosfoglicerato (2,3-BPG, composto che abbassa l’affinità dell’emoglobina con l’ossigeno) che si ha a metà reazione (viene prima aggiunto il gruppo fosfato al C2 e poi tolto quello al C3, quando entrambi i gruppi fosfati sono legati si ha quindi questo intermedio). E’ una reazione reversibile 59 9) ENOLASI a. Il 2-fosfoglicerato viene deidratato (cioè viene tolto H2O) dall’enolasi, a causa di questa deidratazione quindi si ottiene il fosfoenolpiruvato (PEP, piruvato con gruppo fosfato, composto ad alto contenuto energetico proprio perché ha il fosfato). E’ una reazione reversibile 10) PIRUVATO CHINASI a. Il fosfoenolpiruvato viene idrolizzato dalla chinasi (enzima attivato dagli ioni di magnesio e da quelli di potassio), questa chinasi quindi strappa il fosfato al fosfoenolpiruvato facendolo diventare piruvato. Il fosfato strappato si unisce a una molecola di ADP formando la seconda e ultima molecola di ATP della glicolisi. Questa reazione presenta come intermedio l’enolpiruvato. E’ una reazione irreversibile. Questa è l’ultima tappa della glicolisi ed è quella che da come prodotti le 4 molecole di ATP (due per ogni GAP, infatti la fase di recupero energetico avviene due volte), due molecole di piruvato e due molecole di NADH (sempre due perché la fase avviene due volte, ogni esecuzione produce 1 sola molecola di piruvato e NADH) Come si può osservare, le uniche reazioni irreversibili sono la 1, la 3 e la 10, queste tre reazioni, nel processo opposto della glicolisi, cioè la gluconeogenesi, saranno le uniche a variare mentre le altre 7 saranno identiche a quelle elencate in precedenza solo che avverranno in senso opposto in quanto sono reversibili. Le reazioni 7 e 10 sono quelle nelle quali si produce ATP in quanto c’è il trasferimento di un gruppo fosfato dal substrato a una molecola di ADP già presente, questo passaggio prende il nome di fosforilazione a livello del substrato ed è diversa dalla fosforilazione ossidativa dei mitocondri in quanto, quest’ultima, prende il fosfato dalla catena di trasporto degli elettroni e non dal substrato. CAPITOLO 10.3: I DESTINI METABOLICI DEL PIRUVATO Il piruvato, come detto, è dato dall’ultima fase della glicolisi. Questo dopo la glicolisi può prendere diverse strade: in presenza di ossigeno si trasforma in acetil-coenzima-A ed entra nel ciclo di Krebs, poi nella fosforilazione ossidativa e infine diventa CO2 e H2O (anidride carbonica e acqua), tutti questi processi fatti con ossigeno prendono il nome di glicolisi aerobica che va appunto dalla glicolisi fino alla trasformazione in anidride carbonica e acqua del piruvato. Senza ossigeno invece, il piruvato entra nella fermentazione alcolica diventando etanolo (precisiamo però che questo tipo di fermentazione non è fatto dall’uomo in quanto noi non abbiamo l’enzima necessario). Il piruvato presente nel corpo umano infatti, quando c’è poco ossigeno (cioè non c’è la giusta quantità per farlo diventare acetil-CoA), entra nella fermentazione lattica diventando lattato/acido lattico, non entrando quindi nella glicolisi aerobica ma in quella anaerobica, In questa fermentazione è importante sottolineare che il NADH torna ad essere NAD+, cioè torna nella sua forma ridotta al fine di far ripartire la glicolisi. 60 CAPITOLO 10.4: FERMENTAZIONE OMOLATTICA/LATTICA Come detto, quando la quantità di ossigeno è limitata, tipo durante un’attività fisica intensa in un intervallo di tempo ridotto, il piruvato diventa lattato grazie all’enzima lattico deidrogenasi. Questo avviene perché la quantità richiesta di ATP è elevata ma non c’è l’ossigeno a sufficienza per produrlo. Il piruvato diventa lattato, come detto, grazie all’enzima lattico deidrogenasi che gli da i due H strappati al NADH (facendolo quindi tornare alla forma ridotta NAD +). Questa è una reazione reversibile ma non in modo uguale in quanto passare da lattato a piruvato è più difficile (il lattato può comunque tornare a essere piruvato e lo fa con il Ciclo di Cori che avviene nel fegato, cioè l’acido lattico accumulato nel muscolo viene mandato al fegato che lo trasforma in piruvato, questo avviene perché il muscolo non vuole consumare il NAD+ appena prodotto). La reazione complessiva della conversione del glucosio in lattato è: GLUCOSIO + 2 P + 2 ADP 2 LATTATO + 2 ATP + 2 H2O Non c’è una ossidoriduzione netta, si ottiene però, come detto, la rigenerazione del NAD + che mantiene costante il flusso della glicolisi in condizioni anaerobiche (cioè la fa andare avanti). Se questo NAD+ non fosse rigenerato la glicolisi non andrebbe oltre la tappa 5. CAPITOLO 10.5: LA FERMENTAZIONE ALCOLICA Come detto, l’uomo non ha l’enzima adatto per fare questa fermentazione (l’etanolo infatti può essere solo acquisito bevendo alcool). Questa fermentazione alcolica del piruvato comunque, avviene senza ossigeno nel lievito. E’ un fenomeno indispensabile per la rigenerazione del NAD + a partire dal NADH formato nella glicolisi. Questa reazione produce etanolo e CO2 grazie a due reazioni consecutive: 1. La decarbossilazione del piruvato ad acetaldeide ad opera della piruvato decarbossilasi (cioè il piruvato perde CO2) 2. La riduzione dell’acetaldeide ad etanolo ad opera dell’alcol deidrogenasi e la conseguente rigenerazione del NAD+ dal NADH, questo serve a non bloccare la glicolisi (in pratica, questa reazione vede il NADH perdere un H che, insieme ad un altro H già presente, viene assorbito dall’acetaldeide che quindi diventa etanolo) CAPITOLO 10.6: ALTRE VIE DI ALIMENTAZIONE DELLA GLICOLISI (CIOE’ ALTRI MODI PER AVERE GLUCOSIO O INTERMEDI DELLA GLICOLISI) Il glucosio, oltre che dalla dieta, può anche derivare dalla demolizione del glicogeno, o dagli amminoacidi. Gli amminoacidi infatti, non hanno forma di deposito, cioè se vengono ingeriti vengono subito bruciati, sono dei precursori del glucosio quindi possono creare glucosio e far avvenire la glicolisi (di solito si prendono nelle diete povere di carboidrati o quando si vuole fare 61 massa muscolare, si prendono gli amminoacidi cosi vengono usati questi e non le riserve di glicogeno e quindi si aumenta di massa muscolare, viceversa se si vuole dimagrire bisogna fare un’attività aerobica in quanto deve arrivare più ossigeno ai tessuti adiposi perché l’ossigeno brucia i grassi). CAPITOLO 10.7: LA DEMOLIZIONE DEL GLICOGENO Il glicogeno è la forma di deposito del glucosio, cioè tanti glucosi insieme. Per derivare glucosio da esso quindi, bassa staccare il singolo glucosio, questo strappo si fa grazie all’enzima glicogeno fosforilasi. Il glucosio rilasciato può essere normalmente usato come base della glicolisi. CAPITOLO 10.8: REGOLAZIONE DELLA GLICOLISI Come abbiamo visto, gli enzimi della glicolisi possono essere regolati. Nel dettaglio, la glicolisi è regolata: Regolazione allosterica della fosfrofruttochinasi (PKF), questo enzima si attiva se ci sono AMP, fruttosio o 2,6-bifosfato, mentre si inibisce se ci sono ATP, citrato o se diminuisce il pH, cioè se c’è acido lattico nel muscolo Regolazione della piruvato chinasi, enzima che è attivato quando c’è 1,6-bifosfato, inibito invece da ATP e acetil-CoA Regolazione della esochinasi, enzima inibito dal glucosio-6-fosfato (cioè se ho già questo prodotto l’enzima si inibisce al fine di non crearne altro inutilmente, stessa cosa per le regolazioni precedenti) La glicolisi non serve solo a produrre ATP/energia ma anche ad ottenere molecole intermedie di altri processi metabolici (piruvato, glucosio-6-fosfato e 2,3-BPG). CAPITOLO 10.8.1: REGOLAZIONE DELLA GLICOLISI NEL MUSCOLO Nel muscolo a riposo, non avviene la glicolisi in quanto non c’è bisogno di produrre ATP che è già presente in quantità sufficiente, in questo caso quindi viene inibito il PKF e il piruvato chinasi, è una regolazione negativa perché non produce nulla. Nel muscolo in attività invece, PFK e piruvato chinasi sono attivati dall’AMP e dagli altri attivatori elencati nel capitolo precedente. 62 CAPITOLO 10.9: IL DESTINO AEROBICO DEL PIRUVATO Quando c’è ossigeno, come abbiamo visto, il piruvato, dopo le tante reazioni, diventerà anidride carbonica e acqua. Nel dettaglio: 1. Il piruvato diventa acetil-CoA mediante un processo di cinque fasi 2. La reazione globale viene catalizzata dal complesso multienzimatico della piruvato deidrogenasi, costituito da tre diversi enzimi e cinque diversi cofattori. Gli enzimi del complesso multienzimatico sono (con relativi cofattori accanto in corsivo): Piruvato deidrogenasi (E1) + tiamina pirofosfato (TPP) Diidrolipoil transacetilasi (E2) + lipoammide e Coenzima A Diidrolipoil deidrogenasi (E3) + FAD e NAD+ La reazione globale è: PIRUVATO + CoASH + NAD+ acetil-S-Coa + CO2 + NADH L’acetil-CoA entrerà nel ciclo di Krebs producendo NADH e FADH2 usati poi nella fosforilazione ossidativa. CAPITOLO 11: VIE SECONDARIE DI OSSIDAZIONE DEL GLUCOSIO – LA VIA DEL PENTOSIO FOSFATO/DEL FOSFOGLUCONATO/DELL’ESOSIO MONOFOSFATO Oltre che con la glicolisi, il glucosio può essere degradato anche in un altro modo: con la via del pentosio fosfato (nel titolo del capitolo gli altri due nomi sono sinonimi di questo). La scelta su quale percorso sceglie, glicolisi o via del pentosio fosfato, viene fatta secondo le condizioni in cui si trova il corpo. Questa via da come prodotto finale il ribosio-5-fosfato che è un costituente del DNA, RNA ecc., quindi sarà scelta questa via e non la glicolisi per esempio, quando la cellula è in fase di duplicazione. La via del pentosio fosfato ha le seguenti caratteristiche: Produce un tipo diverso di energia, infatti non produce ATP ma poteri riducenti (NADPH) che poi vengono usati per altre funzioni Questo NADPH è il donatore di elettroni nelle biosintesi degli acidi grassi e degli steroli fatte da ghiandole mammarie, tessuti adiposi, corteccia surrenale e fegato Oltre al NADPH, questa via genera anche zuccheri a 5 atomi di C, cioè il ribosio che abbiamo detto, essere il costituente di ATP, di CoA, DNA e RNA Catalizza l’interconversione di zuccheri a 3,4,5,6 e 7 atomi di C 63 Di seguito uno schema riassuntivo della via del pentosio fosfato Come si vede dalla figura, questa via ha due fasi (di seguito però verrà divisa in tre fasi): una non ossidativa e una ossidativa (in questa seconda fase avviene l’interconversione degli zuccheri). Questa via del pentosio-fosfato è una via di degradazione del glucosio-6-fosfato alternativa alla glicolisi che porta alla formazione di NADPH, necessario nelle sintesi riduttive dei processi anabolici. Avviene in tre fasi e la reazione complessiva è la seguente: 3 G6P + 6 NADP+ + 3H2O 6 NADPH + 3CO2 + 2F6P + GAP La fase 1 prevede la produzione di NADPH La fase 2 prevede la isomerizzazione ed epimerizzazione (per epimerizzazione si intende il passaggio da ribosio a xilulosio) La fase 3 prevede il riarrangiamento dello scheletro carbonioso Vediamo nel dettaglio queste tre fasi: FASE 1 – REAZIONI OSSIDATIVE E PRODUZIONE DI NADPH o Si ossida il NADP che diventa NADPH perché prende due H dal C1 del glucosio-6fosfato grazie al glucosio-6-fosfato deidrogenasi, che quindi lo trasforma in 6fosfoglucono-lattone. L’enzima di questa reazione è specifico per il NADP e viene fortemente inibito dal NADPH e non riduce il NAD+ o Avviene poi l’idrolisi dell’estere ciclico, cioè il 6-fosfoglucono-lattone viene idrolizzato dall’enzima 6-fosfogluconolattotasi e passa dalla forma ciclica chiusa a quella aperta/lineare diventando 6-fosfogluconato (aggiunge anche un OH al C5) o C’è poi la decarbossilazione ossidativa che è l’ultima tappa di questa prima fase, il 6fosfogluconato diventa ribulosio-5-fosfato a causa dell’enzima 6-fosfogluconato deidrogenasi che lo decarbossilizza ossidativamente. Questa tappa produce NADPH (2 in totale nella prima fase) e CO2 64 FASE 2 – ISOMERIZZAZIONE O EPIMERIZZAZIONE o In pratica si formano degli epimeri del ribulosio-5-fosfato che diventa ribosio-5fosfato (R5P che è un precursore essenziale nella biosintesi dei nucleotidi, si ha quando si sta duplicando il DNA) in caso di isomerizzazione fatta dal ribulosio-5fosfato isomerasi (cioè cambia la disposizione di tutti i costituenti del ribulosio-5fosfato che si ridispongono e formano questo ribosio), oppure diventa xilulosio-5fosfato (X5P) in caso di epimerizzazione fatta dal ribulosio-5-fosfato epimerasi (per epimerizzazione si intende la ridisposizione dei gruppi di un solo atomo di C, gli altri restano uguali, varia solo la posizione del gruppo OH del C3 in questo caso). Si produce ribosio quando si è in fase di duplicazione di DNA per esempio, altrimenti si farà xilulosio. Le reazioni successive portano alla sintesi degli intermedi della glicolisi (fase 3 di questa via, cioè il riarrangiamento) FASE 3 – RIARRANGIAMENTO SCHELETRICO CARBONIOSO (FASE OSSIDATIVA) o In questa fase, il ribosio-5-fosfato e lo xilulosio-5-fosfato vengono trasformati in fruttosio-6-fosfato e quindi glucosio-6-fosfato mediante delle reazioni reversibili la figura mostra nel dettaglio il riarrangiamento, cioè il variare del numero di atomi di C (le frecce vanno lette in diagonale, cioè si va da X5P a eptulosio-7-P e così via, vedi figura che segue per dettagli). La transchetolasi aggiunge/rimuove 2 atomi di C, la transaldolasi ne aggiunge/rimuove 3. Tutti gli zuccheri che si formano sono degli intermedi della glicolisi quindi si può affermare che questa via si collega alla glicolisi perché produce gli intermedi necessari a quest’ultima. Sia la glicolisi che la via del pentosio fosfato avvengono in contemporanea, o almeno una avviene più volte dell’altra allo stesso momento. 65 Altri dettagli della terza fase e dettagli di come la via del pentosio fosfato interagisce con la glicolisi: 66 CAPITOLO 12: GLUCONEOGENESI La gluconeogenesi è la via opposta alla glicolisi, si passa infatti da piruvato a glucosio. Come sappiamo, la maggior parte delle funzioni del nostro corpo, sono fatte grazie al glucosio che può derivare dalla demolizione del glicogeno (che vedremo più avanti) o assunto dalla dieta. Se invece siamo a digiuno da molto o abbiamo delle particolari patologie, se le scorte di glicogeno sono al minimo in quanto sono già state usate, per mantenere costante la quantità di glucosio nel sangue, il nostro corpo sintetizza glucosio partendo dagli acidi grassi o da altri precursori, con il processo di gluconeogenesi appunto. Questo infatti, è un processo anabolico di sintesi di glucosio a partire da precursori non glucidici (come i prodotti della glicolisi piruvato e lattato (con il ciclo di Cori), gli intermedi del ciclo di Krebs e lo scheletro carbonioso degli amminoacidi [tutti, soprattutto alanina, tranne leucina e lisina]. Un altro precursore è il glicerolo, un derivato dei trigliceridi. Si verifica essenzialmente nel fegato e, in misura minore, nella corteccia surrenale. E’ un processo energeticamente molto dispendiosi e infatti viene fatto solo quando c’è bisogno di farlo (non verrà mai sintetizzato glucosio se c’è glicogeno nel fegato/muscolo o se la quantità ematica è giusta). Tutti i precursori, prima di entrare nella gluneogenesi, si devono trasformare in ossalacetato (che è anche uno degli intermedi del ciclo di Krebs). La gluconeogenesi, è un processo composto da 10 fasi: 7 sono uguali a quelle della glicolisi ma fatte in modo inverso (sono quelle reversibili in quanto sono quasi all’equilibrio perché hanno ∆G°’=0), mentre le restanti tre, sono sempre come quelle della glicolisi (la 1, la 3 e la 10) ma, siccome queste sono irreversibili per questioni energetiche (∆G°’>>0), vengono fatte in modo diverso rispetto a quelle della glicolisi, cioè vengono usati enzimi specifici che sono: Piruvato carbossilasi (usa ATP, presente nella fase 1 della gluconeogenesi quindi serve per fare la 10 della glicolisi in modo opposto) Fosfoenolpiruvato carbossichinasi (usa GTP, presente nella fase 1 della gluconeogenesi quindi serve per fare la 10 della glicolisi in modo opposto) Fruttosio 1,6-bifosfatasi Glucosio-6-fosfatasi Sono proprio queste tre reazioni ad essere regolate allostericamente e covalentemente al fine di regolare appunto il processo. CAPITOLO 12.1: LE TAPPE DELLA GLUCONEOGENESI 1. PIRUVATO CARBOSSILASI (TRASFORMAZIONE DEL PIRUVATO IN FOSFOENOLPIRUVATO, AVVIENE IN DUE FASI) a. Tutta questa tappa avviene nei mitocondri in quanto, l’enzima piruvato chinasi è presente solo in essi. Nella prima parte di questa, il piruvato diventa ossalacetato assorbendo un gruppo carbossilico da uno ione bicarbonato, questa operazione 67 consuma una molecola di ATP ed è fatta dall’enzima piruvato carbossilasi (enzima che presenta un gruppo prostetico, la biotina, vitamina del gruppo B legata covalentemente a un residuo di lisina e che trasporta CO2). b. L’ossalacetato esce dai mitocondri grazie a dei sistemi navetta (da solo infatti, non può uscire perché non può attraversare la membrana mitocondriale). Questi sistemi navetta, trasformano l’ossalacetato in malato (in questa trasformazione il NADH diventa NAD+) in questa forma esce dai mitocondri e, una volta fuori, torna ad essere ossalacetato (in questa trasformazione il NAD+ torna ad essere NADH) c. La seconda parte di questa tappa è la conversione dell’ossalacetato in fosfoenolpiruvato, cosa che avviene grazie all’azione dell’enzima fosfoenolpiruvato carbossichinasi (PEPCK). Questo enzima, usando una molecola di GTP (che diventa GDP + CO) fa questa operazione, operazione che può avvenire sia nei mitocondri che nel citoplasma. 2. FORMAZIONE DEL FRUTTOSIO-1,6-BIFOSFATO (SAREBBERO LE TAPPE DA 9 A 4 DELLA GLICOLISI) a. Si parte dal fosfoenolpiruvato ottenuto nella tappa precedente e si arriva al fruttosio1,6-bifosfato che verrà usato poi nella prima delle restanti tre tappe. Questo passaggio avviene facendo in modo inverso le tappe da 9 a 4 della glicolisi, che sono tutte reversibili in quanto in stato di equilibrio e la loro direzione quindi è regolata dalla concentrazione dei substrati e dei coenzimi. 3. FORMAZIONE DEL FRUTTOSIO-6-FOSFATO (SAREBBE LA 3 DELLA GLICOLISI CHE VARIA PERCHE’ E’ IRREVERSIBILE) E PARTE FINALE CON SINTESI DI GLUCOSIO (TAPPA 1 CHE VARIA) a. Si parte dal fruttosio-1,6-bifosfato e, grazie al fruttosio-1,6-bifosfatasi (FBPasi-1) si arriva al fruttosio-6-fosfato. b. Successivamente, questo fruttosio-6-fosfato, subisce una trasformazione a causa del fosfoglucosio isomerasi (fase 2 della glicolisi fatta in modo opposto) e diventa glucosio-6-fosfato. c. Questo glucosio-6-fosfato diventa glucosio (nella tappa 1 della glicolisi che varia in quanto irreversibile, per varia ovviamente si intende che viene usato un altro enzima rispetto alla glicolisi) grazie all’azione dell’enzima glucosio-6-fosfatasi (enzima presente anche nella glicogenolisi) Per riassumere, il processo di gluconeogenesi è un processo energeticamente molto dispendioso, in quanto utilizza ben 4 ATP, 2 GTP (ovviamente 1 per ogni molecola di piruvato) e 2 NADH. CAPITOLO 12.2: REGOLAZIONE DELLA GLUCONEOGENESI La sintesi del glucosio e la sua degradazione, sono processi regolati reciprocamente (cioè o avviene uno o l’altro in base alle condizioni). Il processo è regolato dai seguenti due enzimi regolatori: Piruvato carbossilasi, enzima allosterico della prima tappa della gluconeogenesi, ha modulazione positiva indotta dall’acetil-CoA (cioè se c’è molto acetil-CoA vuol dire che si sta lavorando) 68 Fruttosio-1,6-bifosfatasi (FBPasi-1), enzima allosterico regolato negativamente da AMP (forma limite dell’ATP, si ha quando si sta lavorando quindi bisogna bloccare questo processo di gluconeogenesi al fine di non sprecare energia, cioè non bisogna sintetizzare ma demolire glucosio per fare ATP), e positivamente da ATP (quando c’è molto ATP infatti, vuol dire che si è a riposo e quindi si può produrre un po’ di glucosio da accumulare). Questo enzima è anche inibito dal fruttosio-2,6-bifosfato in quanto quest’ultimo, attiva la PFK-1 della glicolisi. La concentrazione di questo fruttosio-2,6-bifosfato è regolato dal PFK-2 e dal FBPasi-2, che sono due enzimi regolati da ormoni (glucagone e insulina). CAPITOLO 12.2.1: REGOLAZIONE ORMONALE DELLA GLUCONEOGENESI Come abbiamo detto, nella regolazione della gluconeogenesi, entrano in gioco anche due ormoni: l’insulina (che fa entrare il glucosio nelle cellule, prodotto subito dopo aver mangiato) e il glucagone. Le fasi della regolazione ormonale della gluneogenesi, a causa del glucagone, sono: 1. C’è bassa concentrazione di glucosio nel sangue 2. A causa di questa bassa concentrazione, c’è un aumento della secrezione del glucagone 3. Di conseguenza aumenta la concentrazione di cAMP che il glucagone produce legandosi al suo recettore 4. Questo cAMP attiva le chinasi che fosforilano, facendo quindi aumentare la fosforilazione enzimatica 5. Si attiva quindi la FBPasi-2 e conseguente inattivazione della PFK-2 6. Di conseguenza diminuisce il fruttosio-2,6-fosfato 7. Viene inibito il PFK-1 (enzima della glicolisi che quindi deve essere stoppata) e si attiva il FBPasi (enzima della gluconeogenesi che si deve attivare) 8. Vista l’inibizione degli enzimi della glicolisi e la contemporanea attivazione di quelli della gluconeogenesi, quest’ultima aumenta sensibilmente e viene fatta CAPITOLO 13: METABOLISMO DEL GLICOGENO Il glicogeno è la forma di deposito e inattiva del glucosio, è specifico della sola cellula animale (in quella vegetale infatti abbiamo l’amido), è un polisaccaride composto solamente da alfa-d-glucosio. Ha forma elicoidale, è molto ramificato e si accumula sottoforma di granuli. Questi granuli sono già presenti anche gli enzimi necessari alla sua decomposizione o alla sua sintesi, oltre a proteine regolatrici. E’ presente maggiormente nel fegato (circa il 10%, questo glicogeno viene distribuito come glucosio a tutto il corpo) mentre è meno dei muscoli (circa il 2%, il glicogeno dei muscoli viene usato esclusivamente dal muscolo stesso, non viene distribuito quindi come quello del fegato). E’ una molecola molto “mobile”, cioè viene sintetizzata e degradata in continuazione. La sintesi del glicogeno (glicogenosintesi, fatta dall’enzima glicogenosintasi quando si è a riposo) inizia dalla glicogenina, cioè si parte dal centro della molecola e poi avvengono delle ramificazioni/arborizzazioni ordinate, cioè si ramifica ogni tot numero di glucosi e ogni ramo 69 contiene lo stesso numero di glucosi. Alle estremità delle ramificazioni si attaccano gli enzimi utili alla degradazione e alla sintesi (estremità non riducenti), infatti è proprio li che inizia la degradazione o la sintesi e anche in modo molto rapido. La demolizione del glicogeno si chiama glicogenolisi ed è fatta dall’enzima glicogenofosforilasi quando si è in attività. CAPITOLO 13.1. RUOLO DEL GLICOGENO La presenza del glicogeno, assicura sempre la presenza di glucosio. Questo infatti viene sintetizzato quando si è a riposo e quindi si può accumulare glucosio per poterlo usare in futuro. Quando si fa attività quindi, questo glicogeno viene demolito e torna glucosio. In breve quindi, la funzione del glicogeno è regolare la concentrazione ematica del glucosio (circa 5mM), molecola fondamentale per molte cellule e funzioni. CAPITOLO 13.2: STRUTTURA DEL GLICOGENO Come si vede dalla figura qua sopra, il glicogeno è formato da tanti glucosi scritti in forma ciclica legati tra loro. Le due estremità non riducenti (cioè dove c’è il C4 libero) sono il sito di attacco degli enzimi che degradano o sintetizzano il glicogeno. La ramificazione del glicogeno avviene tra il C1 e il C6 dell’altro glucosio che si legano con un legame 1-6 glicosilico (in figura, il glucosio verde è il primo della ramificazione). L’estremità riducente è quella con il C1 libero. 70 CAPITOLO 13.3: GLICOGENOLISI (CIOE’ LA DEMOLIZIONE DEL GLICOGENO) Nella figura sopra, ogni colore rappresenta una ramificazione. Per essere demolito, il glicogeno subisce l’azione dell’enzima glicogeno fosforilasi (l’attività di questo enzima è regolata sia allostericamente che covalentemente, la sua regolazione è molto importante), un enzima che stacca i primi residui di glucosio della catena (quelli in fucsia, rompe i legami 1-4 tra i vari zuccheri) e li fosforila aggiungendo loro un gruppo fosfato inorganico libero (ecco perché non usa ATP, idrolizza cioè senza usare ATP). I glucosi che si staccano e subiscono questa azione quindi diventano glucosio1-fosfato. Dopo aver staccato quindi i glucosi fucsia, il processo riprende solamente quando l’enzima trasferasi sposta altri glucosi sulla stessa catena in quanto, la fosforilasi riesce a staccare e formare glucosio-1-fosfato solo se i residui da staccare sono distanti almeno 5 residui dal punto di ramificazione. L’ultimo residuo della ramificazione (quello verde con legame 1-6) viene staccato grazie a un enzima deramificante che, prima sposta i glucosi adiacenti ad esso nella “catena di sotto” (sarebbe il fatto della trasferasi detto prima, cioè sposta i tre glucosi attaccati a quello verde, legandoli all’estremità non riducente dell’altra catena con legame 1-4 glicosidico, attività trasferasica) e poi rompe il legame 1-6 glicosidico del glucosio verde, rilasciandolo come glucosio libero non fosforilato (attività glicosidasica). L’enzima deramificante che fa questa duplice funzione è l’alfa-1-4 transglicosidasi e l’alfa-1-6 glicosidasi. I glucosio-1-fosfato che si liberano nella prima parte, sono molto più vantaggiosi energicamente in quanto entreranno in una fase più avanzata della glicolisi, evitando cosi di consumare un ATP che normalmente verrebbe usato per aggiungere il gruppo fosfato a un glucosio libero (in pratica, avendo già il fosfato attaccato a essi, non ne devono assorbire un altro da un ATP che quindi viene risparmiato, salta la fase 1 della glicolisi). Questo glucosio-1-fosfato quindi, entra direttamente nella fase 2 della glicolisi, trasformandosi in glucosio-6-fosfato con una isomerizzazione fatta dalla 71 fosfoglucomutasi (quindi cambiando solo l’ordine dei suoi costituenti, senza usare energia, processo reversibile), di conseguenza, da un glucosio-1-fosfato si otterranno 3 molecole di ATP (se ne producono sempre 4 in totale ma, a differenza del glucosio semplice, questo tipo di glucosio ne usa solo 1 durante tutta la glicolisi quindi c’è un guadagno netto di 3 ATP e non dei classici 2). CAPITOLO 13.4: DESTINI DEL GLUCOSIO-6-FOSFATO Come abbiamo visto, il glucosio-6-fosfato si forma isomerizzando i glucosio-1-fosfato che vengono liberati demolendo il glicogeno. La figura qui di seguito, oltre a mostrare appunto l’origine di questo glucosio-6-fosfato, ci mostra i vari destini che esso può avere: Si osserva quindi che, il glucosio-6-fosfato può: Entrare nella glicolisi e quindi diventare piruvato (e quindi fare il ciclo di Krebs, la fosforilazione ossidativa e “morire” come acqua + anidride carbonica) o lattato a seconda delle condizioni aerobiche o anaerobiche, entra nella glicolisi dalla fase 2, risparmiando come detto, 1 ATP che sarebbe stato usato dalla esochinasi Entrare nel fegato e, tramite l’azione dell’enzima glucosio-6-fosfatasi, presente solo in questo organo, diventare glucosio (GLUCOSIO-6-FOSFATO + H2O GLUCOSIO + P ) ed essere distribuito in tutto il corpo tramite il sangue, è l’operazione che mantiene costante la concentrazione di glucosio nel sangue in quanto, solo il glucosio semplice può entrare nel circolo sanguigno Entrare nella via del pentosio-fosfato e quindi, dopo aver fatto tutte le fasi di questa via, diventare NADH e ribosio (se entra in questa via, ovviamente non va nella glicolisi e viceversa) 72 CAPITOLO 13.5: GLICOGENOSINTESI E SUE FASI Come si può anche intuire dal nome, la glicogenosintesi, è il processo con il quale si crea il glicogeno. Questa sintesi avviene in modo diverso rispetto alla glicogenolisi ed è stata scoperta studiando la Malattia di McArdle che portava ad accumulare glicogeno in quanto, nei pazienti, mancava il glicogeno fosforilasi (primo enzima che entra in gioco della demolizione del glicogeno). Pur mancando questo enzima infatti, i pazienti accumulavano comunque glicogeno, quindi è capito che la sintesi di questo composto non dipende dalla fosforilasi. Nella glicogenosintesi sono coinvolti tre enzimi che convertono il glucosio-1-fosfato in glicogeno (ricordiamo che il glucosio-6-fosfato deriva dal glucosio-1-fosfato che a sua volta deriva dal glucosio semplice). La glicogenosintesi è un processo endoergonico (non spontaneo perché (∆G°’>0)), quindi necessita di una tappa esoergonica al fine di produrre l’energia necessaria. L’energia necessaria viene fornita dall’idrolisi di una molecola ad alto contenuto energetico, l’uridina trifosfato (UTP) che forma UDP-glucosio (questa operazione è fatta dal primo enzima di questo processo di sintesi: l’UDP-glucosio pirofosforilasi). Dopo di che, i vari UDP-Glucosio che si sono formati, vengono uniti con un legame 1-4 glicosilico che vengono uniti, uno alla volta, a una delle estremità non riducenti del glicogeno. Questa operazione è fatta dal secondo enzima di della glicogenosintesi, la glicogeno sintasi. Questo enzima riesce solo ad allungare le catene polisaccaridiche con almeno 7 residui. Questi 7 residui formano un “primer” che viene sintetizzato, sempre a partire da UDP-glucosio, dal terzo enzima della glicogenosintesi: la glicogenina. Le catene lineari ottenute grazie alla glicogeno sintasi, vengono ramificate grazie all’enzima ramificante (amilo-1,4-1,6 transglicosidasi). Questo enzima trasferisce 7 residui alla volta (quelli del primer) attaccandoli al C6 di un altro glucosio (cioè stacca 7 residui dalla catena lineare e li porta “sopra” ad essa, facendo un legame tra il C1 di un glucosio e il C6 dell’altro, vedi figura a 73 lato per dettagli). Ogni punto di ramificazione, si troverà ad almeno 4 residui dal precedente e ogni segmento di 7 residui contiene almeno 11 unità di glucosio. La funzione d’innesco della sintesi del glicogeno è svolta dalla glicogenina, che è una glicosiltrasferasi composta da 2 sub-unità identiche (cioè tutto parte da lei). La glicogenina catalizza l’aggiunta di 8 unità di glucosio all’altra sub-unità, costituendo due corti polimeri iniziali. CAPITOLO 13.6: REGOLAZIONE DEL METABOLISMO DEL GLICOGENO La regolazione della velocità di sintesi e di degradazione del glicogeno (quindi la glicogenosintesi o la glicogenolisi) viene effettuata: Con controllo allosterico, questo perché i due enzimi, glicogeno fosforilasi e glicogeno sintasi che vengono regolati, hanno dei modulatori allosterici in comune e che regolano la loro attività in maniera opposta (cioè se si attiva uno viene inibito l’altro, vedi dopo per dettagli). Con modifiche covalenti reversibili (fosforilazione, quindi fatta da chinasi, e defosforilazione fatta da fosfatasi) degli enzimi interessati nelle reazioni, cioè la glicogeno fosforilasi e la glicogeno sintasi; queste modifiche sono dettate da degli ormoni come l’insulina, l’adrenalina e il glucagone. Entrambe le regolazioni sono approfondite qui di seguito (capitolo 13.6.1 e 13.6.3). CAPITOLO 13.6.1: CONTROLLO ALLOSTERICO DEL METABOLISMO DEL GLICOGENO I modulatori allosterici coinvolti in questo processo sono: Glucosio-6-fosfato (G-6P) ATP AMP Nei muscoli, un aumento di G-6P e di ATP (quindi quando siamo a riposo e non stiamo sprecando energia) attiva la glicogeno sintasi (per fare glicogeno usabile in futuro) e inibisce la glicogeno fosforilasi, fa ciò stabilizzando la forma T, meno attiva, del glicogeno fosforilasi. Sempre nei muscoli, un aumento di AMP (quindi quando stiamo facendo attività e sprecando energia) inibisce la glicogeno sintasi e attiva la glicogeno fosforilasi (infatti non serve glicogeno ma glucosio da demolire per fare ATP), fa ciò stabilizzando la forma R, più attiva, del glicogeno fosforilasi. Nel fegato, il legame del glucosio stabilizza la forma T della glicogeno fosforilasi, quindi inattiva l’enzima non facendo degradare il glicogeno. 74 Come si può vedere quindi, a seconda dell’attivatore/inibitore presente, il glicogeno viene sintetizzato o demolito in glucosio. CAPITOLO 13.6.2: DETTAGLI SULLA GLICOGENO FOSFORILASI La glicogeno fosforilasi, enzima principale della glicogenolisi, è formata da 2 sub-unità uguali ed è presente in due forme: Fosforilasi A (più attiva), quindi in forma rilassata (R) dove ciascuna sub-unità ha un residuo di Ser14 fosforilato Fosforilasi B (meno attiva), quindi in forma tesa (T), identica a quella A ma con i residui di Ser14 non fosforilati CAPITOLO 13.6.3: CONTROLLO ORMONALE DEL METABOLISMO DEL GLICOGENO Il primo processo, quello a destra, è quello spiegato nel capitolo 12.2.1, cioè quello del glucagone. Il riquadro centrale invece analizza i due ormoni iperglicemizzanti (cioè che aumentano la quantità di glucosio nel sangue): Adrenalina, agisce nel muscolo scheletrico e favorisce la glicogenolisi, abbassando la velocità della glicogenosintesi (quindi fa aumentare la produzione di glucosio e non l’accumulo di glicogeno) Glucagone, anche questo ormone aumenta la glicenolisi abbassando la velocità della glicogenosintesi, agisce nel fegato 75 L’unico ormone ipoglicemizzante (cioè che abbassa la quantità di glucosio nel sangue) invece, è l’insulina che si produce dopo ogni pasto, agisce nei muscoli a livello della ghiandola mammaria, e serve a far entrare il glucosio nelle cellule, facendolo quindi uscire dal sangue. Di conseguenza, questo ormone abbassa la glicenolisi e aumenta la velocità della glicogenosintesi. I segnali ormonali, vengono “captati” dall’apposti recettore e si spostano nella cosiddetta “cascata enzimatica del segnale”, passando da una proteina all’altra. Nella figura che segue, viene illustrato il procedimento di come gli ormoni regolano il processo nel fegato (cellula epatica) e nel muscolo. 76 CAPITOLO 14: I POLISACCARIDI – LA CELLULOSA La cellulosa è un polimero lineare, costituito da molecole di beta-D-glucopiranosio (zucchero a 6C), legate tra loro con legami beta-1-4-glicosidici. Svolge nelle piante la stessa funzione che hanno le proteine fibrose nell’uomo, quindi una funzione principalmente strutturale. Dalla sua idrolisi NON si ricava glucosio. CAPITOLO 14.1: L’AMIDO L’amido è costituito da alfa-amilosio ed amilopectina. L’alfa-amilosio è un polimero lineare formato da molecole di alfa-D-glucopiranosio legate tra loro con legami alfa-1-4-glicosidici. Con la sua idrolisi, fornisce molecole di maltosio e/o glucosio. E’ la principale fonte di carboidrati nella dieta umana e non vede mai idrolizzarsi i suoi legami beta-glicosidici. CAPITOLO 14.2: L’AMILOPECTINA L’amilopectina è composta da polimeri lineari di alfa-amilosio, uniti con legami alfa-1-6-glicosidici. E’ uno zucchero molto meno ramificato del glicogeno, infatti la sua ramificazione si verifica ogni 20/30 residui (nel glicogeno invece avviene ogni 8/12 residui). CAPITOLO 15: I LIPIDI Strutturalmente, i lipidi non hanno gruppi funzionali specifici, cioè possono essere formati da tutti i gruppi funzionali, nessuno escluso. La caratteristica principale dei lipidi è la loro insolubilità in acqua e la loro solubilità nei solventi apolari. Le loro funzioni principali sono: Sono la principale forma di conservazione dell’energia, soprattutto in lipidi come grassi e oli derivati (questi infatti danno molta energia al corpo ma in maniera molto più lenta rispetto agli zuccheri che invece ne danno meno ma più velocemente) Funzione strutturale nelle membrane biologiche, funzione fatta dai fosfolipidi e dagli steroli Sono cofattori e trasportatori di elettroni Agenti emulsionanti, cioè fanno stare insieme sostanze che invece, da sole, si separerebbero Messaggeri intracellulari I lipidi si dividono in: Lipidi non fosforilati Lipidi fosforilati o fosfolipidi 77 CAPITOLO 15.1: I GLICEROLIPIDI Da questi glicerolipidi derivano i glicerofosfolipidi, che sarebbero la loro forma fosforilata (cioè con i fosfati). Questi lipidi sono degli esteri, cioè sono formati da un alcol a 3C (il glicerolo appunto) e da un gruppo carbossilico. La parte alcolica non è lipidica e di conseguenza è idrofilica (sappiamo infatti che i lipidi sono idrofobi, in questo caso il gruppo carbossilico rappresenta la parte idrofoba). CAPITOLO 15.2: STRUTTURA DEI LIPIDI Ogni lipide è formato da: Glicerolo (o 1-2-3-propantriolo), zucchero a tre atomi di C, solubile in acqua in tutte le sue conformazioni/proporzioni quindi idrofilo, è la parte alcolica dei lipidi Acidi grassi, cioè acidi carbossilici a lunga catena carboniosa, possono essere sia saturi (non hanno doppio legame ma solo legami singoli, quindi tutti i C sono ibridati sp3) o insaturi (cioè hanno un doppio legame al loro interno in forma cis, cioè gli H sono tutti dallo stesso lato), in natura sono più numerosi quelli a numero pari di atomi di carbonio, è la parte idrofobica dei lipidi, composta minimo da 7C Fosfati (in caso di fosfolipidi) Colina (si lega al fosfato e crea le lectine, lipidi principalmente vegetali), etanolanina o serina (si legano al fosfato, in caso di lipidi animali o detti anche cefaline) Il glicerolo e gli acidi grassi, si uniscono tra loro grazie all’eliminazione di una molecola d’acqua. CAPITOLO 15.2.1: I PRINCIPALI ACIDI GRASSI I principali acidi grassi saturi sono: Acido Laurico, 12C – CH3(CH2)10COOH (10 perché due C sono già fuori, vale anche per quelli dopo) Acido Miristico, 14C – CH3(CH2)12COOH Acido Palmitico, 16C – CH3(CH2)14COOH Acido stearico, 18C – CH3(CH2)16COOH Acido Arachidico, 20C – CH3(CH2)18COOH I principali acidi grassi insaturi, a 18 atomi di C, sono: Acido oleico – CH3(CH2)7CH=CH(CH2)7COOH (come si può vedere, i 14 atomi che restano fuori dalle parentesi e che bisogna distribuire tra i due CH2, si dividono in modo equo; è insaturo quindi ha il doppio legame), è un acido grasso monoinsaturo perché ha un solo doppio legame Acido linoleico, acido grasso di-insaturo in quanto ha due doppi legami nella sua struttura 78 Acido linolenico, acido grasso tri-insaturo in quanto ha tre doppi legami Ricorda, tutti questi acidi grassi insaturi hanno un doppio legame e sono tutti in forma cis, cioè con gli idrogeni del doppio legame (CH=CH) messi allo stesso lato nello spazio, in quanto sono tutti acidi grassi naturali Gli acidi grassi insaturi rendono più fluida la membrana plasmatica, quelli saturi invece la irrigidiscono. CAPITOLO 15.3: GLICERIDI, DIGLICERIDI E TRIGLICERIDI (SONO TUTTI FOSFOLIPIDI) Quelli che a noi interessano sono principalmente i trigliceridi. Come tutti i lipidi ovviamente, anche loro sono formati da glicerolo, da tre acidi grassi in questo caso e da fosfati. I trigliceridi che contengono in prevalenza acidi grassi saturi sono solidi (grassi), mentre quelli contenenti acidi grassi insaturi sono liquidi (oli). Questi derivano dal glicerofosfato (fosfogliceridi) o dalla sfingosinafosfato (sfingomieline). I trigliceridi si accumulano (principalmente come riserva di energia, ma possono anche accumularsi e poi essere usati per costruire le membrane) e chimicamente non sono tossici ne arrecano danni, causano problemi semplicemente perché, quando diventano troppi, finiscono per occupare tutto lo spazio e quindi chiudere le vene ecc. Per disegnare un trigliceride, si parte dalla catena di 3C del glicerolo, vengono rimossi gli OH a tutti e tre atomi di carbonio e, rimuovendo anche un altro idrogeno dell’acido grasso, quest’ultimo si unisce al glicerolo (possono anche essere tre acidi grassi diversi), si aggiungono poi i fosfati al gruppo estereo per indicare la fosforilazione (in breve, un trigliceride si ottiene legando al glicerolo, tre acidi grassi, anche diversi tra loro + tre fosfati e poi una tra colina, serina e etanolammina a seconda che sia una lectina o una cefalina). CAPITOLO 15.4: I FOSFOLIPIDI I fosfolipidi sono dei lipidi anfipatici, cioè hanno una testa polare costituita dal gruppo fosfodiestere e una coda apolare costituita dalle lunghe catene idrocarburiche. CAPITOLO 15.5: IL METABOLISMO DEI LIPIDI Come detto in precedenza, i lipidi rappresentano la principale riserva energetica per gli organismi superiori (danno energia ma in più tempo, per energia immediata si usano zuccheri), oltre ad avere una funzione strutturale in quanto essi sono uno dei principali componenti della membrana plasmatica. Vengono conservati in cellule specializzate: gli adipociti. Da queste cellule, i lipidi, passano al sangue per diffusione passiva secondo gradiente di concentrazione. 79 I trigliceridi, i fosfolipidi e il colesterolo vengono trasportati dal sangue ai tessuti di utilizzo sotto forma di chilomicroni e/o lipoproteine a causa della presenza di acqua nel sangue (varia la forma in base a come sono fatti, infatti le lipoproteine sono quelle con tutti i grassi endogeni, cioè prodotti dal corpo stesso e quindi specifici, sono quelli che vengono usati per fare le membrane plasmatiche, sono sintetizzate a livello epatico/nel fegato; i chilomicroni invece sono quelli che hanno grassi alimentari/esogeni, ovviamente sono formati anche loro da una parte proteica e una lipidica e si formano in seguito alla digestione dei grassi). CAPITOLO 15.5.1: DIGESTIONE DEI LIPIDI ED ASSORBIMENTO La digestione dei lipidi alimentare (il 90% di questi sono trigliceridi) ed il loro assorbimento avviene a livello intestinale. In questi due processi, i lipidi vengono smantellati e poi ricomposti grazie a enzimi (lipasi pancreatica) e sostanze emulsionanti (sali biliari). La lipasi pancreatica idrolizza i legami esterei dei trigliceridi, smantellando i lipidi e rendendo liberi tutti i suoi costituenti (acidi grassi e glicerolo), questo enzima ha una parte idrofila e una parte idrofobica. I sali biliari invece, sono degli emulsionanti, cioè ricompongono i lipidi. Questi sali derivano dal colesterolo e sono sintetizzati nel fegato. I sali biliari inoltre, rappresentano l’unico modo che le cellule epatiche hanno per eliminare l’eccesso di colesterolo. CAPITOLO 15.5.2: TRASPORTO DEI LIPIDI - CHILOMICRONI Come detto, si trasformano in chilomicroni (per entrare nel sangue), i lipidi che contengono i grassi alimentari, quindi la loro parte lipidica varia a seconda della provenienza del grasso che abbiamo ingerito. La trasformazione in chilomicroni degli acidi grassi, avviene nella muscosa intestinale: i lipidi vengono prima riconvertiti in tracilgliceroli e poi impacchettati in chilomicroni appunto, cioè in particelle lipoproteiche che poi vengono rilasciate nel sangue venoso e contengono anche colesterolo, sempre proveniente dalla dieta. Arrivati ai tessuti di utilizzo (muscolo scheletrico e tessuto adiposo), i trigliceridi contenuti nei chilomicroni vengono idrolizzati/degradati dall’enzima extracellulare lipoproteina lipasi. Durante questa fase, gli acidi grassi che si liberano vengono assorbiti e utilizzati dalle cellule, mentre i residui, ricchi di colesterolo vengono trasportati al fegato che li riutilizzerà per altre cose. Per riassumere quindi, nei muscoli scheletrici e nel tessuto adiposo, viene usata solo la parte dell’acido grassa del lipide mentre il colesterolo in più va al fegato che lo usa per fare ormoni steroidei o sali biliari. 80 CAPITOLO 15.5.3: TRASPORTO DEI LIPIDI - LIPOPROTEINE Le lipoproteine, sono la forma finale dei lipidi che contengono dei grassi specifici endogeni. Sono dei complessi lipoproteici e hanno una parte lipidica ben precisa (perché, come detto, contengono dei grassi endogeni sintetizzati dal corpo stesso nel fegato). Le lipoproteine si possono classificare in base alla loro densità, che si misura attraverso la loro velocità di sedimentazione: VLDL: Very Low Density Lipoproteins (densità molto bassa) LDL: Low Density Lipoproteins (densità comunque bassa ma meno delle VLDL, derivano proprio dalle VLDL) HDL: High Density Lipoproteins (alta densità), queste sono le lipoproteine che contengono pochi grassi e portano il colesterolo dove serve, cioè al fegato Maggiore è il contenuto di grassi, minore è la densità. Apolipoproteina: parte proteica del lipide, quasi totalmente senza grassi. CAPITOLO 15.5.4: CATABOLISMO DEGLI ACIDI GRASSI E PREPARAZIONE ALLA BETA-OSSIDAZIONE Questo processo avviene nella matrice mitocondriale e prende il nome di “beta-ossidazione” che è l’insieme di 4 reazioni che si ripetono ciclicamente. La beta-ossidazione ha come risultati/prodotti finali: Acetil-CoA Coenzimi ridotti NADH e FADH2 La beta-ossidazione è preceduta da una fase che avviene nel citoplasma e che prepara il lipide alla beta-ossidazione; questa fase è composta da due operazioni: 1. Attivazione dell’acido grasso (cioè trasformandolo in Acil-CoA), avviene nel citoplasma 2. Trasporto dell’acil-CoA nel mitocondrio mediante un trasportatore specifico Vediamo nel dettaglio queste due fasi: 1. Come detto, l’attivazione dell’acido grasso avviene nel citoplasma ed è catalizzata dall’enzima acil-CoA sintetasi (sintetasi perchè sintetizza usando energia/ATP, vengono infatti idrolizzati due legami ad alto contenuto energetico). In pratica, l’acido grasso si trasforma in Acil-CoA unendosi al CoA stesso, questa stessa unione impedisce all’acido grasso di uscire dalla cellula. E’ una reazione altamente esoergonica. Come si vede dall’immagine che segue, questo è un processo totalmente irreversibile (solo la prima riga è 81 reversibile ma ci si dovrebbe fermare la, cosa che invece non avviene) 2. L’acil-CoA a questo punto, deve essere trasportato nel mitocondrio per entrare nella “betaossidazione” e lo fa usando pochissima energia. Il passaggio dal citoplasma al mitocondrio dell’acil-CoA avviene grazie a un trasportatore particolare: la carnitina. La carnitina, si lega in modo reversibile agli acili attivati nella fase precedente con un legame estereo OH, formando acil-carnitina (in pratica viene rimosso l’acetil-CoA che quindi non passa dall’altra parte) questa unione è catalizzata dall’enzima citoplasmatico carnitina-acil-trasferasi di tipo 1. Questa acil-carnitina passa così nel mitocondrio dove viene ritrasformata in acil-CoA grazie all’enzima carnitina-acil-trasferasi di tipo 2 che, invece di staccare l’acetil-CoA come quello di tipo 1, lo riattacca agli acili, fatto questo passaggio la carnitina torna indietro. 82 CAPITOLO 15.5.4.1: CATABOLISMO DEGLI ACIDI GRASSI – LA BETA-OSSIDAZIONE Dopo aver visto come gli acidi grassi vengono preparati, vediamo ora nel dettaglio il vero processo di demolizione/catabolismo degli acidi grassi: la beta-ossidazione (beta perché tutte e 4 le sue reazioni sono subite dal carbonio beta, cioè il C3). Come detto in precedenza, questo è un processo ciclico formato da 4 reazioni in cui la molecola di acido grasso, entrata nel mitocondrio, viene ridotta di due atomi di C per ogni ciclo (cioè se entra una molecola con 16C come l’acido palmitico, ad ogni beta-ossidazione questo si riduce di 2C quindi diventa a 14C, 12C ecc. fino alla completa demolizione). Come prodotti finali, la beta-ossidazione da: Acetil-CoA, se ne forma una quantità pari al numero di cicli dell’acido grasso interessato (tipo l’acido palmitico che subirà 7 cicli [non 8 perché il numero di cicli si calciola nC/2 -1], produrrà 8 molecole di acetil-CoA perché anche gli ultimi 2C verranno usati, numero cicli +1) FADH2, se ne forma una quantità pari al numero di cicli (quindi nel caso dell’acido palmitico se ne otterranno 7) NADH, se ne forma una quantità pari al numero di cicli Vediamo ora nel dettaglio nel 4 reazioni che compongono la beta-ossidazione: REAZIONE 1 DELLA BETA-OSSIDAZIONE: ACIL-CoA DEIDROGENASI o L’enzima acil-CoA deidrogenasi catalizza la formazione di un doppio legame attraverso una deidrogenazione tra il carbonio alfa e quello beta dell’acil-CoA facendolo diventare trans-∆2enoil-CoA (trans perché gli H del doppio legame sono in posizione opposta tra loro, ∆2 perché il doppio legame interessa il C2). In questa fase si forma il FADH2. E’ una reazione irreversibile. 83 REAZIONE 2 DELLA BETA-OSSIDAZIONE: ENOIL-CoA IDRATASI o L’enzima enoil-CoA-idratasi catalizza l’idratazione del doppio legame dell’enoil-CoA che si era formato nella reazione 1 (ricorda che questa trasformazione avviene solo se l’enoil-CoA è in forma trans). In pratica si aggiunge H2O al doppio legame facendo ossidare il C3. Si forma 3-idrossiacil-CoA. REAZIONE 3 DELLA BETA-OSSIDAZIONE: IDROSSIACIL-CoA DEIDROGENASI o L’enzima idrossiacil-CoA deidrogenasi, catalizza la deidrogenazione del NAD+, proprio in questa fase infatti si produce il NADH. In pratica vengono tolti due H al 3idrossiacil-CoA che cosi diventa beta-chetoacil-CoA. 84 REAZIONE 4 DELLA BETA-OSSIDAZIONE: BETA-CHETOACIL-CoA TIOLASI o Questo enzima, catalizza la rottura del legame tra C-alfa e C-beta, cioè spacca in due il beta-chetoacil-Coa, scinendolo in acetil-CoA e un acil-CoA ridotto di due atomi di carbonio. Questo enzima catalizzante contiene zolfo e proprio grazie a questo riesce a rompere il legame. CAPITOLO 15.5.5: IL BILANCIO ENERGETICO DELLA BETA-OSSIDAZIONE Tutti i prodotti della beta-ossidazione (acetil-CoA, NADH e FADH2) possono portare alla formazione di ATP se proseguono il loro catabolismo nel ciclo di Krebs e nella fosforilazione ossidativa. Il numero massimo di molecole di ATP che possono essere prodotte dalla ossidazione, dipende dal numero di atomi di carbonio dell’acido grasso (come detto infatti, la quantità di prodotti della betaossidazione dipende dal numero di cicli che l’acido grasso fa). Per esempio, nel caso dell’acido palmitico (acido grasso saturo e con 16C), che subisce 7 cicli di beta-ossidazione, si otterrà: 8 Acetil-CoA, se questi vanno nel ciclo di Krebs, dopo tutto il processo produrranno 80 molecole di ATP 7 NADH, che se vengono usati nella fosforilazione ossidativa, portano alla formazione di 17,5 ATP (7 X 2,5) 7 FADH2, che se vengono usati nella fosforilazione ossidativa, portano alla formazione di 10,5 ATP (7 X 1,5) Da questo si può capire che dalla degradazione degli acidi grassi si può ottenere molto ATP, anche più di quello che si ricava dagli zuccheri, solo che per farlo, ci vuole molto più tempo. 85 CAPITOLO 15.6: OSSIDAZIONE DEGLI ACIDI GRASSI INSATURI I processi che abbiamo visto fino ad ora, interessavano solo gli acidi grassi saturi, cioè quelli senza nessun doppio legame. Quando invece si vanno a ossidare acidi grassi insaturi, c’è un problema: questi infatti non generano una forma in trans ma in cis nella reazione 1 della beta-ossidazione e questo blocca la reazione 2 che, come detto, necessità solo di forme in trans. Bisogna quindi trasformare il composto nella forma in trans e lo si fa tramite un enzima particolare, l’enoil-CoA isomerasi che ne cambia la forma tramite isomerizzazione. Questa trasformazione allunga di una reazione l’intero processo che quindi diventa a 5 fasi. I prodotti sono gli stessi di quella degli acidi grassi saturi solo che si produce un FADH2 in meno. Di seguito, l’esempio dell’acido oleico che rende meglio e chiarisce quanto detto: 86 CAPITOLO 15.7: OSSIDAZIONE DEGLI ACIDI GRASSI A CATENA DISPARI Un altro problema nella ossidazione, si ha quando ci si trova davanti ad acidi grassi con un numero dispari di C: sappiamo infatti che, la beta-ossidazione riduce di 2C alla volta, quindi dopo alcuni cicli, si arriva inevitabilmente ad un composto con 3C e che quindi non può andare avanti nel processo. In questo caso, questo prodotto a 3 atomi di carbonio dato dall’ultimo ciclo della beta-ossidazione è il propionil-CoA, cioè un acile a tre atomi di carbonio legato al coenzima A. Questo propionil-CoA viene poi trasformato in succinil-CoA (un intermedio del ciclo di Krebs), mediante tre reazioni che vediamo di seguito nel dettaglio: REAZIONE 1: PROPIONIL-CoA CARBOSSILASI o In questa reazione, il propionil-CoA diventa D-metilmalonil-CoA, processo irreversibile REAZIONE 2: METILMALONIL-CoA RACEMASI o Il D-metilmalonil-CoA diventa L-metilmalonil-CoA, processo reversibile REAZIONE 3: METILMALONIL-CoA MUTASI o Il L-metilmalonil-CoA diventa succinil-CoA, processo reversibile. L’enzima di questa reazione, utilizza come gruppo prostetico un derivato della vitamina B12. In breve, queste tre reazioni, aggiungono un carbonio (con una carbossilazione) al propionil-CoA nella prima tappa, questo carbonio poi viene spostato nelle tappe 2 e 3 al fine di creare il succinilCoA. 87 CAPITOLO 15.8: ULTERIORE DESTINO DELL’ACETIL-CoA PRODOTTO DALL’OSSIDAZIONE DEGLI ACIDI GRASSI Oltre che per produrre ATP, l’acetil-CoA derivante dalla beta-ossidazione, può essere usato per produrre i cosiddetti corpi chetonici, cioè acetone, acetoacetato e beta-idrossibutirraro. Questi corpi chetonici, si creano quando si accumula molto acetil-CoA. Questo processo è chiamato chetogenesi e avviene nelle cellule epatiche. L’acetone, è la forma di espulsione dell’acetil-CoA, cioè quando c’è ne troppo, viene trasformato in questo modo e viene espulso dal corpo tramite le urine L’acetoacetato e il beta-idrossibutirraro invece sono dei carburanti energetici metabolici, usati quando c’è poco glucosio, da cervello, cuore e muscoli. La formazione di questi corpi chetonici (chetogenesi), è illustrata qui di seguito: Come si può vedere, l’acetoacetato è il punto intermedio e si forma unendo tre acetil-CoA insieme. Una volta in questa forma, può diventare beta-idrossibutirraro ne interagisce con il NADH, se invece interagisce con il CO2 diventa acetone è viene espulso in modo irreversibile. Acetoacetato e beta-ibrossibutirraro, prodotti ovviamente nel fegato con la chetogenesi, vanno nel sangue e arrivano ai tessuti periferici dove vengono usati come fonte di produzione alternativa di ATP. Qui l’acetoacetato viene riconvertito in acetil-CoA attraverso le seguenti reazioni: 88 CAPITOLO 16: BIOSINTESI DEGLI ACIDI GRASSI - LIPOGENESI Questa della lipogenesi è una via anabolica molto dispendiosa dal punto di vista energetico, infatti consuma molto ATP e anche del NADPH. Consiste nella sintesi/produzione di lipidi e avviene prevalentemente nel: Fegato Tessuto adiposo Ghiandola mammaria (solo durante l’allattamento) E’ un processo che si verifica nel citoplasma e utilizza acetil-CoA e NADPH come precursori. Il processo quindi, parte dall’acetil-CoA derivato dalla glicolisi e vede aggiungersi alla catena di acido grasso, ogni volta, 2 atomi di carbonio, donate dal malonil-CoA. La biosintesi si arresta alla formazione dell’acido palmitico, cioè una volta che si è arrivati a 16 atomi di carbonio il processo si ferma. Per continuare ad allungare la catena e fare dei doppi legami in essa, servono altri sistemi enzimatici. CAPITOLO 16.1: DIFFERENZE TRA SINTESI E DEGRADAZIONE (CON BETAOSSIDAZIONE) DEI LIPIDI Come si vede dalla figura, le principali differenze tra i due processi sono la localizzazione (cioè dove avvengono), da dove derivano gli acili, quali sono i coenzimi che entrano in gioco e che gli acidi grassi della sintesi sono legati ad una particolare proteina ACP in quanto si trovano nel citoplasma. 89 CAPITOLO 16.2: FASE PRECEDENTE ALLA BIOSINTESI DEGLI ACIDI GRASSI REAZIONE 1 – SINTESI DEL MALONIL-CoA PARTENDO DALL’ACETIL-CoA e da HCO3o La tappa che precede la sintesi dei lipidi è la sintesi del malonil-CoA (che, come abbiamo detto, è il “donatore” degli acili) a partire dall’acetil-CoA della glicolisi. Questa è una tappa irreversibile ed è quella che controlla tutta la sintesi, senza malonil-CoA infatti non si possono costruire i lipidi. La reazione è: ACETILCoA + ATP + HCO3- MALONIL-CoA + ADP + P + H+ Questa reazione è catalizzata dall’enzima acetil-CoA carbossilasi che contiene biotina e lisina. Questo enzima, assorbe un CO2 quando è ancora biotinilenzima e diventa appunto acetilCoA carbossilasi. Sarà proprio questo CO2 (parte verde in figura) che verrà ceduto all’acetil-CoA che diventerà quindi malonil-CoA CAPITOLO 16.3: IL COMPLESSO DELL’ACIDO GRASSO SINTASI Tutte le fasi della biosintesi degli acidi grassi, avviene mediante la ripetizione di 4 tappe (sono 4 tappe opposte a quella della beta-ossidazione) ed è catalizzata dal sistema multienzimatico dell’acido grasso sintasi (FAS, non usa ATP per funzionare), cioè tutte le reazioni di questa via metabolica avvengono all’interno dello stesso gruppo enzimatico e non con enzimi diversi come nel caso del catabolismo dei lipidi. Questo complesso, nei mammiferi, è composto da 7 differenti domini/parti, ciascun dominio è specifico di una reazione. Durante la sintesi dei lipidi, gli intermedi delle reazioni come detto, vengono legati covalentemente, mediante un legame tioestere, a 2 gruppi tiolici (SH), cioè al gruppo SH della proteina ACP, mentre l’altro SH è dato dall’enzima KS. Questo gruppo SH che viene legato è il sito d’ingresso dei gruppi malonici (dei C) durante la biosintesi, cioè si attaccano la. Quindi, prima dell’inizio delle reazioni di condensazione, avviene il trasferimento dei gruppi acilici dal CoA al gruppo SH, in questo momento l’acetil-CoA diventa acetil-ACP e il malonil-CoA diventa malonil-ACP, questa trasformazione è fatta dall malonil/acetil-CoA-ACP trasferasi). 90 CAPITOLO 16.4: FASI DELLA BIOSINTESI DEI LIPIDI Le due sub-unità uguali del complesso enzimatico, legano insieme il malonil-ACP e la catena di acido grasso in accrescimento (l’acil-ACP). Dopo questo processo, seguono le seguenti reazioni (si ripetono in sequenza, vedi dopo per dettagli): 1. 2. 3. 4. Condensazione Riduzione del gruppo carbonilico Deidratazione Riduzione del doppio legame CAPITOLO 16.4.1: FASI DELLA BIOSINTESI DEI LIPIDI NEL DETTAGLIO REAZIONE 1 – CONDENSAZIONE – FORMAZIONE DI ACETOACETIL-ACP o L’enzima condensante beta-chetoacil-ACP sintasi (KS) entra in gioco in questa prima tappa e forma acetoacetil-ACP. La reazione è. ACETIL-ACP + MALONIL-ACP ACETOACETIL-ACP + ACP + CO2 L’atomo di C della CO2 liberata in questa reazione è lo stesso di quello introdotto nella reazione di carbossilazione per la formazione di malonil-CoA REAZIONE 2 – RIDUZIONE DEL GRUPPO CARBONILICO o L’acetoacetil-ACP formato nella reazione 1, subisce la riduzione del suo gruppo carbonilico presente sul C3, diventando beta.idrossibutirril-ACP, questo passaggio è permesso dall’enzima beta-chetoacil-ACP riduttasi (KR) e dal donatore di elettroni NADPH che si trasforma in NADP+ 91 REAZIONE 3 – DEIDRATAZIONE o In questa razione, dagli atomi di carbonio C2 e C3 del beta-idrossibutirril-ACP viene rimossa una molecola di acqua per formare un doppio legame e far diventare il composto trans∆2-butenoil-ACP (∆2 perché il doppio legame è al C2) REAZIONE 4 – RIDUZIONE DEL DOPPIO LEGAME o Per allungare ulteriormente la catena, interviene l’attività trans-acetilasica che legherà un altro gruppo malonile (proveniente da un’altra molecola di malonil-CoA) al gruppo –SH libero dell’ACP. Per fare ciò, viene ridotto/rotto il doppio legame che si era formato nella reazione 3, creando cosi butirril-ACP, processo fatto dall’enzima enoil-ACP reduttasi, anche in questa fase interviene io NADPH Le suddette 4 reazioni si ripetono ciclicamente (aggiungendo quindi 2C alla catena ogni volta) e, al raggiungimento della lunghezza desiderata interviene la quinta attività dell’acido grasso sintasi: la acil-tioesterasi che catalizza il rilascio dell’acido grasso dall’enzima, cioè ferma il processo e “chiude” la catena. 92 CAPITOLO 16.5: STECHIOMETRIA DELLA SINTESI DEGLI ACIDI GRASSI CAPITOLO 16.6: L’ACIDO PALMITICO (16C) Come detto, la sintesi dei lipidi si ferma dopo 7 cicli, cioè l’ultimo acido grasso che è possibile creare è quello che ha 16 atomi di carbonio, cioè l’acido palmitico. Una volta che si è formato si stacca dal complesso enzimatico per permettere la sintesi un altro acido grasso, ad esso infatti non posso attaccarsi altri C. L’acido palmitico può subire reazioni successive da parte di sistemi enzimatici presenti nel reticolo endoplasmatico liscio: Può essere allungato, aggiungendo altri 2C Può subire insaturazione, cioè può vedere inseriti nella sua catena carboniosa, dei doppi legami CAPITOLO 16.7: ACIDI GRASSI ESSENZIALI L’acido linoleico e l’acido alfa-linolenico non possono essere sintetizzati dal nostro organismo e pertanto devono essere assunti, per forza, con la dieta, ecco perchè sono detti essenziali. 93 CAPITOLO 16.8: UTILIZZO DEGLI ACIDI GRASSI Nel fegato, gli acidi grassi possono essere utilizzati: Il metabolismo ossidativo nei mitocondri Sintesi di triacilgliceroli/trigliceridi o fosfolipidi CAPITOLO 17: CICLO DELL’ACIDO CITRICO/DI KREBS Il ciclo dell’acido citrico/di Krebs comprende una serie di otto reazioni che portano all’ossidazione dell’acetil-CoA (prodotto dalla glicolisi, cioè derivano dal piruvato che viene trasformato in esso) a due molecole di CO2 e l’energia liberata viene conservata nei coenzimi ridotti NADH (se ne producono 3 molecole ogni volta) e FADH2 (1 sola molecola prodotta). In questo ciclo si produce anche 1 molecola di GTP. Quindi ricapitolando, questo ciclo, ossidando l’acetil-CoA, produce: 2 molecole di CO2 3 NADH 1 FADH2 1 GTP La reazione globale di questo ciclo è: ACETIL-CoA + 3NAD+ + FAD + GDP + P CoA + 2CO2 + 3NADH + FADH2 + GTP 94 Alcuni intermedi di questo ciclo, sono contenuti in altre vie metaboliche, diverse da quella dei carboidrati (vie sia cataboliche che anaboliche di altre sostanze). Per questo motivo, il ciclo di Krebs è definito come ciclo anfibolico. Tutto questo ciclo, avviene nella matrice mitocondriale. Come si può vedere anche dalla figura, questo ciclo è formato da 8 reazioni che, di seguito, vediamo nel dettaglio. CAPITOLO 17.1: LE FASI CICLO DELL’ACIDO CITRICO/DI KREBS REAZIONE 1 – CITRATO SINTASI o Si parte dall’acetil-CoA che viene condensato/unito con l’ossalacetato per formare il citrato, operazione fatta dall’enzima citrato sintasi. In questa reazione, l’atomo di carbonio dell’acetil-CoA si lega al C2 dell’ossalacetato. E’ una reazione di condensazione altamente esoergonica. Reazione irreversibile. REAZIONE 2 – ACONITASI o L’enzima aconitasi catalizza la reazione di isomerizzazione del citrato che si ridispone e diventa isocitrato, con la formazione dell’inermedio cis-aconitato. La reazione di idratazione del cis-aconitato non catalizzata dall’enzima, formerebbe 4 stereoisomeri, visto che invece interviene l’aconitasi, si forma un solo tipo di stereoisomero. E’ una reazione reversibile. REAZIONE 3 – OSSIDAZIONE DELL’ISOCITRATO AD ALFA-CHETOGLUTARATO E CO2 o In questa tappa, l’enzima isocitrato deidrogenasi catalizza la decarbossilazione ossidativa dell’isocitrato che così diventa alfa-chetoglutarato, con conseguente eliminazione della prima molecola di CO2 (il carbonio di questa molecola appartiene all’ossalacetato) e formazione di NADH. E’ una tappa reversibile 95 REAZIONE 4 – OSSIDAZIONE DELL’ALFA-CHETOGLUTARATO A SUCCINIL-CoA E CO2 o Cosi come nella reazione 3, anche in questa reazione avviene una decarbossilazione ossidativa, stavolta fa trasformare l’alfa-chetoglutarato in succinil-CoA (è un tioestere) e fa ottenere la seconda molecola di CO2 (anche il carbonio di questa molecola è dell’ossalacetato). Questo passaggio è catalizzato dall’enzima alfachetoglutarato deidrogenasi (complesso multienzimatico molto simile al piruvato deidrogenasi, formato da 3 enzimi e 5 cofattori). Sempre in questa fase, avviene il passaggio da NAD+ a NADH. Processo irreversibile. REAZIONE 5 – CONVERSIONE DEL SUCCINIL-CoA IN SUCCINATO o In questa tappa, viene rotto il legame tioestere ad alta energia del succinil-CoA e l’energia che viene rilasciata da questa rottura, viene usata per favorire la fintesi di un legame fosfoanidride sotto forma di GTP, questo processo porta alla formazione di succinato. A fare ciò è l’enzima chiamato succinil-CoA sintetasi (detto anche tiochinasi proprio perché accoppia la rottura del legame tioestere con la sintesi del GTP, altro composto ad alta energia). E’ una fosforilazione a livello del substrato. E’ una reazione reversibile. Dopo questa tappa, l’acetil-CoA è stato ossidato a CO2 e si sono già prodotti 2 NADH e 1 molecola di GTP. Le tre tappe che seguono, sono necessarie per trasformare il succinato in ossalacetato e producono un'altra molecola di NADH più una di FADH2. 96 REAZIONE 6 – OSSIDAZIONE DEL SUCCINATO A FUMARATO o In questa tappa, il succinato viene ossidato e diventa fumarato. Questa tappa è catalizzata dall’enzima succinato deidrogenasi che catalizza appunto, la deidrogenazione stereospecifica del succinato con conseguente formazione di fumarato. In questa tappa viene prodotta l’unica molecola di FADH2 del ciclo di Krebs (si ottiene perché il FAD, legato covalentemente a un residuo di istidina dell’enzima di questa tappa, assorbe elettroni). E’ una tappa reversibile che può essere inibita dal maltonato che ha un CH2 in meno rispetto al succinato. REAZIONE 7 – IDRATAZIONE DEL FUMARATO A MALATO o L’enzima che entra in gioco in questa tappa è la fumarasi che idrata (cioè aggiunge una molecola di acqua) il fumarato e lo trasforma in L-malato. E’ una tappa reversibile e avviene in maniera stereospecifica, cioè si idrata il doppio legame trans del fumarato ma non quello cis del maleato che è un isomero dello stesso fumarato. REAZIONE 8 - OSSIDAZIONE DEL MALATO A OSSALACETATO o Nell’ottava e ultima tappa del ciclo di Krebs, l’L-malato viene ossidato e diventa ossalacetato, operazione fatta dall’enzima malato deidrogenasi. In questa tappa avviene la produzione dell’ultima molecola di NADH. E’ una reazione altamente endoergonica ma è accoppiata alla successiva condensazione dell’ossalacetato con l’acetil-CoA (cioè sarebbe la prima di questo ciclo) che è altamente esoergonica e quindi è possibile farla. 97 CAPITOLO 17.2: BILANCIO ENERGETICO COMPLESSIVO DEL CICLO DELL’ACIDO CITRICO Come abbiamo già visto e detto in precedenza, il ciclo di Krebs avviene nei mitocondri, prende il via dall’acetil-CoA che si ottiene dal piruvato (ottenuto a sua volta dalla glicolisi) prodotto nel citoplasma. Produce 3 NADH, 1 FADH2 e 1 GTP dai quali si ricava un totale di 12 ATP (9 dai 3 NADH, 2 dal FADH2 e 1 dal GTP). A queste 12 molecole però, ne va sottratta una che viene usata nel trasporto attraverso la membrana mitocondriale, quindi per ogni molecola di acetil-CoA vengono prodotti, con il ciclo di Krebs, 11 ATP. CAPITOLO 17.3: REGOLAZIONE DEL CICLO DI KREBS Il ciclo di Krebs, cosi come si vede anche dalla figura, è regolato in quattro punti ben precisi (nella trasformazione da piruvato ad acetil-CoA e nelle sue tre tappe iniziali esoergoniche): Regolazione allosterica del complesso multienzimatico piruvato deidrogenasi, cioè viene inibito o attivato il complesso che trasforma il piruvato in acetil-CoA. o Gli inibitori sono: ATP, Acetil-CoA (fa da inibitore allosterico quindi lo stesso prodotto della reazione) e NADH. Questo complesso può anche essere inibito dalla fosforilazione di alcuni residui di serina. o Gli attivatori sono: Coenzima A, NAD+, AMP, CA2+ 98 Regolazione generale del ciclo dipendente dai livelli di NAD + e da altri indici dello stato energetico della cellula, cioè in base alla quantità e al tipo di substrato presente Reazioni anaplerotiche, cioè reazioni di altri processi metabolici che producono intermedi del ciclo di Krebs (cioè la velocità del ciclo è regolata dalla presenza di intermedi prodotti da altre reazioni che avvengono in parallelo) CAPITOLO 18: LA FOSFORILAZIONE OSSIDATIVA La fosforilazione ossidativa, negli eucarioti, avviene nei mitocondri, nei procarioti invece avviene tutta nella membrana plasmatica. In questo processo, il NADH e il FADH2 vengono ossidati e gli elettroni che si liberano (equivalenti riducenti), vengono trasferiti all’ossigeno molecolare mediante una serie di altri trasportatori di elettroni legati in serie sulla membrana (mitocondriale interna o plasmatica). Parallelamente si produce un eccesso di ioni H+ che vengono rilasciati attraverso la membrana (del mitocondrio o plasmatica). Si genera quindi un gradiente di pH e quindi elettrochimico (∆E) associabile ad una variazione di energia libera: ∆G = -n · F · ∆E L’energia libera ottenuta viene quindi accoppiata alla sintesi di ATP. La fosforilazione ossidativa infatti, è la sintesi di ATP che si verifica in seguito al trasferimento degli elettroni, sottratti durante le ossidazioni, all’ossigeno. CAPITOLO 18.1: IL MITOCONDRIO E LA SUA MEMBRANA La membrana esterna del mitocondrio è permeabile a molecole fino a circa 10kDa, per cui la composizione dello spazio intermembrana è simile a quella del citoplasma (è la parte più esterna del mitocondrio. La membrana interna invece è permeabile solo a O2, CO2 e H2O. E’ costituita per circa il 17% da proteine, queste sono impegnate sia nel trasporto attivo dei metaboliti attraverso la matrice, che nel trasporto degli elettroni (è la membrana delle creste mitocondriali). La membrana interna separa fisicamente il contenuto della matrice da quello del citoplasma, compartimentalizzando le funzioni metaboliche associate. 99 CAPITOLO 18.2: I SISTEMI DI TRASPORTO ATTRAVERSO LA MEMBRANA MITOCONDRIALE A causa della permeabilità controllata/specifica della membrana mitocondriale interna, l’entrata nella matrice mitocondriale o l’uscita da essa dei metaboliti viene mediata da sistemi di trasporto. Le principali sostanze che vengono trasportate attraverso la membrana sono: Piruvato Ossalacetato ADP/ATP, questi vengono trasportati contestualmente da una proteina di membrana (traslocatore ADP/ATP) Malato Fosfato Aspartato NADH (solo gli equivalenti riducenti, quindi solo gli elettroni). Gli equivalenti riducenti del NADH, quindi gli elettroni, vengono trasferiti mediante i sistemi navetta del glicerofosfato o del malato-aspartato. CAPITOLO 18.2.1: IL SISTEMA NAVETTA DEL GLICEROFOSFATO E’ un processo che si ha nel muscolo scheletrico e nel cervello, e si verifica in tre tappe. E’ un sistema che, rispetto a quello del malato-aspartato, trasporta gli equivalenti riducenti dal NADH al complesso 3 e non al complesso 1 come l’altro sistema. Nel dettaglio, in questo sistema, l’enzima 3-fosfoglicerolo deidrogenasi converte il diidrossiacetone fosfato in glicerofosfato, ossidando una molecola di NADH a NAD+. Il glicerofosfato viene poi riconvertito a diidrossiacetone fosfato grazie all’enzima di membrana mitocondriale: il glicerofosfato deidrogenasi che riduce una molecola di FAD a FADH2. Il FADH2 riduce poi il coenzima Q/ubichinone ed entra nella fosforilazione ossidativa. E’ una reazione irreversibile. 100 CAPITOLO 18.2.2: IL TRASLOCATORE ADP-ATP L’ ADP-ATP traslocasi è una proteina di membrana omodimerica. Il sito di legame è unico per ADP ed ATP, questa proteina esiste in due stati conformazionali in cui il sito di legame è rivolto alternativamente verso l’interno o verso l’esterno della matrice mitocondriale. Nella conformazione 1 l’affinità per l’ATP è bassa mentre è alta quella per l’ADP, nella conformazione 2 è esattamente l’opposto (alta per ATP e bassa per ADP). La sua funzione è quella di mediare il trasporto di una molecola di ATP dalla matrice mitocondriale allo spazio intermembrana e di una molecola di ADP dallo spazio intermembrana alla matrice mitocondriale (antiporto, cioè trasporto di due composti diversi allo stesso tempo). La sua azione è associata al processo di fosforilazione ossidativa dei mitocondri. Questo trasporto genera uno squilibrio di cariche negative tra l’interno e l’esterno della matrice mitocondriale; tale squilibrio viene bilanciato dal gradiente protonico generato dalla fosforilazione ossidativa. Lo ione fosfato necessario alla sintesi di ATP viene importato nella matrice da un altro trasportatore che trasferisce questo ione insieme ad uno ione H+, il sistema di simporto (un altro di questi sistemi viene utilizzato per il trasporto del piruvato). CAPITOLO 18.3: TRASPORTO DEGLI ELETTRONI – CONSIDERAZIONI TERMODINAMICHE 101 CAPITOLO 18.4: CATENA DI TRASPORTO DEGLI ELETTRONI Gli elettroni trasportati dai 10 NADH e dai 2 FADH2 ottenuti tramite la glicolisi e il ciclo di Krebs, entrano nella catena di trasporto degli elettroni. In questa catena, gli elettroni passano in 4 complessi enzimatici che contengono circa 10 centri redox con affinità per gli elettroni (E°’) progressivamente crescente. Gli elettroni del NADH e del FADH2 ovviamente, vengono liberati proprio in questi centri redox che li ossidano/riducono a NAD + e FAD. CAPITOLO 18.4.1: IL COMPLESSO ENZIMATICO 1 DELLA CATENA DI TRASPORTO – NADH – COENZIMA Q OSSIDOREDUTTASI Il complesso 1 della catena di trasporto degli elettroni, catalizza il trasferimento degli elettroni dal NADH al coenzima Q/ubichinone. Questo complesso è costituito da 43 catene polipeptidiche e contiene una molecola di FMN (che partecipa alle reazioni redox) e diversi (da 3 a 7) centri ferro-zolfo come cofattori e che partecipano al trasporto di elettroni. Le proteine/cofattori ferro-zolfo sono di due tipi, ma entrambe contengono quattro residui di cisteina che coordinano i complessi ferro-zolfo costituiti da ioni ferro (+3 o +2) e ioni solfuro. La variazione dello stato di ossidazione dello ione ferro permette il trasferimento di un elettrone grazie al sistema coniugato dei legami di coordinazione, ecco perché quindi, lo ione ferro può assumere uno stato di ossidazione intermedio tra +2 e +3. Associato al passaggio degli elettroni (cioè insieme al passaggio degli elettroni che è un processo esoergonico), il complesso 1 trasferisce 4 protoni (H) dalla matrice mitocondriale allo spazio intermembrana (cioè si accumulano protoni H nella membrana). Questo fenomeno comporta una 102 doppia modifica confromazionale del complesso 1 che accetta questi elettroni (cioè cambia forma due volte). CAPITOLO 18.4.2: IL COMPLESSO ENZIMATICO 2 DELLA CATENA DI TRASPORTO – SUCCINATO – COENZIMA Q OSSIDO-REDUTTASI Questo complesso multiproteico è costituito dall’enzima succinato deidrogenasi (è anche un enzima del ciclo di Krebs, contiene FAD), tre centri ferro-zolfo (uno [4Fe-4s] e due [2Fe-2S]) e un citocromob560. I citocromi sono ferro-proteine contenenti gruppi eme, derivati dalla protoporfirina IX. A differenza della mioglobina e della emoglobina, i gruppi eme dei citocromi sono legati covalentemente mediante legami tioetere tra i loro residui vinilici e due residui di cisteine. Il complesso 2, trasferisce gli elettroni dal FADH2 (dal succinato) al coenzima Q. Il coenzima Q quindi, rappresenta il punto di raccolta primario degli elettroni provenienti dal NADH e dal FADH2 (quelli del NADH vengono trasferiti dal complesso 1). Il coenzima Q è inoltre associato alla navetta del glicerofosfato per cui gli elettroni del NADH citoplasmatico (quello della glicolisi) producono una molecola di ATP in meno. Ricorda: questo complesso 2 NON opera dopo il complesso 1 ma fornisce semplicemente un altro ingresso alla catena. CAPITOLO 18.4.3: IL COMPLESSO ENZIMATICO 3 DELLA CATENA DI TRASPORTO – COENZIMA Q-CITOCROMOc OSSIDO-REDUTTASI Questo complesso contiene un centro ferro-zolfo ([2Fe-2S]) e tre citocromi (due citocromob e un citocromoc1). Questo complesso trasferisce gli elettroni dal coenzima Q/ubichinone al citocromoc, che è un citocromo solubile. Gli elettroni poi passano dal complesso 3 al complesso 4 sulla superficie della membrana mitocondriale interna. 103 CAPITOLO 18.4.4: IL COMPLESSO ENZIMATICO 4 DELLA CATENA DI TRASPORTO – CITOCROMOc OSSIDASI Questo complesso contiene quattro centri redox: due sono citocromi (citocromoa e citocromoa3) e due ioni rame. Catalizza il trasferimento di quattro elettroni provenienti da quattro molecole di cictocromoc diverse secondo la relazione: 4 CITOCROMOC (Fe2+) + 4H+ + O2 4 CITOCROMOC (Fe3+) + 2 H2O Catalizza quindi le ossidazioni (con acquisto di 1 elettrone) di 4 molecole consecutive di citocromoc ridotto e la contemporanea riduzione di una molecola di O2. Questo complesso, trasferisce gli elettroni dal citocromoc all’ossigeno molecolare, riducendolo ad H2O. CAPITOLO 18.5: FOSFORILAZIONE OSSIDATIVA – TEORICA CHEMIO-OSMOTICA I complessi 1, 3 e 4 generano un gradiente di pH tra l’interno e l’esterno della membrana mitocondriale con un pH maggiore all’interno del mitocondrio. Nella teoria chemio-osmotica, sviluppata da Mitchell, è proprio il gradiente elettrochimico generato dal gradiente di pH che alimenta la sintesi di ATP tramite la forza motrice protonica. E’ stato calcolato che occorre un gradiente di circa tre protoni per fornire la giusta energia libera per la sintesi di una molecola di ATP. La sintesi dell’ATP viene catalizzata dall’enzima ATP sintasi costituita da due componenti definiti F1 e F0; questo enzima viene anche definito F1-F0 ATPasi. SPIEGAZIONE IN BREVE DA WIKIPEDIA [La teoria prevede che mentre gli elettroni "scendono" lungo la catena di trasporto, gli ioni H+ presenti nella matrice vengono trasportati attivamente nello spazio intermembrana. Si genera così una differenza di concentrazione degli H+ sui due versanti della membrana interna mitocondriale. A causa di questo gradiente di concentrazione, gli ioni H+ tendono a rientrare per diffusione. Dato che la membrana è impermeabile, per attraversarla essi hanno bisogno di una proteina di trasporto: l'ATP sintasi, un complesso enzimatico che catalizza la sintesi dell'ATP a partire da ADP e fosfato. In tal modo il rientro degli ioni fornisce alla reazione di sintesi dell'ATP l'energia necessaria. In pratica è il fatto dell’ATP-sintetasi che gira, causa ioni che entrano uno alla volta, visto in biologia.] 104 CAPITOLO 18.5.1: FOSFORILAZIONE OSSIDATIVA – F1-F0 ATPasi E’ una proteina multimerica transmembrana costituita da diverse sub-unità. Ha due unità funzionali denominate F1 e F0. La componente F0 è costituita da un canale protonico transmembrana e da uno stelo di collegamento all’altra componente. Questa componente è insolubile in acqua. La componente F1 è costituita da una proteina estrinseca di membrana rivolta verso la matrice mitocondriale, è dissociabile dalla componente F0 ed è solubile in acqua. Una volta solubilizzata però, perde la capacità di sintetizzare ATP e acquista la capacità di idrolizzarlo (cioè scinderlo) in ADP + P. La sintesi dell’ATP di questa proteina, viene accoppiata al passaggio di protoni H attraverso il suo canale transmembrana (il fatto che gli ioni H scendono uno alla volta) che fa cambiare la forma della sua parte catalitica. Questa parte catalitica è costituita da tre protomeri identici, contenenti due sub-unità ciascuno. Questi protomeri possono avere tre conformazioni: Stato O, conformazione aperta e con affinità molto bassa per i ligandi e cataliticamente inattiva Stato L, conformazione più chiusa che lega debolmente i ligandi e cataliticamente inattiva Stato T, conformazione chiusa con alta affinità per i ligandi e cataliticamente attiva La sintesi dell’ATP avviene in tre tappe: 1. ADP e P si legano alla conformazione L 2. Tre cambi conformazionali che richiedono energia: trasformano un dimero dalla forma L alla forma T, un altro dimero passa dalla forma T alla forma O, l’ultimo dimero passa dalla forma O a quella L (in poche parole, cambiano conformazione in senso anti-orario, vedi immagine sotto) 3. L’ATP viene sintetizzato a livello della forma T e viene rilasciato dal dimero nella forma O, unendo ADP e P. CAPITOLO 18.5: FOSFORILAZIONE OSSIDATIVA – REGOLAZIONE La fosforilazione ossidativa è regolata in base alle richieste energetiche cellulari. Le concentrazioni di ATP e ADP regolano la velocità di trasferimento degli elettroni attraverso la catena respiratoria, tramite una serie di controlli coordinati sulla respirazione, sulla glicolisi e sul ciclo dell’acido citrico (cioè in base a quanto ATP o ADP c’è, questa aumenta o diminuisce di velocità). 105 CAPITOLO 19: METABOLISMO DEGLI AMMINOACIDI / AZOTATO Si osserverà soprattutto che fine fa la parte amminoacidica degli amminoacidi in quanto la parte carboniosa entra o nel metabolismo degli acidi grassi o in quello degli zuccheri. Particolare attenzione va al catabolismo degli amminoacidi. Il metabolismo degli amminoacidi, è un complesso di reazioni di sintesi e di degradazione durante le quali, gli amminoacidi, vengono assemblati come precursori delle proteine o vengono degradati per ottenere energia metabolica. Qui a lato, uno schema riassuntivo del metabolismo degli amminoacidi. Gli amminoacidi sono una classe di composti che non hanno forma di deposito e che quindi non si accumulano, quindi o formano proteine o vengono trasformati in metaboliti comuni e quindi fornitori di energia per altre vie metaboliche (non vengono accumulati gli amminoacidi anche perché possono essere tossici). Il gruppo amminico viene modificato in urea e quindi non è più pericoloso, anche se in grande quantità. La fonte principale degli amminoacidi sono le proteine ingerite con la dieta o quelle che non “servono” più alla loro funzione (cioè enzimi, immunoglobuline ecc. che non servono più e che quindi sono degradate e ridotte a singoli amminoacidi al fine di non farle diventare tossiche). Dall’idrolisi delle proteine si ottengono infatti, amminoacidi che possono essere direttamente riutilizzati come tali o degradati per produrre energia ed essere quindi usati in altre vie metaboliche. La degradazione/catabolismo delle proteine che non servono più, può avvenire mediante tre processi: Degradazione lisosomiale (proteine cellulari), avviene mediante i lisosomi Degradazione ubiquitina dipendente (proteine cellulari), avviene grazie all’ubiquitina, degradazione più specifica, questo processo può essere alterato da patologie Digestione gastrointestinale (proteine esogene della dieta), avviene o nello stomaco a livello gastrico quindi, o nell’intestino 106 Ognuno di questi tre processi, ha enzimi specifici e adattati a rompere il legame peptidico in quella specifica zona del corpo. Gli amminoacidi sono la terza fonte di energia, infatti è più facile usare zuccheri e lipidi. Per far entrare gli amminoacidi nelle cellule muscolari ci vuole l’insulina, quindi indirettamente ci vogliono gli zuccheri. Rifacendosi alla figura di pag.106, si osserva che: Quando dividiamo gruppi amminici (NH4) dagli scheletri carboniosi, questi ultimi sono chetoacidi ed entreranno sempre nel ciclo di Krebs, creando una reazione anaplerotica, cioè alimenta il ciclo di Krebs usando intermedi di altre reazioni Il gruppo NH4 invece può fare la sintesi di altri amminoacidi o entrare nel ciclo dell’urea, che è una doppia ammide nella quale sono contenuti due gruppi ammidici, è non tossica e viene poi espulsa tramite i reni, il ciclo dell’urea si intreccia con il ciclo di Krebs Arginina e aspartato invece possono anche non passare nel ciclo dell’urea ma entrare direttamente nel ciclo di Krebs CAPITOLO 19.1: DEGRADAZIONE LISOSOMIALE Questo processo degrada tutte le proteine che le cellule assumono per endocitosi, avviene nei lisosomi che hanno le proteasi, cioè gli enzimi che sono in grado di rompere il legame peptidico, questi enzimi hanno un pH ottimale intorno a 5 che li rende inattivi a pH neutro (cioè non succede niente se si rompe il lisosoma). Questo processo non usa ATP. CAPITOLO 19.2: DEGRADAZIONE UBIQUITINA-DIPENDENTE E’ un meccanismo di degradazione che non avviene con i lisosomi e che usa ATP (usa 1 ATP per ogni legame peptidico da rompere). In questo processo è coinvolta l’ubiquitina (proteina di piccole dimensioni) e un complesso multienzimatico detto proteasoma. Vengono degradate solo proteine segnalate dall’ubiquitina che si lega alle proteine da demolire (formano proteine ubiquitinate, è qui che si usa l’ATP), le “etichetta” e quindi queste poi vengono demolite dal complesso enzimatico (una proteina può anche essere segnalata più volte). Questo processo, se alterato, provoca malattie anche gravi perché vengono segnalate e distrutte proteine errate. Le proteine ubiquitinate vengono degradate, come detto, dal proteasoma, cioè da un complesso enzimatico costituito da una parte centrale cilindrica e da due altri parti che si trovano all’estremità. Il proteasoma è costituito da 28 sub-unità proteiche/proteine di due tipi, denominate alfa e beta. Queste sub-unità sono organizzate in 4 anelli di sette sub-unità ciascuno. La proteina ubiquitinata entra da una parte ed esce come amminoacidi singoli dall’altra (è meglio dire che produce frammenti polipeptidici composti da circa 8 peptidi che poi saranno ridotti nel citosol), cioè attraversa tutto il complesso cilindrico. 107 CAPITOLO 19.3: DIGESTIONE GASTROINTESTINALE Questo processo, come detto, ha due fasi: una gastrica (soprattutto grazie alla pepsina) e una intestinale (favorita dagli enzimi pancreatici come la tripsina, la chimotripsina ed elastasi). Interessa gli amminoacidi provenienti dalle proteine introdotte con la dieta. Gli enzimi coinvolti sono le endopeptidasi (che riconoscono solo sequenze specifiche e rompono partendo dal mezzo) o le esopeptidasi (rompe partendo dall’estremità). Gli amminoacidi liberi vengono assorbiti a livello della muscosa intestinale, trasferiti al ciclo sanguigno che li trasporta ai tessuti di utilizzo, essenzialmente fegato (organo generico) e muscoli (che li useranno per le fibre di miosina ed actina). Anche in questa fase, lo scheletro carbonioso ed il gruppo amminico dell’amminoacido hanno destini diversi. Il gruppo amminico viene escreto sotto forma di urea, acido urico o ammoniaca (in base al tipo di animale: urea dell’uomo, acido urico tipico degli uccelli, l’ammoniaca invece è fatta dai pesci). Il gruppo amminico può essere tenuto se viene trasformato in glutammato. La degradazione degli amminoacidi avviene essenzialmente all’interno delle cellule epatiche. La prima tappa di questa degradazione prevede la rimozione del gruppo alfa-amminico, un processo che si compone in due passaggi: Formazione del glutammato mediante transamminazione Deamminazione ossidativa del glutammato con produzione di ammoniaca e alfachetoglutarato Nella reazione di transamminazione si ha il trasferimento del gruppo alfa-amminico da un amminoacido ad un alfa-chetoacido (essenzialmente ossalacetato o piruvato) e questo forma un amminoacido diverso (forma cioè glutammato, aspartato o alanina) ed un nuovo chetoacido. Gli enzimi che catalizzano queste reazioni vengono chiamati transamminasi/amminotrasferasi e sono enzimi citoplasmatici (cioè quasi tutti gli amminoacidi vengono trasformati, se cedono il loro gruppo amminico all’alfa-chetoglutarato, in glutammato e la parte che ha ceduto il gruppo amminico diventa un chetoacido generico). Come si vede, il gruppo amminico viene espulso come urea. Sch.carb: destino anabolico o catabolico 108 CAPITOLO 19.3.1: LA REAZIONE CATALIZZATA DALLE TRANSAMMINASI In molte reazioni, l’accettore del gruppo amminico è l’alfa-chetoglutarato (chetoacido con 5 atomi di carbonio). Questa reazione è reversibile (cioè se accoppio un amminoacido e un chetoacido succede l’opposto) e tutte le amminotrasferasi/trasnamminasi utilizzano come cofattore il piridossalfosfato (PLP), un derivato della vitamina B6. Per transamminazione del piruvato si ottiene alanina. Il gruppo amminico viene prima legato all’enzima, questo legame fa uscire l’alfa-chetoacido che ha ceduto il gruppo, poi dopo entra il secondo chetoacido che accetta quel gruppo e si modifica in glutammato con questa transamminazione, meccanismo quindi ordinato perché uno cede e un altro acquista. Solo due amminoacidi non posso fare questa cosa, non si possono transamminare: il glutammato (ovviamente) e la prolina che infatti non ha gruppo amminico. Il risultato delle reazioni di transamminazione è raccogliere i gruppi amminici di diversi amminoacidi su un unico tipo di prodotto: il glutammato. Il glutammato poi, dovrà cedere a sua volta questi gruppi amminici per permettere la loro escrezione e questo lo fa con la deamminazione ossidativa e avviene nel fegato grazie all’enzima glutammato deidrogenasi. I gruppi amminici saranno rilasciati nel fegato sotto forma di ammoniaca. CAPITOLO 19.3.2: DEAMMINAZIONE OSSIDATIVA DEL GLUTAMMATO Questa reazione avviene nel citoplasma degli epatociti (nel fegato) ed è catalizzata dall’enzima glutammico deidrogenasi che porta alla rimozione di uno ione ammonio ed alla ossidazione dell’atomo di carbonio a cui esso era legato. Utilizza o NAD+ o NADP+ come accettore di elettroni, a seconda della condizione di riposo o lavoro della cellula (NADP a riposo, NAD a lavoro/in contrazione). Il ∆G°’ di questa reazione è 30kJ/mole, ma l’enzima funziona in vivo in condizioni vicine all’equilibrio (∆G = 0, in base alle concentrazioni dei reagenti) pertanto le concentrazioni relative dirigono la reazione. Lo ione ammonio viene poi convertito in urea per essere escreto. La reazione inversa è importante per la rimozione dell’ammoniaca molto tossica, soprattutto a livello nervoso, in quella inversa si usa solo NADPH. La reazione va verso destra o sinistra in base allo stato fisiologico del corpo. R Ricorda: questo cedere i gruppi amminici da parte del glutammato, libera ammoniaca. 109 CAPITOLO 19.3.3: TRASPORTO DELL’AMMONIACA FORMATA IN ALTRI TESSUTI L’ammoniaca è una specie molto tossica, pertanto non può essere trasportata liberamente nel circolo sanguigno, quindi bisogna de-tossificarla per farla arrivare al fegato e renderla anche solubile, quindi bisogna trasformarla. Questa trasformazione avviene grazie al ciclo glucosio-alanina/di Cori (ammoniaca prodotta nel muscolo) L’ammoniaca prodotta dalla degradazione degli amminoacidi nel muscolo (cioè dal metabolismo muscolare), viene trasportata sotto forma di alanina, quindi non tossica, formata a partire dal piruvato grazie all’enzima alanina amminotrasferasi, che è un isoenzima muscolare. L’ammoniaca, si può anche trasformare anche in lattato se c’è NADH, sempre partendo dal piruvato. In questi modi quindi trasportiamo lo scheletro carbonioso del piruvato al fegato che lo utilizza per fare altre cose. Nel fegato quindi, il gruppo amminico dell’alanina viene trasferito a un alfa-chetoglutarato, formando glutammato e piruvato grazie all’alanina-amminotrasferasi, che è un isoenzima epatico (il glutammato fornirà ammoniaca, il piruvato formerà glucosio con la gluconeogenesi). Il glutammato formerà quindi ammoniaca grazie all’azione dell’enzima glutammico deidrogenasi, mentre il piruvato potrà formare glucosio attraverso la gluconeogenesi. L’ammoniaca prodotta negli altri tessuti, essendo anch’essa tossica, deve essere trasformata per de-tossificarla. Questa operazione è fatta dalla glutammina sintetasi che combina l’ammoniaca prodotta nei tessuti e il glutammato, formando cosi glutammina. Questa trasformazione richiede ATP e avviene in due tappe che sono descritte qui di seguito: RIASSUMENDO: L’AMMONIACA (TOSSICA E NON PUO’ ANDARE COSI NEL SANGUE): DAL MUSCOLO AL FEGATO SI TRASPORTA COME ALANINA E FA IL CICLO GLUCOSIOALANINA DAGLI ALTRI TESSUTI AL FEGATO VIENE TRASPORTATA COME GLUTAMMINA NEL FEGATO DIVENTA UREA (CICLO DELL’UREA) E POI ESCRETATA CON I RENI 110 CAPITOLO 19.4: IL CICLO DELL’UREA E’ un processo ciclico che porta alla fissazione degli ioni ammonio tossici (gruppi ammidici) con formazione di urea che viene poi eliminata con le urine. La reazione complessiva è la seguente: Dalla figura si vede che serve per forza aspartato. Da questo ciclo si forma il fumarato che è un intermedio del ciclo di Krebs, oltre ovviamente all’urea. Sono presenti due gruppi amminici, uno viene della deamminazione del glutammato e un altro da glutammato (NH2). E’ un processo che non consuma energia in quanto il fumarato viene utilizzato nel ciclo di Krebs e ciò permette di recuperare energia, anzi è un ciclo leggermente vantaggioso in termini energetici. Nell’urea viene messa anche CO2. CAPITOLO 19.4.1: LE REAZIONI DEL CICLO DELL’UREA Questo processo ciclico è formato da 5 reazioni: due mitocondriali e tre citosoliche, quindi c’è cooperazione tra citosol e mitocondrio. REAZIONE 1 – CARBAMMIL-FOSFATO SINTETASI o Viene sintetizzato carbammil-fosfato usando ATP. L’ATP infatti interagisce con il bicarbonato, formando carbonil-fosfato, successivamente nel carbonil-fosfato l’ammoniaca (NH3) sostituisce il fosfato creando il carbammato, alla fine una seconda molecola di ATP fosforila il carbammato formando proprio il carbammil-fosfato che è ad alto contenuto energetico anche se non viene utilizzato per sintetizzare direttamente ATP. Avviene nel mitocondrio. L’enzima di questa reazione è ovviamente il carbammil-fosfato sintasi e, come visto, usa 2 ATP che è presente nel mitocondrio, cosi come tutti gli altri reagenti REAZIONE 2 – ORNITINA TRANSCARBAMMILASI o In questa reazione, si trasferisce il gruppo carbammilico all’ornitina, un amminoacido non proteico prodotto dal ciclo dell’urea stesso, creando citrullina. L’enzima di questa 111 reazione è l’ornitina trans-carbamillasi. Questa reazione avviene nel mitocondrio. Le altre reazioni avvengono nel citoplasma quindi la citrullina deve essere trasportata fuori dal mitocondrio (mentre l’ornitina viene fatta entrare con lo stesso meccanismo di trasporto ma in maniera opposta, cioè fa uscire citrullina e porta dentro ornitina che è fondamentale per il ciclo dell’urea) REAZIONE 3 – ARGININ-SUCCINATO SINTETASI o In questa reazione, si condensa la citrullina con una molecola di aspartato, formando arginin-succinato (arginina legata a succinato, fatto dall’enzima arginin-succinato sintetasi), consuma ATP (questo viene degradato ad AMP+2P). Questa reazione avviene nel citosol e l’urea acquista il secondo atomo di azoto (cioè il secondo gruppo amminico). Il pirofosfato viene idrolizzato dalla pirofosfatasi, quindi come detto, si idrolizzano due legami dell’ATP (quindi si consumano 2 ATP) REAZIONE 4 – ARGININ-SUCCINASI o In questa reazione si idrolizza l’arginin-succinato, grazie all’azione dell’enzima arginin-succinasi, e diventa arginina e fumarato. Il fumarato viene trasformato prima in malato (grazie alla fumarasi) e poi in ossalacetato (grazie al malato deidrogenasi). Gli enzimi nelle parentesi sono gli stessi del ciclo di Krebs ma stavolta sono citoplasmatici; dal malato si produce NADH, mentre l’ossalacetato può generare aspartato grazie all’azione della transamminasi. L’argnina invece, rappresenta il precursore immediato dell’urea e quindi non bisogna transamminarla. REAZIONE 5 – ARGINASI o Nella reazione finale di questo ciclo, si ha l’idrolisi dell’arginina in urea e ornitina. L’ornitina può riprendere questo ciclo venendo trasportata nel mitocondrio. L’urea invece, che è molto solubile in acqua, viene rilasciata nel ciclo sanguigno che la trasporta ai reni dove viene escretata tramite urina. Come visto, in questo ciclo si consumano 4 molecole di ATP, però questa energia usata viene recuperata in quanto si producono parallelamente NADPH (grazie al glutammico deidrogenasi) e NADH (grazie al malato deidrogenasi) che possono generare 5 molecole di ATP, cioè vengono prodotti intermedi del ciclo di Krebs o di altre vie metaboliche. Questo si ottiene degradando lo scheletro carbonioso degli amminoacidi usati in questo ciclo. Si distinguono quindi due classi di amminoacidi: 112 Glucogenici, gli scheletri di questi vengono degradati a piruvato, chetoglutarato, succinilCoA o ossalacetato, quindi in tutti intermedi del metabolismo degli zuccheri Chetogenici, vengono degradati ad acetil-CoA o ossalacetato, quindi intermedi del metabolismo dei grassi Quindi la parte carboniosa degli amminoacidi può portare alla produzione di energia sotto forma di ATP o essere utilizzata per la sintesi altre molecole. CAPITOLO 19.5: BIOSINTESI/ANABOLISMO DEGLI AMMINOACIDI Gli amminoacidi si classificano in essenziali e non essenziali in base al fatto che siamo in grado di sintetizzarli da soli o meno. Gli amminoacidi non essenziali vengono sintetizzati attraverso vie semplici che partono da quattro intermedi metabolici comuni: piruvato, ossalacetato, chetoglutarato e 3-fosfoglicerato. Le vie metaboliche per la sintesi degli amminoacidi essenziali, presenti solo nelle piante, sono state perse durante l’evoluzione quindi bisogna introdurli con la dieta. Il grado di essenzialità varia a seconda della fascia di età. CAPITOLO 20: IL TESSUTO MUSCOLARE Il tessuto muscolare è un tessuto altamente specializzato che ha la funzione di garantire i movimenti volontari ed involontari dell’organismo. Sulla base di caratteristiche strutturali, funzionali e di localizzazione, si può classificare questo tessuto, in tre tipi: Muscolo scheletrico/striato (volontario) Muscolo cardiaco (involontario) Muscolo liscio (involontario) 113 CAPITOLO 20.1: IL TESSUTO MUSCOLARE SCHELETRICO/STRIATO Le caratteristiche morfologiche di questo tipo di tessuto sono: Ha cellule allungate e fusiformi Ha molti nuclei periferici Ha striature evidenti Sincizio: cioè quando molte cellule si uniscono e garantiscono la funzionalità del muscolo, in questo modo viene assicurata una propagazione dell’impulso nervoso, quindi fare contrazioni alla massima velocità e con elevata precisione. L’organizzazione strutturale quindi è molto regolare: fibrille al centro, nuclei e mitocondri nelle strutture adiacenti L’organizzazione del muscolo è di tipo gerarchico: Lo stato più esterno si chiama epimisio Un epimisio è formato da permisio Un perimisio è formato da endomisio Un endomisio è formato da miofilamenti Un microfilamento è formato da miofibrille Ogni miofibrilla è formata da sarcomeri CAPITOLO 20.1.1: STRUTTURA DI UNA MIOFIBRILLA Come detto, l’organizzazione regolare ed intervallata delle miofibrille nei sarcomeri è data dalla disposizione di due miofilamenti. Essi sono costituiti da diverse proteine tra cui le due più abbondanti sono: Miosina, caratterizza il filamento spesso, sono disposti al centro del sarcomero Actina, caratterizza il filamento sottile, questi filamenti sono disposti parallelamente, partono dalle linee Z e non arrivano al centro del sarcomero Le zone più scure della banda A del sarcomero, sono dovute alla sovrapposizione dei due filamenti. La zona meno intensa presenta solo filamenti spessi. CAPITOLO 20.1.2: CARATTERISTICHE STRUTTURALI DELLE PROTEINE CONTRATTILI – LA MIOSINA La miosina è una proteina oligomerica a sei sub-unità: nella sua struttura quaternaria si identificano due catene pesanti identiche e due catene leggere differenti. Le catene pesanti sono costituite da una porzione fibrosa contenente lunghe catene ad alfa-elica che si intrecciano tra loro e da due porzioni globulari a cui si legano le due coppie di catene leggere. 114 Le porzioni fibrose di molecole diverse di miosina, tendono ad aggregarsi lasciando verso l’esterno le porzioni globulari (cioè tutte le teste fuori mentre si intersecano le code) formando cosi i filamenti spessi. Un filamento spesso mediamente contiene 400 molecole di miosina. Le porzioni globulari nel filamento spesso sporgono in modo regolare e sono responsabili di legami crociati con i filamenti sottili durante la contrazione. Queste porzioni infatti hanno elevata affinità per l’actina, cioè le teste che vanno verso l’esterno si legano con l’actina. CAPITOLO 20.1.3: CARATTERISTICHE STRUTTURALI DELLE PROTEINE CONTRATTILI – L’ACTINA L’actina contenuta nelle fibre muscolari può essere actina G o actina F (questa deriva dalla forma G in presenza di ioni di magnesio, ha struttura elicoidale). Ogni monomero di actina G, può legare una molecola di ATP o ADP. All’actina f si possono legare altre due proteine (presenti in quantità nettamente minore): Tropomiosina, costituita da due sub-unità differenti (alfa e beta) che avvolgono l’actina F e le danno la forma Troponina, è formata da tre sub-unità diverse: o Troponina T (TnT), lega la tropomiosina o Troponina I (TnI), impedisce il legame tra actina e miosina inibendo l’attività ATPasica o Troponina C (TnC), presenta elevata affinità per gli ioni calcio Ogni molecola di tropomiosina interagisce con sette monomeri di actina G. L’interazione actina-tropomiosina, in condizione di riposo, “nasconde” i siti di legame dell’actina per la miosina. La contrazione muscolare prevede la dissociazione del complesso, cioè lo “smascheramento di questi siti di legame”, e si basa sul legame di ioni di calcio (Ca++) alla troponina C, che vengono liberati in seguito alla propagazione dell’impulso nervoso. CAPITOLO 20.1.4: ALTRE PROTEINE CONTRATTILI STRUTTURALI Titina: proteina molto grande che si dispone parallelamente ai filamenti di actina e miosina, è coinvolta nell’assemblaggio e nel mantenimento della struttura delle fibrocellule Nebulina: regola la lunghezza dell’actina F Alfa-actinina: esistono 4 isoforme e la isoforma 3 viene espressa nelle fibre veloci. Evidenze sperimentali riportano la correlazione tra l’iper-espressione di tale isoforma e le performance dei velocisti (cioè se ho questa espressione sono più adatto alla corsa, doping genico). 115 CAPITOLO 20.2: “SLIDING FILMENT THEORY” DELLA CONTRAZIONE MUSCOLARE La contrazione muscolare si basa su un meccanismo di scorrimento reciproco dei filamenti spessi e sottili presenti nei sarcomeri. A riposo, un sarcomero è lungo 2,5 um. In seguito alla contrazione la lunghezza dei sarcomeri si accorcia fino a circa 1um. Il meccanismo di contrazione prevede che le teste della miosina si spostano lungo i filamenti di actina provocando cosi l’accorciamento. La sequenza di eventi che si verificano sono: Rilascio degli ioni calcio dalle cisterne nel sarcoplasma e legame alla TnC Legame dell’actina alla miosina e induzione dell’attività ATPasica. ADP e P restano legati “Power Stroke” a carico delle teste dell’actina in seguito alla dissociazione del P, cioè quando il fosfato viene rilasciato cambia la conformazione delle teste Scambio ADP-ATP per riottenere la forma attiva della miosina. Provoca anche il distacco della miosina dall’actina Trasporto degli ioni calcio nelle cisterne In pratica, il passaggio chiave, è la dissociazione del gruppo fosfato, quindi la liberazione di energia avuta con lo staccamento di questo fosfato rompendo il legame. L’energia liberata dall’idrolisi dell’ATP viene utilizzata per modifiche conformazionali a carico della miosina. CAPITOLO 20.3: CLASSIFICAZIONE DELLE FIBRE MUSCOLARI Esistono due tipi di muscoli striati: Muscoli rossi attività lenta e continua Muscoli bianchi attività veloce e di potenza Tale classificazione è basata sull’esistenza di due tipi diversi di fibre muscolari. Le fibre lente di tipo 1 a bassa velocità ossidativa: provvedono alla fintesi dell’ATP per via aerobica mediante fosforilazione ossidativa mitocondriale Le fibre rapide di tipo 2 ad alta velocità glicolitica: producono ATP in modo anaerobico attraverso la via glicolitica che può essere alattacida o lattacida. I diversi muscoli striati presentano diversa composizione di tali fibre muscolari. Questa composizione può variare anche in base a età, sesso, allenamento, condizione fisiche o doping. CAPITOLO 20.4: SVILUPPO DEL TESSUTO MUSCOLARE Nell’embrione, il tessuto muscolare, si sviluppa nel mesoderma (dermomiotomo) a partire da cellule chiamate mioblasti. 116 Da alcuni mioblasti (cellule staminali), si ottengono cellule allungate polinucleate detti miotubi, in cui i nuclei si trovano ancora in posizione centrale. Altri rimangono quiescenti e saranno utilizzate sia nell’accrescimento muscolare, sia nella rigenerazione del tessuto muscolare danneggiato. Il controllo del differenziamento, viene effettuato dalla miostatina, un membro della famiglia del fattore di crescita trasformante beta (TGF-beta), che esercita un effetto bloccante la crescita muscolare. Mutazioni nel gene per la miostatina, provocano una iperplasia muscolare (doping genico). CAPITOLO 20.5: FONTI ENERGETICHE DELLA CONTRAZIONE MUSCOLARE La sintesi dell’ATP per la contrazione muscolare avviene essenzialmente mediante tre meccanismi diversi: Meccanismi anaerobici alattacidi, basati sull’utilizzo di ATP e fosfocreatina Meccanismi anaerobici lattacidi, basati sull’utilizzo dell’idrolisi parziale del glucosio (fermentazione omolattica) Meccanismi aerobici, basati sull’utilizzo di substrati (carboidrati, acidi grassi o amminoacidi), metabolizzati in presenza di O2. Questo tipo di meccanismo si verifica nel tessuto muscolare prevalentemente formato da fibre rosse, ricche di mitocondri e ben vascolarizzate La prevalenza/utilizzo di tali meccanismi dipende dal tipo di muscolo scheletrico e/o dal lavoro da esso svolto. CAPITOLO 20.5.1: MECCANISMI ANAEROBICI ALATTACIDI Esistono due meccanismi di sintesi di ATP Alattacido: Sintesi di ATP da fosfocreatina Sintesi di ATP da attività miochinasica CAPITOLO 20.5.1.1: FOSFOCREATINA E CREATINA La fosfocreatina viene sintetizzata a partire dalla creatina. La creatina (Cr) viene a sua volta sintetizzata a livello epatico e renale (oltre a quella proveniente dalla dieta mangiando carni rosse), a partire da tre amminoacidi: arginina, glicina e metionina, è un composto azotato non proteico. Nel nostro corpo sono presenti circa 2g/KG di cretina ed il suo turnover giornaliero è di circa 2 grammi. Il muscolo scheletrico contiene circa il 95% di creatina e circa i 2/3 sono sotto forma di fosfocreatina. 117 La creatina viene trasportata ai tessuti di utilizzo (muscolare e cerebrale) dove viene fosforilata e trasformata in fosfocreatina (PCr) ad opera dell’enzima creatina chinasi (CK) ed a spese dell’ATP (consumando 1 ATP in caso di reazione inversa), in grado di catalizzare anche la reazione inversa. La creatina viene invece eliminata a livello renale sotto forma di creatinina che viene sintetizzata a partire dalla fosfocreatina. CAPITOLO 20.5.1.2: SINTESI DI ATP DA FOSFOCREATINA I livelli di fosfocreatina nel muscolo sono 3-5 volte superiori a quelli di ATP. Nel corso di uno sforzo intenso ma protratto nel tempo, quindi anaerobico, per pochi secondi, per mantenere costanti i livelli di ATP, la fosfocreatina viene rapidamente convertita in creatina. Tamponamento energetico temporale: nella fase di recupero, le scorte di fosfocreatina vengono recuperate in tempi più lunghi (2-3 minuti), attraverso i meccanismi ossidativi aerobici. CAPITOLO 20.5.1.3: SINTESI DI ATP DA ATTIVITA’ MIOCHINASICA L’enzima miochinasi, catalizza la sintesi di ATP in base alla reazione: Questa reazione contribuisce a mantenere relativamente costante la quantità di ATP. Inoltre, nella cellula: ATP > ADP >> AMP Quindi, durante uno sforzo muscolare di breve durata, il consumo di ATP porterà ad una produzione più significativa di AMP. Pertanto, l’AMP è un ottimo segnale energetico per la cellula, infatti quando esso è presente in grandi concentrazioni, significa che la cellula è in debito energetico. L’AMP inoltre, è anche un attivatore di due enzimi coinvolti nel metabolismo dei carboidrati: Fosfofruttochinasi, enzima della glicolisi (PFK-1) Glicogeno fosforilasi, enzima della glicogenolisi Attraverso meccanismi più complessi, aumentate quantità di AMP sono in grado di incrementare l’ingresso di acil-CoA all’interno del mitocondrio, provocando quindi un aumento del flusso dei substrati della beta-ossidazione, processo quindi di lipolisi. La PFK-1, catalizza la fosforilazione ATP-dipendente del fruttosio-6-fosfato in fruttosio-1-6bisfosfato (tappa limitante della glicolisi), nella fase di investimento energetico della glicolisi (cioè quando si usa energia). L’attivazione allosterica di questo enzima, provoca un aumento della velocità di tutta la via glicolitica. Si definisce carica energetica: 118 Il numeratore rappresenta la quantità di ATP presente e potenzialmente ottenibile dall’ADP. La carica energetica quindi, rappresenta la frazione molare di ATP potenzialmente disponibile sul contenuto totale di nucleotidi adenilici. In condizioni fisiologiche, la carica energetica è di circa 0,85, cioè la maggior parte dei nucleotidi adenilici si trova sotto forma di ATP. Le condizioni di carica energetica più bassa attivano il catabolismo ossidativo per rigenerare ATP. CAPITOLO 20.5.2: MECCANISMI ANAEROBICI LATTACIDI Questi meccanismi, si attivano dopo pochi secondi dall’inizio di un esercizio intenso, cioè dopo che la concentrazione di fosfocreatina si è molto ridotta. In tali condizioni il muscolo utilizza le riserve di glicogeno per produrre, attraverso la glicolisi anaerobica, l’ATP. Infatti, durante un esercizio intenso, la velocità di trasporto dell’ossigeno e la bassa velocità di ossidazione dei substrati del ciclo di Krebs nella fosforilazione ossidativa, non sono compatibili con la velocità di utilizzo di ATP a livello muscolare. Pertanto, il piruvato, invece di entrare nel ciclo di Krebs (sotto forma di Acetil-Coa), viene ridotto ad acido lattico con la fermentazione omolattica che rigenera il NAD ossidato e impedisce il blocco della glicolisi. La resa energetica per mole di glucosio è di 2 moli di ATP per il glucosio libero, ed è di 3 moli di ATP per il glucosio proveniente dal glicogeno. Questo meccanismo di sintesi di ATP, caratterizza gli sforzi muscolari intensi e di durata oltre i 4 secondi. La formazione di acido lattico produce un aumento di H+ che inibisce la PFK-1 bloccando quindi la produzione di ATP anaerobica lattacida, al fine di ridurre la fatica/potenza. L’abbassamento del pH viene controbilanciato dai sistemi tampone cellulari inorganici o organici. In particolare, nelle cellule muscolari, questo effetto viene attribuito al di-peptide carnosina (alanina-istidina). Inoltre, il lattato, insieme allo ione H+, viene trasportato all’esterno della cellula dai trasportatori dei monocarbossilati (MCT) per poter essere utilizzato da altri tessuti per destini diversi quali la glucogenesi (nel fegato) e l’ossidazione (nel muscolo cardiaco). Quindi l’attività muscolare può essere monitorata determinando la quantità di lattato rilasciato. 119 CAPITOLO 20.5.2.1: I TRASPORTATORI DEI MONOCARBOSSILATI (MCT) Questi, catalizzano la diffusione facilitata del lattato (secondo gradiente) ma anche di altri monocarbossilati come il piruvato, i corpi chetonici ed altri chetoacidi. Questi trasportatori, sono una famiglia di 9 membri e le isoforme MCT1 e MCT4 sono quelle che si trovano nel tessuto muscolare. In particolare, MCT1 è più espresso nelle fibre di tipo 1 (quindi nei muscoli rossi, bassa velocità ossidativa), mentre l’MCT4 si trova in nelle fibre di tipo 2 (quindi muscoli bianchi, alta velocità glicolitica). In alcuni casi, la quantità di MCT1 aumenta nei muscoli rossi con l’allenamento (resistenza). CAPITOLO 20.6: POTENZA E CAPACITA’ LATTACIDA La potenza lattacida è la quantità di ATP prodotto nell’unità di tempo con il meccanismo lattacido; questa potenza è importante nelle prove di corsa veloce e breve in quanto, proprio a causa della breve durata, non si raggiungono i valori critici del pH intracellulari e non si esauriscono le scorte di glicogeno. La capacità lattacida invece, è la quantità di ATP formato se tutti i substrati disponibili fossero consumati; questa non è valida in quanto si può avere una riduzione di potenza prima che il substrato sia esaurito completamente. La capacità lattacida dipende da componenti muscolari e non muscolari. Le componenti muscolari sono: Elevate concentrazioni di sistemi tampone intracellulari Capacità delle fibre muscolari di lavorare a pH critici Diffusione rapida di lattato e H+ dalla fibra (MCT) Tamponamento della quantità di H+ nei liquidi extracellulari Metabolismo del lattato nelle fibre di tipo 1 (muscolo) Le componenti non muscolari sono: Tamponamento a livello ematico Eliminazione del lattato ematico negli organi addetti CAPITOLO 20.7: UTILIZZAZIONE DEL LATTATO In seguito all’immissione nel torrente circolatorio, la rimozione del lattato può avvenire mediante: Ciclo di Cori (glucogenesi da lattato) Ossidazione nel tessuto muscolare scheletrico Shuttle fibre bianche fibre rosse 120 Ossidazione nel tessuto muscolare cardiaco CAPITOLO 20.8: MECCANISIMI AEROBICI DI SINTESI DI ATP Nelle prove di lunga durata ma di intensità non massimale (tipo la maratona), l’energia viene fornita dall’ossidazione aerobica di carboidrati, acidi grassi e di amminoacidi. Dal catabolismo di questi substrati viene prodotto NADH e FADH2 nel ciclo di Krebs che poi, grazie alla fosforilazione ossidativa, sintetizzeranno ATP. CAPITOLO 20.8.1: TRASPORTATORI DEL GLUCOSIO – GLICEMIA 5mM Questi trasportatori sono delle proteine trans-membrana di 5 tipi diversi (GLUT1-5) in base alla loro espressione tissutale. La loro efficacia nel trasportare il glucosio, viene valutata in base al valore di KT. Questo valore rappresenta la concentrazione di glucosio a cui si misura metà della velocità massima di trasporto. Rappresenta quindi una misura dell’affinità per il glucosio (valori piccoli caratterizzano un’efficienza di trasporto più alta). GLUT1 e GLUT 3: ubiquitariamente espressi, sono coinvolti nel trasporto continuo del glucosio (KT = 1mM) GLUT2: nel fegato e nelle cellule beta del pancreas, è coinvolto nella captazione del glucosio per la glicogenosintesi (KT = 15-20 mM) 121 GLUT4: nel muscolo, è coinvolto nell’ingresso del glucosio per la sua ossidazione (KT = 5mM), la sua espressione aumenta in seguito alla secrezione di insulina GLUT5: nell’intestino, trasporta glucosio nel circolo sanguigno CAPITOLO 20.9: REGOLAZIONE METABOLISMO GLUCIDICO E LIPIDICO 122 CAPITOLO 20.10: POTENZA, CAPACITA’ E RESA ENERGETICA 123 ALTRE DEFINIZIONI UTILI Contrazione muscolare: Cosa porta il muscolo a contrarsi? Quando il muscolo si contrae produce ormoni anabolici, che a loro volta producono messaggeri chimici che inviano il segnale fino al nucleo. Qui avviene la sintesi proteica per produrre la miosina (Proteina dei muscoli scheletrici, si differenzia dall’insulina per la forma strutturale degli amminoacidi). Inoltre, per aumentare la massa muscolare (massa magra, mentre la massa grassa è costituita dai lipidi) c’è bisogno delle proteine nobili; in questo modo il muscolo può utilizzare queste ultime in carenza di carboidrati Metabolismo umano: il nostro è un metabolismo ossidativo (respirazione cellulare), reazioni cataboliche (sostanze che vengono degradate per estrarre energia chimica) e reazioni anaboliche (piccole molecole che si convertono in molecole progressivamente più grandi, sintesi molecolare). L’equilibrio del metabolismo è detto omeostasi Biomolecole semplici: sono composti organici in quanto la sua derivazione è di tipo naturale, hanno sempre il carbonio, lo sono gli amminoacidi, i monosaccaridi, le basi azotate. Sono inorganici invece se manca il carbonio, lo sono acqua e Sali minerali Biomolecole complesse: composte da più biomolecole semplici, sono biopolimeri (lo sono le proteine, i polisaccaridi ecc.) Basi azotate: adenina, citosina, guanina, timina e uracile Isotopi: stesso numero di protoni, varia il numero di neutroni Reazione esotermica: sviluppa calore, contenuto energetico dei prodotti inferiore a quello dei reagenti Reazione endotermica: formazione dei prodotti accompagnata da calore, contenuto energetico dei prodotti superiore a quello dei reagenti Glicolisi: è l’unica via catabolica che non dipende dalla presenza dell’ossigeno. Sangue arterioso: sangue ossigenato, rosso Sangue venoso: sangue non ossigenato ma ha CO2 e protoni con esso, blu Sport Anaerobico: nello sforzo non richiede ossigeno per degradare il glucosio, produce acido lattico Glucosio: sintetizzato a partire dal glicogeno che è la sua forma di deposito Zimogeno: enzima non attivo, presente nel corpo in forma inattiva Gli antinfiammatori “distruggono” anche gli anticoagulanti in quanto, gli agenti infiammatori e questi anticoagulanti hanno mediatori chimici in comune, ecco perché, a lungo andare, gli antinfiammatori possono provocare ulcere allo stomaco o dolori intestinali dopo la loro assunzione Fosforilare: operazione fatta dalle chinasi Defosforilare: operazione fatta dalle fosfatasi (sia questa operazione che la fosforilazione sono controllate da ormoni) 124 Proteine con regolazione allosterica: tutte le proteine regolate in questo modo, hanno una forma T e una forma R proprio come l’emoglobina Differenza tra NAD e FAD: cambia la provenienza degli H Sintasi: sintetizzare senza usare energia Sintetasi: sintetizzare usando energia 125 126