Pag. 1 a 175
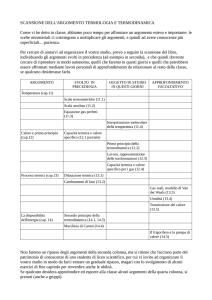
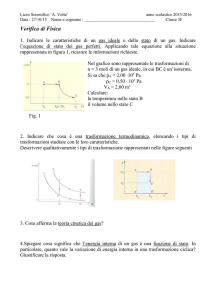
Termodinamica
Introduzione
Il presente capitolo contiene diversi argomenti che, nati separatamente, sono poi
stati unificati dalla teoria atomica. La teoria atomica nasce, come è ben noto,
nell’antica Grecia. La filosofia presocratica, sviluppatasi nelle colonie greche
dell’Italia meridionale e dell’attuale Turchia, affronta i problemi fondamentali
dello spazio, del tempo, del moto e della costituzione della materia. Aristotele
riprenderà questi temi e accetterà alcune delle soluzioni proposte dai presocratici e
ne respingerà altre. Secondo Empedocle1 (490-430 AC) medico di Agrigento, il
mondo era composto da quattro elementi: terra, acqua, aria e fuoco. L’etere,
sostanza incorruttibile, era riservata a riempire gli spazi superiori. Probabilmente
si tratta della prima presa di coscienza dell’esistenza di tre stati di aggregazione
della materia (solido, liquido, gassoso) e del ruolo che il calore, cioè il fuoco
(concepito come una sostanza), aveva nei passaggi di stato. Notiamo che
Empedocle si doveva rendere conto della necessità di considerare anche forze
oltre che materia, forze che lui chiama astio e concordia. Se pensiamo alla nostra
teoria atomica (dunque in un ambito diverso da quello di Empedocle), possiamo
forse tradurre questo nell’idea che, se gli atomi (o le molecole) vengono
compressi, si sviluppano forze repulsive che si oppongono alla compressione, ma
se tentiamo di allungare un materiale allora si sviluppano forze attrattive che si
oppongono all’allungamento. Aristotele riprende la sua teoria nella fisica,
(“Meglio sarebbe, allora, assumere principi meno numerosi, anzi finiti, come fa
Empedocle” Aristotele, Fisica, BUR, pag. 13). A causa della sua enorme
autorevolezza, le idee di Aristotele saranno accettate per secoli, fino all’avvento
della fisica moderna.
I primi filosofi che invece propugnarono l’idea dell’esistenza degli atomi furono
Democrito di Abdera in Tracia (460-430 AC) e Leucippo di Mileto in Asia
Minore (approssimativamente metà del quinto secolo AC). Le loro idee, riprese in
forma di poema, furono divulgate da Tito Lucrezio Caro (Pompei, 94 AC-Roma
50 AC) nel suo “De Rerum Natura”. In sostanza la filosofia greca nella sua analisi
preliminare del problema della costituzione della materia, non vede che due
1
…e in mezzo porterò questo tema degli elementi non generati,
il fuoco e l’acqua e la terra e l’immenso culmine dell’aria,
che mai non hanno inizio né termine alcuno,
e l’astio rovinoso, da parte, e la concordia conciliatrice.
Di qui tutte le cose che furono e saranno e le cose che sono…
Empedocle, Poema fisico e lustrale, a cura di C. Gallavotti, Oscar Mondadori, pag. 7
Cap. 5 – Termodinamica e calorimetria
possibili idee o la materia è un continuo divisibile all’infinito o la materia ha una
costituzione atomica.
L’atomo di Democrito ha però una forma e perciò una dimensione non nulla,
finendo col costituire esso stesso un continuo all’interno del proprio volume2. Una
teoria coerentemente atomica avrebbe dovuto invece considerare gli atomi
puntiformi. In affetti al giorno d’oggi noi chiamiamo “atomi” dei costituenti della
materia che non sono affatto indivisibili (“atomo” (ατοµοσ) viene dal greco
τεµνειν =tagliare che, con l’aggiunta di un’alfa privativa, dà “che non si può
tagliare”). Essi sono una struttura/aggregato di sub-costituenti. Ultimamente però
si è arrivati ai veri atomi: particelle, come gli elettroni e i quark, che sono appunto
puntiformi. Sono esse i veri atomi. Va detto che Democrito non poteva non
concepire i propri atomi se non dotati di volume e forma: nessuna altra
caratteristica (massa, carica elettrica, spin) poteva distinguerli. In aggiunta la
difficoltà con atomi puntiformi è che non si poteva costruire una materia estesa
con particelle puntiformi. Comunque ciò lo espose alle critiche di Aristotele, che
attaccò Democrito proprio su questo punto. Al giorno d’oggi i nostri “atomi”
indivisibili (leptoni e quark, tutte particelle puntiformi) possono essere distinti
tramite la massa, la carica ed altri numeri quantici senza cioè, fare ricorso a
proprietà geometriche. I costituenti di una particella, per esempio un protone,
sono i quark puntiformi, questi una volta legati fra loro costituiscono un insieme
che non può essere penetrato, respingendo ogni altro particella che tenti di
penetrare nello stesso volume. Un’ulteriore differenza tra gli atomi democritei e
quelli dei giorni nostri sta nel fatto che gli atomi di Democrito erano eterni, i
nostri “atomi” indivisibili possono essere creati (sia pure in coppia) o distrutti (sia
pure in coppia), mentre di eterno non c’è che un principio fondamentale, un
impalpabile concetto: quello di energia, questa sì eterna.
In conclusione, la filosofia presocratica ci ha lasciato in eredità il conflitto tra due
concezioni del mondo: una continua e una atomica Occorre anche dire che la
filosofia greca ci ha lasciato in eredità l’idea di legge fisica. Si può per esempio
leggere la spiegazione delle piogge e del ciclo dell’acqua o quella dei terremoti
data da T. L. Caro per apprezzare, come per la prima volta, il terremoto è
attribuito a cause naturali e non all’ira di Zeus contro qualcuno (De Rerum Natura
libro sesto, pag. 533): è nata la fisica!. 3
In tempi moderni, Galilei intuì che il calore poteva essere collegato col moto
molecolare. I. Newton (1642-1727) fu certamente un sostenitore dell’atomismo al
punto da insistere che anche la luce era formato da corpuscoli4. Anche il suo
2
…E poiché essi esistono, bisogna riconoscere che sono solidi ed eterni
T. L. Caro De rerum natura, Luca Canali, BUR pag. 119
3
Cfr Carlo Rovelli, Che Cos’è la Scienza, Oscar Mondadori, (2014), cap. 2.
4
“All things being consider’d, it seems probable to me, that God in the Beginning form’d
Matter in solid, massy, hard, impenetrable, moveable particles of such Size and Figures,
2
Cap. 5 – Termodinamica e calorimetria
rivale Robert Hook (1635-1703) applicò alla teoria dell’elasticità l’idea che la
materia era fatta di atomi. Nella sua opera la “Micrographia”(1664) scrisse che il
calore era “nothing else but a very brisk and vehement agitation of the parts of a
body”. Eventualmente D. Bernoulli (1700-1782) lavorò sulla teoria cinetica dei
gas, ma il suo lavoro (Hydrodinamica del 1738) fu ignorato.
Un altro convinto sostenitore della teoria atomica fu Pierre Gassendi (1592-1656).
Egli scrisse che “non è per nulla contraddittorio che dalla superficie dei corpi si
stacchino continuamente certi efflussi di atomi, in cui rimangono costanti la
posizione e l’ordinamento che sussistevano nei corpi stessi”5. In effetti egli
sostenne anche l’atomismo dello spazio e del tempo.
Egli entrò in polemica con gli aristotelici e in particolare contro la loro obiezione
“all’atomismo che la materia è divisibile all’infinito, perché comunque ogni
corpuscolo, per quanto piccolo possa essere pensato, consiste pur sempre di parti
distinte l’una dall’altra e per ciò divisibili”6. A questo la fisica moderna risponde
che “gli atomi” sono puntiformi.
L’atomo era un’idea il cui tempo era arrivato.
All’inizio furono chimici come Gay-Lussac, Lavoisier, Proust, Dalton, Avogadro
ed altri ad applicare l’ipotesi atomica alla chimica e dimostrarne la fecondità in
quell’ambito.
La teoria atomica eventualmente vincerà e le teorie del continuo dovranno essere
messe da parte. Il continuo cacciato dalla porta rientrerà però dalla finestra con le
onde. Il mondo non è fatto di soli atomi, ma anche da campi di forza che sono un
continuo spaziale. La scoperta che la descrizione delle particelle puntiformi deve
essere fatta usando sia il concetto di punto materiale che quello d’onda imporrà
una descrizione del mondo (almeno quello subatomico) che usa due termini,
apparentemente antitetici, ma considerati ottimisticamente complementari, di
oggetto esteso e di oggetto puntiforme.
Sulla base della teoria atomica, possiamo descrivere il comportamento dei gas e la
loro fisica, parleremo dunque di “teoria cinetica dei gas”. Con la teoria atomica
riusciremo anche a spiegare il calore e la temperatura e capire i fondamenti della
termodinamica, che noi applicheremo quasi esclusivamente ai gas, anche se la
definitiva accettazione dell’idea atomica da parte della comunità scientifica avrà
come ovvia conseguenza lo studio dei liquidi e dei solidi in termini atomici. Ci
occuperemo della viscosità dei gas, della loro diffusione e del trasporto del calore,
nonché del passaggio della corrente elettrica. Lo studio dei moti browniani con la
teoria di A. Einstein e i lavori sperimentali di J. Perrin e R. A. Milikan, chiudono
and with such other Properties…and that these primitive Particles are …so very hard, as
never to wear…”
Newton, Opticks, Dover publications, pag. 400
5
Cassirer, Ernst, Storia della filosofia moderna, Einaudi, pag. 46.
6
Gianfranco Cantelli, Enciclopedia UTET:
3
Cap. 5 – Termodinamica e calorimetria
il periodo storico di scetticismo sull’idea dell’esistenza degli atomi. Lo studio dei
calori specifici dei gas condurrà comunque a problemi risolubili solo con la
meccanica quantistica.
Ci occuperemo anche della termodinamica della radiazione elettromagnetica con
lo studio del cosiddetto “corpo nero”.
Iniziamo con lo studio del calore e della temperatura partito dalle sensazioni
individuali di caldo e di freddo e poi evoluto verso una teoria quantitativa delle
cause che sono alla base di queste sensazioni. Due concetti distinti vanno
esaminati, differenziati e resi misurabili: la temperatura ed il calore. Esamineremo
poi le leggi dei gas, in particolare dovremo esaminare il fatto che un gas esercita
una pressione sulle pareti del suo contenitore. E’ possibile spiegare la pressione
con lo scambio di quantità di moto tra molecole e pareti, cioè con gli urti sulla
parete? Dimostreremo che ciò è in effetti possibile. Armati di questo successo,
esamineremo la distribuzione delle velocità secondo Maxwell e le applicazioni
della teoria alla spiegazione di fenomeni come la viscosità, il trasporto del calore
e la diffusione. Ci occuperemo poi del primo e del secondo principio della
termodinamica e vedremo che anche questi hanno una naturale spiegazione nella
ipotesi atomica. In particolare la spiegazione dell’irreversibilità dei fenomeni
termodinamici sarà un vero trionfo per la teoria cinetica dei gas Si comprenderà
insomma l’origine microscopica del calore e l’evoluzione dei sistemi rispetto alla
loro temperatura, al calore ceduto o ricevuto ed a diversi altri parametri
termodinamici.
I concetti legati alla termodinamica hanno un’applicabilità vastissima che va dai
motori termici alla cosmologia, da semplici esperienze quotidiane a profonde
implicazioni scientifiche e filosofiche. In particolare il continuo aumento della
entropia decide il fatto che il tempo scorre solo in un senso: non si torna indietro.
La meccanica non conosce in quale verso scorre il tempo, la termodinamica sì.
Forse vale la pena di concludere questa introduzione con alcune considerazioni
sulla fisica e sul suo significato. Le sue problematiche sono ancora quelle
proposte quasi tremila anni fa dalla filosofia greca e le risposte, sempre più
sofisticate, oscillano sempre tra due opposti concetti proposti dalla stessa filosofia
greca. Tutti gli studi su problemi singoli e apparentemente limitati sono in realtà
tasselli che vanno nella direzione di costruire il puzzle del mondo fisico.
Purtroppo nel nostro paese, l’idealismo di certi filosofi, tra cui B. Croce, hanno
respinto la fisica nel novero delle scienze tecniche, insomma in un secondo piano
culturale che non è davvero meritato. Solo nel secondo dopoguerra la fisica
italiana sembra aver ripreso slancio. Non ci resta che augurarci che questo slancio
continui, ma dobbiamo ricordarci che il tentativo di riproporre la fisica, e la
scienza in genere, come subordinata alle scienze umanistiche e quindi limitarla al
ruolo di conoscenza tecnica continua a ritornare: basta notare quanti studenti
conoscono le leggi di Keplero e il loro ruolo nel passaggio dal Medio Evo a quello
4
Cap. 5 – Termodinamica e calorimetria
moderno. Non dimentichiamoci che il fanatismo religioso è sempre in agguato
dietro l’angolo. Non dimentichiamoci dello sciagurato incendio della biblioteca di
Alessandria e dell’omicidio di Ipazia. Anche le critiche verso Aristotele,
sicuramente giustificate nel complesso, dovrebbero essere rivolte con altrettanto
vigore a quelli che hanno usato Aristotele come filosofo “ufficiale”.
Infine un’ultima considerazione sul rapporto tra fisica e matematica. La
matematica è il linguaggio della fisica. Questa affermazione di Galilei è
ineccepibile. Tuttavia c’è chi tende a presentare la fisica come una sorta di
matematica applicata. La fisica cerca di costruire un’immagine del mondo e
dunque usa la matematica come suo linguaggio per uno scopo che non è affatto
quello della matematica pura. E’ un po’ come confondere il mestiere del
romanziere e quello del linguista. Studiare la lingua è molto diverso dallo scrivere
I Promessi Sposi. Non dimentichiamo poi l’aspetto sperimentale della fisica. Si
può teorizzare quel che si vuole, ma l’esperimento ha un potere di veto: può
falsificare qualunque teoria e se questo succede la teoria, per quanto ineccepibile
matematicamente ed esteticamente ammirabile. deve essere abbandonata. Questo
non accade in matematica!
Rimane comunque assai oscuro il perché la matematica sia il linguaggio della
fisica. Rimane cioè oscuro perché le previsioni della matematica siano così spesso
corrette ed aprano un fronte nuovo. Le previsioni della teoria quantisticarelativistica di Dirac fecero scoprire l’antimateria prima sulla carta che in
laboratorio. Perché la matematica ha questo enorme potere di predizione? Sembra
quasi che la Natura si debba assoggettare alla logica matematica. Mi pare che
nessuno abbia ancora una risposta. Giusto uno dei misteri ancora aperti.
1. La temperatura
Tutti noi abbiamo sperimentato il contatto con un corpo caldo o freddo ed
abbiamo una conoscenza intuitiva di cosa voglia dire “caldo” o “freddo”. Occorre
però tradurre i concetti di “caldo” e “freddo” in un’operazione di misura che dia
un risultato numerico ed associ al concetto primitivo un numero. Questo, anche al
fine di evitare l’elemento soggettivo (quello che è caldo per una persona può non
esserlo per un’altra).
Si tratta quindi di paragonare il nostro sistema (il corpo) ad un altro corpo
standard (il termometro) che sia altrettanto caldo, ma che sia tarato in modo da
fornire una temperatura. C’è in questo procedimento una vaga rassomiglianza con
la misura del tempo, dove viene paragonata l’evoluzione del sistema meccanico in
esame (una cometa che si avvicina al Sole, per esempio) con il numero di cicli di
un orologio. Vi è però anche una grande differenza, per il fatto che il termometro
deve essere portato in equilibrio termico con il sistema del quale si vuole misurare
la temperatura. S’intende anche che, perché abbia senso misurare la temperatura
5
Cap. 5 – Termodinamica e calorimetria
di un sistema, questo deve essere in equilibrio termico, deve, cioè, essere
caratterizzato da un’unica temperatura al suo interno. Detto ciò, occorre trovare
un effetto legato alla variazione di temperatura di un corpo che sia misurabile in
termini di parametri fisici noti, in modo da poter usare questo corpo come
termometro.
Mettendo a contatto corpi a temperatura diversa, questi si portano alla stessa
temperatura (postulato zero della termodinamica) e, dunque, mettendo a contatto
il termometro ed il sistema del quale si vuole misurare la temperatura, il
termometro si porta alla stessa temperatura del corpo: misurando il parametro
fisico prescelto del termometro, si conoscerà allora la temperatura del termometro
e perciò quella del sistema. Si può obiettare che, se non si sa misurare la
temperatura, allora non si può affermare che “mettendo a contatto corpi a
temperatura diversa, questi si portano alla stessa temperatura”. Vero, ma non del
tutto, perché è comunque chiaro che, mettendo una pietra in un catino d’acqua
bollente, la pietra diventerà calda anzi bollente come l’acqua. Se l’impressione
qualitativa non convincesse, allora - come primo passo verso la definizione
operativa della temperatura - occorrerà dimostrare che i corpi in contatto si
portano alla stessa temperatura, cosa non difficile perché si può notare che il
“termometro” evolve allo stesso stato se messo in contatto con l’acqua o con la
pietra dopo che questa è stata immersa nell’acqua per un po’ di tempo. Più
insidiosa potrebbe essere l’obiezione che, mettendo la pietra nell’acqua, la
temperatura finale della pietra e dell’acqua sono sì la stessa, ma non quella che
l’acqua aveva all’inizio. Ne seguirebbe che un’operazione di misura modifica
sempre la grandezza “temperatura” che vogliamo misurare. La ragione sarebbe
naturalmente che la temperatura di un corpo dipende da un qualcosa che il corpo
ha e che dovrebbe parzialmente cedere al termometro, perché questo si porti
all’equilibrio termico. Questa è una considerazione giusta, ma si può verificare
che tale effetto può essere reso piccolo a piacere. Si prenda, per esempio,
dell’acqua come sistema del quale si vuole misurare la temperatura e si definisca
un termometro, come un’asticella metallica della quale possiamo misurare la
lunghezza. È facile verificare che l’asticella immersa in un bagno d’acqua
bollente cambia la sua lunghezza. È facile anche verificare che la lunghezza
dell’asticella lasciata immersa nell’acqua mentre quest’ultima viene riscaldata,
cambia in modo continuo. Si possono allora considerare uguali le temperature di
due corpi (per esempio due masse d’acqua), se la nostra asticella acquista la stessa
lunghezza quando è messa a contatto e portata in equilibrio con i due corpi. Si
scaldino adesso esattamente nello stesso modo due masse diverse d’acqua, in
modo da poterle ragionevolmente supporre alla stessa temperatura, e si mettano a
contatto termico con il termometro. L’asta raggiunge la stessa lunghezza? Se si
notano delle differenze, si può però verificare che la lunghezza finale del
termometro tende ad un valore preciso che non dipende più dalla quantità
6
Cap. 5 – Termodinamica e calorimetria
d’acqua. Si possono allora interpretare questi fatti, affermando che il qualcosa,
chiamiamolo il “calore”, che l’acqua cede al termometro è una frazione sempre
più piccola del suo calore iniziale e quindi la temperatura finale del sistema acqua
più termometro tenderà alla temperatura iniziale dell’acqua. In questo modo ci si
trova a definire due concetti: la temperatura, come indice del calore (vedremo che
dovremo dire “energia interna”) posseduto da un corpo, ed il calore stesso.
Del resto è facile affermare che la temperatura è solo un indice del calore
posseduto da un corpo. La quantità di combustibile che dobbiamo bruciare per
riscaldare un litro od un metro cubo di acqua non è la stessa. Se il riscaldamento
avviene a causa della fiamma che è stata accesa, allora si può affermare che la
quantità di calore che abbiamo fornito, e che quindi l’acqua possiede, dipenderà
dalla massa d’acqua.
Effettuando degli esperimenti comparativi, per vedere se il riscaldare alla stessa
temperatura dell’acqua o piuttosto un altro liquido comporti cessioni di quantità di
calore diverse, permette alla fine di definire la quantità di calore Q ceduta alla
massa m per variare la sua temperatura di ∆T, grazie alla formula: Q = cm∆T , dove
c è un coefficiente specifico della sostanza, detto “calore specifico”, che
rappresenta la quantità di calore da cedere alla massa unitaria della sostanza
perché aumenti la sua temperatura di un grado.
Da notare che:
• Per essere significativa questa definizione ha bisogno della definizione di una
scala termometrica, che è peraltro arbitraria a questo punto. Come esempio di
scale arbitrarie si consideri quella Celsius o centigrada: si ponga la differenza
di temperatura tra un miscuglio di acqua e ghiaccio in equilibrio termico e
l’acqua in ebollizione, pari a 100 gradi. Si prenda, per esempio, un capillare
pieno di mercurio e si noti la lunghezza della colonnina di mercurio, quando il
termometro è in equilibrio termico con il miscuglio acqua-ghiaccio e con
l’acqua bollente. Si divida poi l’intervallo di lunghezza in cento parti uguali e
si otterrà il grado Celsius. Allo stesso modo si possono definire le scale
Fahrenheit e Réaumur (0 e 80 gradi agli stessi punti della scala Celsius).
• Si tenga presente che le quantità di calore conosciute sono esclusivamente
quelle scambiate: non si conosce il calore realmente posseduto dal corpo: si sa
solo che gli si è ceduta, o si è ricevuta da esso, una quantità di calore, pari a
quella precedentemente definita: Q = cm∆t . Lo stesso vale per la temperatura:
non si ha una definizione di temperatura assoluta, si misurano solo delle
variazioni di temperatura rispetto ad uno zero arbitrario.
• Molti termometri sono, in effetti, basati sulle variazioni di lunghezza di un
corpo in funzione della temperatura. In pratica, per piccole variazioni della
temperatura, si può assumere che la lunghezza vari linearmente con la
temperatura stessa. Si tratta, in realtà di una prima approssimazione resa
7
Cap. 5 – Termodinamica e calorimetria
possibile dal fatto che la funzione l=l(t) può essere sviluppata in serie di
potenze con l’espansione troncata al primo ordine: l (t ) = l 0 + αl 0 ∆t , dove α è
detto “coefficiente di espansione lineare” e rappresenta la variazione relativa
della lunghezza dell’asta per grado di temperatura: α = ∆ l .
l0 ∆t
In pratica, tanto le misure di temperatura che quelle di calore vengono effettuate
con delle tecniche che sono state sviluppate lungo un periodo di tempo abbastanza
lungo. Tali tecniche sono state raffinate attraverso una serie di accorgimenti volti
a ridurre dispersioni e perdite di calore, utilizzando materiali isolanti e tecniche di
coibentazione, a tenere in conto i calori assorbiti o ceduti agli apparecchi, a
misurare i calori specifici dei vari materiali usati, ecc...
Non si possono descrivere qui tutti gli
termometro
apparecchi ed i termometri che sono stati
sviluppati nel corso del tempo. Ci si
accontenti di sapere che vi sono dei
termometri basati sulla scala Celsius e
Parete
sull’espansione dei materiali in funzione
interna
della temperatura e che esistono degli
vuoto
apparecchi, detti “calorimetri” che
H 2O
vengono utilizzati per misurare le
quantità di calore trasferite.
Parete a
Un esempio di calorimetro è nella fig. 1.
specchio
Si tratta di un contenitore cilindrico con
un coperchio. Attraverso un foro
praticato nel coperchio, si può introdurre
un termometro all'interno del contenitore.
Si abbia anche uno strumento che
Fig. 1: Calorimetro.
permetta di mescolare ed agitare
eventuali liquidi presenti nel contenitore.
Si supponga di voler misurare il calore trasferito ad un corpo, aumentandone la
temperatura di ∆t . Si può riempire il cilindro di acqua ed immergervi un corpo a
temperatura t 2 fino a che la sua temperatura scenda al valore t1 .
Questa sarà anche la temperatura finale dell’acqua.
Pertanto la quantità di calore ceduta all’acqua dal corpo sarà data da Q = cm∆t . E
questa quantità sarà anche uguale alla quantità di calore ceduta all’acqua
Q = c H O m H O ∆t H O .
2
2
2
In questo modo, eguagliando le due quantità di calore, è possibile misurare il
calore specifico del corpo, una volta che se ne sia misurata la massa pesandolo:
8
Cap. 5 – Termodinamica e calorimetria
c=
c H 2O m H 2O ∆t H 2O
m∆t
. Per completare la misura occorre conoscere il calore
specifico dell’acqua. La cosa più logica da fare è di definirlo come unitario: la
quantità di calore unitaria, la “caloria”, è la quantità di calore che dobbiamo
cedere alla massa unitaria di acqua perché la sua temperatura vari di un grado
centigrado. Dunque: c H O = 1 . Si parla di piccola o di grande caloria a seconda che
2
la massa d’acqua scelta nella definizione sia il g o il kg.
La quantità C = cm , dove m è la massa del corpo e c è il calore specifico, viene
chiamata “capacità termica” del corpo. Per realizzare un buon calorimetro
occorre, come si è detto, fare attenzione a diversi effetti che possono
compromettere la misura di interesse. Prima di tutto, occorre ridurre i fenomeni di
dispersione del calore: se una quantità sostanziale di calore attraversa le pareti del
cilindro e viene persa, la temperatura finale del sistema corpo + acqua non
dipenderà solo dalle masse e dai calori specifici in gioco. Ne segue che occorre
conoscere in dettaglio le quantità di calore che, per una ragione o per l’altra,
vengono perse. Si devono inoltre misurare vari coefficienti che misurano la
capacità di una sostanza di trasmettere il calore e tenerne conto nella
progettazione e nell’uso dei calorimetri. Per esempio, è noto che la trasmissione
del calore avviene attraverso vari meccanismi. Uno è l’irraggiamento: ogni corpo
irraggia onde elettromagnetiche e quindi perde calore. In genere, però, assorbe
anche onde elettromagnetiche dal mondo esterno. All’equilibrio, si crea un
bilanciamento tra il calore che viene perso e quello che viene acquisito.
L’irraggiamento da parte di un corpo caldo è un fenomeno familiare: basta
mettere la mano in prossimità di un radiatore acceso. Se vogliamo tenere conto di
questo effetto e ridurne l’importanza, occorrerà dotare il cilindro di una doppia
parete: quella interna irraggerà verso quella esterna che rifletterà l’infrarosso
indietro alla prima parete. La seconda parete deve essere uno specchio! È in
questo modo che si fabbricano i “thermos”. Se poi si fa il vuoto tra le due pareti,
allora non ci sarà un materiale che trasmette il calore per “convezione”, tra le due
pareti. La “convezione” in questo caso avverrebbe nel modo seguente: l’aria si
riscalda e si muove tra la parete calda e quella fredda cedendo calore alla parete
fredda. Senza materiale tra le due pareti non si verifica neppure il trasporto di
calore per “conduzione”, che vuol dire calore trasportato attraverso un corpo.
Dunque si è imparato a costruire un contenitore per calorimetria studiando i
meccanismi di trasferimento del calore.
2. Le leggi dei gas
Con il termine di gas si intende uno stato di aggregazione della materia in cui il
materiale non ha né forma né volume fissi. In effetti, un gas è un insieme di
9
Cap. 5 – Termodinamica e calorimetria
molecole mono o poli atomiche con un’interazione reciproca debole o nulla. Le
molecole che costituiscono il gas sono libere di allontanarsi l’una dall’altra,
occupando tutto il volume messo a disposizione ed assumendo pertanto la forma
del contenitore. Se l’interazione tra le molecole è nulla e se le molecole sono tanto
piccole da potersi considerare a volume nullo, allora il gas è detto “ideale”. La
nozione di gas ideale discende più da una legge di comportamento che dalle
proprietà delle interazioni o del volume delle molecole: tuttavia le due cose
finiscono con l’identificarsi. La prima legge dei gas ideali dice che il prodotto del
volume per la pressione è costante se il gas è mantenuto a temperatura costante:
PV = cost a t = cost (legge di Boyle e Mariotte, 1616)
Si prenda dunque un
cilindro con un pistone
Pistone
(fig. 2) e si comprima il
gas. Si inserisca un
termometro, in modo da
misurare la temperatura
Termometro (t)
del gas. Si noterà che la
V
Manometro (P)
temperatura non rimane
costante. E' noto che,
Gas
gonfiando una ruota di
bicicletta, il pneumatico
e la pompa si riscaldano.
.
È
possibile
però
Fig. 2: Legge di Boyle e Mariotte.
assicurarsi un buon
contatto termico con
l’esterno e fare in modo
che lo scambio di calore
con l’esterno mantenga
la temperatura costante.
Che questo avvenga realmente, può essere verificato con un termometro. Il
volume, inoltre, è facilmente misurabile, essendo direttamente correlato alla
posizione del pistone. Il volume del gas è, infatti, il prodotto dell’area della base
interna del cilindro per l’altezza, misurata a partire dalla base inferiore interna del
cilindro.
Si deve adesso definire cosa si intende con il termine “pressione”.
È noto a tutti che, se si gonfia un palloncino, l’aria che abbiamo immesso nel
palloncino (o nel pneumatico, appunto) esercita una forza sulle pareti del
palloncino stesso: questa forza che si esercita contro le forze elastiche della
gomma è direttamente misurata dall’estensione del palloncino. L’aria esercita
10
Cap. 5 – Termodinamica e calorimetria
dunque una pressione. Per pressione si intende il rapporto tra la forza esercitata
sulle pareti e l’area delle pareti. È possibile, inoltre, che la forza esercitata dipenda
dalla posizione: si definisce quindi pressione il rapporto tra forza ed area
dF
infinitesime, P =
. Attenzione! Col termine “pressione” si intende uno scalare,
dS
dunque la forza qui considerata ha una direzione fissa: quella perpendicolare alla
parete.
Un manometro è uno strumento che viene sottoposto a forze di pressione che
provocano lo spostamento di un ago su di un quadrante. Un comune barometro
aneroide è un esempio di manometro: un piccolo cilindro metallico al cui interno
è stato fatto il vuoto, viene deformato dalla pressione esercitata dall’aria. Lo
spostamento della parete deformata viene trasmesso, amplificato mediante un
sistema di leve, ad un ago che si sposta lungo un quadrante.
Un metodo, forse più diretto, per
Gas
Aria
misurare la pressione di un gas ed in
particolare quella atmosferica, consiste
nel disporre di una colonna liquida in un
tubo ad U (fig. 3). La pressione esercitata
dal gas sulla superficie libera del liquido
in uno dei rami della U spingerà il
liquido verso l’alto nell’altro ramo della
h
U: il dislivello h nei due rami misura
proprio la pressione del gas. Il ramo non
in contatto con il gas può essere lasciato
aperto e dunque soggetto alla pressione
di quel gas che chiamiamo atmosfera e
che è costituito al 20% di O2 e per il resto
.
di N2. Oppure può essere chiuso avendo
Fig. 3: Tubo ad U.
fatto il vuoto sopra il liquido. Se il tubo è
aperto si misurerà la differenza di
Aria
pressione tra i due rami e dunque la
pressione
del
gas
relativamente
Vuoto
all’atmosfera.
Viceversa è possibile chiudere uno dei
rami del tubo, riempirlo di liquido e poi
raddrizzare la U in modo che al di sopra
del liquido vi sia il vuoto. Il vuoto non
h
sarà comunque assoluto, giacché il
liquido evapora e dunque nello spazio al
di sopra del liquido rimarrà comunque il
vapore (saturo) del liquido. Se l’altro
11
.
Fig. 4: Barometro di Torricelli (a
sifone).
Cap. 5 – Termodinamica e calorimetria
ramo rimane aperto verso l’atmosfera,
allora il dislivello misura direttamente la
pressione atmosferica (fig. 4).
Un apparato di questo tipo è detto
barometro di Torricelli. Come è noto, la
pressione atmosferica ammonta a circa 1
kg / cm2 .
Per non avere una colonna troppo alta nel barometro si utilizza uno dei liquidi più
pesanti: il mercurio (Hg). Un altro vantaggio del mercurio è quello di avere una
tensione di vapore saturo molto bassa.
Vediamo adesso com’è correlata l’altezza della colonna liquida alla pressione.
Se si prende una colonna di liquido di densità ρ e di area di base S, il peso della
colonna sarà: mg = Vρg = hSρg . Dunque la pressione sarà: P =
mg
= hρg (Legge
S
di S. Stevin 1548-1620). Poiché la densità del mercurio è di 13 , 6 g / cm 3 , risulta
che l’altezza della colonna di mercurio corrispondente ad un’atmosfera standard
al livello del mare (a 00C ed a 450 di latitudine) è di 760mm di Hg il che produce
una pressione di 1,013bar. Il bar è l’unità di pressione corrispondente a
10 N / cm 2 ≈ 1kg / cm 2 .
Si deve ricordare tuttavia che l’unità standard di pressione nel sistema SI è il
N / m 2 , detto pascal (Pa). Ne segue che la pressione di un’atmosfera corrisponde a
1, 013 ⋅ 10 5 Pa .
Spesso si usa come unità di misura la pressione di un’atmosfera, chiamandola
semplicemente “atmosfera” o atm. Si usa anche il “mm di Hg”, altrimenti detto
“torricelli” ed abbreviato in Torr, che è la pressione esercitata da una colonna di
mercurio alta 1mm. In acqua (densità = 1 g/cm3), un’atmosfera corrisponde
all’incirca a 10m.
Naturalmente, poiché la pressione al suolo dipende dall’altezza della colonna
d’aria atmosferica, più in alto si sale, più la pressione atmosferica si abbassa. Un
barometro può quindi essere usato anche come altimetro. La pressione diminuisce
con l’altezza anche perché l’accelerazione di gravità diventa più piccola
all’aumentare della distanza dal centro della Terra. Questo tuttavia è un effetto
piccolo, che di solito si trascura. Non è invece trascurabile la variazione della
densità atmosferica (si veda più avanti) qualora si calcoli la variazione della
pressione con l’altitudine. Per quanto oggigiorno la cosa possa sembrare
abbastanza logica e semplice, occorre ricordare che il vuoto realizzato da
Torricelli nel suo barometro risultò piuttosto sorprendente all’epoca per tutti
coloro che avevano pensato che il vuoto non potesse esistere. Questo fatto inflisse
12
Cap. 5 – Termodinamica e calorimetria
sicuramente un’ulteriore sconfitta ai principi dell’aristotelismo∗. Secondo
Aristotele, lo spazio non aveva una realtà indipendente dai corpi. Erano i corpi ad
essere dotati di estensione e dunque a produrre lo spazio: così uno spazio vuoto
risultava essere un controsenso. Questa idea era in contrasto con quella degli
atomisti (Democrito, Lucrezio) che immaginavano invece il vuoto tra gli atomi.
Nel momento in cui però si è stati capaci di realizzare il vuoto nello spazio
superiore del tubo, si è avuta una prova che Aristotele sbagliava e gli atomisti
avevano ragione. Esiste uno spazio indipendente dai corpi nel quale i corpi
possono essere contenuti. Per Aristotele tuttavia, la negazione del vuoto aveva
anche il senso di richiedere che lo spazio potesse essere il supporto di fenomeni
come la luce. Lo spazio dunque veniva
definito, in assenza di corpi fatti di materia ordinaria (inclusa l’aria), dall’etere
che veniva al contempo a essere quel corpo la cui estensione generava lo spazio
∗
Un’interessante rassegna storica si può leggere in: M. Mamiani, Storia della scienza
moderna, Laterza (1998), pag. 165. Per conoscere le idee di Aristotele sul vuoto, si legga:
Aristotele, Fisica in Opere, Vol. 3, Laterza (1995), pag. 86 e seguenti.
13
Cap. 5 – Termodinamica e calorimetria
ed il mezzo “luminifero”. Come si è visto, l’esistenza dell’etere venne invocata
anche da Maxwell come supporto del campo elettromagnetico e poiché l’etere
sembrava essere un assoluto, avrebbe potuto generare, nel senso aristotelico, lo
spazio assoluto invocato da Newton. L’esperimento di Torricelli dimostra che è
possibile realizzare uno spazio vuoto di materia ordinaria. L’esperimento di
Michelson chiarirà, quasi tre secoli dopo, che l’etere luminifero non esiste.
Einstein, in seguito, eliminerà il concetto di spazio assoluto. In conclusione:
avevano ragione gli atomisti. Qualche anno dopo (1654) Otto von Guerricke,
inventata la pompa da vuoto, fece un esperimento in cui due emisferi di rame
furono fatti combaciare e svuotati dell’aria. Molti cavalli, attaccati a ciascun
emisfero, per quanto tirassero, non furono capaci di separare i due emisferi.
Ritornando alle leggi dei gas, la legge di Boyle e Mariotte può riscriversi nella
forma più completa (legge dei gas perfetti):
PV = nRT
dove n è il numero di moli di gas, R=8,31J/0K è detta “costante dei gas” e T è la
temperatura assoluta o Kelvin, della quale si parlerà più estesamente. Per ora la
considereremo una scala di temperature come quella Celsius, ma con lo zero
spostato di 273,15 gradi sotto lo zero Celsius.
Fu scoperto infatti da A. Avogadro (1776-1856) che il volume V0 di una mole di
un gas qualunque, alla pressione P0 di un’atmosfera ed a temperatura ambiente (T0
=273 0K) - le cosiddette “condizioni standard” - è sempre di 22,414litri o
22 , 414 ⋅ 10 −3 m 3 .
Ciò vuol dire che la quantità: R =
P0V 0
è indipendente dal gas ed
T0
8,31 J/0K. Ricordiamo inoltre che in una mole di gas ci sono
N A = 6 , 06 ⋅ 10 23 molecole. NA è noto come “numero di Avogadro” e fu misurato da
J. Perrin nel ‘900.
uguale a
14
Cap. 5 – Termodinamica e calorimetria
Esperimento degli emisferi di Magdeburgo.
Nel 1802 fu inoltre scoperto da L. Gay-Lussac (1778–1850) che l’espansione a
pressione costante di un gas in funzione della temperatura obbedisce alla legge:
V = V 0 (1 + αt ) , dove t è la temperatura Celsius; α è il coefficiente di espansione e
1 0 −1
C . Se riscaldiamo il
risulta essere essenzialmente uguale per tutti i gas: α =
273
gas a volume costante vale una legge simile per l’aumento di pressione:
P = P0 (1 + αt ) , con lo stesso valore di α . Ne segue che, se riscaldiamo un volume
V0 di gas facendolo passare dalla pressione P0 alla pressione P a volume costante
e poi lo facciamo espandere a temperatura costante, si avrà:
PV = V0 P0 (1 + αt ) = V0 P0
273 + t
T
, dove abbiamo indicato con T la nuova
= V0 P0
273
T0
scala di temperature empiriche definita sopra e con T0 il valore corrispondente allo
zero gradi centigradi in questa scala.
15
Cap. 5 – Termodinamica e calorimetria
Per la legge di Gay-Lussac, cambiando pressione o volume del gas, si avrà
dunque:
PV
PV
= 0 0 = R . Ne segue che, per una quantità di gas pari a n moli,
T
T0
dovrà essere: PV= nRT.
Per ultimo vogliamo enunciare la legge di Dalton. Se mescoliamo
n1 ...nk
moli di
k gas perfetti nello stesso volume V , ciascuno di essi avrà la “pressione
parziale” Pk =
nk RT
. La pressione totale sarà semplicemente la somma delle
V
pressioni parziali: PV = (n1 + ... + nk )RT = nRT .
Vale la pena di ricordare che in tempi moderni, l’idea che esistessero atomi e
molecole fu resuscitata dalle due leggi: una detta delle proporzioni semplici (J
Proust, 1720) che asserisce che “quando due o più elementi reagiscono per
formare un certo composto, essi si combinano secondo proporzioni di massa
definite e costanti” e l’altra detta delle proporzioni multiple (J. Dalton) che
asserisce che “le quantità di un elemento che si combinano con una ben definita
quantità di un altro per formare diversi composti sono fra loro in un rapporto
intero e piccolo”. Queste due leggi trovano una spiegazione facile nell’idea
atomica.
Tornando al problema della variazione della pressione con l’altitudine sul livello
del mare, consideriamo un’applicazione dell’equazione dei gas perfetti.
Facciamo allora l’ipotesi che l’atmosfera abbia tutta la stessa temperatura e
vediamo come la pressione cambia con l’altitudine. Se passando da una altezza h
ad h+ dh , a densità ρ (h) , la pressione cambia di dP = − gρ (h) dh , allora possiamo
AP
AP
dh , che,
sostituire ρ (h) =
e avere l’equazione differenziale: dP = −g
RT
RT
integrata, dà:
P(h) = P0 e
−
h
h0
, con
h0 =
RT
gA
(=
8, 31 ⋅ 300
9, 81 ⋅ 30 ⋅ 10 3
= 8, 47 ⋅ 10 3 m )
a T = 300 K e A = 3 0 per l'aria Poiché però la colonna d’aria non è realmente
isoterma eccetto che per le prime centinaia di metri, questa equazione ha limitato
valore; per h piccoli la possiamo sviluppare in serie e concludere che:
P ( h ) = P0 (1 −
h
).
h0
Due precisazioni:
16
Cap. 5 – Termodinamica e calorimetria
•
Il segno meno nell’espressione:
dP = − gρ ( h) dh
all’aumentare di h, la pressione cala. Dunque:
•
è necessario perché,
dP
<0.
dh
AP
è un altro modo di esprimere l’equazione dei gas perfetti.
RT
Infatti il numero di moli n che appare nel membro di destra è il rapporto tra la
massa del gas e il suo peso molecolare A, rendendo esplicito questo rapporto,
m
dividendo ambo i membri per V e ponendo infine ρ = , si ottiene:
V
La formula ρ (h) =
h
−
m
m PA
PV = nRT = RT ⇒ ρ = =
. Si noti che la formula P(h) = P0 e h0 si può
A
V RT
riscrivere P (h) = P0e
Boltzmann) e m =
−
−
gAh
RT
A
NA
= P0e
−
gAhN A
N A RT
= P0e
−
gmh
kT
in cui k =
R
NA
(Costante di
è la massa di una molecola. Infine si può scrivere:
U(h)
kT ,
P0e
in cui U(h) è l’energia potenziale di una molecola nel campo
gravitazionale costante.
C’è poi da aggiungere che un gas si comporta come gas perfetto fino a che la sua
densità non è troppo alta. All’aumentare di quest’ultima si noteranno delle
deviazioni dal comportamento indicato dalla legge di Boyle ed occorrerà
utilizzare la legge di Van der Waals (1837-1923), vedi oltre, che differisce dalla
legge dei gas perfetti per due termini:
an2
(P + 2 )(V − nb) = nRT 7
V
I gas che non obbediscono alla legge dei gas perfetti, sono i vapori, ai quali si è
già accennato. Se si riscalda un liquido, si osserva che una parte di esso evapora
lentamente, passa cioè alla fase gassosa. Se il vapore che si forma viene tenuto in
un volume chiuso al di sopra del liquido, ad un dato momento il liquido non
evaporerà più. Ciò accade quando la pressione o tensione del vapore raggiunge un
7
4
3
3
Il “covolume” b rappresenta il volume delle molecole: b = N A πρ , con
ρ
il raggio
molecolare. Si può facilmente vedere che si tratta di un numero piccolo confrontando la
densità di un liquido e di un gas che sono normalmente nel rapporto 10 − 3 . Nel liquido le
molecole sono a contatto, dunque il loro volume è prossimo a b, nei gas è V .
17
Cap. 5 – Termodinamica e calorimetria
valore massimo, detta “tensione di vapore saturo”8. Perché si abbia ulteriore
evaporazione, occorre che la temperatura aumenti ancora. Quando la temperatura
raggiunge un valore per cui la tensione di vapore saturo è uguale alla pressione
esterna, allora l’evaporazione si trasforma in ebollizione. Ebbene i vapori non
obbediscono alla legge dei gas, perché, se compressi, una volta che hanno
raggiunta la tensione di vapore saturo, non possono fare altro che condensarsi. Del
resto ogni gas può essere liquefatto, se la sua temperatura è al di sotto di un valore
tipico di ogni gas e chiamata” temperatura critica”. In questo caso, occorre
prestare attenzione perché un gas può essere portato in prossimità della
saturazione dove un ulteriore aumento di pressione porta alla condensazione.
Dunque occorre tenersi lontano dalla saturazione. Se però il vapore viene portato
ad una temperatura abbastanza alta rispetto a quella di ebollizione, cioè superiore
ad un temperatura critica, allora esso non condensa più, indipendentemente dalla
pressione alla quale lo si sottopone. Un gas in queste condizioni può più
facilmente comportarsi come un gas perfetto. Gas molto lontani che a temperatura
ambiente sono molto lontani dalla temperatura critica sono: l’ossigeno, l’azoto, i
gas nobili, ecc...
Vogliamo aggiungere che con un gas perfetto si può realizzare un termometro, per
esempio, mantenendo il gas a volume costante e misurando la variazione di
pressione con la temperatura. Il termometro così realizzato non dipende dal gas
scelto.
Ogni passaggio di stato, per esempio passaggio da liquido a gassoso o viceversa,
comporta l’assorbimento o l’emissione di una quantità di calore, detta calore
latente di quel passaggio di stato. Ci sarà allora un calore latente di fusione o
evaporazione, ecc… Durante l’ebollizione la temperatura del liquido rimane
costante. Stessa cosa dicasi per la fusione. Durante la liquefazione di un vapore la
pressione è costante. Vediamo che succede usando delle isoterme, cioè usando il
piano P − V e tracciando le curve a T = costante.
P
A
B
V
8
Il rapporto tra la tensione del vapore d’acqua presente in un ambiente e quella del vapore
saturo alla stessa temperatura è chiamata normalmente “umidità relativa” che è misurata
dai comuni igrometri.
18
Cap. 5 – Termodinamica e calorimetria
Questo è l’andamento della curva a temperatura costante, se la temperatura è
inferiore ad un certo valore di temperatura detta” temperatura critica”.
A destra del punto B, il gas è un vapore, che raggiunge, muovendosi verso
sinistra, la saturazione appunto nel punto B. Nel tratto B-A, il vapore è saturo e
con una sua frazione liquefatta sempre maggiore muovendosi verso A. tentando di
aumentare la pressione si ottiene solo un aumento della frazione liquefatta. Il
tratto B-A è isobaro ovvero a pressione costante. Se il valore di questa pressione è
superiore a quella esterna, abbiamo l’ebollizione. Al punto A, il vapore è
completamente liquefatto. Ogni aumento di pressione conduce solo a variazioni di
volume trascurabili, ovvero il liquido è incomprimibile. Al di sopra della
temperatura critica, la curva è la curva indicata dall’equazione di stato del gas e il
gas non può essere liquefatto. Durante il tratto A-B calore (latente di
evaporazione) viene assorbito, nel tratto B-A calore (latente di liquefazione) viene
rilasciato.
3. Le origini atomiche del calore e del comportamento dei gas
In effetti, non è difficile farsi un quadro chiaro della fenomenologia della
temperatura, del calore e del comportamento dei gas. Storicamente, fu proprio
così che l’idea dell’esistenza degli atomi fu definitivamente accettata: grazie alle
spiegazioni che essa forniva ai fenomeni termici e termodinamici. Il punto
principale è capire come l’esistenza degli atomi sia legata a quella quantità che
abbiamo chiamato “calore”. Dunque, un gas è un insieme di particelle
essenzialmente non interagenti fra loro che chiamiamo molecole. Le molecole
saranno in moto e potranno essere rappresentate da punti materiali dotati di
energia cinetica e quantità di moto. Le velocità sono molto diverse fra loro ed
orientate in ogni possibile direzione. Le molecole si urteranno e cambieranno
direzione, energia cinetica e quantità di moto ad ogni collisione∗. Nel suo libro “la
rivoluzione dimenticata” che tratta della scienza nel periodo alessandrino, Lucio
Russo nota che (pag 346) “ il termine gas è una deformazione fiamminga del
greco χαοσ (caos) e fu introdotto nel XVI secolo da J.B. Van Helmont, cui
∗
Scrive Tito Lucrezio Caro:
“Certamente, infatti, gli atomi non si sono disposti
ciascuno nell’ordine proprio per un loro disegno sagace,
né certo pattuirono quali moti essi avrebbero impresso;
ma poiché in mille modi diversi, sbalzati dagli urti,
senza posa si aggirano nel vuoto da tempo infinito,
e provano ogni genere di moto e ogni tipo di unione...
Tito Lucrezio Caro, La natura delle cose, BUR (1997), pag. 149.
19
Cap. 5 – Termodinamica e calorimetria
evidentemente era ben nota sia la natura caotica dei gas sia l’origine greca di tale
nozione”.
È possibile da un quadro di questo genere ricavare l’equazione dei gas perfetti ed
un’interpretazione del calore e della temperatura? La risposta è positiva. I primi
calcoli di questo genere furono eseguiti da Bernoulli (1720) poi da Joule (per
molecole urtanti la parete perpendicolarmente e tutti con la stessa velocità, pari
alla velocità media), furono poi estesi da R. Clausius al caso di molecole urtanti
la parete con qualunque angolo, ma tutte con la stessa velocità pari alla velocità
media. Clausius ottenne l’equazione dei gas perfetti e al posto del volume usò il
volume meno il covolume V −b. Seguiamo prima il ragionamento di Clausius,
modificandolo cioè non tenendo in conto il covolume e introducendo la
distribuzione delle velocità che, come vedremo, risulta inutile.
Se il Parete
nostro gas è contenuto in un recipiente di volume V, le molecole di cui è
composto urteranno continuamente le pareti
pn
del recipiente, trasferendogli una certa
Molecola
quantità di impulso. In media la quantità
totale di moto trasferita alle pareti sarà zero,
sommando su tutte le pareti, ma ciascuna
θ
superficie elementare dS del recipiente
pf
riceverà un impulso; è facile vedere che la
quantità di moto trasferita ad ogni collisione
è: dp = 2 p n , dove p n è la componente
− pn
normale alla parete dell’impulso della
molecola urtante. Dalla fig. 5, infatti, è chiaro
Fig. 5: Urti contro le pareti.
che:
p f − pi = p n − (− p n ) = 2 p n . Ciò segue dalla
p
i
ν
θ
formula della teoria delle collisioni:
vcosθ
p1 = p0
m1
m2
(cosθ + cos2 θ + 22 − 1)
m1 + m2
m1
che impone che il modulo finale dell’impulso
p1
Fig. 6: velocità nell’urto
sia uguale al modulo iniziale
p0 ,
se la
massa m2 →∞, come deve essere per un
muro o una parete. Inoltre si assume qui che
la reazione del muro sia puramente
ortogonale al muro e dunque la componente
della quantità di moto finale parallela al muro
rimanga la stessa dopo la collisione, per
20
Cap. 5 – Termodinamica e calorimetria
mancanza di forze che potrebbero cambiarla.
Ne segue che la componente normale al muro
deve essere la stessa di quella iniziale, ma col
verso cambiato.
Pertanto ciascuna collisione dovuta ad una particella di impulso p = m v
contribuirà al momento trasferito per unità di tempo (dunque alla forza)
sull’elemento di superficie dS con la quantità: 2mvn=2mvcosθ (fig. 6).
La pressione totale si può calcolare dal numero per unità di tempo di collisioni
dovute alle molecole con velocità compresa tra v e v+dv , che supponiamo essere
una frazione f(v) del totale N, ad un angolo compreso tra θ e θ+dθ rispetto alla
parete. Sommando (integrando), poi, su tutti i valori di v e θ; si avrà:
dP =
dF N
2πd (− cosθ )
= f (v)dv
2mvcosθv cosθ
dS V
4π
I diversi termini rappresentano (si veda fig. 6):
N
f (v)dv è il numero di molecole presenti in ogni volumetto con velocità v
•
V
2πd (− cosθ )
•
è la frazione dell’angolo solido dalla quale provengono le
4π
molecole che urtano la parete ad un angolo θ. Il prodotto di questi due primi
fattori dà il numero di particelle con velocità v ed angolo θ. Di queste
particelle, quelle contenute nel cilindretto di volume:
• dSvdt cos θ , urteranno dS nel tempuscolo dt , trasferendo un impulso pari a:
• 2mvcosθ ad ogni collisione.
Occorre moltiplicare questi fattori tra loro e integrare su tutti gli angoli d’impatto
e su tutte le velocità possibili.:
P=
N
V
∞
∫
0
π
2
∫
f ( v ) mv 2 dv cos 2 θd ( − cos θ ) =
0
1 N
3V
∞
∫ f (v)mv
0
2
dv =
N
1
nm A v 2 , dove n è il
3
V
numero di moli di gas presenti nel recipiente: n =
PV =
N
. Infine abbiamo
NA
1
1
nmN A v 2 = Nmv 2 . Notiamo che qui si assume una semplice interazione
3
3
con la parete consistente in un rimbalzo come una pallina di gomma contro un
muro. Ciò non corrisponde al vero: le molecole vengono assorbite dalla parete e
riemesse dopo un certo periodo di tempo. Quello che importa è che l’impulso
medio portato alla parete venga mediamente fuori con la componente normale
21
Cap. 5 – Termodinamica e calorimetria
invertita. Se del resto così non fosse il contenitore si muoverebbe a causa delle
collisioni del gas sulle parete, ciò che sarebbe in violazione del terzo principio
della dinamica. Dunque stiamo estendendo alle singole collisioni ciò che è
sicuramente vero in media.
Comunque un modo di evitare completamente questa idea del rimbalzo delle
molecole sulla parete è il seguente. Prendiamo un piano all’interno del gas che lo
divida in due parti una destra e una sinistra. L’impulso che la parte sinistra cede
alla parte destra per unità di tempo ed area sarà dato da:
N
2π d ( − cos θ )
f ( v ) dv
mv cos θ v cos θ , che è la formula precedente tranne il
V
4π
fattore due. La parte destra tuttavia deve cedere la stessa quantità d’impulso a
quella sinistra, se siamo all’equilibrio. Dunque la quantità di moto a sinistra del
N
2π d ( − cos θ )
piano sarà cambiata di 2 volte
f ( v ) dv
mv cos θ v cos θ e
V
4π
ritroviamo la stessa pressione di prima. Stavolta tuttavia la abbiamo calcolata
all’interno del gas piuttosto che sulla parete.
Joule aveva trovato precedentemente la stessa espressione per il prodotto p V
con un ragionamento un po’ più elementare. Egli considerò un contenitore cubico
di spigolo l . Un terzo delle particelle si muovono lungo ciascun asse coordinato,
assi presi paralleli agli spigoli. Il tempo di andata a ritorno da una parete a quella
2l
v
opposta è: t = . Pertanto ciascuna molecola urta una parete
volte al
v
2l
secondo9, scambiando con la parete un impulso pari a 2mv . Pertanto la quantità
1
v
, che
di moto scambiata con una parete per unità di tempo è: F = N 2 mv
3
2l
possiamo riscrivere: F =
3
2 l
1
1N 2 2
Nmv
=
mv l . Ovvero F = 1 N mv 2 o
3
3l
l 3V
S 3V
1
mv 2 .
3
È facile identificare quest’espressione con la legge dei gas perfetti, a patto di
1
2
ammettere che sia: mNA v = RT . Inoltre possiamo definire la velocità quadratica
3
infine possiamo scrivere: PV = N
9
Si noti che questo ragionamento è valido solo se le particelle sono puntiformi. Se così
non è, allora le molecole urtano continuamente altri atomi e prendono molto tempo ad
arrivare all’altra parete.
22
Cap. 5 – Termodinamica e calorimetria
media come: v 2 = 3 R T = 3kT , dove abbiamo definito la “costante di
N Am
m
Boltzmann” k come: k = R = 1,37 ⋅ 10 − 23 J / 0 K .
NA
La necessità di avere la temperatura costante nella legge di Boyle e Mariotte
2
discende, come è evidente, dal fatto che v cambia al variare della temperatura.
1 2
Possiamo aggiungere che l’energia media delle molecole del gas ε = mv , è
2
3
funzione della temperatura, secondo la relazione: ε = kT (= 0,035eV a 0°C).
2
Questa formula viene interpretata dicendo che ad ogni grado di libertà, cioè
1
dimensione, compete l’energia media ε = kT e quindi il fattore 3 viene dal
2
fatto che una molecola può muoversi lungo tre assi (tre gradi di libertà). Tale
affermazione è nota come “teorema di equipartizione dell’energia” (J. C.
Maxwell).10
Questa formula connette l’energia cinetica media delle molecole con la scala delle
temperature di Kelvin, dunque ci consente un’interpretazione microscopica della
temperatura in termini di energia media o energia totale - l’energia media
moltiplicata per N - del gas. Più alta la temperatura, più alta è l’energia del
sistema gas. Allo zero Kelvin, l’energia totale è zero.
A titolo informativo, diciamo che, se si sciolgono n moli di un soluto in una
soluzione di volume V, Le molecole del soluto si comportano come un gas
perfetto di volume V e daranno quindi luogo ad una pressione supplementare
(detta pressione “osmotica”) pari a quella di un gas perfetto di n moli nel volume
V. La costante R è esattamente quella dei gas perfetti. Ad esempio sciogliendo 10g
di zucchero di canna (A=342) in un litro di acqua si ottiene una pressione
osmotica di 0,691 Atmosfere.
Per futura referenza, notiamo che la formula PV = N
1
mv 2 può riscriversi nella
3
1 Nm 2 1
Nm
densità del gas, formula questa che
v = ρ v 2 ,con ρ =
3 V
3
V
ricorda quella della resistenza di un gas all’avanzamento di un corpo.
forma: P =
10
Tale teorema vale per i termini di energia quadratici (vedi oscillatore armonico più
avanti). Una dimostrazione generale si trova in J. Jeans An introduction to the kinetic
theory of gases p.260.
23
Cap. 5 – Termodinamica e calorimetria
Esiste un altro modo di dimostrare che la temperatura è connessa all’energia
media delle molecole: il teorema del viriale di Clausius. Applichiamo il secondo
principio della dinamica
ad una molecola di massa m :
m
d2x
dt 2
= Fx ⇒ mx
d2x
dt 2
= xFx e simili per le coordinate
y e z . Utilizziamo adesso la
2
2
2
2
2
relazione: d ( x ) = ( dx ) 2 + x d x per ottenere: − m ( dx ) 2 = − m d ( x ) + xFx e,
2
2
2
dt
2
dt
dt
dt
2
dt
sommando sulle tre coordinate, otteniamo:
−
m d2
dy
dz
dx
( x 2 + y 2 + z 2 ) + xFx + yF y + zFz = − m ( ) 2 + ( ) 2 + ( ) 2 = − mv 2 .
2
2 dt
dt
dt
dt
Sommando adesso su tutte le molecole, il primo termine a sinistra va a zero.
Questo perché siamo all’equilibrio e dunque la quantità x + y + z sommata su
tutte le molecole deve essere fissa e la sua derivata rispetto al tempo è nulla.
( xFx + yFy + zFz ) = − mv2 che possiamo
Rimaniamo dunque con la relazione:
2
∑
anche
∑
i
scrivere:
ri ⋅ Fi
= −2 H
∑ r ⋅ F = −2H .
i
i
i
2
2
∑
Mediando
sul
tempo,
abbiamo:
. La quantità a sinistra si chiama “viriale”. La quantità a
destra è il doppio dell’energia cinetica totale delle molecole. Calcoliamo il viriale.
Questo riceve tre contributi: 1. dalle molecole non in collisione ad un istante
scelto 2. dalle molecole che sono in collisione tra loro 3. dalle molecole in
collisione con le pareti. Il viriale va calcolato facendo ipotesi sul tipo di forze cui
il sistema è soggetto. Nel nostro caso: il primo contributo è zero perché non ci
sono forze agenti sulle molecole, il secondo è anche zero perché la forza esercitata
da una molecola su un’altra durante una collisione è uguale e contraria a quella
che la seconda esercita sulla prima e le molecole hanno la stessa coordinata.
Rimane da calcolare il contributo delle collisioni contro le pareti. La forza
infinitesima esercitata dall’urto delle molecole su una superficie infinitesima della
parete è in media, data dalla pressione: dF = − PndS . Integrando sulla
superficie,
∇ ⋅r =
abbiamo:
∑ r ⋅ dF
∫S
∫
V
= −P r ⋅ ndS = −P ∇⋅ rdV = −3PV .
∂ x ∂y ∂z
+
+
= 3 . In conclusione troviamo:
∂ x ∂y ∂z
∑
i
ri ⋅ Fi
Poiché
= −3PV = −2 H
.
A questo punto, se ammettiamo che valga il teorema di equipartizione
dell’energia (vedi la fine del prossimo paragrafo): <
>=
=
,
sostituendo, troviamo l’equazione dei gas perfetti, ovvero ammettendo che
l’equazione dei gas perfetti sia vera, troviamo: < >=
=
. Se si
24
Cap. 5 – Termodinamica e calorimetria
fanno ipotesi sulle forze intermolecolari (qui poste uguali a zero) dal teorema del
viriale, si possono ottenere altre equazioni di stato, come l’equazione di Van der
Walls (vedi Boato, lezioni di termodinamica, pag. 98). Notiamo anche che la
quantità ∑ ri ⋅ Fi è l’energia potenziale media del sistema per forze derivabili
i
da
un
∑
=
i
potenziale
∑
ri ⋅ Fi = −
∑ U (r )
i
i
(r
dU
) r = ri
dr
del
= =−
∑
. La quantità
∞
l’integrale
i
centrale
i
(
∑
tipo:
i
(
d
( rU ( r )) r = ri +
dr
d
( rU ( r )) r = ri
dr
( n > 1)
=
infatti:
∑ U (r )
i
i
possiamo calcolarla come
+∞
d
∫−∞ dr (rU (r)) = rU (r) −∞ = 0 . Il teorema del viriale (Clausius, 1820)
sostiene in conclusione che: l’energia potenziale media del sistema è il doppio di
quella cinetica (cambiata di segno). Abbiamo visto, per fare un esempio, che, nel
caso di moti centrali ( = 1) e per orbite circolari (in cui le energie sono
costanti), l’energia potenziale vale il doppio dell’energia cinetica cambiata di
segno.
1
Notiamo infine che la formula PV = N mv 2 , può essere riscritta:
3
v=
3 PV
=
Nm
3P
ρ
con
ρ
densità del gas. Si può paragonare questa formula a
quella della velocità del suono (vedi note sulle onde) v =
γP
. Per un gas
ρ
biatomico (aria) il rapporto tra i calori specifici a pressione e volume costante vale
7
γ = = 1, 4 . Il rapporto tra le due velocità vale.1,46. In parole la velocità
5
quadratica media è del 46% più grande di quella del suono.
Confortati da questi successi, possiamo procedere per vedere come l’ipotesi
atomica ci dica ancora di più sul nostro gas.
4. La teoria cinetica dei gas
Il calcolo che, come si è visto, ci ha fatto pervenire al notevole risultato di avere
un’interpretazione microscopica della temperatura, è basato sull’idea di avere una
distribuzione di velocità. È vero che, secondo la fisica classica, basta conoscere le
condizioni iniziali del sistema per calcolare le traiettorie di tutte le molecole ad un
25
Cap. 5 – Termodinamica e calorimetria
certo istante, ma come si fa a calcolare la posizione e la velocità ad ogni istante di
un numero di molecole dell’ordine del numero d’Avogadro, cioè ≈ 10 23 ?
Si è allora concettualmente passati ad una maniera diversa di studiare i processi
termodinamici. Si utilizza adesso il metodo statistico: medie, fluttuazioni, ecc...:
un metodo molto diverso da quello deterministico Newtoniano. L’iniziatore del
metodo statistico e l’antesignano dell’uso delle statistiche nella teoria dei gas fu J.
C. Maxwell. I suoi argomenti sono riportati qui di seguito. La distribuzione di
Maxwell si può ricavare col seguente ragionamento (J. C. Maxwell 1856,
comunicazione alla British Association)11. In tutto quel che segue, assumeremo
che si tratti di un solo gas, cioè che le caratteristiche delle molecole, in particolare
la massa, siano le stesse.
4a La distribuzione delle velocità secondo Maxwell
dn
= g (v x , v y , v z ) dv x dv y dv z = 4πg (v x , v y , v z )v 2 dv
N
, che rappresenta il rapporto tra il numero di particelle con componenti
1. Cerchiamo la funzione:
della velocità pari a vx , vy , vz dn e il numero totale di particelle del gas
N . Questo rapporto dà anche la probabilità che una particella specifica
abbia le componenti della velocità pari a vx , vy , vz . Quando si dice che le
componenti della velocità di una particella sono pari a
vx , vy , vz , si
intende che le componenti della velocità siano rappresentate da un punto
contenuto in un piccolo (infinitesimo) cubetto dello spazio della velocità
(cioè lo spazio con assi vx , vy , vz ) di spigoli
dvx , dvy , dvz .
2. Richiediamo che essa si possa scrivere come il prodotto di tre probabilità:
g(vx , vy , vz ) = f (vx ) f (vy ) f (vz ) , dove si è ammesso che le probabilità
per le tre componenti devono essere la stessa funzione f ( x ) , giacché i
tre assi sono fisicamente equivalenti. Abbiamo anche ammesso che le
probabilità di avere una certa
certa
vx
sia indipendente da quella di avere una
v y o vz : questa assunzione giustifica il fatto che la probabilità si
sia scritta come il prodotto delle tre probabilità, di avere la componente
11
Riportato in J. Jeans - An introduction to the Kinetic Theory of gases Appendix 1.
26
Cap. 5 – Termodinamica e calorimetria
della velocità sull’asse X pari a v x moltiplicata per quella di avere la
componente della velocità lungo l’asse Y pari a vy moltiplicata per quella
di avere la componente della velocità lungo l’asse Z pari a v z .
Assumiamo in più che ciascuna componente abbia un valore compreso tra
− ∞ e + ∞ , anche se questa assunzione in effetti comporta che l’energia
cinetica di una molecola possa essere infinita. Si intenda che le
componenti possono assumere valori molto grandi.
Ricordiamo
che
deve
essere:
3
+∞
+∞
g ( v x , v y , v z ) dv x dv y dv z = f (v x ) dv x = 1
−∞
−∞
∫
∫
ovvero
che
+∞
∫−∞ f (vx )dvx = 1 cioè che
f ( v x ) sia “normalizzata” ad 1. La probabilità
totale deve, infatti, essere uguale ad 1. Sostituiamo allora a f ( v x ) la
+∞
f (vx ) /
∫−∞ f (vx )dvx = Af (vx )
che è sicuramente normalizzata ad 1 e
similmente per le altre due componenti. Con la condizione aggiuntiva
che: g(vx ,0,0) = A f (vx ) e simili. Da questa si deduce che f (0) = 1
3. Richiediamo che sia f ( v x ) = f ( − v x ) , il che ci porta a scegliere
3
f = f (vx2 ) , per usare il rasoio di Occam.
4.
Richiediamo che la g (vx , v y , vz ) = g (v 2 ) , cioè che la distribuzione
dipenda solo dal modulo della velocità e non da come quel modulo sia
ottenuto in termini di componenti.
Ne segue che: g (v 2 ) = A3 f (vx2 ) f (v 2y ) f (vz2 ) . Ma per due componenti uguali a
zero, g ( v 2 ) = A 3 f ( v 2x ) e simili. Dunque g = A 3 f , cioè la dipendenza funzionale
di g dal suo argomento è la stessa di f . Ne discende che se
vx2 + v2y + vz2 = costante , Allora anche . g (v 2 ) = A3 f (vx2 ) f (v 2y ) f (vz2 ) = costante .
Differenziando,
si
ha:
2
df ( v y )
df (v x2 )
df ( v z2 )
f ( v 2y ) f ( v z2 ) dv x + f (v x2 )
f (v z2 ) dv y + f (v x2 ) f (v 2y )
dv z = 0 per
dv x
dv y
dv z
2vxdvx + 2vydvy + 2vzdvz = 0 . Dividendo la prima per
f (vx2 ) f (v2y ) f (vz2 )
27
Cap. 5 – Termodinamica e calorimetria
2
df ( v x2 )
1 df ( v y )
1 df ( v z2 )
dv
+
dv
+
dv z = 0
x
y
f ( v x2 ) dv x
f ( v 2y ) dv y
f ( v z2 ) dv z
1
otteniamo:
Moltiplicando per la seconda per un coefficiente arbitrario β e sommando, si ha:
(
2
df ( v x2 )
1 df ( v y )
+
2
βv
)
dv
+
(
dv y + 2 βdv y ) +
x
x
f ( v x2 ) dv x
f ( v 2y ) dv y
(
df (vz2 )
df (vz2 )
+ 2 βvz f (vz2 ) = 0 che è certo soddisfatta se
+
2
βv
)
dv
=
0
.:
z
z
dvz
f (vz2 ) dvz
1
1
e solo se le tre equazioni:
2
df (vx2 )
1 df ( v y )
+
2
=
0
,
βv
+ 2 βv y = 0 ,
x
f (v x2 ) dvx
f ( v 2y ) dv y
1
df (vz2 )
+ 2 βvz = 0 sono soddisfatte. Si vede che le funzioni che risolvono
f (v z2 ) dvz
1
−βv2x
queste equazioni sono degli esponenziali: e
2
2 −av2
2
y −avz
e
g(v2 ) = e−av = e−avx e
,... e si ricava subito che
= f (vx2 ) f (v2y ) f (vz2 ) che, come si vede, soddisfa
2
2
2
2
tutte le condizioni richieste. In conclusione: g(v ) = Af (vx ) Af (v y ) Af (vz ) = ,
−βv2y
= Ae−βvx Ae
2
Ae−β z = A3e−βv dove le due costanti A e β
v2
2
si ottengono,
imponendo normalizzazione e valore dell’energia media, come faremo poco più
m
. Quello che abbiamo
avanti. Per ora anticipiamo che β risulta uguale a
2kT
ottenuto è che f (v x , v y , v z ) = Ae
−
mv 2
2 kT
= Ae
−
εc
kT
in cui ε c è l’energia cinetica delle
molecole. E’ facile, ricordando l’andamento della densità di un gas in un campo
esterno,
capire
che
la
formula
si
estende
a
f ( x, y, z , v x , v y , vz ) = Be
−
ε c +U ( x , y , z )
kT
= Be
−
ε
kT
nel caso di presenza di campo esterno
con energia potenziale U = U ( x , y , z ) .
4b La distribuzione delle velocità secondo Boltzmann
Il metodo di Boltzmann parte dalla considerazione delle conseguenze delle
collisioni molecolari. Si noti che nella trattazione di Maxwell, il fatto che le
molecole siano puntiformi o no non è particolarmente rilevante: il processo
attraverso il quale le molecole del gas raggiungono lo stato di equilibrio non è
28
Cap. 5 – Termodinamica e calorimetria
preso in esame. Boltzmann assume che le molecole siano sfere rigide di raggio
dato. La differenza di attacco del problema sta nel fatto che Maxwell è interessato
al sistema in equilibrio: all’equilibrio le collisioni hanno già sortito l’effetto di
portare il sistema nello stato finale e diventano irrilevanti. Boltzmann è
interessato a descrivere l’evoluzione del sistema fino all’equilibrio, evoluzione
durante la quale le collisioni giocano un ruolo centrale.
Ciascuna molecola ha tre componenti della velocità, dunque è rappresentata da un
punto nello spazio delle velocità. Lo stato del gas è rappresentato da una nuvola di
N punti in tale spazio. Se prendiamo un volumetto infinitesimo dv x dv y dv z ad un
determinato istante troviamo un numero n di molecole. Tale numero cambia nel
tempo perché, a causa delle collisioni, alcune particelle passano da un volumetto
(o cella) ad un altro. Se alcune molecole passano dalla cella in considerazione ad
un’altra (si intenda che il loro punto rappresentativo passa da una cella ad
un’altra) con una diminuzione della popolazione (cioè del numero n), altre
molecole passano nella cella in considerazione a causa di altri urti che le fanno
migrare da una cella diversa alla cella in considerazione. Il numero n dunque
rimarrà o no costante in conseguenza del bilanciarsi dei due processi. In questo
modo, la funzione f = f ( v x , v y , v z ) sarà in generale anche funzione del tempo
f = f (v x , v y , v z , t )
e raggiungerà uno stato stabile, cioè con popolazione stabile in
∂f
=0.
∂t
Procediamo allora a calcolare il numero di molecole che per unità di tempo
migrano fuori o dentro la cella stabilita, facciamo la differenza tra i due numeri e
vediamo sotto quali condizioni (cioè per quale forma della funzione
∂f
= 0 . In realtà, la derivata che ci
f = f ( v x , v y , v z ) ) si ha uno stato stabile per cui
∂t
interessa è la derivata totale della distribuzione f ( x , y , z , w x , w y , w z , t ) :
ogni cella, dopo un lasso di tempo. L’equilibrio sarà raggiunto quando
df
∂f
=
+
dt
∂t
∂f
∑ ∂x
i
wi +
i
∂ f Fi
i m
∑ ∂w
i
. Come si vede, il secondo e terzo addendo non
sono necessariamente nulli. Tuttavia il secondo termine a destra
∂f
∑ ∂x
i
wi
sarà
i
nullo, se la distribuzione è la stessa in ogni punto del gas, cioè se è indipendente
dalle coordinate. L’ipotesi che all’equilibrio la distribuzione sia indipendente dal
punto spaziale che si considera è detta “ipotesi del caos molecolare”. Il terzo
termine ∑ ∂ f Fi infine è nullo se le forze esterne (per esempio un campo
i
∂w i m
gravitazionale cui il gas è soggetto) e le forze intermolecolari sono nulle. Se
tuttavia queste ipotesi non sono vere, allora occorre prendere in considerazione
29
Cap. 5 – Termodinamica e calorimetria
anche questi due termini e la richiesta di essere all’equilibrio deve esprimersi con
l’annullamento della derivata totale e non di quella parziale.
Consideriamo la collisione tra due particelle i cui vettori velocità siano
(molecole di tipo A) e
wx,w y,wz
v x , v y , vz
(molecole di tipo B). Decidiamo pure di fissare
la direzione ( θ ) della congiungente tra i centri al momento della collisione (linea
d’impatto) e la velocità relativa v − w . La conservazione dell’energia, della
quantità di moto e l’avere fissato la direzione della linea d’impatto definiscono le
velocità delle particelle in uscita dalla collisione. Chiamiamole v x , v y , v z e
wx,w y,wz
. Il numero di collisioni (di tipo
α
) che avvengono tra i due tipi di
molecole nell’unità di tempo e per unità di volume è dato allora dall’espressione:
v − w f (w) f (v)σ(wx , wy , wz )dwxdwydwz cosθdΩ a norma di quanto detto al
paragrafo sulla diffusione di Rutherford. Abbiamo semplificato le notazioni e
scritto f = f (v ) invece di f = f ( v x , v y , v z ) e simili. La sezione d’urto qui usata
σ (w x , w y , w z )
potrà essere nel caso più semplice la sezione d’urto tra sferette
rigide (vedi cap.4). d Ω è l’angolo solido infinitesimo all’interno del quale si
trova la direzione d’arrivo rispetto alla linea d’impatto. Se integriamo su tutte le
possibili velocità delle molecole di tipo B, otteniamo il numero di tutte le
possibili collisioni che le molecole di tipo A subiscono nell’unità di tempo e
dunque il numero di molecole di tipo A che lasceranno il loro volumetto
infinitesimo nell’unità di tempo e volume, chiamiamo questo numero Z e
dove
abbiamo:
Z = ∫ v − w f ( w ) f ( v )σ ( w x , w y , w z )dw x dw y dw z cos θ d Ω
l’integrazione è da fare su tutte le quattro variabili.
Con lo stesso metodo possiamo calcolare il numero di particelle R che, per unità
di tempo e volume, entrano nel volumetto scelto. Partiamo stavolta col definire lo
stato finale dato dalle velocità v x , v y , v z e w x , w y , w z . Lo stato iniziale deve allora
essere definito dalle componenti delle velocità
v x , v y , vz , w x , w y , w z
e l’angolo θ
In maniera analoga a prima il numero di particelle che saltano nella celle prescelta
saranno: R = ∫ v − w f ( w ) f ( v )σ dw x dw y dw z cos θ d Ω . La variazione di popolazione
nella nostra cella sarà allora: Z − R . Occorre adesso ricordare che lo stato finale
deve avere componenti v x , v y , v z per la particella di tipo A, mentre le particelle di
tipo B possono essere qualunque. Poiché le equazioni di conservazione fissano le
componenti finali in funzione di quelle iniziali e viceversa, possiamo fissare le
componenti finali v x , v y , v z , per ottenere appunto una particella nella cella
30
Cap. 5 – Termodinamica e calorimetria
inizialmente considerata e fissando così le componenti
variabili le
wx,w y,wz
v x , v y , vz ,
che risulteranno funzione delle
l’integrazione sulle tre variabili
wx,w y,wz
mentre lasciamo
wx,w y,wz
. Di qui
vista nella formula precedente, che
può essere cambiata in un’integrazione sulle variabili
wx,w y,wz
, introducendo lo
jacobiano della trasformazione. Risulta tuttavia che questo jacobiano vale
dwx dw y dwz
dwx dw y dwz = dwx dw y dwz ,
semplicemente 1, in formule dwx dw y dwz =
dwx dw y dwz
perché risulta che lo jacobiano
dwx dw y dwz
dwx dw y dwz
= 1 . Ne segue che possiamo scrivere:
∫ v − w f ( w ) f (v )σ dw dw dw cos θ d Ω
Z − R = ∫ v − w ( f ( w ) f ( v ) − f ( w ) f ( v ))σ dw
R=
x
y
e
z
x
dunque
dw y dw z cos θ d Ω . Possiamo
allora concludere che la derivata temporale della funzione
f = f (v x , v y , v z )
df
= ∫ v − w ( f ( w ) f ( v ) − f ( w ) f ( v ))σ dw x dw y dw z cos θ d Ω
dt
Ora se vogliamo che la popolazione della nostra cella sia costante, possiamo
eguagliare semplicemente a zero tale derivata e la derivata è certamente zero, se
f ( w ) f ( v ) − f ( w ) f (v ) = 0 . Questa condizione è però solo sufficiente, ma non
necessaria, perché l’integrale può essere nullo se l’integrando cambia segno da
qualche parte ottenendosi così contributi positivi e negativi all’integrale con un
possibile risultato nullo. Più avanti vedremo che non è così. Per ora vediamo
quale è la conseguenza di porre: f ( w ) f (v ) − f ( w ) f (v ) = 0
Ricordiamo che deve essere per la conservazione dell’energia:
w 2 + v 2 = w 2 + v 2 ⇒ w 2 = w 2 + v 2 − v 2 . Senza perdita di generalità, poniamo:
f (v ) = eϕ ( v ) ,
2
f (w) = eϕ(w ) , f (v ) = eϕ ( w ) ,
2
2
f (w) = eϕ(w ) . Si noti che
2
abbiamo posto l’argomento della funzione uguale al modulo quadro della velocità
come maniera più semplice di realizzare il fatto che il segno delle componenti
della
velocità
deve
essere
irrilevante.
Possiamo
riscrivere
f ( w ) f ( v ) − f ( w ) f (v ) = 0
nella
forma
logaritmica
ln( w ) + ln( v ) = ln( w ) + ln( v ) ⇒ φ(w ) + φ(v ) = φ(w + v − v ) +φ(v ) e
differenziare a turno
rispetto a v, a w e a v . Otteniamo:
d
d
d
d
e
φ (v 2 ) =
φ (w 2 + v 2 − v 2 ) ;
φ (w 2 ) =
φ (w 2 + v 2 − v 2 )
dv
dv
dw
dw
2
2
2
2
2
2
31
Cap. 5 – Termodinamica e calorimetria
d
d
Per
cui
è:
φ (w 2 ) =
φ (w 2 + v 2 − v 2 )
dv
dv
d
d
d
φ (v 2 ) =
φ (w 2 ) =
φ ( v 2 ) Queste eguaglianze tra funzioni di variabili
dv
dw
dw
indipendenti possono essere vere se e solo se esse sono uguali ad una stessa
d
1
costante, cioè se:
e simili, da cui otteniamo che
φ (v 2 ) = − 2
dv
a
φ (v ) = −
2
ν2
a
2
+ ln A3 con ln A 3 costante d’integrazione e simili. Ne segue che:
f (v x , v y , v z ) = A e
3
−
v 2x + v 2y + v z2
a2
. Le costanti verranno calcolate più avanti,
imponendo la conservazione dell’energia totale e la normalizzazione del sistema.
Abbiamo così trovato ancora la distribuzione delle velocità che risulta identica a
quella trovata da Maxwell con tutt’altri argomenti.
La dimostrazione che la condizione f ( w ) f (v ) − f ( w ) f (v ) = 0 è non solo
sufficiente, ma anche necessaria procede definendo la funzione
H = ∫ f ( u ) ln f ( u )du x du y duz , calcolandone la derivata rispetto al tempo
dH
df
= (1 + ln f ( u)) dux du y duz =
dt
dt
∫
=
e
∫ v − w (1 + ln f (u))( f ( w ) f (v ) − f (w ) f (v ))σ dw dw dw
x
dimostrando
che
tale
derivata
si
y
può
z
cos θ d Ωdux du y duz
scrivere
nella
forma
dH
f (v ) f ( w )
dudvdw v − w σ dw x dw y dw z cos θ d Ω
= ( f ( w ) f ( v ) − f ( w ) f ( v )) ln
dt
f (v ) f ( w )
∫
il cui integrando è necessariamente negativo: conseguentemente l’integrale è
sempre negativo. Questo dimostra allo stesso tempo che la funzione H decresce
sempre e che la quantità f ( w) f (v ) − f ( w) f (v ) , se non è nulla è sempre dello
stesso segno. Il fatto che il segno sia sempre uguale esclude la possibilità che
l’integrale possa essere nullo senza che sia f ( w ) f (v ) − f ( w ) f (v ) = 0 . Ciò implica
che la condizione f ( w ) f (v ) − f ( w ) f (v ) = 0 sia necessaria e sufficiente, ovvero
che la distribuzione di Maxwell-Boltzmann non sia una distribuzione possibile,
ma l’unica distribuzione possibile.
In aggiunta però abbiamo trovato che la funzione H = ∫ f ( u ) ln f ( u )du x du y duz
decresce sempre ovvero che H = − ∫ f ( u ) ln f ( u )du x du y du z cresce sempre. La
funzione −H sarà identificata con l’entropia del sistema più avanti, quando
avremo studiato il secondo principio della termodinamica.
32
Cap. 5 – Termodinamica e calorimetria
Possiamo adesso ricavare la cosiddetta equazione del trasporto di Boltzmann.
L’equazione del trasporto di Boltzmann serve a calcolare l’andamento temporale
della densità di punti nello spazio delle fasi f ( x , y , z , w x , w y , w z , t ) col tempo. Ha
parecchie applicazioni tra cui la diffusione dei neutroni nei reattori nucleari. La
otteniamo semplicemente scrivendo a sinistra la derivata totale della
f ( x , y , z , w x , w y , w z , t ) , a destra mettiamo l’espressione di tale derivata che
abbiamo appena calcolata a partire dalla teoria delle collisioni:
∂f
∂f
∂f Fi
wi + ∑
+∑
=
∂t
i ∂x i
i ∂w i m
= ∫ v − w ( f ( w ) f ( v ) − f ( w ) f ( v ))σ cos θ d Ωdw x dw y dw z
Possiamo vedere che all’equilibrio questa equazione è equivalente all’equazione
di continuità. All’equilibrio infatti l’integrale a destra è zero. Assumiamo poi che
il sistema non sia soggetto a forze esterne: ∑ ∂f Fi = 0. Abbiamo così che:
i
∂w i m
∂f
∂f
wi = 0 Se integriamo sulle variabili velocità e se le collisioni non
+∑
∂t
i ∂xi
cambiano il numero di particelle per unità di volume, cioè se n( x , y , z , t ) non
dipende dal tempo n = n( x , y , z ) , si ha:
∂f
∂f
∫ ( ∂t + ∑ ∂x
i
i
+
∂f
∫ ∑ ∂x
wi )dw x dw y dwz =
∂
∂t
∫ f ( x, y, z, w , w , w )dw dw dw
x
z
x
y
z
wi dw x dw y dwz = 0
i
i
cioè: ∂ n + ∑ ∂ n w i = 0 , ovvero: ∂n + ∂
∂t
j = nw
y
i
∂x i
∂t
∂x i
∑ nw
i
=0
o ancora :
i
∂n
+∇⋅ j = 0
∂t
Che è appunto l’equazione di continuità.
4c La distribuzione delle velocità secondo la meccanica statistica
Gli argomenti che seguono sono in parte stati introdotti in fisica da J. W. Gibbs
che fu il fondatore della meccanica statistica.
Innanzitutto il sistema in studio viene rappresentato in uno spazio matematico
speciale. Ogni particella si trova in uno stato ben definito, tutte le volte che si
siano specificati le coordinate e l’impulso della particella stessa. Dunque
prendiamo uno spazio a sei dimensioni, tre per la posizione e tre per la quantità di
33
Cap. 5 – Termodinamica e calorimetria
moto: in questo spazio un punto è rappresentativo dello stato dinamico di una
particella. In verità, una molecola non è equiparabile ad un punto materiale
perché, essendo un oggetto composito, esistono necessariamente altri gradi di
libertà e dunque altre “coordinate” necessarie per descriverne lo stato interno. Qui
si trascurerà questo fatto, limitandosi alle sei coordinate precedentemente
descritte.
Se si vuole dunque rappresentare un sistema di N particelle, si ha bisogno di uno
spazio a 6N coordinate, dove ogni punto rappresenterà uno stato del sistema
attraverso le coordinate e le quantità di moto istantanee di ogni particella. Questo
spazio è detto “spazio delle fasi” ( γ space). Nella nostra trattazione attuale è però
meglio usare uno spazio a sei coordinate, anche esso detto “spazio delle fasi” ( µ
space) nel quale il sistema sarà rappresentato da N punti, uno per ogni particella.
Consideriamo adesso che non si è necessariamente interessati a sapere dove si
trova il punto rappresentativo di una molecola, piuttosto può interessare sapere in
quale volumetto ∆ x ∆ y ∆ z ∆ p x ∆ p y ∆ p z tale punto si trovi. Dividiamo allora lo spazio
delle fasi in m volumetti e distribuiamo le molecole (o piuttosto i loro punti
rappresentativi) in questi volumetti Ogni distribuzione rappresenterà uno stato del
sistema di molecole. Evidentemente si possono distribuire le N molecole su m
volumetti in molti modi diversi. Dal punto di vista macroscopico, però, questi stati
non sono tutti distinguibili, giacché non ha importanza quale molecola occupi
quale volumetto: le molecole non possono essere distinte, nel senso, appunto, che
l’essere una o un’altra molecola in uno specifico stato non dà luogo ad effetti
macroscopicamente distinguibili.
Si può pensare che nel volumetto iesimo, siano rappresentati, in effetti, gli stati
non di una, ma di più molecole, diciamo n i . Dunque si avranno n 1 punti nel
volumetto ( ∆ x ∆ y ∆ z ∆ p x ∆ p y ∆ p z ) 1 ,…, n k punti nel volumetto ( ∆ x ∆ y ∆ z ∆ p x ∆ p y ∆ p z ) k e
∑n = N . In tal caso, lo stato del sistema è definito assegnando il numero di
i
i
molecole il cui stato è rappresentato all’interno del volumetto iesimo. È chiaro
allora che esiste un numero H di stati microscopici diversi che corrispondono
allo stesso stato macroscopico. Più precisamente: tutti quegli stati che si
ottengono permutando le posizioni delle diverse molecole tra i diversi volumetti
sono uguali. Il numero di stati indistinguibili fra loro, dati gli
ni , è:
H =
N!
n1 !...n k !
(numero degli allineamenti). A questo punto, assumiamo che la probabilità P di
trovare il sistema in un certo stato, è proporzionale al numero di modi
microscopici di realizzare quello stato, cioè: P = cost ⋅ H .
Possiamo esemplificare questa affermazione considerando un semplice caso che si
incontra di frequente. Supponiamo di avere due insieme di palline, uno bianco ed
34
Cap. 5 – Termodinamica e calorimetria
uno nero. Possiamo estrarre a sorte da un contenitore una pallina bianca o una
nera con la stessa probabilità pari a
1
. Immaginiamo di estrarre dieci palline.
2
Avremo così una sequenza del tipo bnnbnbbnnb, in cui le lettere stanno a
indicare il tipo di pallina estratto. La probabilità complessiva di una qualunque
sequenza specificata sarà uguale a:
probabilità
1
P = ( )n , sarà cioè il prodotto di 10 volte la
2
1
di ciascuna estrazione (bianca o nera) Per inciso, ricordiamo che
2
2 10 = 1 024 . Dunque qualunque sequenza ha la stessa probabilità di essere
estratta. Ciò sembra logico: non ci aspettiamo che una particolare sequenza esca
con più frequenza di un’altra. Forse è un po’ più sorprendente se diciamo che
anche le sequenze bbbbbbbbbb o nnnnnnnnnn hanno la stessa probabilità delle
altre sequenze. Eppure è così perché ciascuna volta che estraiamo una pallina la
probabilità che sia bianca o nera è la stessa e il caso non ha memoria di quello che
è stato estratto prima. Chiariamoci le idee. Se quello che vogliamo sapere è se una
sequenza con 5 palline nere e 5 palline bianche è più probabile di una contenente
solo palline bianche o solo palline nere, la risposta è sì. Ciò non è dovuto però al
fatto che una specifica sequenza con 5 palline bianche e 5 palline bianche sia più
probabile di una sequenze con tutte palline uguali Ciò è dovuto al fatto che si può
realizzare una sequenza con 5 palline e bianche 5 nere in molti più modi che una
sequenza monocolore. Una sequenza monocolore può essere realizzata in un solo
modo, cioè estraendo ogni volta una pallina del colore di quella precedentemente
estratta; viceversa una sequenza con 5 palline di un colore e 5 dell’altro può
realizzarsi per esempio con sequenze del tipo: nbnbnbnbnb, oppure nnbbnnbbnb,
etc… In quanti modi H dunque possiamo realizzare una sequenza con 5 palline
di ciascun colore? Il calcolo combinatoriale ci dà il risultato: H =
N = n1 + n2 , formula che, nel caso in esame, diventa:
H=
N!
, con
n1 ! n2 !
10!
= 252 . In
5!5!
conclusione ci sono 252 modi di realizzare una sequenza con 5 palline di ciascun
colore e uno solo di realizzare una sequenza con palline tutte dello stesso colore.
Prendiamo questa affermazione come a significare che estrarre 5 palline di
ciascun colore è 252 volte più probabile che estrarle tutte uguali. In effetti
possiamo dire che non stiamo calcolando la probabilità di avere tutte palline
uguali o 5 palline bianche e 5 palline nere, stiamo però calcolando le probabilità
relative. In effetti se vogliamo calcolare la probabilità vera dobbiamo dividere per
il numero complessivo di sequenze possibili (detta “normalizzazione”, in questo
35
Cap. 5 – Termodinamica e calorimetria
caso 1024) Per esempio, abbiamo che la probabilità di avere 5 bianche 5 nere è:
252/1024=0,25=25%. Ai fini del calcolo della probabilità massima, questa
normalizzazione è inutile e la mettiamo da parte, chiamando H =
N!
la
n1 ! n2 !
probabilità. Possiamo allora vedere l’andamento della probabilità in funzione del
rapporto fra i numeri n1 / n2 . Eseguendo il calcolo, otteniamo la seguente tabella
n1
n2
H
0
1
2
3
4
5
6
7
8
9
10
10
9
8
7
6
5
4
3
2
1
0
1
10
45
120
210
252
210
120
45
10
1
36
Cap. 5 – Termodinamica e calorimetria
H
n1
Come era ovvio la tabella dà risultati simmetrici:4b6n è equiprobabile a 4n6b.Il
massimo si ottiene per 5n5b. Si noti che, come è logico, in ogni riga è
n1 + n2 = 10 = N , giacché il numero totale di palline deve essere costante in ogni
sequenza.
Possiamo immaginare di riformulare questo esempio, prendendo palline tutte
dello stesso colore che mettiamo a caso con eguale probabilità in due secchielli
diversi. Quale è la probabilità di averne finalmente n 1 nel primo secchiello e n 2
nel secondo secchiello? La discussione procede come prima e ci fa giungere alla
stessa tabella: il massimo si ottiene nel caso di 5 palline in ciascun secchiello.
Immaginiamo adesso di fare diventare il numero di secchielli molto grande N ,
ciascuno con una popolazione n1, n2 ... , in più chiamiamo i secchielli “celle nello
spazio delle fasi µ ” e ci troviamo nel caso considerato all’inizio con una tabella
con un numero molto grande di righe e di colonne. Per quali valori della
popolazione si trova il massimo? Questo è il prossimo problema.
Alla identificazione della quantità H con la probabilità di una specifica
ripartizione delle molecole tra le celle dello spazio delle fasi si può giungere in un
altro modo, con ragionamento tipico della meccanica statistica.
37
Cap. 5 – Termodinamica e calorimetria
Torniamo allo spazio delle fasi ( γ space) a 6N coordinate. Ciascun punto
rappresenta uno stato del sistema (non della singola molecola). Prendiamo un
insieme (detto “ensemble”), di sistemi tutti identici (stessa energia, numero di
particelle…), ma non nello stesso stato. A ciascun sistema dell’ensemble
corrisponderà un punto rappresentativo nel γ space. Prendiamo sistemi che hanno
la stessa energia totale E entro un intervallino infinitesimo dE (ensemble
microcanonico). Ammettiamo che, all’interno del volumetto dello spazio γ
limitato dalle due superfici definite dalle condizioni E =costante E + dE =
costante, la densità di punti sia costante, mentre sia nulla all’esterno e dividiamo
l’intero volumetto in volumetti
dVγ , ognuno contenente un punto. Gli stati
microscopici che rappresentano uno stesso stato macroscopico saranno in numero
di H , come prima. Esisteranno allora H volumetti
dVγ con un punto che
rappresenta quello stato macroscopico. Scegliendo un volumetto qualsiasi si avrà
una probabilità proporzionale ad H di trovare lo stato con ni in dVi (dello
spazio µ ) e dunque un certo stato macroscopico che così avrà anche lui una
probabilità pari ad H . La figura in basso dovrebbe aiutare a capire quanto detto.
Nella figura abbiamo tracciato una superficie (definita dalla condizione =
).
In realtà si intende che tale superficie ha uno spessore (dovuto al fatto che
≠ 0)e
pertanto rappresenta un volume che chiamo V. Il volume V è stato diviso in volumetti,
due dei quali (quelli col punto nero) contengono stati definiti, diversi microscopicamente,
ma uguali macroscopicamente. La probabilità che un sistema dell’ensemble, scelto a
caso, sia nello stato definito è data dal rapporto di due al numero totale dei volumetti. In
pratica possiamo dire che la probabilità di trovarsi in quello stato macroscopico è
proporzionale a due.
38
Cap. 5 – Termodinamica e calorimetria
Passiamo adesso a cercare quale è lo stato macroscopico più probabile ovvero a
cercare il massimo della funzione H al variare degli ni . Noi possediamo
naturalmente una tecnica di ricerca dei massimi di una funzione attraverso la
derivazione, ma tale tecnica è inapplicabile a funzioni di numeri interi. Per ridurre
il calcolo alla tecnica che ci è nota. Cominciamo col trovare il massimo della
grandezza ln H , piuttosto che di H : questo non fa alcuna differenza sul piano
concettuale, giacché la funzione ln è una funzione crescente del suo argomento,
tuttavia potremo in questo caso usare l’approssimazione di Stirling. Il massimo
della probabilità dovrà essere ottenuto con la condizione che l’energia totale ed il
numero totale di molecole del sistema siano costanti. Dovranno allora valere le tre
equazioni:
d ln H = 0
dni = 0
dN =
i
dU =
ε i dni = 0
i
∑
∑
Utilizzando il metodo dei moltiplicatori arbitrari di Lagrange, si moltiplichi la
seconda equazione per α e la terza per β , e le si sottragga alla prima:
d ln H − α dN − β dU = 0 . Si calcoli adesso il differenziale di
ni
ln H nelle variabili
e si sostituisca. La formula di Stirling, che vale per n grande, come
supporremo essere tutti gli
ni
asserisce che: ln( n!) ≈ n ln n − n . Da questo segue:
39
Cap. 5 – Termodinamica e calorimetria
∏ n1!) = −∑d (lnn !) = −d ∑(n ln(n ) − n ) = −∑ln(n )dn .
d ln H = d ln(
i
i
i
i
i
i
i
i
i
i
i
Notare che si è utilizzata la condizione:
∑dn =dN = 0 .
i
Sostituendo:
i
∑ dn (ln(n ) + α + βε ) = 0 .
i
i
i
i
Perché questa relazione sia vera, a prescindere dai dni , deve essere:
ln( n i ) + α + βε i = 0 per ogni ε i . Svolgendo questa relazione, si trova:
ln(ni ) +α + βεi = 0 ⇒ln(ni ) = −α − βεi ⇒ ni = e−αe−βεi = NAe−βεi . Naturalmente deve
essere: N =
∑n
i
i
= AN
∑e βε
−
⇒ A=
i
i
1
∑e βε
−
i
.
i
Sostituendo, si ottiene: n i =
Ne
− βε i
∑e
− βε i
⇒ fi =
ni
= Ae − βε i , che è chiamata la
N
i
“statistica di Maxwell-Boltzmann, come si è visto. Passando adesso al continuo
troveremo in un volume infinitesimo d τ = dxdydzdp x dp y dp z un numero di
particelle::
1
dn = NCe−βε dxdydzdpx dpy dpz Con C = +∞
.
∫e
− βε
dxdydzdpx dp y dpz
−∞
Notiamo che i nostri punti si trovano in una corona sferica nello spazio degli
impulsi compresa tra le sfere di raggi p e p + dp . Se passiamo a coordinate
sferiche nello spazio delle velocità, il corrispondente volumetto dello spazio delle
2
fasi sarà dVsf = dxdydz4πp dp , la frazione di particelle in esso presenti sarà:
e − βε dxdydz 4πp 2 dp
e − βε dxdydzv 2 dv
dn
che può essere integrata sul volume
= +∞
= +∞
N
e − βε dxdydz 4πp 2 dp
e − βε dxdydzv 2 dv
∫
∫
0
0
− βε 2
immediatamente. Si ha: dn = + ∞e v dv , da cui si può ricavare l’energia media:
N
∫e
− βε
v 2 dv
0
40
Cap. 5 – Termodinamica e calorimetria
+∞
dn 1 ∫0
ε
∫0 N = 2 m +∞
+∞
∫
e
e
mv 2
2
−β
mv
2
−β
4
v dv
2
v 2 dv
1
= m
2
+∞
−1
βm
∫ v d (e
3
+∞
mv 2
2
)
0
+∞
∫
0
−β
e
−β
mv
2
2
v 2 dv
∫ve
2
3
=
2β
0
+∞
∫
0
e
−β
−β
mv
2
mv 2
2
3
2β
=
2
v 2 dv
0
3
1
da cui, poiché l’energia media è: ε = kT , risulta β =
.
2
kT
Calcoliamo adesso la costante C =
1
+∞
∫e
− βε
. Prima si integri
dxdydz 4πp 2 dp
0
−∞
sull’intero volume V
del gas, ottenendo: C
−1
=
∫
4πVp e
2
−
β
2m
p2
dp . L’ultimo
0
+∞
integrale si trova sulle tavole e si ha:
∫x e
2 − ax 2
dx =
1 π
4a a
(mentre è
0
+∞
∫e
− av 2
dv =
0
1 π
2 a
12
), da cui: C −1 = V (2πmkT ) 3 . Si ha dunque finalmente:
ε
−
dn
1
=
e kT dxdydz 4πp 2 dp ”
N V ( 2πmkT ) 3
+∞
“normalizzata”, cioè:
Si noti che questa distribuzione è
dn
∫ N = 1 , come deve essere. Calcoliamo adesso da questa, la
0
+∞
I=
12
∫−∞ e
π
=
a
+∞
−ax 2
∫0 e
+∞
∫−∞ e
dx ⇒ I =
−ar 2
2
π
2ardr =
a
+∞
∫0
−ax2
+∞
dx
∫−∞ e
−ay 2
+∞
dy =
∫0 e
π
π
e dt = ⇒ I =
⇒
a
a
−t
−ar 2
+∞
∫0 e
dxdy =
−ax2
dx =
1 π
2 a
R. Feynman Lectures on Physics pag. 40-6 del vol 1. Il secondo integrale si fa per parti:
+∞
I=
∫
−∞
2
z2e−az dz =
+∞
∫
−∞
zd (
−1 −az2
1
e )=
2a
2a
+∞
∫
−∞
2
e−az dz =
1 π
⇒
2a a
+∞
∫0
2
z2e−az dz =
1 π
4a a
41
Cap. 5 – Termodinamica e calorimetria
distribuzione delle velocità calcolata per la prima volta da J. K. Maxwell.
Riscriviamo la precedente formula in coordinate polari, integriamo sul volume e
sostituiamo p = mv . Otteniamo così la distribuzione di velocità di Maxwell:
mv2
3
dP(v) 1 dn
m 2 2 − 2kT
=
= 4π (
) ve
, il cui grafico viene riportato in fig. 7, e quella
dv
N dv
2π kT
di Boltzmann:
p
−
−
dn
4π p2dp
m3
dP ( p) =
e 2 mkT =
e
=
3
N
(2π kT )
(2π mkT )3
2
m ( v 2x + v 2y + vz2 )
2 kT
dv x dv y dv z .
0,006
0
tratteggiata T=273 K
0,005
0
continua T=200 K
dN/dv
0,004
0,003
0,002
0,001
0,000
0
1000
2000
3000
4000
5000
velocità (m/s)
Fig. 7: Distribuzione di Maxwell.
Calcolando il valore di
3
P(v) = 4π (
v
per cui si annulla la derivata della funzione
2
mv
m 2 2 − 2kT
) v e
, si ottiene il valore più probabile della velocità. Risulta:
2πkT
42
Cap. 5 – Termodinamica e calorimetria
1
2
ε p = mv2p = kT .
2 kT
, ovvero risulta che l’energia più probabile è:
m
vp =
Possiamo anche scrivere che:
−
1
P ( p) =
e
(π v p )3
m ( v 2x + v 2y + vz2 )
2 kT
dv x dv y dvz Per esempio, per una molecola di O2 a
0°C, la velocità più probabile è pari a 375m/s. Si può confrontare questo valore
con la velocità di fuga dal campo terrestre come suggerito al par. 5 del Cap. 4.
+∞
8kT
= 476m / s per
πm
∫0 vP(v)dv =
Si possono poi calcolare, la velocità media v =
0°C . Lasciamo allo studioso lettore il compito di verificare questi due
N2 a
1
2
ultimi risultati. Vorremmo anche far notare che: ε = mv 2 =
1
2
ε = mv 2 =
1 8kT 4kT
=
m
e
2 πm
π
3kT
non sono evidentemente uguali. Del resto anche l’altra possibile
2
+∞
definizione di energia media: ε =
∫ εP (ε )dε = kT
non dà lo stesso risultato delle
0
due precedenti; è vero tuttavia che quest’ultima coincide con l’energia “più
1
probabile” ε p = mv2p = kT . C’è da dire comunque che tutte e tre le definizioni
2
danno un valore di ≈ kT .
Dimostriamo adesso esplicitamente il teorema di equipartizione dell’energia, il
quale dice che ad ogni termine dell’energia quadratico in una coordinata o in
momento, va attribuita l’energia media
. Scriviamo per l’energia media delle
molecole:
ε = ∫ε
dn
=
N
−∞
1
(2π mkT ) 3
∫
( p x2 + p 2y + pz2 )
2m
−∞
−∞
+∞
p2
e
−
ε
kT
p2
dp x dp y dpz =
+∞
p2
y
z
x
−
−
−
1
( ∫ p x2 e 2 mkT dp x ∫ e 2 mkT dp y ∫ e 2 mkT dpz +
(2π mkT ) 3 2 m −∞
−∞
−∞
1
=
−∞
+∫ e
−∞
−
p x2
2 mkT
+∞
dp x
∫
−∞
2
y
pe
−
p 2y
2 mkT
+∞
dp y
∫e
−∞
−
pz2
2 mkT
−∞
dpz + ∫ e
−∞
−
p x2
2 mkT
+∞
dp x
∫e
−∞
−
p 2y
2 mkT
+∞
dp y
∫
2
z
pe
−
pz2
2 mkT
−∞
Si noti che ciascun integrale è calcolato tra - ∞ e + ∞ come deve essere per una
componente che può variare appunto tra tali limiti, a differenza di un modulo che
43
dpz )
Cap. 5 – Termodinamica e calorimetria
può essere solo positivo. Le tre triple integrazioni (in realtà prodotti di tre integrali
separati) sono tutte uguali e del tipo:
−∞
∫
x 2 e − ax dx
2
−∞
+∞
∫
+∞
e − ay dy
2
−∞
∫e
− az 2
a=
dz , con
−∞
1
, utilizzando le formule degli
2mkT
integrali date prima otteniamo per ciascuno dei tre addendi:
1
( 2πmkT ) 3
=
−∞
1
2m
3
∫
2
−∞
−∞
1 2mkT
2m 2
1
( 2πmkT )
∫
+∞
+∞
∫
x 2 e − ax dx e − ay dy e − az dz =
2
2
−∞
2πmkT 2πmkT 2πmkT =
1
kT
2
In conclusione, l’energia media è la somma di tre addendi ciascuno uguale a
1
kT . Possiamo facilmente generalizzare il procedimento al caso di un sistema a
2
n gradi di libertà: , … ,
con momenti coniugati : # , … , # , se l’energia è una
funzione lineare quadratica delle coordinate e dei momenti: (alcuni dei
coefficienti potrebbero essere nulli):
+∞
data da: ε =
∫ εe
−∞
+∞
∫e
−
−
n
2n
1
n +1
ε = ∑ ai qi2 + ∑ ai pi2 . L’energia media è
ε
kT
dq1 , ..., dqn , dp1 , ..., dpn
.
ε
kT
dq1 , ..., dqn , dp1 , ..., dpn
−∞
che si può dividere in una serie di 2 termini, ognuno dei quali, dopo le
semplificazioni tra numeratore e denominatore, si può scrivere nella forma:
+∞
ai
εi =
∫qe
2
i
−∞
+∞
∫e
−
−
a i qi2
kT
a i qi2
kT
dqi
(ovvero lo stesso con #% al posto di
% ).
dqi
−∞
Proviamo ad integrare ciascuno di questi termini per parti e troviamo:
+∞
εi =
a q2
i i
1
d − kT
e
dqi
− kT ∫ qi
2 −∞ dqi
+∞
∫e
−∞
−
ai qi2
kT
dqi
a q2
− i i
1
= kT −qi e kT
2
dqi
= 1 kT
+ −∞
2
+∞
aq
2
− i i
kT
−∞
e
dq
i
∫−∞
+∞
+∞
∫e
−
ai qi2
kT
44
Cap. 5 – Termodinamica e calorimetria
Poiché ci sono 2 di questi termini troviamo: ε = 2 n
1
kT .
2
1
kT
2
, in cui è il numero di gradi di libertà. In molti casi riguardanti i gas, i termini di
energia potenziale (quella dipendente dalle coordinate) mancano e la formula si
1
riduce a ε = n kT , come nell’esempio precedente. Un esempio in cui invece
2
esiste l’energia potenziale è quello di un sistema statistico di oscillatori armonici.
Ripetiamo il calcolo esplicito per un insieme di oscillatori armonici
unidimensionali e dimostriamo che, se invece di particelle libere avessimo degli
oscillatori armonici lineari, troveremmo che: ε = kT , avendo l’oscillatore
armonico due contributi all’energia: quello cinetico e quello potenziale:
1
1
1
ε = mv 2 + mω 2 x 2 = m(v 2 + ω 2 x 2 ) , mediamente uguali.
2
2
2
In conclusione, l’energia media è la somma di 2 addendi ciascuno uguale a
∫
ε =
−
1
m (v 2 + ω 2 x 2 ) e
2
m ( v 2 +ω 2 x 2 )
2 kT
dpdx
Vsf
∫
Vsf
∫p e
2
=
m ( v 2 +ω 2 x 2 )
−
2 kT
e
dpdx
1 π
1 4a a 1
=
+ mω 2
2m 1 π
2
2 a
+∞
1
2m
0
+∞
∫
−
p2
2 mkT
p2
−
2
mkT
e
+∞
dp
+
1
mω 2
2
dp
0
1 π
4a ' a '
1
2
π
=
∫x e
2
0
+∞
∫e
−
−
mω 2 x 2
2 kT
mω 2 x 2
2 kT
dx
=
dx
0
1
1
2mkT 1
2kT
kT kT
+ mω 2
=
+ mω 2
=
+
= kT
2
4am
4a '
4m
4
2
2
mω
a'
Si può pensare di calcolare l’energia media in un altro modo. Consideriamo lo
spazio delle fasi degli oscillatori armonici. L’energia di un oscillatore è:
E=
1 2 1
p + mω 2 x 2 , che riscritta nella forma:
2m
2
1
1
p2 +
mω 2 x 2 = 1 ,
2mE
2E
rappresenta un’ellisse nello spazio delle fasi di semiassi: a = 1
ω
2E
e b=
m
2 mE
,
la cui area è:
S=
∫
pdx = πab = 2πE / ω =
E
ν
. Consideriamo adesso una sequenza di queste ellissi
di area h, 2h, 3h… in modo che l’intercapedine tra due ellissi di contorno sia
45
Cap. 5 – Termodinamica e calorimetria
sempre h, avremo: S n =
∫
pdx = nh =
En
ν
⇒ E n = nhν (notare l’identità di questa
condizione con quella di Bohr per l’atomo d’idrogeno. Bohr estende in effetti
questa condizione ad un sistema periodico con due gradi di libertà) per i valori
dell’energia corrispondenti alle varie ellissi. Il valore medio dell’energia per gli
oscillatori i cui punti rappresentativi si trovino tra due ellissi d’ordine n e n+1,
E − En
1
= nhν + hν .
sarà E = E n + n+1
2
2
Calcoliamo adesso l’energia media di tutti gli oscillatori di frequenza ν , senza
fare prima il limite per h → 0 . Sostituiamo all’integrale la somma sui valori medi
appena calcolati:
∑ nhνe
ε=
∑e
− βnhν
n
− βnhν
+
hν
d
=−
ln(
2
dβ
∑e
n
− βnhν
)+
hν
d
=−
ln(
2
dβ
∑ x ) + h2ν =
n
n
n
=−
d
hν
d
dx hν
hν hν e − β h ν hν
1
1
ln(
)+
=
ln((1 − x))
+
=−
( − hν x ) +
=
+
dβ 1 − x
dx
dβ
2
2
1− x
2 1 − e − βhν
2
hν
con x = e − β hν . Si noti che se h → 0 , ε → kT e che, se T → 0 , ε →
.
2
p
E3
E2
E1
x
h
h
h
4d. Metodi sperimentali per verificare la distribuzione di Maxwell
I primi esperimenti per verificare la distribuzione di Maxwell- Boltzmann, furono
eseguiti guardando non a molecole di un gas, ma agli elettroni emessi per effetto
46
Cap. 5 – Termodinamica e calorimetria
termoionico da un metallo caldo (vedi Fisica II) O. W. Richardson and F. C.
Brown (1908), da Schottky e infine da Germer (1925)13.
Un esperimento con cui si può verificare la distribuzione di Maxwell è mostrato
nella prossima figura (Lammer, 1929).
Fascetto di
molecole
Un fascetto di molecole di mercurio ben collimato viene indirizzato su un
selettore di velocità cioè un sistema costituito da due ruote con due settori aperti e
sfasati di ∆ϕ fra loro, che ruotano con velocità angolare ω nella direzione
indicata dalla freccia. La sorgente è una fiala riscaldata dall’esterno con un lungo
becco d’uscita a tubo di sezione S per collimare le particelle in uscita. Se il tempo
impiegato dalle molecole a raggiungere la seconda ruota è t , la molecola passerà
attraverso il settore aperto nella seconda ruota, se ∆ϕ = ωt ovvero se:
t = d / v = ∆ ϕ / ω , indicando con v la velocità incognita delle molecole e d la
distanza fra le ruote. Dunque: v = d ⋅ ω / ∆ ϕ . Variando la ω , si possono
selezionare tutte le velocità. Contando poi il numero di molecole di velocità ben
definita che passano per unità di tempo, si può produrre un istogramma che dà la
distribuzione di Maxwell. In realtà, il numero di particelle, per unità di tempo, che
passano attraverso le due ruote ed, eventualmente, colpiscono lo schermo di fondo
è dato da:
dn
dn
dn
dn
= j ⋅ ndS =
v ⋅ ndSdv =
vdSdv , in cui j =
dvv è la densità di corrente di
dt
Vdv
Vdv
Vdv
particelle dotate di velocità pari a
v
(la direzione è per tutte la stessa: l’asse del
tubo di uscita). Come si vede, la quantità che contiamo sullo schermo
dn
non è
dt
mv 2
uguale a
13
−
dn
≈ v 2 e 2 kT dv , cioè la distribuzione di Maxwell, a causa di un
V
Per ulteriori dettagli vedere J. Jeans – An introduction to the theory of gases – pag. 124
47
Cap. 5 – Termodinamica e calorimetria
mv 2
−
dn
dn
ulteriore fattore v .Ciò che misuriamo, dunque, è:
=v
≈ v3e 2 kT dv . Occorre
dt
V
dunque correggere i dati per questo fattore v extra.
Una variante fu eseguita da I. Z. Zartmann (1931) usando un cilindro rotante.
Un altro metodo (Ornestein and van Wyck, 1932) di misurare la distribuzione di
Maxwell è di usare un gas (He) i cui atomi, eccitati da una scarica elettrica,
emettono luce. Così vedremo le righe dello spettro di emissione (vedi atomo di
Bohr al cap.4) allargate dall’effetto Doppler (vedi cap. 2 del prossimo corso) a
formare una banda a causa del moto delle sorgenti. Dallo spostamento della
frequenza rispetto a quella centrale, possiamo dedurre la velocità della particella
che ha emesso e, misurando la luminosità totale ad ogni frequenza, otteniamo il
numero relativo di atomi che nel fascio hanno quella velocità. Il metodo fu anche
applicato alla radiazione da una stella per conoscerne la temperatura superficiale.
Le molecole del gas, sorgente della luce, hanno moti a caso dovuti all’agitazione termica.
3
2
dn m 2 mv
La statistica di Maxwell-Boltzmann ci dice che:
e 2 kT dv x dv y dv z è la
=
n 2π kT
frazione di molecole per unità di volume aventi velocità tra
l’effetto
Doppler,
ν = ν 0 (1 +
preso
l’asse
Z
come
vx e vx + dvx
asse
di
etc…Ora per
mira,
risulta:
c
v
v cos θ
dν . Sostituiamo nella statistica
) = ν 0 (1 + z ) ovvero: dvz =
ν0
c
c
e integriamo su
vx
3
e su
v y , per ottenere:
mv 2
1
mc2 (ν −ν 0 )2
(ν −ν 0 )
−
dn m 2 2π kT 2kTz
1
m 2 c
2 kTν 02
2σ 2
e
dv
e
d
e
=
=
ν
=
z
n 2π kT m
2πσ
2π kT ν 0
kT ν 0
kT 1
con σ =
. Come si vede, si ottiene una gaussiana: la riga non è più
=
m c
m λ0
2
infinitamente sottile, ma ha una distribuzione in frequenza attorno alla frequenza centrale
che è
gaussiana. Prendiamo il vapore di Sodio,
λ0 = 550⋅ 10−9 m , ν0 = 5,4⋅1014 s−1 ,
A = 23 ⇒ m = 3,84⋅ 10−26 kg ,
risulta: σ = 1,07⋅10
9 −1
s
e
σ
= 2 ⋅ 10−6 .
ν0
Come si vede la larghezza è comunque assai piccola rispetto alla frequenza centrale.
48
Cap. 5 – Termodinamica e calorimetria
Un terzo metodo sfrutta il moto di caduta degli atomi emessi da una sorgente
uguale a quella del primo metodo. Lo schema chiarisce il metodo.
Gli atomi emessi dalla sorgente non sono mai perfettamente collimati e
abbandoneranno la sorgente con un piccolo angolo θ verso l’alto, dunque avranno
una traiettoria parabolica come un proiettile. Essi sono costretti a passare
attraverso un foro nel punto y = 0 x = d , il che vuol dire che selezioniamo atomi
con una “gittata” pari a d = 2
v0 x v0 y
g
. L’atomo colpirà lo schermo di fondo dove
1
viene conteggiato ad una quota negativa pari a y = − gt 2 + v0 yt , con t = l , cioè
2
v0 x
v l
v v
a y = − 1 g ( l ) 2 + 0 y . Usando questa relazione e d = 2 0 x 0 y
2
v0 x
v0 x
g
possiamo
calcolare le due componenti e dunque il modulo della velocità
Y
l
schermo
sorgente
X
d
y
iniziale v 0 in funzione di y. Otteniamo: v0 =
1
2
l 2 (d − l )2 + y 2 d 2
g
. Notare
yl ( d − l )
che y e d − l sono entrambi negativi. Naturalmente il volo degli atomi deve
avvenire in un vuoto spinto per evitare collisioni con le molecole d’aria che ne
modificherebbero la traiettoria.
5. Frequenza di collisione e cammino libero medio
Un’obiezione che fu mossa alla teoria cinetica dei gas fu che, se le molecole
fossero puntiformi come è implicito nell’equazione di stato dei gas perfetti, allora
il forte odore di un liquido appena stappato in un angolo di una stanza dovrebbe
essere percepito dall’altro lato della stanza immediatamente, date le velocità con
49
Cap. 5 – Termodinamica e calorimetria
cui si spostano le molecole. Abbiamo infatti visto che le velocità molecolari
medie sono dell’ordine di centinaia di metri al secondo. Del resto nessuna
collisione molecolare interverrebbe a rallentarne il movimento, giacché molecole
puntiformi non hanno alcuna possibilità di collidere: unici urti possibili sarebbero
quelli contro le pareti del contenitore. Clausius introdusse (1858) allora l’idea che
le molecole avessero un volume non nullo. Si noti che questa ipotesi non è
coerente con l’equazione di stato dei gas perfetti che andrà modificata per dare, ad
esempio, l’equazione di Van der Waals, come si è già detto.
5a Frequenza di collisione cammino libero medio
Clausius modificò il procedimento già visto precedentemente (che era stato
inventato da lui stesso modificando con l’introduzione degli angoli la precedente
N
dimostrazione di Joule) usando al posto del fattore
, come abbiamo visto
V
N
con b la somma dei volumi delle molecole,
precedentemente, il fattore
V −b
ovvero il “covolume”.
Clausius immaginò le molecole come sferette di raggio R0. Si noti altresì che, in
questo modo, non si tiene in conto il fatto che le molecole, oltre ad essere dotate
di un volume, potrebbero essere dotate di un’azione reciproca a distanza. Nella
trattazione che segue, noi calcoleremo la frequenza di collisione e il cammino
libero medio dando a R0. il significato di raggio molecolare, ma, se esistessero
forze a distanza, R0. avrebbe piuttosto il significato di un raggio d’azione e non
necessariamente indicherebbe una dimensione caratteristica della molecola.
Calcoliamo adesso la frequenza con la quale una molecola batte contro un’altra
all’interno del volume di gas. La molecola avrà una sezione trasversale pari a
S = π R 02 . Tuttavia occorre considerare che entrambe le molecole hanno raggio R0 ,
ne segue che, se il centro di una molecola entra nel cerchio centrato su un'altra e
di raggio 2 R 0 , si avrà una collisione. Se le molecole stessero tutte ferme tranne
una, quest’ultima vedrebbe un’area coperta per unità di area pari al numero di
molecole per unità di volume n =
N
moltiplicata per la sezione trasversale del
V
cerchio di raggio 2 R 0 e per il cammino fatto dalla nostra molecola nell’unità di
tempo. Poiché questa è la probabilità di collisione per unità di tempo, ovvero il
numero dm di collisioni nel tempo dt (vedi la discussione fatta per lo
“scattering” di Rutherford al cap. 4 delle dispense, prendere il flusso di particelle
entranti pari a 1 con velocità v ,
N
il numero di bersagli e la sezione d’urto pari
V
50
Cap. 5 – Termodinamica e calorimetria
2
a σ = 4π R0 ), abbiamo che il numero di collisioni nel tempuscolo dt
dm =
è
N
N
4π R02vdt , ne segue che la frequenza di collisione è: f = 4π R02v , con
V
V
ovvio significato dei simboli. In realtà, poiché le altre molecole non stanno ferme,
occorre usare la velocità relativa fra le due molecole che si urtano. Mediando14
abbiamo: f =
N
N
4π R02 vrel = 4π R02 2v . Chiaramente il numero di molecole per
V
V
unità di volume, può, almeno nel caso di un gas perfetto, essere riscritto in altri
modi:
p=
π
N RT
N
N
p
p
p
8 kT
=
kT ⇒
=
⇒ f =
4π R02 2 v =
4π R02 2
= 16 pR02
NA V
V
V
kT
kT
kT
πm
mkT
Per quanto riguarda il cammino libero medio λ, ( 1 / f è evidentemente il tempo
medio tra collisioni) abbiamo: λ = v =
f
kT
p 4π
R02
v
vrel
=
kT
4π
2 pR02
=
1
4 2π R02 n
. La
formula appena ottenuta è calcolata usando la distribuzione di Maxwell che come
v
1
=
= 0, 707 , il ragionamento di Clausius, che non
vrel
2
conosceva ancora la distribuzione delle velocità, usava il fattore v = 3 = 0, 75 .
vrel 4
si è detto, prevede che
Vediamo quale era questo ragionamento. Supponiamo che tutte le molecole del
gas abbiano velocità v1 , mentre la molecola in esame abbia velocità v 2 .
Calcoliamo il modulo di v2 − v1 . Abbiamo vrel = v2 − v1 = v12 + v22 − 2v1v2 cos θ
dove θ è l’angolo tra i due vettori velocità. Poiché i moduli delle velocità sono
presi costanti, questa quantità dipende solo dall’angolo θ e poiché la probabilità
d Ω sin θ dθ
=
di avere un angolo θ è
(dopo integrazione sull’angolo ϕ abbiamo
4π
2
che:
π
π
sin θ 2 2
1
v1 + v2 − 2v1v2 cosθ dθ =
(v12 + v22 − 2v1v2 cos θ )3/2
0
2
6v1v2
vrel =
∫0
vrel =
1
v
4
( v1 + v2 ) 3 − ( v1 − v2 ) 3 che, per v1 = v2 , dà:
= . Nel caso di uso
6 v1v2
vrel 3
cioè:
della distribuzione di Maxwell occorre tenere anche in conto il fatto che le
velocità non sono fisse, il che dà un calcolo più lungo.
14
Il calcolo della velocità relativa media è in Loeb, kinetic theory of gases, p. 95 e in E.
Bloch The kinetic theory of gases , p. 167.
51
Cap. 5 – Termodinamica e calorimetria
Se prendiamo un fascio di molecole e lo facciamo viaggiare attraverso un gas
rarefatto, le molecole del fascio colpiranno le molecole del gas e saranno perse; il
−x
numero di molecole nel fascio diminuiranno esponenzialmente: n ( x ) = n0 e l
(vedi discussione sul decadimento dei muoni al cap. 1), come si ricava dalla
1
l
ipotesi che dn( x) = − n( x )dx e il cammino che mediamente viene fatto dalle
molecole del fascio è: λ = l . dn ( x ) è il numero di collisioni di un fascio di n ( x )
particelle in un bersaglio di lunghezza dx cambiato di segno ed è pertanto:
dn( x ) = n( x )
N
N
σ dx , ne segue che: λ = l = σ
V
V
−1
che è identica alla formula
3
. Dunque si può misurare il
4
cammino libero medio, misurando l’attenuazione di un fascio che si muove in un
N
, cioè la densità del gas, si ottiene la sezione d’urto σ ,
gas stazionario. Noto
V
ovvero il raggio molecolare.
ricavata precedentemente a parte il fattore
5b La viscosità
Dalla teoria cinetica dei gas si può anche dedurre il coefficiente di viscosità, che
abbiamo incontrato precedentemente nello studio dell’esperimento di Millikan,
1
mv , dove v è la velocità media, nota dalla distribuzione di Maxwell.
η=
3 2 4π R02
Si può vedere che,
λ (m)
2 R0 ( m )
v (m / s)
H2
1 1, 0 6 ⋅ 1 0 − 8
2, 7 5 ⋅ 1 0 − 1 0
1705,03
1, 5 4 ⋅ 1 01 0
0,84⋅10−5(0,835)
He
17 , 5 5 ⋅10 −8
2 ,1 8 ⋅ 1 0 − 1 0
1205,64
6, 8 5 ⋅1 0 9
1,875⋅10−5(1,86)
N2
5, 89 ⋅ 1 0 − 8
3, 7 7 ⋅ 1 0 − 1 0
8, 08 ⋅ 10 9
1, 5 9 ⋅ 1 0 − 5
O2
6, 25 ⋅ 10 − 8
3, 6 6 ⋅ 1 0 − 1 0
475,67
426,26
6 , 8 2 ⋅1 0 9
1,89⋅10−5(1,89)
f (s−1)
η(kg ⋅ s−1m−1)
52
Cap. 5 – Termodinamica e calorimetria
misurato il coefficiente di viscosità η e, nota la massa m della molecola15, si
possono ottenere il raggio molecolare e quindi il cammino libero medio. Diamo di
seguito alcuni valori della velocità media, cammino libero medio e frequenza di
collisione a T = 273K , p = 1013hPa e
N
= 2,69 ⋅1025 m−3 per alcuni gas (i numeri
V
tra parentesi sono valori misurati).
La definizione del coefficiente di viscosità si ottiene considerando un piano sulla
superficie di un fluido in movimento laminare lungo un asse X. Detto
dv
il
dz
Z
gradiente della
Superficie del liquido
v
Y
X
velocità in direzione (Z) perpendicolare al moto, si ha una forza di trascinamento
che il piano esercita su quello contiguo di superficie dxdy pari a: dF = η
dv
dxdy
dz
(formula di Newton). Questa formula definisce appunto il coefficiente di viscosità
η . Per il valore della viscosità dell’aria vedere cap. 4 (esperimento di Millikan).
La viscosità di un liquido può essere misurata con vari apparecchi, per esempio il
viscosimetro di Ostwald, con cui si misura il tempo di deflusso del liquido
attraverso un capillare tenuto verticalmente. E’ utile soprattutto per misurare
viscosità relative. Si tratta di far defluire in un capillare verticale un volume Q di
liquido, usando la legge di Poiseuille (vedi app. 2) che lega il flusso
volumetrico16 φ alla differenza di pressione agli estremi del capillare ∆p = ρ gh(t )
4
e alla viscosità: φ = π r ∆ p con l e
8 ηl
15
r
lunghezza e raggio del tubo.
Per inciso, la massa della molecola si ottiene dividendo il peso molecolare per il numero
−3
23
−26
di Avogadro. Per esempio, abbiamo: mN2 = 28 ⋅10 kg / 6,06 ⋅10 = 4, 24 ⋅10 kg
−27
mH2 = 3,30 ⋅10−27 kg , mO2 = 5,28 ⋅10−26 kg , mHe = 6,6 ⋅10 kg .
16
Se si vuole il flusso di massa occorre moltiplicare per la densità.
53
Cap. 5 – Termodinamica e calorimetria
A
B
Normalmente, il liquido riempie il rigonfiamento di destra, ma può essere aspirato
nel rigonfiamento di sinistra con una pompetta di gomma fino al livello A. Si
lascia poi discendere il liquido attraverso il tubetto (capillare) di sinistra fino al
livello di equilibrio B , in modo che la quantità di liquido Q defluita sia uguale
al volume del rigonfiamento di sinistra. L’intero sistema deve essere termostatato.
t1
∫
Si misura il tempo di deflusso. Il volume di fluido Q = φ (t )dt defluisce in un
0
t1
∫
tempo t1 dato da: Q = φ (t )dt =
0
t1
t1
0
0
π r4
π r4
π r4
ρ g h(t )dt =
ρ ght1 , in cui
∆pdt =
8 ηl
8 ηl
8 ηl
∫
∫
tutte le variabili sono costanti strumentali tranne ρ e η. Facendo defluire la
stessa quantità di due liquidi diversi e misurando i due tempi di deflusso t1 e t 2 ,
si ha:
ρt
ρ t
π r4
π r4
ρ1 ght1 =
ρ 2 ght 2 ⇒ 1 1 = 2 2 . Note le densità, dai tempi di deflusso si
8 η1 l
8 η2l
η1
η2
ottiene il rapporto tra le viscosità dei due liquidi, ovvero la viscosità di uno
rispetto ad un liquido di riferimento. Il rapporto η / ρ si chiama la viscosità
cinematica.
Un altro metodo usa la legge di Stokes. Lasciamo cadere una sferetta di raggio r
e massa m nel liquido, misuriamo la velocità finale v∞ che, come abbiamo visto
discutendo l’esperimento di Millikan, dipende da η e otteniamo:
v∞ =
mg
mg
.
⇒η =
6πη r
6π rv∞
Un ultimo apparecchio consiste in due cilindri coassiali; nell’intercapedine tra i
due cilindri si pone il liquido in misura e si mette in rotazione il cilindro esterno.
54
Cap. 5 – Termodinamica e calorimetria
Il cilindro interno ruota di un angolo θ caricando una molla a spirale. Dal valore
di θ si può ricavare la viscosità.
L’unità di misura della viscosità è il Poiseuille ( 1Pl = 1Pa ⋅ s = 10Poise ).
5c L’equazione di Fick, la diffusione e la conduzione termica
Consideriamo una quantità fisica Q conservata, trasportata dalle molecole. E’
dQ
, moltiplicando le
dV
N
densità di molecole per la quantità di Q che ciascuna trasporta ρQ = q . Se la
V
∂ρ Q
quantità è conservata deve valere l’equazione di continuità:
+ ∇ ⋅ jQ = 0 . La
∂t
possibile definire una densità di questa quantità: ρQ =
quantità jQ è nota come densità di corrente e serve a calcolare il flusso della
quantità attraverso una superficie. L’equazione appena scritta si interpreta nel
senso che il flusso uscente (entrante) da (in) una superficie chiusa S produce una
diminuzione (aumento) della densità della quantità in esame all’interno della
superficie stessa. La densità di corrente è: jQ = ρQv .
∫S jQ ⋅ ndS
rappresenta
quanta Q fluisce dentro o fuori S nell’unità di tempo. Per calcolare il vettore jQ
possiamo usare lo stesso procedimento usato nel calcolare la pressione sulla
parete di un contenitore. Se v è il modulo della velocità, f (v ) la densità di
r
1−
probabilità di una certa v e e l ( con l il cammino libero medio) la probabilità
l
che la particella attraversi la superficie provenendo da una distanza pari a r
possiamo scrivere il flusso di Q attraverso la superficie elementare jQ ⋅ ndS ,
nella forma (vedere discussione sul calcolo della pressione):
1
jQ ⋅ n =
4π
=
1
v
2
−r
π
el
∫
0
l
+∞
∫ dΩ ∫
Ω
0
e
−r
l
1
ρQ f (v)v ⋅ ndvdr =
l
4π
∫ρ
Ω
e
Q
−r
l
l
+∞
dr cos θ d Ω
∫ f (v)vdv =
0
dr ρQ (θ ) cos θ d (− cos θ )
55
Cap. 5 – Termodinamica e calorimetria
θ
ϕ
n
Se la ρQ è uguale ai due lati della superficie, l’integrale dà evidentemente zero,
come si vede integrando su cosθ . Se la ρQ dipende dalla direzione di arrivo della
molecola, cioè se la ρQ non è costante ai due lati di dS , chiamiamo ρ Q
= ρ Qs
, la
densità delle particelle che vengono dal lato sinistro di dS e ρ Q D la densità di
quelle che vengono da destra. Notare che per le particelle che vengono da sinistra
π
π
è: 0 < θ < per le altre è < θ < π . Pertanto l’integrale diventa:
2
2
−
r
−r
π /2
π
l
1
e
el
jQ ⋅ n = v
dr ρQS cosθ sin θ dθ +
dr ρQD cosθ sin θ dθ . Nel secondo
2
l
l
0
π /2
π
integrale possiamo porre θ = +θ ' . Sostituendo e integrando, abbiamo:
2
∫
∫
−r
π /2 − r
π /2
l
1
e
el
jQ ⋅ n = v
dr ρQS cos θ sin θ dθ −
dr ρQD cos θ 'sin θ ' dθ ' =
2
l
l
0
0
∫
1
= v
2
La
π /2
∫
0
e
−r
l
l
∫
dr ( ρQS − ρQD ) cos θ d (− cos θ )
differenza
( ρ QS − ρ QD ) = − ( ρ QD − ρ QS ) = − (
(ρQS − ρQD )
d ρQ
dn
è
) x = 0 ∆x . La quantità
chiaramente
∆x è 2r cosθ per
particelle provenienti dalla stessa distanza r da lati opposti della superficie, sotto
56
Cap. 5 – Termodinamica e calorimetria
lo stesso angolo θ .
d ρQ
è la derivata di ρQ rispetto alla normale n alla superficie
dn
elementare dS . Infine sostituiamo nell’integrale e abbiamo, integrando su r :.
0
v d ρQ
1
jQ ⋅ n = −
2l cos2 θ d (− cosθ ) = − vl∇ρQ ⋅ n
2 dn
3
∫1
Ne segue che jQ = − 13 vl∇ρQ . Poiché la conservazione della quantità Q richiede
che −
∂ρ Q
∂t
= ∇ ⋅ jQ , si ha:
∂ρ Q
∂t
= 13 vl ∆ρQ , ovvero posto
G = 13 vl ,
∂ρ Q
∂t
= G ∆ρ Q che
è l’equazione di Fick17.
Applichiamo l’equazione di Fick adesso a tre casi:
1. Viscosità. Prendiamo la quantità Q = mu , cioè l’impulso lineare di uno
degli strati di fluido definiti nel moto laminare descritto precedentemente.
La quantità di impulso lineare che passa attraverso ogni piano per unità di
dF
), per quanto detto, é:
dS
dF
1
∂(mu )
∂ (u )
1
1
= j = − nvl
= = −η
⇒ η = nvlm = ρ vl , ( ρ densità) da
dS
3
∂z
3
3
∂z
tempo ed unità di superficie (cioè
cui:
η =
1
3
nmvl =
mv
3 2 4π
R 02
.
In
questo
caso:
mG = η ⇒ G =
η
m
,
∂ (mu ) η
= m ∆( mu ) .
∂t
2. Diffusione. Prendiamo il numero di molecole di un tipo in un fluido come
quantità Q . ρQ è la concentrazione n . Il flusso di particelle per unità di
tempo e unità di superficie è: j = − 13 vl
Infine: D
∂2n
∂x
2
=
∂n
∂n
= −D
, da cui: D = 13 vl .
∂x
∂x
∂n
.
∂t
3. Trasmissione del calore. Q è l’energia media 32 kT ρQ = n 32 kT è la
densità
di
energia:
j = − 13 nlv 23 k
∂T
∂T
= −χ
,
∂x
∂x
da
cui
χ = 12 knvl
57
Cap. 5 – Termodinamica e calorimetria
(coefficiente di trasmissione del calore), presa la ε = 32 kT , otteniamo che
v =
3
m
kT e dunque: χ =
1
2
knl
3
m
kT
18
. Se usassimo la statistica di B.
invece del teorema di equipartizione dell’energia, avremmo: v = 8kT ⇒
πm
χ = knl
1
2
8
πm
kT (vedi più avanti analogo commento sulla conducibilità
elettrica). Infine χ
∂ 2T
∂x
2
=
∂T
.
∂t
Dal punto di vista macroscopico si ha che, dato un volumetto infinitesimo dV
limitato da una superficie dS , la variazione di temperatura nel volumetto a seguito
T
del passaggio di calore è data da − C ρ ∂ dV = j ⋅ ndS = ∇ ⋅ jdV , con C
∂t
calore specifico e
ρ
∂T
= κ ∆ T . Posto
∂t
infine: ∆ T = 1 ∂ T .
χ ∂t
Cρ
j = −κ∇T . Sostituendo:
la densità. Del resto abbiamo:
χ=
κ
= coefficiente di trasmissione del calore, si ha
Cρ
5b. Esempi
La soluzione dell’equazione di Fick in coordinate cartesiane si può scrivere come
il prodotto di tre funzioni, ciascuna con una dipendenza su una variabile spaziale
oltre che sul tempo: ρ ( x, y , z , t ) = ρ1 ( x, t ) ρ 2 ( y , t ) ρ3 ( z , t ) .La cosa si può dimostrare
come è già stato fatto per l’equazione di Poisson: sostituendo la soluzione come
prodotto di tre funzioni, dividendo per ρ ( x, t ) e separando l’equazione in tre,
notando che ci sono tre pezzi ognuno funzione di una sola variabile e che pertanto
i tre pezzi devono essere separatamente uguali a zero. Si ottengono così tre
equazioni (di Fick) nelle tre funzioni ognuna delle quali è una Gaussiana, si
ottiene perciò come soluzione finale: ρ ( r , t ) =
18
1
3
(4π Dt ) 2
e
−
r2
4 Dt
+ C con C valore
Valgono anche qui le stesse considerazione fatte a proposito della conducibilità elettrica
di un metallo, visto come un gas di elettroni: si ottiene la stessa formula per la
conducibilità termica.
58
Cap. 5 – Termodinamica e calorimetria
asintotico della densità. Facciamo vedere che ρ ( x , t ) =
soluzione di ciascuna di queste tre equazioni: D
dρ
dt
=
1
4π D
e
2
− x
4 Dt
(
−1
2 t 3/ 2
+
x2
4 Dt5/2
)
;
∂2ρ
∂x 2
=
∂2n
∂x
1
4π D
2
e
=
2
− x
1
3
(4π Dt ) 2
e
−
x2
4 Dt
+ C è la
∂n
, calcolando le derivate:
∂t
4 Dt
(
−1
2 Dt 3/ 2
+
x2
4 D 2 t 5/ 2
).
La
soluzione è dunque una gaussiana che nell’origine ( x = 0 ) all’istante iniziale. È
infinitamente alta e stretta, ma che poi si allarga col tempo. Questa soluzione
chiaramente descrive bene la diffusione di una goccia d’inchiostro che si allarga
in un catino d’acqua. Descrive però anche quello che succede, ad esempio, se si
porta istantaneamente un’estremità di una sbarra ad alta temperatura. L’aumento
di temperatura si espande nel tempo lungo la sbarra. A situazione stazionaria
∂T
= 0 , da cui si deduce che la
∂t
∂x
T −T
distribuzione di temperatura sarà lineare: T ( x) = 2 1 x + T1 , se gli estremi della
l
sbarra, di lunghezza l , sono mantenuti alle temperature T1 e T2 .
raggiunta, l’equazione sarà
∂ 2T
2
= 0 , perché
6. Velocità di deriva in un campo elettrico e legge di Ohm
Se nel nostro gas esistono degli ioni (elettroni o ioni positivi) sottoposti all’azione
di un campo elettrico E , questi si muoveranno, collidendo dopo un cammino
libero medio λ, nella direzione del campo elettrico con velocità media, detta
“velocità di deriva”, W . La velocità di deriva è naturalmente sovrapposta al moto
termico ordinario che però ha componente media nulla in ogni direzione.
Proviamo a calcolare il cammino libero medio:
+∞
λ=
∫
0
1 eE 2 dP
1 eE
t
dt =
2 m
dt
2 m
+∞
∫t
0
2
dP
dP
1 eE 2
dt =
t , dove abbiamo indicato con
dt
dt
2 m
la densità di probabilità di collisione tra t e t+dt e con t 2 , il tempo quadratico
medio tra due collisioni successive. Si noti che, nella formula che dà lo spazio
percorso tra due collisioni, abbiamo posto a zero la velocità (media) iniziale (
1 eE 2
1 eE 2
t , invece di v0 t +
t ). E’ vero che, in media, v 0 è zero, potendo
2 m
2 m
essere sia positiva che negativa con eguale probabilità. La velocità di deriva sarà
59
Cap. 5 – Termodinamica e calorimetria
t2
allora: W = λ = 1 eE
. Per calcolare la velocità di deriva, occorre allora
2 m
t
t
calcolare il rapporto tra il tempo quadratico medio e il tempo medio tra due
collisioni successive. Per far questo, abbiamo bisogno di calcolare prima di tutto
la
dP
. Se indichiamo con N = N (t ) il numero di ioni che al tempo t dopo l’ultima
dt
collisione non hanno subito ancora collisioni, questo numero sarà decresciuto di
dN = −
N
dt , dove ammettiamo che il numero di ioni che collide tra t e t+dt è
τ
proporzionale al numero N e al tempuscolo dt e che 1 / τ sia la costante di
proporzionalità, similmente a quanto fatto nel caso del decadimento radioattivo.
Evidentemente, 1 / τ deve essere proporzionale alla sezione d’urto di collisione di
uno ione su una molecola, sezione d’urto che sarà, a bassa velocità, puramente
elastica, mentre ad energie superiori sarà essenzialmente inelastica, poiché lo ione
−t
ha abbastanza energia da eccitare o ionizzare l’atomo. Ne segue che: N (t) = N0 e τ
−t
dP
dN
1
=
= e τ . Da questa distribuzione possiamo calcolare le due
, dunque:
dt N 0 dt τ
medie richieste:
+∞
t =
∫
0
dP
tdt =
dt
∫
∫
0
tdt =
τ
0
dP
t2
dt =
dt
In conclusione:
+∞
τ
e
+∞
t2 =
t
+∞ −
∫
−t
d (−e τ
0
∫
t2
0
t2
t
eτ
τ
)tdt =
∫
−t
d ( −τe τ
−t
) dt =
+∞
dt =
∫
0
= 2τ ⇒ W =
−t
+∞
−τ e τ
=τ e
0
−t
+∞
+∞
0
+ ∞ −t
∫
+∞
t 2 d (−e τ ) = 2 e τ tdt = 2τ
0
dP
∫ dt tdt = 2τ
2
.
0
e
eE
τ ⇒ W = µE . Il coefficiente µ = τ è noto
m
m
come “mobilità” dello ione nel gas (Townsend).
Un altro risultato è che la densità di corrente: j = ne W , con n =numero di ioni per
unità di volume, è: j = neW =
ne 2τ
E = σE , che è la legge di Ohm.
m
ne 2τ ne 2 λ
e 2 nλ
=
=
La conducibilità é: σ =
(Drude). La resistività
m
m v
3km T
sarà così:
ρ=
1
σ
=
3km T
. Usando non il
e 2 nλ
teorema di equipartizione
60
Cap. 5 – Termodinamica e calorimetria
dell’energia,
+∞
v=
σ=
ma
∫0 vP(v)dv =
la
8π kT
,
m
statistica
di
usando
Boltzmann,
questa
abbiamo
trovato
espressione
si
che:
ottiene:
ne 2 λ
e 2 nλ
= π
che differisce dalla formula precedente per un
m v
2 2mkT
fattore numerico. Da notare che la formula vale anche in un metallo, in cui
sostituiamo al moto degli ioni positivi quello degli elettroni liberi. Il metallo dal
punto di vista della teoria della conduzione elettrica e termica è da considerarsi
come un gas di elettroni liberi contenuti in un recipiente (teoria di Drude) ed in
collisione col reticolo cristallino. In effetti il modello di Drude parte
dall’equazione del moto degli elettroni sotto l’azione del campo elettrico esterno:
dW
m
m
m
= eE − W , il moto contiene anche una forza di “attrito” − W
dt
τ
τ
proporzionale alla velocità di deriva. Si vede che, in assenza di campo elettrico, la
velocità di deriva si annulla esponenzialmente con costante di tempo τ, detta “
dW
tempo di rilassamento”. In condizione di stazionarietà, invece,
= 0 , che
dt
restituisce la legge di Ohm. Se poniamo τ =∞, abbiamo invece che:
2
dj ne
dW
E , che è la prima equazione di London. Essa
=
m
= eE e dunque:
dt m
dt
descrive la densità di corrente in un conduttore con conducibilità infinita ovvero
resistività nulla (superconduttore).
Poiché
neλ
sono indipendenti dalla temperatura (vedi pag. 25),
coll’aumentare della temperatura, come:
σ∼
1
T
σ
si riduce
. In realtà l’andamento
1
. Vedremo come stanno le cose usando la
T
statistica di Boltzmann ed eventualmente quella di Fermi Dirac nel cap. 3 di
Fisica Generale 2.
Il lavoro fatto sulle cariche in un conduttore di sezione dS e lunghezza dl
(volume: dSdl ):
sperimentale è più simile a σ ∼
61
Cap. 5 – Termodinamica e calorimetria
dL = n(dSdl )eEdl = neWdSEdldt ⇒ jdSdVdt = idVdt ⇒
dL
= idV . Integrando sulla
dt
lunghezza del conduttore, otteniamo una potenza: P = iV = i 2 R =
V2
R
che è la
causa del riscaldamento del conduttore, essendo essa dissipata sul reticolo
cristallino del metallo. Questo riscaldamento è noto come effetto Joule.
Il cammino libero medio sarà infine: λ =
eE 2
1
τ e la frequenza di collisione: f =
m
τ
.
La velocità di deriva misurata in funzione del campo elettrico è mostrata nei
grafici in basso. Nel caso del CO 2 la linearità è evidente. Nel caso della mistura
Argon-isobutano, come nella maggior parte dei gas, la linearità dura fino ad un
certo campo massimo, poi la curva si abbassa ed eventualmente la velocità rimane
più o meno costante o, come si dice, “satura”. Dunque la nostra teoria ha valore
solo a campi bassi. La deviazione dalla linearità è facilmente spiegata.
Consideriamo la costante τ ; essa è tale che il numero di collisione per unità di
dx
lunghezza di cammino è: dN = N , con v velocità delle particelle. Ne segue
vτ
che, utilizzando il formalismo del cap.4 sulla diffusione di Rutherford, si può
mettere in relazione la costante τ con la sezione d’urto di collisione ionedx
1
= NMσ dx ⇒ = Mσ v con M = numero di molecole
molecola, σ : dN = N
vτ
τ
e
e 1
per unità di volume. Troviamo così: µ = τ =
, ovvero che la mobilità
m
m Mvσ
dipende dalla sezione d’urto ione-molecola. Se questa è funzione del campo
elettrico, non ci possiamo più aspettare una mobilità costante. In realtà, la sezione
d’urto è funzione dell’energia dello ione a sua volta funzione del campo elettrico.
Dunque ciò che succede è che all’aumentare del campo elettrico l’energia dello
ione al momento della collisione è mediamente più alta, la sezione d’urto aumenta
e abbassa il valore della mobilità, producendo una deviazione dalla linearità della
curva W − E .
62
Cap. 5 – Termodinamica e calorimetria
63
Cap. 5 – Termodinamica e calorimetria
Qui occorre fare una pausa per riflettere sul significato di “sezione d’urto”. Nel
caso dell’esperimento di Rutherford, la sezione d’urto ha un senso puramente
geometrico: essa rappresenta l’area trasversale di ogni atomo entro cui deve
entrare una particella alfa per essere deviata. Anche nel caso atomico abbiamo
considerato la sezione d’urto come una sezione trasversale della molecola entro
cui un’altra particella deve passare perché si abbia una collisione. In realtà,
occorre dire che il campo coulombiano del nucleo si estenderebbe fino
all’infinito, tuttavia per parametri d’impatto superiori al raggio atomico il campo
elettrico viene posto uguale a zero, perché gli elettroni atomici esattamente
compensano la carica nucleare e il campo viene bruscamente posto uguale a
zero19. Come sappiamo, per carica totale uguale a zero, il campo non è
necessariamente nullo, ma possono esistere dei termini di dipolo e quadrupolo che
formano un campo di frangia esterno all’atomo. Anche così, la sezione d’urto
divergente dovuta al campo colombiano, ora schermato dagli elettroni esterni,
dσ
non diverge più
diventa meno divergente e la sezione d’urto totale σ =
dΩ
∫
Ω
come nel caso coulombiano. Non è pensabile del resto che la sezione trasversale
cambi con l’energia del proiettile: dunque il concetto di sezione d’urto passa ora
da un concetto puramente geometrico a quello di un coefficiente che determina la
probabilità di collisione. La transizione è dovuta al fatto che oltre alla possibilità
di una collisione elastica, con conservazione dell’energia cinetica totale, si apre la
possibilità di una collisione in cui la struttura atomica sia modificata dall’urto con
conseguente assorbimento di energia che non apparirà più nella forma di energia
cinetica, ma come energia interna all’atomo o molecola e verrà emessa più tardi
come luce. Si aprono nuove possibilità o come si dice “nuovi canali”. Il canale
inelastico, a differenza di quello elastico, può dipendere dall’energia del proiettile
perché il trasferimento d’energia all’atomo sarà funzione della struttura
dell’atomo o della molecola che subisce l’urto e quindi della possibilità di
assorbire energia. Dobbiamo allora considerare una sezione d’urto totale pari alla
somma della sezione d’urti elastica e di quella inelastica: σ t = σ e + σ i ,
complessivamente funzione dell’energia.
Nei campi della fisica atomica, nucleare e subnucleare, esistono molti più canali,
ovvero i canali inelastici sono multipli con produzione di particelle secondarie
(come la luce cui si menzionava poche righe prima). Il concetto di sezione d’urto
19
Tuttavia nel caso dei plasmi, cioè dei gas ionizzati, le collisioni si devono trattare come
collisione ione-ione e torna il problema della divergenza della sezione d’urto differenziale.
Anche qui occorre trovare una distanza massima tale che, se il parametro d’impatto supera
questa distanza, ci sia un “taglio” . Al di sopra di qualche distanza il campo coulombiano
non è più tale. Non è difficile immaginare che per distanze dell’ordine dell’interdistanza
tra gli atomi il campo visto da uno ione non è più puramente coulombiano.
64
Cap. 5 – Termodinamica e calorimetria
però rimane valido, malgrado l’aumentare della complessità. Ramsauer misurò la
variazione della sezione d’urto con l’energia (o la velocità) di un elettrone che si
muove in un gas. Elettroni ottenuti per effetto fotoelettrico da una lastrina di
metallo, vengono accelerati da un campo elettrico e raggiungono una certa
velocità proporzionale alla radice quadrata del potenziale usato. Gli elettroni
vengono immessi in un tubo circolare contenente il gas scelto alla pressione scelta
e fatti circolare con l’aiuto di un campo magnetico costante perpendicolare alla
sezione mediana del tubo. Alcune fenditure eliminano gli elettroni distanti
dall’orbita centrale e dunque con energie diverse da quelle originalmente prodotte
con il potenziale acceleratore a causa di collisioni con gli atomi. La carica persa
dal fascio e raccolta sulle pareti e sulle fenditure viene misurata. In questo modo,
misuriamo la frazione di elettroni persi per collisione dal fascetto e, dunque la
sezione d’urto al variare di pressione ed energia.
Sulla caratteristica della curva W − E di saturare si basano certi strumenti usati
nella fisica nucleare e subnucleare: le camere a deriva (“drift”). Un filo (o anche
molti fili disposti parallelamente) vengono posti ad alta tensione positiva rispetto
ad un involucro a massa. Quando una particella ionizzante passa attraversa il gas
contenuto nella camera, ionizza un certo numero di atomi ( ∼ 10/ cm ). Sotto
l’azione del campo elettrico, gli elettroni prodotti dalla ionizzazione si muovono
verso il filo. Poiché il campo in prossimità del filo è molto alto, gli elettroni
acquisiscono una tale energia da produrre molta ionizzazione secondaria che, a
sua volta, produce altra ionizzazione (valanga). Questa carica produce un segnale
elettronico, che ci dice che una particella è passata attraverso la camera. Tuttavia,
se siamo in grado di misurare il tempo impiegato dalla carica per muoversi dal
punto di produzione a quello di raccolta (filo), la conoscenza della velocità di
deriva ci consente di dire a quale distanza dal filo la carica è stata prodotta e
dunque dove è passata la particella primaria. La risoluzione ottenibile è
tipicamente dell’ordine di 100µm . Un esempio di camera a deriva è dato nella
figura più avanti.
La risoluzione è limitata dalla diffusione degli ioni.
Vediamo cosa è la diffusione. Prendiamo un gran numero di N ioni tutti generati
in un punto. Per raggiungere il filo essi devono percorrere una distanza d sull’asse
congiungente il punto di generazione e il filo. Deve dunque essere d = Wt + x ,
dove t è il tempo di impiegato a raggiungere il filo e x è la proiezione del
cammino dovuto all’agitazione termica lungo l’asse di deriva. E’ chiaro che il
valore medio di x sulle diverse particelle sarà zero, ma sulla singola particella
esso non è zero. Sarà così: 0 = Wδ t + δ x , ovvero ci sarà, in media quadratica, una
fluttuazione del tempo di deriva misurato collegata al valore quadratico medio di
x . Il fatto che la particella abbia un moto di deriva non influenza il valore
quadratico medio di x .
65
Cap. 5 – Termodinamica e calorimetria
Immaginiamo dunque di eliminare il moto di drift e ci troviamo con un insieme di
particelle che, generate in un punto, se ne allontanano diffondendo. La diffusione
obbedisce all’equazione della diffusione di Fick:. La soluzione di questa
dN
equazione è una gaussiana:
=
dx
N
4π Dt
e
−
x2
4 Dt
+ C , come abbiamo già visto. La
sua deviazione standard è: σ = x = 2Dt . Il coefficiente D è noto come
“coefficiente di diffusione”. Dunque uno ione, soggetto ad un moto di deriva
verso il filo, avrà una traiettoria non retta, ma un moto a zig zag (“random walk”)
più lungo (breve) a causa dell’agitazione termica, cioè della diffusione. Poiché
tale allungamento (accorciamento) fluttua, anche la misura del tempo di drift
fluttua, generando un errore statistico sulla determinazione del punto di
generazione dello ione, ovvero della distanza dal filo a cui è passata la particella
2
ionizzante, pari a σ = 2 Dd .
µE
66
Cap. 5 – Termodinamica e calorimetria
Nella figura sopra: un campo elettrostatico è formato con fili a differenti
potenziali (grading wires). Altri fili (sense wires) raccolgono gli ioni formatisi per
il passaggio di radiazione ionizzante (track). L’immagine di sopra mostra le
equipotenziali e quella di sotto le traiettorie degli ioni (elettroni), traiettorie che
sono indistinguibili dalla linee di forza. Lo sfalsamento tra loro dei sense wires
67
Cap. 5 – Termodinamica e calorimetria
serve per poter dire se la traccia è passata a sinistra o a destra del piano dei
sense wires stessi.
La tabella in basso dà la mobilità e il coefficiente di diffusione per tre ioni (che si
muovono nello stesso gas) in condizioni normali.
Gas
Coeff. di diff. ione positivo D+ ( Mobilità µ+ ( m 2 ⋅ s − 1 ⋅ V − 1 )
m2 / s )
H2
0, 34 ⋅ 10 − 4
1 3 ,0 ⋅ 1 0 -4
He
0, 26 ⋅ 10 − 4
1 0 ,2 ⋅ 1 0 -4
O2
−4
0, 06 ⋅ 10
2,2 ⋅ 10 -4
Particelle macroscopiche, ma di piccole dimensioni ( a =raggio della particella
∼ 1µ m ) diffondono nello stesso modo e
D è dato da:
D=
kT
1
, con
N A 6πη a
η
coefficiente di viscosità. Misurando D si può misurare il numero di Avogadro
(Perrin).
7. I moti browniani
Nel De Rerum Natura, Lucrezio si esprime così:
Osserva infatti, ogni volta che raggi trapelano
e infondono la luce del Sole nell’oscurità delle stanze:
vedrai molti corpi minuscoli vorticare
in molteplici modi nel vuoto nella luce stessa dei raggi
…
Danno inizio al movimento di per sé gli elementi basilari delle cose;
indi quei corpuscoli formati da una piccola struttura
…
Così il moto sale dalle particelle elementari e a poco a poco
Giunge ai nostri sensi, così che si scorga
Il movimento anche dei corpi che possiamo vedere alla luce del sole,
pur se non appaia con evidenza per quali impulsi si produca.20
20
De rerum natura p. 167
68
Cap. 5 – Termodinamica e calorimetria
E’ una descrizione perfetta dei moti browniani.
L’esistenza degli atomi non è stata creduta da tutti gli scienziati fino agli inizi del
‘900, malgrado i grandi progressi che l’idea della loro esistenza faceva fare alla
chimica ed alla fisica. Il problema era che nessuno aveva mai visto un atomo:
troppo piccolo per essere visto! Persino Max Planck nel 1900 dubitava ancora
della loro esistenza. E’ curioso notare che, malgrado il fatto che nessuno aveva
visto il calorico o l’etere luminifero (cioè l’etere che si supponeva supportasse le
vibrazioni che costituivano la luce), tanti credessero in queste due sostanze che si
è poi dimostrato non esistere. Un modo per rendere evidenti i moti atomici era
quello di osservare al microscopio una particella grande abbastanza da essere vista
e piccola abbastanza da subire pochi urti da parte delle molecole del mezzo nel
quale la si poteva immergere, in modo che le fluttuazioni degli urti da un lato
potessero essere viste sotto forma di improvvisi moti nella direzione opposta,
proprio come quelli degli atomi. Nel 1828 il botanico Robert Brown osservò
appunto tali moti di granuli di polline (diametro di pochi µ m ) in una soluzione
acquosa. La teoria completa dell’effetto fu elaborata nel 1905 da A. Einstein. Gli
esperimenti di Perrin e Millikan ne dimostrarono l’esattezza. Da quel momento in
poi anche i più acerrimi nemici dell’idea di atomo, come il grande chimico
lettone-tedesco Friedrich Wilhelm Ostwald, deposero le armi.
Abbiamo studiato la teoria della diffusione, descriviamo adesso la teoria di
Einstein dei moti browniani. Abbiamo dunque che, trattando una particella
macroscopica come una grossa molecola essa diffonde dal punto di origine
viaggiando una distanza x distribuita secondo una gaussiana. Rimane il problema
di calcolarne la σ = 2Dt cioè D in funzione dei parametri delle particelle e del
fluido.
La nostra particella obbedisce all’equazione classica della meccanica:
dv
= − µ v + F (t ) , dove la F (t ) è la forza generata dagli urti. Dunque:
dt
dv
µ
v(t )
m
= − v + a(t ) = −
+ a(t ) , τ =
è chiamato tempo di rilassamento.
dt
m
τ
µ
m
Proviamo ad integrare questa equazione, moltiplicando l’equazione per
x
dv
v(t )
= −x
+ xa (t ) e,
dt
τ
d 2 x2
dt 2
= 2( v 2 + x
sfruttando
le
relazioni:
x (t ) v (t ) =
x:
2
1 dx
;
2 dt
dv
dv 1 d 2 x 2
1 d 2 x2
1 dx 2
)⇒ x
=
− v 2 , troviamo:
− v2 = −
+ xa (t )
dt
dt 2 dt 2
2 dt 2
2τ dt
d 2 x2
. Mediando si ha:
dt 2
+
2
2kT
1d x
2
, la cui soluzione
= = 2 v + 2 xa(t ) =
m
τ dt
69
Cap. 5 – Termodinamica e calorimetria
è:
x(t )2 =
−t
2kT 2 t
τ − (1 − e τ ) ,
m
τ
che,
per
t >> τ ;τ << 1 ,
dà:
2kT m 2 µ 2kT
kT
t =
t = σ 2 = 2Dt ⇒ D =
.
Notare
che:
2
m µ m
µ
µ
kT
kT
v 2 + xa (t ) =
, perché: v2 =
a norma dei risultati ottenuti in meccanica
m
m
statistica, tenuto in conto il fatto che abbiamo qui un problema unidimensionale, e
perché: x a ( t ) = 0 , per ogni valore di x deve essere, infatti, nulla la media delle
x(t )2 =
accelerazioni a ( t ) .
Tale teoria è verificabile sperimentalmente: si tratta di misurare la distanza
raggiunta in media dalle particelle in funzione del tempo.
8 L’equazione di Van der Waals
Nello scrivere l’equazione dei gas perfetti, abbiamo fatto due approssimazioni: a)
la dimensione delle molecole è stata trascurata e b) abbiamo assunto che le
molecole non interagiscano tra loro che all’atto di una collisione. L’assunzione a)
è ovviamente falsa: nella discussione sulla frequenza di collisione e/o nel calcolo
del cammino libero medio, abbiamo visto che occorre pensare alle molecole come
delle sferette di raggio R0 del valore di alcune unità in 10 − 10 m . Se consideriamo
una mole di gas, ovvero un numero di Avogadro di molecole, il volume totale di
4
3
queste molecole sarà: V = π R03 N A e, se abbiamo
n
moli, allora il loro volume
4
3
totale (covolume) sarà: V = n π R03 N A = nb . Ne segue che, poiché nell’equazione
dei gas perfetti all’aumentare delle pressione, il volume si riduce senza limiti,
occorrerà correggere tale equazione introducendo al posto del volume la quantità
V − nb , assumendo che il gas non possa essere compresso ad un volume al di sotto
del covolume.
Nell’assunzione b), c’è il ragionamento che le forze tra molecole non possano che
essere di origine elettrica, le molecole sono tuttavia neutre e ne segue che non
esistono forze coulombiane tra di esse. Tuttavia le molecole, pur essendo neutre,
possono avere un momento di dipolo. Tra dipoli interagenti ci sono forze attrattive
e, dunque, ciascuna molecola è dotata di un’energia potenziale oltre che di
un’energia cinetica. Questo comporta che il termine di pressione nell’equazione
dei gas perfetti vada modificato in modo proporzionale al quadrato della densità
70
Cap. 5 – Termodinamica e calorimetria
ρ∼
2
n
. Da tutto ciò segue l’equazione di Van der Waals: ( p + a n )(V − nb ) = nRT
V
V2
, in cui i coefficienti a e b dipendono dal gas e di cui mostriamo un grafico per
l’elio in basso, confrontandolo col comportamento di un gas perfetto. I parametri
sono ricavati nel modo discusso più oltre. In effetti possiamo ricalcolare il raggio
molecolare R0 a partire da b per i gas menzionati in precedenza e di cui
conosciamo il raggio molecolare R0 (ricavato dalle misure di cammino libero
medio) e confrontare tali valori.
2 R0 ( m )
H2
1, 37 ⋅ 10 − 10
−10
b (misurato in m3)
R0 ( m )
2, 6 6 1 ⋅ 1 0 − 5
1, 74 ⋅ 10 − 10
2, 37 ⋅ 10
−5
1, 6 7 ⋅ 1 0 − 1 0
He
1, 0 9 ⋅ 1 0
N2
1, 88 ⋅ 10 − 10
3, 9 1 3 ⋅ 1 0 − 5
1, 97 ⋅ 10 − 10
O2
1, 83 ⋅ 10 − 10
3,1 8 3 ⋅ 1 0 − 5
1, 8 4 ⋅ 1 0 − 1 0
Vediamo che almeno gli ordini di grandezza sono corretti. Nel calcolare il raggio
molecolare, occorre però tenere in conto che, se mettiamo le sferette
rappresentanti gli atomi a contatto, rimangono degli spazi morti. Così, disegniamo
tante sferette di diametro 2 R 0 a contatto e circoscriviamo ogni sfera con un
cubetto di spigolo 2 R 0 . Il volume totale delle sferette starà a quello dei cubetti
come il volume di una sferetta starà a quello del cubetto circoscritto:
4
π R03
4
1
3
=
π ≃ . Nel calcolo della tabella precedente abbiamo già assunto che
3
3⋅8
2
(2 R0 )
occorresse moltiplicare il volume di una sferetta per due. In formule:
4
b = N A 2 ⋅ π R03
3
Osserviamo anche che i valori del covolume sono effettivamente trascurabili
rispetto al volume molare che a TPV normali è di 22,4141 litri. In altre parole
l’approssimazione del gas perfetto è ragionevole, in condizioni normali e fino a
quando il covolume non diventa importante.
71
Cap. 5 – Termodinamica e calorimetria
Per il biossido di carbonio vediamo nella figura che segue l’andamento della
p = p (V ) , per varie temperature. Si vede che esiste una temperatura (temperatura
critica o TC ) al di sotto della quale nasce un comportamento della curva in cui si
osservano un minimo ed un massimo locali. Chiaramente tale comportamento ha
poco senso, in effetti, c’è un tratto in cui, aumentando il volume, la pressione
aumenta. Tuttavia in questa zona, il gas in realtà liquefa. Dunque: riducendo il
volume al di sotto di un certo valore, se siamo al di sotto della temperatura critica,
il gas liquefa e, durante la liquefazione, la pressione rimane costante e la
trasformazione sarà rappresentata da un segmento orizzontale, piuttosto che il
massimo e il minimo dalla curva di van der Waals. La pressione corrispondente al
volume VC alla temperatura critica, che nel caso del CO2 è TC = 304 K , sarà
chiamata PC .
72
Cap. 5 – Termodinamica e calorimetria
Il valore di PC si può calcolare in funzione dei due parametri
punto, di coordinate PC , VC , TC deve essere vero che: (
(
∂2 p
)T = costante = 0 : p = −
∂V 2
∂2 p
∂V
2
= −6
an2
V
4
+
2nRT
(V − nb)3
an 2
V2
nRT
+
;
V − nb
.
La
da
prima
cui:
a
e b . In effetti al
∂p
)T =costante = 0 e
∂V
∂p
an2
nRT
=2
−
3
∂V
V
(V − nb)2
eguagliata
a
zero
e
dà:
2an2(V − nb)2 − nRTV3 = 0 e la seconda −6an2 (V − nb)3 +V 4 2nRT = 0 . Da queste
due equazioni, ponendo T = TC
e V = VC ricaviamo: VC = 3 nb , TC =
(dall’equazione di van der Waals) PC =
e a=
a
27b
2
8a
e
27 Rb
. Da cui possiamo ricavare: b =
RTC
8PC
27 (RTC )2
, ovvero: misurando le coordinate termodinamiche critiche si
64 PC
conoscono
parametri;
a
a
e b . Il grafico precedente è stato fatto per il CO2 , usando i
= 0, 364 J ⋅ m 3 / m oli 2 e b = 0, 0 4 2 67 ⋅ 1 0 − 3 m 3 / m o le , ricavati dalla
73
Cap. 5 – Termodinamica e calorimetria
conoscenza sperimentale di PC , VC , TC . A questo punto, su ogni isoterma a
temperatura inferiore a quella critica possiamo tirare un segmento orizzontale dal
volume a cui comincia la liquefazione a quello in cui la liquefazione del gas è
completa e chiederci, se è possibile determinare il volume e dunque la pressione a
cui comincia la liquefazione. Riprendiamo il grafico relativo alla CO2 alla
temperatura T = 273K . Consideriamo una trasformazione nella forma di un ciclo
(vedi oltre) che partendo dal punto A ritorni al punto A usando parte della curva di
van der Waals come indicato in figura (ACDBA). Alla fine del ciclo sia l’energia
interna che l’entropia devono essere uguali al loro valore iniziale:
δQ 1
Q
δ Q = == 0 . Ne segue che non essendo stato scambiato
∆U = ∆S =
=
T
T
T
calore e non essendo variata l’energia interna, il lavoro fatto durante il ciclo è
zero. Dunque il segmento orizzontale deve essere posto ad una altezza tale che il
lavoro fatto durante il picco e quello fatto durante la valle siano uguali e di segno
contrario e diano dunque un lavoro totale uguale a zero.
∫
∫
Si può derivare l’equazione di van der Waals usando il teorema del viriale, come
detto precedentemente. Il teorema del viriale, come già discusso, trovava:
3
1
∑ i ri ⋅ Fi = − 2 H , ovvero 2 nRT = − 2 i ri ⋅ Fi dove la somma si intende
estesa a tutte le particelle e a tutte le forze e la media fatta su un intervallo
temporale adeguatamente lungo. Si tratta allora di calcolare il secondo membro,
sotto le nuove ipotesi che le molecole non siano puntiformi, ma siano sferette con
∑
74
Cap. 5 – Termodinamica e calorimetria
un loro raggio R0 e che tra loro esista una forza attrattiva. Un primo contributo al
viriale viene dalle collisioni contro le pareti. Questo contributo è stato calcolato
−3PV .
già
ed
era
uguale
a
Abbiamo
così:
3
3
1
nRT = PV −
2
2
2
∑ r ⋅F
i
i
⇒ nRT = PV −
i
1
3
∑ r ⋅F
i
i
i
, dove la somma deve
essere estesa alle interazioni tra le molecole, interazione che non era considerata
nella derivazione dell’equazione di stato del gas perfetto.
La somma −
1
3
∑ r ⋅F
i
i
i
, può calcolarsi nel modo seguente. Supponiamo che la
forza21 sia derivabile da un potenziale U = U ( r ) , cioè: F = −
dU
. Inoltre
dr
R0 m
) , r > R0 (potenziale attrattivo<0) e +∞ per r < R0
r
(potenziale repulsivo>0). Sostituiamo la somma con un integrale. Le molecole
prendiamo U (r ) = −U0 (
U (r )
N − kT
e
con cui una data molecola interagisce hanno densità pari a
, dove
V
l’esponenziale di Boltzmann garantisce che stiamo tenendo in conto l’azione delle
forze sulla densità. Il numero di molecole in una corona sferica centrata sulla
U (r )
N − kT
e
4π r 2dr .
molecola scelta e di raggio incluso tra r
V
Nel sostituire nella somma, dobbiamo moltiplicare nuovamente per N , per tenere
in conto tutte le possibili molecole e dividere per 2 onde evitare di contare un
termine due volte. In conclusione:
e r + dr sarà: dN =
1
−
3
∑
N2
ri ⋅ Fi = 2π
i
3V
N 2 kT
= 2π
3V
+∞
∫0
+∞
∫0
U (r )
dU − kT
N 2 kT
r
e
dr = 2π
dr
3V
3
+∞
∫0
U (r )
−
d
r
(−e kT ) =
dr
3
U (r )
−
d
r
(1 − e kT ) . Tenere presente che l’aggiunta di 1 sotto la
dr
3
derivata nell’integrando non modifica l’integrale. Integriamo per parti:
1
−
3
∑
1 3N 2 kT
ri ⋅ Fi = −2π
i
3 V
+∞
−
∫0 r (1− e
2
U (r )
kT )dr
=−
nRT
B , con
V
21
Notare che l’esistenza di forze intermolecolari è alla base della spiegazione dei
fenomeni di tensione superficiale e della capillarità.
75
Cap. 5 – Termodinamica e calorimetria
R0
B = 2π N (
∫
r 2 (1 − e
−
+∞
U (r )
U (r )
−
kT ) dr +
r 2 (1 − e kT )dr )
∫
0
.
Abbiamo
così:
R0
nRT = PV − nRT
B
B
. Ovvero: PV = nRT (1 + ) . Il primo integrale è immediato
V
V
perché all’interno della sferetta il potenziale è infinito e dunque l’esponenziale è
zero. Il secondo integrale può essere calcolato facilmente nella gamma di
temperature per cui sia U ( r ) << kT . In tal caso possiamo scrivere
U (r)
−
e kT
≈ 1−
U (r)
kT
, allora abbiamo:
R3
B = 2π N ( 0 +
3
+∞
∫
r (1 − e
2
−
U (r )
kT ) dr )
R0
R 3 2π N
U 0 R0m
= 2π N 0 −
kT
3
+∞
∫
R0
R3
= 2π N ( 0 +
3
+∞
∫
R0
R 3 2π N
U (r )
r
dr ) = 2π N 0 −
kT
kT
3
2
+∞
∫
r2
U 0 R0m
R0
R3
2π N
aN
r 2− m dr = 2π N 0 −
U 0 R03 = nb −
3 ( m − 3) kT
N A2 kT
3
2π N A2
B
U0 R03 . In conclusione: PV = nRT (1 + ) , ovvero :
con b = 2π N A R0 e a =
(m − 3)
V
3
PV = nRT (1 +
1
a
1
a 2
( nb −
n )) = nRT (1 + nb ) −
n V ⇒
V
RT
V
V2
1
1
n2
= nRT (1 + nb ) ≅ nRT / (1 − nb ) ⇒ P (1 + a
)(V − nb ) = nRT
V
V
V2
.
Si noti che perché le cose tornino è necessario che sia m > 3 .
76
rm
dr =
Cap. 5 – Termodinamica e calorimetria
Andamento del potenziale di Lennard-Jones. Si tratta di un potenziale
intermolecolare ottenuto per via teorica con la forma: U ( r ) = 4ε R0 − R0 .
12
r
6
r
Il comportamento di tale potenziale descrive bene una molecola praticamente
incompressibile: il potenziale praticamente diverge a r = R0 ed è negativo per
r > R0 . Si noti che l’andamento attrattivo in
1
ha un esponente maggiore di tre
r6
come richiesto nella precedente dimostrazione. Tale andamento si può giustificare
come segue. Prendiamo l’energia di interazione tra due dipoli molecolari;
d1 ⋅ d 2
d ⋅ nd ⋅ n
l’energia
media
sarà
−3 1 32 ) ;
3
r
4πε 0 r
1 d1d 2
U =
( cos θ − 3 cos θ1 cos θ 2 ) . L’angolo medio tra i dipoli si
4πε 0 r 3
U=
1
(
calcola come sarà fatto per il campo in un mezzo materiale (vedere oltre) e dà
cos θ =
1
d1d 2
4πε 0 3kTr 3
che
fornisce
il
contributo
del
primo
termine:
d1d 2 1 d1d 2
1 2 (d1d 2 ) 2
=
, con l’andamento previsto
(
)
4πε 0 r 3 4πε 0 3kTr 3
4πε 0 3kTr 6
B
U = 6 . Il secondo termine dà un contributo uguale, ma tre volte più grande e
r
1 2 (d1d 2 ) 2
B
di segno opposto. In conclusione: U = − 6 con B = −2(
)
4πε 0
3kT
r
U =
1
(Keesom “allignment energy”). Esiste anche un secondo termine dovuto ai dipoli
indotti da una molecola polare (Debey-Falkenhagen) induction energy con lo
stesso andamento in funzione della distanza. Comunque entrambi i termini sono
in genere dominati dalle forze di London anche note come “dispersion energy”.
Un semplice modellino è il seguente (ripreso da C. Kittel Introduction to solid
state physics). Consideriamo le due molecole come due oscillatori armonici con le
due cariche (nucleo e nuvola elettronica) oscillanti linearmente. La loro energia è:
p12 1 2 p22 1 2
+ kx1 +
+ kx2 cui va aggiunta però l’energia d’interazione che
2m 2
2m 2
k
modificherà la frequenza di oscillazione iniziale ω0 =
. L’interazione si
m
H0 =
compone di quattro pezzi: attrazione positivo-negativo e negativo-positivo e
77
Cap. 5 – Termodinamica e calorimetria
repulsione positivo-positivo e negativo-negativo. Facendo un modello lineare,
abbiamo per questo secondo termine:
H1 =
e2 1
1
1
1
e 2 x1 x2
2
+
−
−
≃
−
, per x1 , x2 << r
4πε 0 r r + x1 − x2 r + x1 r − x2
4πε 0 r 3
r
+
x2
+
-
-
x1
1
1
ε ε2
= (1 − + 2 + ...) .
r +ε r
r r
p2 1
p2 1
e 2 x1 x2
l’energia totale è:
H = 1 + kx12 + 2 + kx22 − 2
2m 2
2m 2
4πε 0 r 3
Ricordare che
Così
L’espressione dell’hamiltoniana (cioè l’energia), si può riscrivere, ponendo
x1 + x2
x − x2
p + p2
p − p2
, xa = 1
, ps = 1
e pa = 1
, nella forma:
2
2
2
2
p2 1
pa2 1
2e 2
2e 2
2
H = s + (k −
)
x
+
+
(
k
+
) xa2 . Da questa si vede che il
s
3
3
2m 2
4πε 0 r
2m 2
4πε 0 r
xs =
sistema è ancora equivalente a due oscillatori che, a causa dell’interazione
reciproca, hanno cambiato frequenza di oscillazione: da
ω± =
k
2e 2
1 2e 2
1
2e 2
±
=
ω
(1
±
−
(
) 2 + ...)
0
3
3
3
m 4πε 0 mr
2 4πε 0 mr 8 4πε 0 mr
ω0 =
quantistica)
E=
e
diviene
ℏω
1
1
1
2e
ℏω + + ℏω + = 2 ℏω 0 − 0 (
)2
3
2
2
2
8 4πε 0 mr
complessiva dell’energia pari a: ∆E = U = −
a
espandendo
nell’ultimo passaggio la radice quadrata in serie di potenze di ε =
L’energia di due oscillatori era dunque pari a E = 2
k
m
2e 2
.
4πε 0 mr 3
1
ℏω0 (vedi meccanica
2
dopo
l’interazione
2
con
una
variazione
ℏω0
2e 2
B
(
)2 = − 6
3
8 4πε 0 mR
r
78
Cap. 5 – Termodinamica e calorimetria
L’andamento repulsivo
1
deriva dal principio di esclusione di Pauli ed è un
r12
problema quantistico. Alla repulsione tra atomi contribuisce anche la repulsione
elettrostatica tra cariche simili (nucleo-nucleo, elettrone-elettrone)
Notiamo infine che l’equazione PV = nRT (1 +
nella forma: ( P +
a
V
2
n 2 )V = nRT (1 +
1
a
(nb −
n)) , si può riscrivere
V
RT
1
nb ) , da cui si vede bene che dal calcolo è
V
emerso un termine di pressione aggiuntivo, termine che deriva dall’integrazione
sullo spazio esterno alla molecola dove le forze sono attrattive. Il gas è soggetto
dunque ad una pressione totale che deriva parzialmente dalle pareti, che è quella
che misuriamo con un manometro, e parzialmente dall’attrazione delle altre
molecole che si oppongono così all’espansione del gas. Evidentemente il termine
nRT
1
nb deriva dall’integrazione sullo spazio della molecola dove le forze sono
V
repulsive e dunque ha a che fare con il volume della molecola e la sua
incompressibilità.
9. Resistenza del mezzo all’avanzamento di un corpo
Abbiamo visto che nel caso di una sfera di raggio r che si muova in un gas di
viscosità data da η, la resistenza dello stesso all’avanzamento è data dalla legge di
Stokes: F = − 6πη rv e abbiamo usato questa formula nell’esperimento di Millikan,
dove le goccioline d’acqua sono quasi esattamente delle sfere a causa delle forze
di coesione interna. Questa formula vale solo a bassa velocità. A più alta velocità
la resistenza è data da una formula dovuta a I. Newton: F = −CρSv , in cui C è
un coefficiente dipendente dalla forma del corpo, S è la sezione trasversale e ρ è
la densità del fluido in cui si muove il corpo. La giustificazione data da Newton è
approssimativa ed è basata sul fatto che la forza esercitata dal fluido è
proporzionale alla variazione della quantità di moto ceduta dal corpo al fluido e
quindi è data dalla massa di fluido spazzato in un secondo dal corpo ( S ρ v )
moltiplicata per la velocità v .
Possiamo cercare di calcolare la resistenza all’avanzamento del corpo nel fluido
dovuta alle collisioni molecolari, ora che abbiamo a nostra disposizione la teoria
cinetica dei gas.
2
v0
79
−vx
X
vx
Cap. 5 – Termodinamica e calorimetria
Abbiamo visto che sulla parete di un contenitore la pressione esercitata dal gas è
dF N
2πd (− cosθ )
= f (v)dv
2mvcosθv cosθ , ovvero:
data dall’espressione: dP =
dS V
4π
dP =
dF
N
= ρ v2 f (v)dv cos2 θ d (− cos θ ) , con ρ =
m ; abbiamo integrato su
dS
V
ϕ e
semplificato un fattore 2. La precedente formula è stata stabilita in un sistema in
cui il gas (ovvero il suo contenitore) era fermo. Adesso pensiamo ad una
superficie piatta che si muove nel gas. La formula precedente vale ancora,
premesso che si usi per la velocità v ' = v + v0 ,in cui la velocità v0 è la velocità
della superficie rispetto al mezzo in cui si muove. Allineeremo la velocità v0 con
l’asse X. Dunque abbiamo:
dF
dP =
= ρ v '2 f ( v ) dv cos 2 θ d ( − cos θ ) = ρ v x '2 f ( v ) dvd ( − cos θ )
dS
Abbiamo:
v'x = vx −v0 ,
v'y = vy ,
v'z = vz .
La
funzione
2
f (v) = 2π (
m 3 − 2mvkT 2
) e v dv va allora posta nella precedente formula:
2π kT
m(v2
−
m 3
dP = 2π (
) ρ vx '2 e 2 kT dv ' v 2 d (− cos θ ) . Normalmente il fattore
2π kT
davanti alla radice quadrata sarebbe 4π non 2π ; tuttavia occorre tenere conto
del fatto che le particelle che hanno un certo modulo della velocità, ma si
allontanano dal disco non contano: questo abbassa la probabilità di avere un certo
modulo della velocità e di muoversi nel semispazio giusto di un fattore 2. Per
ottenere la pressione totale, adesso integriamo:
PS =
m 3
(
) ρ
2π kT
+∞
π
+∞
∫ v' ∫e
2
0
−
mv 2
2 kT
v 2 dv 2π d ( − cos θ ) =
0
π
2
mv
−
m 3
2
2
= (
) ρ ∫ v ' cos θ ∫ e 2kT v2d Ωdv =
2π kT
0
0
80
Cap. 5 – Termodinamica e calorimetria
=
+∞
2
mv
−
m 3
(
) ρ ∫ ( v x + v0 ) 2 e 2 kT dv x dv y dv z =
2π kT
−∞
+∞
+∞
+∞
ma
ma
+∞ 2 − ma
−
−
m
2
2 kT
2 kT
=
ρ ∫ vx e dvx + 2v0 ∫ vx e dvx ∫ +v0 ∫ e 2kT dvx =
2π kT −∞
−∞
−∞
−∞
2
2
2
1
= ρ v2 + 2v02 . Si tenga conto del fatto che
3
dispari),
che
=
+∞
∫
−∞
m
2π kT
statica
+∞
∫ve
2
x
−
m ( v x − v0 ) 2
2 kT
dv x = v x2 =
−∞
che
mv 2
+∞
y
−
m
e 2 kT dv y = ∫
2π kT
−∞
dipende
+∞
∫ve
x
−
ma 2
2 kT
dv x = 0 (la funzione è
−∞
2
mv
− z
m
e 2 kT dv z = 1
2π kT
e
che
1 2
v . Il risultato ci dice che c’è una pressione
3
dalla
velocità
quadratica
media(
1 2 kT
RT Nm RT nNAm nRT
v ρ= ρ=
=
=
delle molecole e una
3
m
NAm V NAm V
V
pressione che dipende dal quadrato della velocità ( v0 ) con cui si muove il disco
nel gas e quindi una forza resistente pari a
accordo col risultato di Newton.
R = −ρ Sv02 sulla superficie S , in
9. Il primo principio della termodinamica
Abbiamo cominciato dunque a vedere che, nel quadro dei fenomeni legati alla
temperatura e al calore, l’energia delle molecole sembra svolgere un ruolo
importantissimo. In effetti, il primo principio del la termodinamica è il principio
generalizzato di conservazione dell’energia, principio esteso ai sistemi
termodinamici e includente tanto il calore che il lavoro fatto dal sistema.
Cominciamo da quest’ultimo.
Si consideri un cilindro con un pistone mobile (fig. 8), senza attrito. Il cilindro
contenga un gas perfetto a pressione P. Il volume è indicato da V. Si può rilasciare
il pistone e il gas si espanderà contro la pressione inferiore dell’atmosfera o contro
una forza esterna. Si calcoli il lavoro fatto dal pistone. Secondo la definizione data
al Cap. 2, il lavoro infinitesimo è: dL = F ⋅ d s . Poiché la forza esercitata contro il
81
Cap. 5 – Termodinamica e calorimetria
pistone è PdS , perpendicolarmente al pistone e dunque nella stessa direzione
dello spostamento infinitesimo ds , il lavoro sarà dL = PdSds= PdV .
Si può a questo punto integrare, ma si deve stare attenti a come effettuare questa
integrazione: che tipo di trasformazione sta avvenendo nel gas?
Per esempio, può accadere, ed è il caso più semplice, che la trasformazione dal
volume iniziale a quello finale, avvenga a pressione costante. In questo caso
l’integrazione è banale e dà: L = P(V − V0 ) . Tuttavia la trasformazione può
avvenire in molti altri modi.
Consideriamo il piano P-V. Ogni punto in questo piano rappresenta uno stato
possibile del nostro gas ed una curva rappresenta la sequenza di stati
successivamente assunti dal gas, ovvero, la sua evoluzione.
P, V
Fig. 8: Cilindro con pistone mobile.
In fig. 9, è rappresentata l’espansione a
temperatura costante di un gas perfetto.
Nello stesso grafico, la retta orizzontale
rappresenta un’espansione a pressione
costante.
Il lavoro fatto durante l’espansione è
l’area sotto la curva, limitata dal volume
iniziale e da quello finale.
Si faccia il calcolo del lavoro prodotto in una trasformazione isoterma da un gas
che si espande da un volume iniziale Vi ad un volume finale Vf:
Vf
L=
∫
Vi
22
Vf
∫
pdV = nRT
Vi
Vf
dV
=nRT ln
, utilizzando l’equazione dei gas perfetti22.
V
Vi
Si noti che il lavoro su una isoterma per un gas che obbedisce all’equazione di Van der
Waals è: L = nRT ln
V f − nb
Vi − nb
+ an2 (
1 1
− ) . Il primo termine cambia poco, ma si trova
V f Vi
un secondo termine che rappresenta il lavoro fatto contro le forze d’attrazione
intermolecolari. In un’espansione contro forze esterne nulle il primo termine sarà zero, ma
il secondo no. L’energia interna, somma di cinetica e potenziale, non cambia, ma quella
cinetica si ridurrà e dunque per un gas reale, in un’espansione contro il vuoto si osserverà
82
Cap. 5 – Termodinamica e calorimetria
Durante un’isobara, il lavoro sarà:
L = p (V
f
− Vi )
.
È possibile naturalmente riscaldare o raffreddare il gas, cioè fornire o togliere
delle quantità di calore dQ al gas durante l’espansione o compressione.
2
P (N/m )
10000
8000
Fig. 9: Isoterma ed isobara di un gas perfetto.
6000
4000
2000
0
0
2
4
6
8
10
3
V(m )
Si assegni al lavoro fatto dal sistema sull’esterno il segno positivo, ed il segno
negativo al lavoro fatto sul nostro gas dall’esterno. Si convenga pure di assegnare
il segno positivo al calore ceduto al gas e quello negativo al calore sottratto al gas.
La questione che si deve affrontare a questo punto è che cosa sia effettivamente il
calore. La risposta a questa domanda fu trovata da Joule, il quale si rese conto che
il calore non era altro che energia in transito dal sistema o verso il sistema e riuscì
a misurarne il rapporto con l’energia meccanica, il cosiddetto “equivalente
meccanico della caloria”: 1caloria = 4,18 Joule .
Tutti sappiamo che, gonfiando un palloncino od un pneumatico, si verifica un
riscaldamento del gas. Questo va interpretato nel senso che il lavoro meccanico
che facciamo per comprimere il gas non è scomparso, ma si ritrova sotto la forma
di calore.
Possiamo per esempio far cadere un oggetto dalla scrivania al pavimento. Sulla
scrivania l’oggetto ha energia potenziale. Appena prima di colpire il suolo questa
comunque una riduzione della temperatura a differenza di un gas perfetto. Vedi
dipendenza dell’energia interna dal volume più avanti.
83
Cap. 5 – Termodinamica e calorimetria
energia potenziale si è trasformata in energia cinetica: dopo l’urto però l’oggetto è
fermo sul pavimento. Dove è finita l’energia? Sembra scomparsa, ma ci
potremmo accorgere che, in effetti, una parte dell’energia ha deformato o rotto
l’oggetto, ma una parte è servita a scaldarlo.
Pensiamo allora a fare cadere un peso, ma in modo tale che l’energia potenziale
del peso si trasformi in energia cinetica e che questa scaldi un liquido in un
calorimetro, in modo tale da poter misurare esattamente quanto calore si è
sviluppato. Poiché il lavoro fatto dalla forza di gravità è noto, allora dalla misura
del calore sviluppato si potrà stabilire il rapporto d’equivalenza tra energia e
calore. Rimanendo questo rapporto costante in ogni esperimento di conversione
del lavoro in calore, se ne deduce che il calore è effettivamente una forma di
energia.
Acqua
P=mg
Fig. 10: Esperimento di Joule.
A questo punto si ricordi che – come è stato detto precedentemente - dal punto di
vista atomico il gas ha una certa energia interna, pari alla somma delle energie
cinetiche di tutte le molecole che lo compongono. Ci si può subito rendere conto
allora, della possibilità di stabilire un principio generalizzato di conservazione
dell’energia, utilizzando come parti la cui si somma si conserva il lavoro fatto da
o sul gas, e l’energia interna alla quale si va ad aggiungere o sottrarre il calore che
viene dato o ceduto dal sistema. In conclusione, si avrà:
84
Cap. 5 – Termodinamica e calorimetria
− L + Q = ∆U
Questa eguaglianza, lavoro più calore uguale alla variazione di energia interna,
che esprime il principio generalizzato di conservazione dell’energia, costituisce il
“primo principio delle termodinamica”.
Torniamo adesso all’esperimento di Joule.
Dalla fig. 10, si vede bene quale sia l’idea del mulinello di Joule. Il peso cade e fa
girare l’asse; le alette attaccate all’asse agitano l’acqua che deve passare per lo
stretto passaggio tra le alette mobili e quelle fisse saldate al contenitore del
calorimetro. L’acqua si scalda e si può misurare la quantità di calore sviluppata
dall’agitazione dell’acqua direttamente dalla variazione di temperatura dell’acqua.
L’energia che è andata a scaldare l’acqua è: E = mgh , con h pari all’altezza di
caduta del peso. L’equivalente meccanico della caloria J risulta di 4,18Joule/cal,
come già detto. Dal punto di vista molecolare è molto facile capire che succede
quando si comprime un gas e questo si scalda. Prendiamo il solito sistema
cilindro-pistone. Pensiamo il pistone in moto, con velocità v0 , mentre comprime
il gas. La componente della velocita lungo l’asse di movimento nel sistema del
pistone è :
vx + v0 . Dopo l’urto la molecole si allontana dal pistone colla stessa
velocità rispetto al pistone e perciò con velocità vx = vx + 2v0 rispetto al cilindro.
La
variazione
di
energia
cinetica
sarà
pertanto:
v
1
1
m(vx + 2v0 )2 − mvx 2 = 2mvx v0 + 2mv02 = 2mv0vx (1 + 0 ) ≈ 2mv0vx
per
2
2
vx
v0
<< 1. Aumento di energia cinetica vuol dire aumento di temperatura come
vx
sappiamo. Stesso se facciamo espandere il gas da cui segue un raffreddamento.
Vediamo alcune applicazioni importanti. Come sappiamo il calore specifico è la
quantità di calore necessaria a fare aumentare la temperatura della massa unitaria
di una sostanza di un grado. Particolarmente per i gas, però, questa definizione va
completata specificando sotto quali condizioni avviene il riscaldamento. E’
evidente infatti che, se il riscaldamento avviene a volume costante, non vi è lavoro
fatto sull’esterno dal gas in espansione. Se, al contrario, il riscaldamento avviene a
pressione costante, allora si verifica un aumento di volume e dunque viene fatto
del lavoro verso l’esterno.
Possiamo così definire due calori specifici, uno a pressione costante c P , ed uno a
volume costante c v . Quale sia la relazione tra questi due calori specifici, lo
possiamo stabilire usando il primo principio della termodinamica e l’equazione
85
Cap. 5 – Termodinamica e calorimetria
dei gas perfetti. Riscaldiamo il nostro gas a pressione costante e per il primo
principio abbiamo: dL + dU = PdV + dU = δQ . Si divida tutto per la variazione di
δQ
dV dU
+
= ( ) P =const . Dall’equazione dei gas si ha:
temperatura dT : P
dT
dT
dT
PdV + VdP = PdV = nRdT .
Prendiamo n =1 (si parlerà quindi di calori molari Cp e CV), per cui: P
Sostituendo: R +
CP −
dV
= R.
dT
δQ
dU
= ( ) P =const . Il termine a destra è ovviamente C p :
dT
dT
dU
dU
= R . Del resto
dT
dT
è il calore specifico a volume costante, infatti,
riscaldando il gas a volume costante, non c’è lavoro sull’esterno e la quantità di
calore ceduta al gas va interamente ad aumentare l’energia interna: dunque il
calore specifico a volume costante non è altro che la variazione dell’energia
interna per variazione di grado di temperatura. Per cui risulta che la differenza tra
i calori molari è uguale alla costante dei gas: C P − C V = R (relazione di Meyer).
Si può adesso fare anche il caso di una trasformazione a volume costante - nessun
lavoro viene fatto sull’esterno - e ottenere dal primo principio che dQ = dU . Si
δQ dU 3 kN A dT 3
dT
CV =
=
=
= R(
divida
per
e
si
ha
che:
dT
dT
2
dT
2
( = 1 2, 4 7 1 J / ( m o le ⋅ K ) ) = 2, 982cal / ( mole ⋅ K ) ), dove l’ultimo passaggio è
giustificato dal fatto che in una mole di gas ci sono esattamente NA molecole e che
3
3
kT : U = N A kT . Questa formula ignora
2
2
l’esistenza di gradi di libertà aggiuntivi oltre quelli traslazionali e pertanto vale
solo per gas monoatomici per i quali: γ = 1,67 . Possiamo anche scrivere:
ciascuna ha un’energia media ε =
γ =
CP
1
1
f +2
, con
= ( fR + R ) / ( fR ) =
CV
2
2
f
f
numero dei gradi di libertà. Nel
calcolo dei calori molari il principio di equipartizione gioca, dunque, un ruolo
essenziale23
Si noti che per un gas biatomico esistono tre gradi di libertà oltre quelli relativi al
moto del baricentro della molecola. Prendiamo la retta che unisce i centri dei due
atomi: la sua direzione è definita da due angoli , θ e φ , Inoltre le due molecole
23
Come si vede, lim γ = 1 . Ora gli atomi hanno molti livelli, come è testimoniato dalle
f →∞
righe ottiche, dunque dovrebbero avere γ ≈ 1 , Ciò non risulta vero, perché il contributo di
questi livelli è trascurabile a causa del fatto che difficilmente essi possono essere eccitati.
86
Cap. 5 – Termodinamica e calorimetria
possono oscillare lungo quest’asse. Considerando in aggiunta solo i due gradi di
5
libertà θ e φ , l’energia media della molecola passa a kT . Per il sesto grado di
2
libertà, esisterà non solo un’energia cinetica, ma anche un’energia potenziale:
ciascuna contribuisce un termine
diventa così
1
kT per un totale di kT .Il calore specifico
2
7
R . Consideriamo la formula quantistica dell’oscillatore armonico:
2
2
1
Kθ
ε = hν + θ , con θ = hν . Il contributo al calore molare sarà dU = R θ / T ,
θ
2
k
dT
T
eT
e −1
contributo che passa da zero a T = 0 ad R per T → ∞ . Pertanto il calore specifico di
un gas biatomico sarà uguale a
5
7
R a bassa temperatura e a R ad alta temperatura.
2
2
(grafico da Wikipedia)
Un’importante categoria di trasformazioni sono le trasformazioni adiabatiche:
Esse sono le trasformazioni durante le quali non ci sono scambi di calore con
l’esterno: dQ=0. Per queste trasformazioni si può stabilire un’importante
equazione. In una trasformazione adiabatica si ha: dL = −dU , cioè:
PdV = − dU = − C V dT . È possibile eliminare la variazione di temperatura usando
l’equazione dei gas:
87
Cap. 5 – Termodinamica e calorimetria
PdV + VdP = RdT = (C P − C V ) dT = −C V (1 −
CP
) dT = − C V (1 − γ ) dT = − dU (1 − γ ) ,
CV
dove si è indicato con γ il rapporto tra i due calori molari.
Utilizzando il primo principio, si sostituiscano:, dL=PdV=-dU. Dunque si avrà:
Valori di γ da confrontare con le predizioni del primo principio (da J. Jeans An
introduction to the kinetic theory of gases, 1962 pag. 279)24.
Si osservi l’ottimo accordo tra il valore di γ sperimentale e quello teorico per i gas
monoatomici. In effetti l’accordo è tanto buono che appare evidente che gli unici gradi di
libertà che hanno “assorbito” energie sono quelli traslazionali. In ogni caso non c’è
nessuna traccia di energia da associare ai gradi di libertà interni. Per esempio, sappiamo
che l’elio è formato da un nucleo e due elettroni: è immaginabile che a seguito delle
collisioni, l’energia degli elettroni dovrebbe aumentare, cioè una parte delle collisione
dovrebbe essere anelastica. La spiegazione di questo strano fatto venne solo con la
88
24
Cap. 5 – Termodinamica e calorimetria
PdV + VdP = (1 − γ )PdV ⇒ VdP = −γPdV ⇒
relazione,
si
ottiene
finalmente:
ln
dP
dV
= −γ
.
P
V
Integrando
P
V
= ln( ) −γ ,
P0
V0
cioè:
questa
PV γ = costante.
Utilizzando l’equazione dei gas si ottiene anche: TV γ −1 = costante e P1−γ T γ =
costante.
Torniamo adesso al problema di determinare l’andamento della pressione in
funzione dell’altitudine sul livello del mare. Riprendiamo l’espressione
dP = − gρ ( h) dh dal paragrafo precedente e supponiamo che la colonna d’aria sia
adiabatica, cioè non scambi calore con l’aria adiacente.
È vero, infatti, che l’aria è un cattivo conduttore di calore. Dall’equazione appena
stabilita sulle trasformazioni adiabatiche, risulta evidentemente che:
Pρ −γ = P0 ρ0−γ ,
sostituendo
1
Integrando:
∫x
precedente,
abbiamo:
−
P γ
P
) ρ 0 dh ⇒ x γ dx = −αdh , dove: α = g ρ 0 / P0 e x =
P0
P0
dP = − gρdh = − g (
x
nell’espressione
1
−1
γ
dx =
1
γ
−1
x
γ −1 ∫1
d (x
γ
+1
h
∫
) = − α dh , si ottiene infine:
0
γ
P = P0 (1 − αh
1 γ − 1 ρ0
γ −1 γ −1
) che, posto:
g ⇒ h0 = 2, 7 ⋅ 104 m , ( γ = 7 )
=
γ
h0
P0
γ
5
dà:
meccanica quantistica e il riconoscimento che le energie interne sono quantizzate e grandi
rispetto a quelle scambiate nelle collisioni.
89
Cap. 5 – Termodinamica e calorimetria
γ
P ( h) = P0 (1 −
7
h γ −1
h
2
) = P0 (1 −
)
2, 7 ⋅ 104
h0
F1
100000
Pressione (Pa)
80000
60000
40000
20000
0
0
2000 4000 6000 8000 10000 12000 14000 16000 18000 20000
Altitudine (m)
1
5
h
h
2
)
ρ ( h ) = ρ 0 (1 − ) γ −1 = ρ 0 (1 −
h0
2, 7 ⋅ 10 4
3
Densità (kg/m )
F1
0
0
5000
10000
15000
20000
25000
30000
Altitudine (m)
90
Cap. 5 – Termodinamica e calorimetria
T (h) = T0 (1 −
h
h
) = T0 (1 −
)
h0
2, 7 ⋅ 104
F2
300
250
Tempertura (K)
200
150
100
50
0
0
5000
10000
15000
20000
25000
30000
Altitudine (m)
−γ
−γ
Le ultime due equazioni si ottengono usando Pρ = P0 ρ0 e l’equazione dei gas
perfetti. Da notare che h 0 è il valore di h per cui la pressione, la densità e la
temperatura si annullano, non il valore di h per cui P = P0 .
Un’applicazione simile si ha nel caso del gas che circonda un buco nero o una
stella di neutroni. L’interesse di questo problema sta nel fatto che nello spazio
interstellare esiste un gas di protoni con una densità corrispondente ad un protone
a metro cubo. In prossimità di un buco nero, il gas si condensa e si riscalda
raggiungendo temperatura molto grandi presso il raggio di Schwartzschild (pag.
156 delle dispense). In tali condizioni, il gas emette raggi X e γ . Un astronomo
terrestre che guardi nella direzione giusta del cielo, vedrà un punto da cui
provengono raggi X e γ , senza una sorgente luminosa (stella). Data la massa del
buco nero si possono calcolare i valori di pressione, temperatura e densità del gas.
Una situazione del genere è stata trovata negli anni ’70 nella costellazione del
Cigno (Cygnus X1) dove una stella visibile ruota intorno ad una compagna
invisibile con una massa pari a sei volte quella solare (non può essere né una nana
bianca, né una stella di neutroni). Quel punto è una sorgente di raggi X.
Assumendo che la massa dominante si quella del buco nero, abbiamo una g non
costante:
91
Cap. 5 – Termodinamica e calorimetria
g =G
M
h2
,
invece
dP = −G
differenziale:
g = 9 ,81 m / s 2
di
M
h
2
.
Sostituendo,
ρ ( h ) dh = −GM ρ ( h )
dh
h2
,
otteniamo
che
l’equazione
possiamo
integrare
−γ
γ
−
facilmente, utilizzando la relazione adiabatica: Pρ = P0 ρ0 .
1
1
−
P
dh
GM
dh
dP = − GM ( ) γ ρ 0 2 ⇒ x γ dx = −
ρ 0 2 , con x = P / P0 . Integriamo
P0
h
P0
h
e abbiamo:
GM 1
x − 1) =
ρ
(
P
h
γ −1
γ
γ −1
γ
0
, da cui: x
γ −1
γ
= 1+
0
γ − 1 GM 1
ρ
e,
γ P0 0 h
γ
γ
γ −1
γ −1
γ − 1 GM
γ − 1 GM
1
1
ρ0 ⇒ P = P0 1 +
ρ0 . Posto
infine, x = 1 +
h
h
γ P0
γ P0
γ − 1 GM
h0 =
ρ :
γ P0 0
γ
h γ −1
abbiamo la risposta: P = P0 1 + 0 .
h
Volendo, possiamo completare l’esercizio, calcolando la densità e la temperatura
γ
−γ
−
del gas in funzione della distanza dal buco nero h . Dalla relazione Pρ = P0 ρ0 ,
P
h
sostituendo la pressione in funzione di h , abbiamo: ρ = ρ 0 ( )1 / γ = ρ 0 1 + 0
P0
h
1
γ −1
e, dall’equazione dei gas perfetti: T = PV = A P = A P0 1 + h 0 .
nR
R ρ
R ρ0
h
Importanti sono le precedenti considerazioni nell’applicazione allo studio della
struttura delle stelle. Si esamini il caso di una massa gassosa, autogravitante,
soggetta cioè alla propria gravitazione. Evidentemente la massa tenderà a
contrarsi, cioè a ridursi di volume sotto l’attrazione della propria gravità; così
facendo però si comprimerà e si riscalderà generando una pressione interna che
tenderà a bilanciare la gravitazione. Fu proprio questo, in effetti, il meccanismo
proposto da Helmholtz, a metà dell’Ottocento, per spiegare l’emissione della luce
da parte del Sole e delle stelle: il lavoro fatto dalla gravitazione comprime il gas
delle stelle e lo riscalda al punto da fargli emettere luce. Si trovò che questo
92
Cap. 5 – Termodinamica e calorimetria
meccanismo non bastava a spiegare la lunga vita del Sole∗, che venne valutata da
Lord Kelvin intorno a 20 milioni d’anni. Solo nella prima metà del secolo appena
finito, si comprese che il meccanismo aveva a che fare con le leggi della fisica
nucleare. La compressione del gas da parte della gravitazione innalza la
temperatura a valori tali da far si che si inneschino delle reazioni di fusione di
nuclei leggeri (H e He). Questo in quanto, ad alte temperature (107K), le energie (
3
ε = kT ) dei nuclei sono occasionalmente tali da superare la repulsione
2
coulombiana (cioè sono sulla “coda” della distribuzione di Boltzmann), anche, in
verità, con l’aiuto dell’effetto “tunnel” scoperto da G. Gamow per spiegare il
decadimento alfa (vedere il capitolo sulla meccanica quantistica). Queste reazioni
producono calore che bilancia l’attrazione gravitazionale, prevenendo il collasso
della stella e dandole una vita di miliardi di anni, invece che di centinaia di
milioni di anni∗∗.
Consideriamo una massa gassosa a simmetria sferica (fig. 11). Lo strato di
molecole nella corona sferica compresa tra il raggio r e r+dr, verrà attratta dalla
massa all’interno della sfera di raggio r, M(r), in accordo con la legge della
gravitazione universale.
r+dr
F
r
M(r)
ρ(r)
In un certo senso, questa corona sferica è
il coperchio del contenitore sferico di un
gas, coperchio che comprime il gas, non
perché spinto verso il centro dall’esterno,
ma perché attirato dalla massa dentro il
contenitore. Naturalmente sarà:
Fig.11: Massa gassosa a simmetria
sferica.
∗
La vita del Sole doveva essere almeno uguale a quella della Terra e la vita della Terra era
valutata a molto più di 20 milioni d’anni dai geologi che la stimavano dai tempi di
formazione delle rocce. Inoltre 20 milioni d’anni era una stima incompatibile con la teoria
dell’evoluzione di Darwin.
∗∗
Per un’interessante storia degli sviluppi dell’astrofisica dagli inizi, vedere il Cap. 7 di A.
Masani, Astrofisica, Editori riuniti (1984). Nel testo di fisica Corso di fisica medica di
Oswaldo Casali (1878), si riportava la teoria che il riscaldamento del Sole fosse dovuto
alla continua caduta di asteroidi sulla sua superficie (si veda l’esercizio 9 al Cap. 2).
93
Cap. 5 – Termodinamica e calorimetria
r
M (r ) =
∫ 4πz ρ (z)dz ,
2
con
ρ
0
densità del gas. La forza che la corona
sferica esercita sull’interno sarà quindi:
dF = − g ( r ) ρ ( r ) dV = −G
M (r )
r
2
4πr 2 ρ ( r ) dr = −G
M (r )
r2
ρ ( r ) Sdr , per cui la pressione
sarà:
r
∫
M (r )
dP
1
= − g (r ) ρ (r ) = −G 2 ρ (r ) = −G 2 ρ (r ) 4πz 2 ρ ( z )dz .
dr
r
r
Usando
l’equazione
0
Pρ
−γ
= P0 ρ0−γ
,
possiamo
scrivere
r
dP
1
= −G 2 ( ρ 0 P0−1/ γ ) 2 P1/ γ ( r ) ∫ 4π z 2 P1/ γ ( z ) dz . Se risolviamo questa equazione
dr
r
0
integro-differenziale (almeno numericamente) otteniamo la funzione P (r ) , che
dipende dai parametri ρ 0 e P0 , e le due altre funzioni: ρ ( r ) = ρ 0 (
T ( r ) = T0
P (r ) 1 / γ
)
P0
e
A P (r )
.
R ρ (r )
Imponendo che a r = R S (=raggio della stella), si abbia ρ ( R S , ρ 0 , P0 ) = 0 e che:
MS =
∫ ρ (r )dV (= massa della stella), troviamo due equazioni che ci consentono
V
di esprimere pressione e densità al centro della stella in funzione della massa e del
raggio della stella stessa.
Studiando questa equazione, si raggiunge la conclusione che vi sono due
parametri liberi per ogni soluzione, parametri che possono essere scelti come la
massa M e raggio R della stella. Ne segue che data una massa, il raggio e dunque
la grandezza della stella possono cambiare e questo fa sì che la stella possa
evolvere eventualmente contraendosi lentamente mano a mano che si esaurisce il
combustibile nucleare. Se ha una massa piccola (meno di 1,4 volte la massa
solare), la stella si riduce ad una nana bianca. Se invece la sua massa è maggiore,
essa è destinata a divenire una stella di neutroni. Per masse maggiori a circa 2
volte la massa solare, la stella diverrà un “buco nero”.
94
Cap. 5 – Termodinamica e calorimetria
Un altro caso interessante si ha se: ρ = cost . È il caso dei pianeti. Per esempio, si
scelga Giove. La densità non è esattamente costante, ma cambia poco: quella
media è 1,3 g / cm 3 , quella massima è: 4 ,3 g / cm 3 e, dunque, possiamo considerarla
4 3
2
costante. La soluzione sarà: M (r) = πr ρ e P ( r ) = P0 − π G ρ 2 r 2 . Per
3
3
r = RS
4
3
si ha: 0 = P0 − G ρ ( πρ RS )
3
Gρ MS
1
= P0 −
Da cui si vede che la
2 RS
2 RS
pressione centrale P0 , determinata dal raggio R S = 7 ⋅ 10 7 m e dalla densità o,
alternativamente, dalla massa totale M S = 2 ⋅ 10 27 kg , risulta essere dell’ordine di
10 7 Atm . Questa pressione determina un’energia media al limite di quella
necessaria per ridurre il materiale di Giove in gas ionizzato (plasma). Se, in altre
parole, la sua massa fosse stata un po’ più grande, Giove sarebbe divenuto una
stella, il sistema solare avrebbe una stella binaria come si osserva in altri casi, per
esempio la splendida Sirio ha una compagna Sirio B, molto meno luminosa.
La ricerca di buchi neri e stelle di neutroni si persegue cercando sorgenti di raggi
X, come spiegato prima.
10. Il secondo principio della termodinamica
Durante il diciottesimo secolo, nel Regno Unito ed in Francia, così come in tutto
il resto d’Europa, si sviluppò un forte interesse per l’utilizzo del calore nella
realizzazione di motori da applicare ai trasporti (treni e navi), ma anche per altri
sistemi, come pompe ecc... Il primo motore a vapore fu sviluppato da T.
Newcomen (1663-1729) e dal suo assistente Calley per prosciugare, azionando
una pompa, l’acqua che ristagnava sul fondo delle miniere. La loro prima
macchina risale al 1716.
Gli sviluppi successivi sono dovuti a J. Watt (1736-1819) e sono del 1769.
Occorre dire che, sin dai tempi della classicità alessandrina, i greci costruirono
semplici macchine che producevano lavoro a partire dal calore (eolipila di Erone).
Sulla spinta di questi interessi tecnico-pratici, si pose il problema di quale fosse la
maniera migliore di sfruttare il calore ed, in particolare, se esistesse un limite, più
stringente di quello imposto dalla conservazione dell’energia, sul suo
sfruttamento.
Il secondo principio della termodinamica è proprio relativo a questo limite e, pur
essendo inizialmente nato come affermazione sui limiti di sfruttamento del calore,
divenne poi un principio con conseguenze cosmologiche.
La costruzione di una macchina che utilizzava il calore, passò attraverso la
scoperta e l’uso di caldaia, cilindro e pistone (probabilmente un’evoluzione della
pentola con coperchio) e vari marchingegni meccanici, come le ruote, le bielle, la
95
Cap. 5 – Termodinamica e calorimetria
distribuzione, ecc... Cilindri e pistoni devono tornare al punto di partenza dopo un
giro completo della ruota cui sono connessi dalla biella, altrimenti sono
inservibili: dunque occorrono macchine cicliche, cioè tali che il gas o vapore
incluso nel cilindro si espanda sì, ma torni poi a contrarsi e a riacquisire la
configurazione iniziale. Parliamo di macchine cicliche, macchine, cioè, nelle quali
il gas effettua un ciclo completo.
Si deve a questo punto fare una breve digressione e definire che cosa si intende
per trasformazione, dunque anche per trasformazione ciclica, o ciclo, reversibile.
Per trasformazione reversibile, si intende una trasformazione il cui ciclo possa
realizzarsi in un senso, ma anche nel senso opposto.
La grande differenza tra trasformazioni reversibili e non reversibili sta nel fatto
che in una trasformazione reversibile tutte le variazioni sono infinitesime, mentre
in quelle irreversibili sono finite. Per far passare calore da una sorgente ad un
corpo, per esempio un gas, abbiamo bisogno di differenze finite di temperature tra
corpo e sorgente.
In una trasformazione reversibile, possiamo immaginare di ridurre piccole a
piacere le differenze di temperatura e di rendere enormemente lungo il tempo
occorrente per gli scambi di calore. Con differenze nulle però, il senso della
trasformazione diventa meno ovvio: il calore potrà fluire dalla sorgente al gas, ma
potrà anche essere fatto fluire all’inverso dal gas alla sorgente.
Se prendiamo il piano P-V che abbiamo già usato per calcolare il lavoro fatto da o
su un gas che cambia di volume, un ciclo sarà rappresentato da una curva chiusa,
come in fig. 12, dove il gas partito da uno stato, rappresentato dal punto A, vi
ritorna dopo un ciclo.
Di cicli se ne possono
A
immaginare quanti se ne
P
vogliono,
comprendenti
isoterme, espansioni a volume o
pressione costante, espansioni
adiabatiche.
Tali
cicli
rappresentano, in modo più o
meno
idealizzato,
il
funzionamento delle macchine a
dei
motori
a
V vapore,
combustione interna e di varie
macchine ideali.
Fig. 12: Ciclo di un gas sul piano P-V.
La rappresentazione che si
utilizza qui prescinde da attriti e
da altre miserie pratiche: si vuole
96
Cap. 5 – Termodinamica e calorimetria
capire infatti quali sono i limiti
di utilizzo della macchina.
Gli attriti potranno poi sempre essere ridotti, migliorando i materiali od il sistema
di lubrificazione. Dunque alla macchina a vapore corrisponderà il ciclo teorico di
Rankine, a quella a benzina il ciclo Otto, ecc... Un parametro critico sarà il
rendimento η della macchina: quantità che indica quale frazione del calore
disponibile è stata trasformata in lavoro utilizzabile. La questione è molto
rilevante, come sappiamo bene. Paghiamo il combustibile - legna, carbone,
petrolio...- che bruciando produce calore: delle calorie prodotte quante se ne
perdono? Una parte del calore va letteralmente in fumo: bruciando gas per
riscaldare una pentola d’acqua, la fiamma sotto il fondo della pentola la riscalda,
ma il gas che lambisce i fianchi della pentola e sale in alto non è a temperatura
ambiente e, dunque, porta via una parte del calore che abbiamo pagato. Questo
problema però rientra nel novero dei problemi pratici: è vero che una parte del
calore prodotto nella combustione viene perso ancora prima di entrare nella
macchina, ma, di quello che è stato assorbito dalla macchina, quale parte viene
effettivamente trasformato in lavoro? Il rendimento η dunque, rappresenta la
frazione di calore assorbita dal fluido termodinamico e trasformata in lavoro
meccanico. La distinzione tra calore ceduto al fluido termodinamico e prodotto
nella combustione, naturalmente, svanisce nel caso di motori a combustione
interna, dove è lo stesso fluido termodinamico a bruciare e dunque a riscaldarsi.
Questo meccanismo chiaramente evita certe perdite di calore e fa preferire le
macchine a combustione interna (motori a scoppio e motori Diesel) a quelle a
combustione esterna (macchine di Stirling, macchine a vapore). Diciamo anche
che il ciclo di una macchina a combustione interna è un ciclo solo in senso molto
lato: il fluido in realtà può bruciare una sola volta! Fatta questa premessa ci si
può domandare perché mai non si possa trasformare (sempre in un ciclo!) tutto il
calore assorbito dalla sorgente termica in lavoro: il primo principio dopotutto non
lo vieta. Il fatto è che nessuno ha mai ideato una macchina ciclica in grado di
trasformare tutto il calore assorbito in lavoro: c’è sempre una seconda fase nella
quale, per riportare il sistema allo stato iniziale, occorre assorbire lavoro e
soprattutto cedere del calore ad una sorgente fredda. Il gas di scarico di un motore
deve essere liberato nell’atmosfera per tornare allo stato iniziale. Dunque una
macchina ciclica viene sempre idealizzata come una macchina che opera tra due
temperature: quella di una sorgente calda a temperatura TC e quella di una
sorgente fredda a temperatura TF. Dalla sorgente calda la macchina preleva calore,
ne trasforma parte in lavoro meccanico e, per chiudere il ciclo, torna a cedere una
parte del calore alla sorgente fredda. Se le considerazioni precedenti sono vere, è
possibile enunciare il secondo principio della termodinamica, nella forma che gli
hanno dato Kelvin e Planck:
97
Cap. 5 – Termodinamica e calorimetria
Non tutto il calore ceduto da una sorgente può essere trasformato in lavoro
meccanico da una macchina ciclica.
Una macchina operante tra due sorgenti a diverse temperature, fu immaginata da
Sadi Carnot e descritta nel suo libro del 1824 Rèflexions sur la puissance motrice
du feu∗. Il ciclo è quello illustrato in fig. 15.
P
1
Q1 T1
2
4
Q2 T2
3
V
Fig. 15: Ciclo di Carnot.
Le due curve 1→ 2 e 3 → 4 sono
isoterme a temperatura T1 e T2: è a
queste temperature che il gas
assorbe ed emette il calore. Le altre
due curve sono adiabatiche.
Calcoliamo il rendimento della
macchina di Carnot. Il lavoro fatto
dal fluido termodinamico sarà pari
alla differenza tra il calore assorbito
e quello restituito alla sorgente
fredda. Dovendo infatti il fluido
avere la stessa energia interna
all’inizio ed alla fine del ciclo, il
lavoro può solo venire dal calore
assorbito, tolto, beninteso, quello
ceduto alla sorgente fredda.
L
Q
T
Poichè L = Q1 − Q2 , il rendimento sarà: η = 1 − 2 = 1 − 3→ 4 = 1 − 2
Q1
L1→ 2
T1
V3
)
V4
. Si
V
ln( 2 )
V1
ln(
può facilmente mostrare che il rapporto tra i logaritmi vale 1, usando le equazioni
dei gas perfetti e quella delle trasformazioni adiabatiche. Si ha:
1. p 1V1 = p 2 V 2
2.
3.
p2V2γ = p3V3γ
p 3V 3 = p 4 V 4
γ
γ
4. p1V1 = p4V4
Partendo dalla 3. e usando le altre abbiamo:
∗
Sadi Carnot, La potenza motrice del fuoco, Tessere, CUEN, 1996.
98
Cap. 5 – Termodinamica e calorimetria
p Vγ
V Vγ
V3
V
V
V
p
V
= 4 = ( 3 ) γ 1 1γ = ( 3 ) γ 2 1γ ⇒ ( 3 ) γ −1 = ( 2 ) γ −1 ⇒
V4
p3
V4
V
V
V1
p 2V 2
V1V 2
4
4
V3
V
) = (γ − 1) ln( 2 )
V4
V1
V
V
da cui in fine: ln( 3 ) / ln( 2 ) = 1 . Pertanto:
V4
V1
⇒ (γ − 1) ln(
η = 1−
T2
. Possiamo dimostrare la
T1
stessa cosa in un modo diverso: partiamo dalla relazione vera per le trasformazioni
adiabatiche:
deriviamola
e
otteniamo:
T 2V 3γ −1 = T1V 2γ −1 ,
γ −1
( γ − 1)T 2 V 3γ − 2 d V 3 = ( γ − 1)T1V 2γ − 2 dV 2 ⇒ ⇒ T2V3
Integrando si ha infine: ln
dV3
dV3 dV2
dV2
.
= T1V2γ −1
⇒
=
V3
V2
V3
V2
V3
V
= costante, in altre parole il rapporto 3 non cambia
V2
V2
spostando il punto V 2 lungo l’isoterma a T1 e dunque deve essere uguale a
Possiamo anche notare che la precedente relazione
V4
.
V1
dV3 dV2
si può riscrivere:
=
V3
V2
V2 dV3
T P dV
− 1 = 0 ⇒ 1 3 3 − 1 = 0 , usando l’equazione dei gas perfetti o infine:
V3 dV2
T2 P2 dV2
T1 P3 dV3
− T2 P2 = 0 .
dV2
Esiste un’altra maniera, equivalente, di enunciare il secondo principio della
termodinamica, dovuta a R. J. E. Clausius (1822-1888):
E’ impossibile realizzare una trasformazione il cui unico risultato finale sia di
trasferire calore da una sorgente più fredda ad una più calda.
Questo non vieta che si possa trasferire calore da una sorgente fredda ad una
calda: ogni frigorifero domestico fa appunto questo. Stabilisce, però, che, per
farlo, occorre effettuare del lavoro.
È facile vedere che l’enunciato di Kelvin–Planck e quello di Clausius sono
equivalenti. Per farlo occorre, però, illustrare il funzionamento di una macchina
frigorifera.
Si può far funzionare all’inverso la macchina di Carnot. Così facendo, il lavoro
cambia di segno: quello che veniva fatto sul mondo esterno è adesso assorbito dal
mondo esterno. La quantità di calore Q1 prima assorbita è adesso ceduta e
viceversa quella prima ceduta, Q2, è adesso assorbita. In conclusione, si avrà
utilizzo di lavoro fatto dall’esterno per trasferire una certa quantità di calore dalla
99
Cap. 5 – Termodinamica e calorimetria
sorgente fredda a quella calda. Una macchina frigorifera si potrà dunque
realizzare invertendo un qualunque ciclo reversibile. Il punto rappresentativo della
trasformazione si muoverà in senso antiorario invece che orario sul piano delle
variabili di stato25.
A questo punto si può dimostrare l’equivalenza dei due enunciati. Si dimostra
prima che, se l’enunciato di Kelvin–Planck è falso, lo è anche quello di Clausius.
Successivamente si dimostra che, se l’enunciato di Clausius è falso, lo è anche
quello di Kelvin–Plank. Di conseguenza, se uno è vero lo sarà anche l’altro.
Si utilizzi un sistema costituito da una macchina che produce lavoro tra due
sorgenti a temperatura T1 e T2. Tra le stesse due sorgenti si faccia funzionare una
macchina frigorifera.
Se l’enunciato di Kelvin–Planck è falso, è possibile far funzionare la macchina
ciclica & usando il calore Q estratto da una sola sorgente e producendo il lavoro
L=Q. Quindi la macchina frigorifera ' potrà, utilizzando il lavoro L, trasferire il
calore Q1 = L + Q 2 = Q + Q 2 dalla sorgente fredda a quella calda. Alla fine di un
ciclo, il sistema macchina più frigorifero saranno nello stesso stato ed una certa
quantità di calore sarà fluita dalla sorgente fredda a quella calda. Dunque
l’enunciato di Clausius è anch’esso falso.
Se, d’altra parte, l’enunciato di Clausius è falso, allora è possibile trasferire una
quantità di calore Q2 dalla sorgente fredda a quella calda, usando una macchina &
che usi lavoro zero. La macchina ' (fig. 16 b) potrà allora funzionare tra le due
sorgenti assorbendo la quantità di calore Q1 dalla sorgente calda e cedendo la
quantità di calore Q2 a quella fredda. Sarà quindi stato prodotto del lavoro ( a
sole spese della sorgente calda che ha ceduto la quantità di calore ) − ) , mentre
quella fredda ha prima ricevuto, ma poi ceduto la quantità di calore Q2. Dunque
anche l’enunciato di Kelvin – Planck è falso.
)
Fig. 16a. Se è falso l’enunciato
di Kelvin-Plank, è falso
l’enunciato di Clausius
&
(=)
'
)+)
25
)
Per una macchina frigorifera il rendimento (o coefficiente di performance COP) è il
rapporto η =
Q1
, cioè il rapporto tra calore estratto dalla cella frigorifera diviso per il
L
lavoro speso per estrarlo. Attenzione: può essere maggiore di 1.
100
Cap. 5 – Termodinamica e calorimetria
(
)
Fig. 16 b. Se è falso l’enunciato
di Clausius, è falso l’enunciato
di Kelvin-Plank
C
)
M
)
)
L’enunciato di Kelvin-Planck mette in evidenza come non sia possibile navigare
utilizzando il solo calore estratto dal mare, oppure volare sfruttando il calore
dell’aria. Occorre sempre una seconda sorgente fredda. Nelle parole di Carnot “La
produzione di potenza motrice è dovuta non ad un consumo reale di calore, ma al
suo trasporto da un corpo caldo ad un corpo freddo...Vedremo tra poco che questo
principio è applicabile ad ogni macchina messa in moto dal calore. Secondo
questo principio, non è sufficiente produrre calore per generare potenza motrice; è
necessario procurarsi del freddo: senza di esso il calore sarebbe inutile.” (La
potenza motrice del fuoco, pag. 10).
Il secondo principio della termodinamica vieta, in sostanza, il moto perpetuo di
una macchina che operi usando l’energia (cioè il calore) sottratta ad una sola
sorgente. Tale moto perpetuo non è vietato dal primo principio della
termodinamica che invece esclude solo che una macchina possa produrre
l’energia che le serve per funzionare.
L’enunciato di Clausius mette in evidenza come la tendenza spontanea del calore
sia quella di fluire dalla sorgente calda a quella fredda: se si vuole ottenere
l’inverso occorre spendere lavoro. Dunque esiste una tendenza naturale nei
processi termodinamici: mentre nella meccanica, abbiamo visto che l’inverso di
un processo è un processo egualmente realizzabile, si vede che invece in
termodinamica i processi hanno un verso spontaneo ben preciso.
Si può, a questo punto far vedere che tutte le macchine reversibili, operanti tra
due sorgenti a temperatura T1 e T2 e funzionanti secondo un ciclo costituito da
due isoterme ed altre due trasformazioni omologhe, rigenerative hanno lo stesso
rendimento dato dalla formula ormai familiare: η = 1 −
T2
(teorema di E.
T1
Reitlinger, 1873). Con l’aggettivo “rigenerativo” deve intendersi che le quantità di
calore scambiate nell’andare da una isoterma all’altra e viceversa sono uguali e
pertanto i lavori fatti nell’andare da un’isoterma all’altra e viceversa sono uguali,
giacché la variazione di energia interna nell’andare da T1 a T 2 o da T 2 a T1 ,
sono certamente uguali perché dipendono solo dal salto di temperatura. In tal caso
il calore scambiato in una trasformazione (per esempio nell’andare dalla
temperatura maggiore a quella minore) può essere conservato in una parte della
macchina (chiamata appunto “rigeneratore”) da cui può essere ripreso nella
101
Cap. 5 – Termodinamica e calorimetria
trasformazione inversa (nell’andare da quella a temperatura inferiore a quella
maggiore). La macchina funziona così solo con due sorgenti esterne.
LM − Lc
M
Q’1
Q2
LC
T1
T2
Q2
Q1
C
Fig. 17: Ciclo di Carnot con seconda
macchina reversibile ciclica.
Consideriamo il sistema costituito da
un ciclo di Carnot C più una seconda
macchina reversibile M ciclica
operante tra le stesse sorgenti (fig. 17).
Facciamo funzionare M come motore
e C come refrigeratore. Supponiamo
che la macchina di Carnot e la
macchina M siano progettate in modo
che la quantità di calore ceduta Q2 da
M alla temperatura inferiore sia uguale
alla quantità di calore che la macchina
di Carnot sottrae alla stessa sorgente.
diventa
In tal modo la sorgente a
inutile e può essere eliminata.
Se la macchina M ha efficienza maggiore, abbiamo: η c = 1 −
Q2
Q
< ηM = 1 − 2 ,
Q1
Q '1
da cui si deduce che Q1 < Q '1 . Il lavoro fatto dal motore è: (, = )′ − ) > ) −
) = (. . Dunque possiamo usare una parte del lavoro (, pari a (. per fare
funzionare il refrigeratore C e la differenza (, − (. rimane come lavoro prodotto.
In conclusione abbiamo costruito una macchina ciclica che produce lavoro col
calore sottratto ad una sola sorgente, in violazione del secondo principio.
Se, viceversa, la macchina ha efficienza inferiore a quella di Carnot, posso
invertire i ruoli nella precedente discussione e trovare allo stesso modo una
violazione del secondo principio. Dunque, le efficienze devono essere uguali.
In realtà occorre fare diverse precisazioni a quanto detto. Quali sono le macchine
termiche cicliche operanti con due sorgenti solamente, a parte, s’intende, la
macchina di Carnot?
In realtà non ce ne sono. La ragione sta nel fatto che le due curve che collegano le
due isoterme fanno per forza passare il fluido attraverso una continua variazione
di temperatura che, a parte il caso di trasformazioni adiabatiche (Carnot), implica
l’uso di una infinità continua di sorgenti con le quali il fluido entra in contatto e
scambia calore. Ne segue che il precedente teorema non ha significato perché
paragona le macchine di Carnot con se stesse. Tuttavia, possiamo limitarci a
trasformazioni per cui le quantità di calore scambiate durante i passaggi dalla
isoterma superiore a quella inferiore e viceversa siano uguali ( a parte il segno).
Questi cicli si chiamano “rigenerativi”. Come abbiamo già detto, con l’aggettivo
102
Cap. 5 – Termodinamica e calorimetria
“rigenerativo” intendiamo appunto che le quantità di calore scambiate per andare
da un’isoterma all’altra, dunque che i lavori fatti, giacché la variazione di energia
interna è la stessa, sono uguali. In tal caso il calore scambiato in una
trasformazione (per esempio nell’andare dalla temperatura maggiore a quella
minore) può essere conservato in una parte della macchina (chiamata appunto
“rigeneratore”) da cui può essere recuperato nella trasformazione inversa
(nell’andare dalla temperatura inferiore a quella superiore). In effetti è
ragionevole in tal caso ignorare queste quantità di calore e usare solo le quantità
di calore scambiate sulle isoterme, come per la macchina di Carnot, nel definire il
rendimento. In tal senso una macchina rigenerativa è una macchina che funziona
solo con due sorgenti esterne di calore e il rendimento è il solito:
V2
Q
V1
. L’ultimo passaggio è giustificato esattamente come
η = 1− 2 = 1−
V3
Q1
T1 ln
V4
T2 ln
nel caso del ciclo di Carnot. Chiaramente il rendimento di queste macchine è
V2 V3
= . Vediamo sotto quali
V1 V4
uguale a quello della macchina di Carnot, se
condizioni questa uguaglianza è vera. Evidentemente stiamo considerando il caso
V3
∫ pdV = costante al variare del punto V , infatti l’uguaglianza dei lavori
in cui
2
V2
V3
V4
V2
V1
∫ pdV = ∫ pdV
deve essere vera comunque si scelgano i punti
V1
e
V2 ;
inoltre: V4 = V4 (V1 ) e V3 = V3 (V2 ) ; la forma esatta di questa funzione dipende
dalla curva che unisce le due isoterme. Deve così essere:
V3
df (V3 ) dV3 df (V2 )
d
d
pdV = 0 =
( f (V3 (V2 )) − f (V2 )) =
−
in cui la
dV2 V
dV2
dV3 dV2
dV2
∫
2
df (V )
funzione f è la primitiva di p : p =
. Possiamo scrivere dunque che:
dV
103
Cap. 5 – Termodinamica e calorimetria
dV3
T dV T
V dV T
− P2 = 0 ⇒ 3 3 − 2 = 0 o infine: 2 3 = 2 . La stessa cosa
dV2
V3 dV2 V2
V3 dV2 T3
V1 dV4 T1 T2 V2 dV3
= = =
deve però valere all’altro estremo della isoterma:
.
V4 dV1 T4 T3 V3 dV2
P3
dV3
V V
dV2
La condizione diventa così: 1 3 =
, che dà la condizione cercata, se
V4 V2 dV4
dV1
dV3 dV4
=
= a ⇒ V3 = aV2 e V4 = aV1 . Questa condizione è soddisfatta dalle
dV2 dV1
trasformazioni politropiche che sono trasformazioni del tipo: PV x = costante.
Tale classe di trasformazioni contiene le adiabatiche ( x = γ ), le isobare ( x = 0 ),
le isoterme ( x = 1 ) e le isocore (riscrivendo P1/ xV = costante e prendendo x=∞
). Infatti:
x
PV
= costante ⇒ T V
1
x −1
= costante
⇒T
x −1
V = costante. Ovvero
1
1
1
⇒ T3 V3 = T2 V2 ⇒ V3 =
x−1
x−1
T2
x−1
1
T3
V2 , che dà il risultato cercato, se poniamo:
x−1
1
a=
T2
x−1
1
T3
. Possiamo infine verificare che il lavoro fatto durante la politropica
x−1
V3
∫ PdV non dipende dalla scelta di V :
2
V2
V3
∫
V2
V3
V3
1− x
PV x 1−x
PV
V 1− x
PdV = PV V dV = PV
= 2 2 (V3 − V2 ) = 2 2 (a1− x − 1)
1− x V 1− x
1− x
V
x
2 2
∫
−x
x
2 2
2
2
che è costante perché muovendosi
V2
sull’isoterma il prodotto
PV
2 2 = costante.
Naturalmente, se usiamo due isocore (ciclo di Stirling) a = 1 e se usiamo due
isobare, a =
T2
.
T3
104
Cap. 5 – Termodinamica e calorimetria
Possiamo così concludere che esistono altre macchine reversibili operanti fra due
sorgenti, ovvero che il teorema di Reitlinger è giustificato.
Concludendo: l’efficienza della macchina di Carnot è la massima possibile. Ogni
altra macchina reversibile operante tra le stesse temperature ha lo stesso
rendimento della macchina di Carnot. Le macchine reali (cioè non reversibili)
possono solo avere rendimento inferiore.
Dimostrato questo, possiamo finalmente precisare anche la scala delle
temperature assolute. E’ evidente da quanto detto che, se fosse possibile avere una
sorgente allo zero della scala Kelvin, allora l’efficienza di una qualunque
macchina termodinamica diverrebbe unitaria.
Ne segue che: lo zero della scala Kelvin è la temperatura di una sorgente fredda
tale che una qualunque macchina reversibile operante con questa sorgente fredda
ed una qualunque altra sorgente ha rendimento termico pari a uno.
Se è impossibile avere rendimento pari ad 1, allora non sarà possibile avere una
sorgente allo zero assoluto ovvero sarà impossibile raffreddare un corpo fino allo
zero assoluto: ci si può avvicinare quanto si vuole, senza però mai raggiungerlo.
Questa affermazione costituisce il terzo principio della termodinamica o teorema
di Nernst ed è, in realtà, una conseguenza del secondo principio..
È anche possibile eseguire una misura di temperatura usando un ciclo
termodinamico operante tra una temperatura fissa T0 di riferimento (per esempio
quella della mistura di acqua e ghiaccio, già usata per la scala Celsius) e quella del
corpo di cui si vuole misurare la temperatura T. Si avrà: T = T0
Q
. Dunque,
Q0
misurando le quantità di calore scambiate da una macchina ciclica reversibile
funzionante tra la sorgente di riferimento a temperatura T0 ed il corpo, si misurerà
la temperatura del corpo stesso.
11. L’entropia
Esiste una terza maniera, formulabile per via matematica, di esprimere il secondo
principio della termodinamica ed è ancora dovuta a Clausius. Essa asserisce che in
una trasformazione ciclica:
δQ
≤0
T
il segno di eguaglianza essendo valido solo per trasformazioni reversibili.
In questa relazione, δQ è la quantità infinitesima di calore assorbita. Ricordiamo
che stiamo parlando di cicli in cui il punto rappresentativo dello stato del sistema
∫
105
Cap. 5 – Termodinamica e calorimetria
si muove in senso orario (ciclo motore). Se il punto si muove in senso antiorario
(ciclo frigorifero), occorrerà cambiare di verso alla disuguaglianza precedente.
Notiamo che, se il segno di eguaglianza è valido, cioè nel caso di trasformazioni
B
reversibili, allora la funzione: S ( B) = S ( A) +
δQ
∫T
è una funzione di stato, avendo
A
l’integrale un valore indipendente dal cammino usato per calcolarlo (cfr. la teoria
del potenziale scalare in elettrostatica). La funzione S, che è dunque definita a
meno di una costante, è nota come “entropia” ed è calcolabile solo nel caso di
trasformazioni reversibili.
La dimostrazione del teorema di Clausius si può ottenere considerando un sistema
S che subisce trasformazioni cicliche assorbendo e cedendo quantità di calore
δ Q i (positive o negative) da n sorgenti a temperatura T i (fig. 17b) Con ciascuna
sorgente possiamo collegare una macchina di Carnot ('% ), funzionante come
motore o come refrigeratore, in modo tale che la quantità di calore scambiato con
l’iesima sorgente sia −δ Qi per ciclo; in questo modo ciascuna sorgente si troverà
alla fine del ciclo ad avere scambiato complessivamente un calore nullo. Tutte le
macchine di Carnot funzionano con l’altra sorgente a temperatura T ' ,
scambiando con questa delle quantità di calore δ Qi ' . Poiché per ciascuna
macchina di Carnot vale :
δ Qi
Ti
=
scambiate da S , vale la relazione:
δ Qi '
T'
∑
i
, si trova che, per le quantità di calore
δ Qi
Ti
∑δQ '
i
=
i
. La quantità
T'
∑δ Q ' deve
i
i
però essere uguale, alla fine di un ciclo, al lavoro fatto dal sistema complessivo
S con le n macchine di Carnot. Tuttavia quest’ultimo deve essere uguale a zero
perché il sistema complessivo è equivalente ad una sola macchina ciclica operante
dalla sola sorgente a temperatura T ' . Ne segue che ∑ δ Q i = 0 . Passando al caso
i
Ti
di un numero infinito di sorgenti con cui vengono scambiate infinite quantità
δQ
= 0.
infinitesime di calore si trova:
T
δQ
< 0 . Si può
Infine per una trasformazione non reversibile si ha sempre:
T
capire questo, considerando che le temperature delle sorgenti calde ( δQ > 0 )
saranno superiori e quelle della sorgenti fredde ( δQ < 0 ) inferiori a quelle del
fluido. Questo minimizza i termini positivi nella somma e massimizza invece
quelli negativi. Essendo infatti la trasformazione irreversibile i salti di temperatura
∫
∫
106
Cap. 5 – Termodinamica e calorimetria
saranno finiti e non infinitesimi, cioè le temperature delle sorgenti saranno un po’
più grandi o un po’ più piccole di quelle del sistema.
/)
'
Fig. 17b
/)1
/)
/)
/)
S
/)
/)′
'
'
/)′
/)′
′
Y
B
irreversibile
reversibile
A
X
Poiché per un sistema reale ci
si muoverà sempre lungo una
trasformazione irreversibile,
passando da uno stato A ad
uno stato B, per associare a
questo cambiamento di stato
una variazione di entropia
occorre
utilizzare
una
qualunque
trasformazione
reversibile che conduca dallo
stesso stato iniziale allo stesso
stato finale.
Consideriamo un ciclo che,
partendo da A, arrivi a B e poi
ritorni ad A.
Fig. 18: Ciclo con trasformazione reversibile
ed irreversibile.
Si immagini di effettuare un mezzo ciclo in maniera reversibile ed un mezzo ciclo
in modo irreversibile (fig. 18).
A
Nel mezzo ciclo reversibile, si può calcolare la variazione di entropia
∫ dS ,
B
B
nell’altro mezzo ciclo (irreversibile ) si può calcolare
δQ
∫T
(
δQ
T
non è un
A
107
Cap. 5 – Termodinamica e calorimetria
differenziale esatto in questo caso!), ed infine si possono confrontare le due
quantità
usando
la
relazione
di
Clausius.
Si
ottiene:
B
δQ
A
∫T ∫
+
A
B
B
dS < 0 ⇒
δQ
B
∫T ∫
A
<
B
dS ⇒S ( B ) − S ( A) >
A
δQ
∫ T . Si noti che se invertiamo il
A
ciclo, si ottiene la stessa relazione:
A
δQ
B
∫T ∫
+
B
δQ
+
A
δQ
∫ T . Si noti, inoltre che, se invertiamo le due
A
“reversibile”
e
“irreversibile”
B
∫T ∫
B
dS > 0 ⇒ S ( B) − S ( A) ≥
A
etichette
A
B
A
dS < 0 ⇒ S ( A) − S ( B) ≥
nella
figura,
allora
otteniamo:
δQ
∫ T , cioè che si vada da A a B o viceversa, non
B
importa: l’entropia aumenta ancora. Anche qui invertendo il ciclo si ottiene lo
stesso risultato.
Questo porta a concludere che:
Se un sistema è isolato e dunque non scambia calore con l’esterno ( δ Q = 0 ),
allora l’entropia aumenta sempre o al più resta costante: S ( B) − S ( A) ≥ 0 .
A questo punto, è chiaro che l’entropia serve a misurare l’irreversibilità delle
trasformazioni reali: in una trasformazione non reversibile, infatti, un sistema
isolato procede sempre verso una situazione di maggiore entropia.
Dunque un sistema isolato, raggiunto lo stato di massima entropia, non potrà
ulteriormente evolvere.
Calcoliamo adesso l’entropia di un gas perfetto monoatomico come funzione di
V e T.
Dal 1° principio:
dV
δ Q = dU + pdV = nCV dT + pdV = n(CV dT + RT
)=
V
3
δQ
3
dV
3 dT dV
= nR( dT + T
)⇒
= nR(
+
) ⇒ S = kN0 ln(VT 2 ) + costante
2
V
T
2 T
V
Inoltre per l’entropia di un gas perfetto si hanno le seguenti relazioni:
∂S
∂S
δ Q dU p
dS = ( )V dU + ( )U dV =
=
+ dV
∂U
∂V
T
T T
∂S
1 ∂S
p
⇒ ( )V = ,( )U =
∂U
T ∂V
T
108
Cap. 5 – Termodinamica e calorimetria
12. Interpretazione statistica dell’entropia
Abbiamo visto che la condizione di equilibrio statistico del sistema si raggiunge
quando la quantità H =
N!
, (o meglio,
n1 !...n k !
ln H ) che è proporzionale alla
probabilità che il sistema sia in quello stato dinamico, abbia un massimo ovvero
differenziale nullo: d ln H = 0 . In maniera equivalente anche per la quantità
1
1
S = k ln H = −k ln
dS = kd ln H = −kd ln = 0
sarà vero che:
con:
H
H
1
ln
=
( ni ln( ni ) − ni ) − ln N ! =
ni ln( ni ) − N − ln N ! =
ni ln( ni ) + C
H
i
i
i
∑
∑
∑
con C una costante che ignoreremo. Passando al continuo, riscriviamo S usando
+∞
la regola:
∑i ni ln(ni ) → ∫ dvx dvy dvz ln( dvx dvy dvz dV )dvx dvy dvz , con
dn
dn
0
3
mv 2
dn
m 2 − 2 kT
= N(
) e
2π kT
dv x dv y dv z dV
26
, con N =
N0
il numero di molecole per
V
unità di volume, formula che possiamo riscrivere anche (integrando sul volume):
3
mv2
m 2 − 2kT
dP(v)
dn = N0 (
) e
4π v2 dv = N0
dv ,. Otteniamo:
2π kT
dv
26
In realtà, n i è un numero di molecole, mentre a destra appare un numero per unità di
volume dello spazio delle fasi. Per evitare questo problema, potremmo dividere per le
unità di misura. Detta h , la dimensione delle cellette minime ∆p x ∆x , ∆p y ∆y , ∆p z ∆z , il
volume dello spazio delle fasi può calcolarsi in unità di h 3 . Abbiamo allora:
2
3 mv
h3
dn
h2 2 − 2kT
ni →
= N(
) e
, naturalmente in meccanica quantistica h è
m3 dvx dvy dvz
2π mkT
da identificare con la costante di Planck. In questo caso, si ottiene per l’entropia:
3
V 4π emE 2
S = kN 0 ln
da cui si può dedurre l’equazione dei gas perfetti. (M.
3
h 3N 0
Planck, - the theory of heat radiation, Dover p. 130)
109
Cap. 5 – Termodinamica e calorimetria
+∞
→
∫
mv 2
3
m 2 − 2 kT
N 0 P(v)(ln( N (
) e
))dv .
2πkT
Continuando:
0
1
ln = N 0
H
+∞
∫0
3
P(v)(ln( N (
3 +∞
m 2
= N 0 (ln( N (
) )
2πkT
∫
m 2 mv 2
) −
))dv =
2π kT
2kT
+∞
m
P (v)dv −
2kT
0
∫ v P(v)dv) =
2
0
3
2
3
m
m
m
1
v 2 ) = N 0 (ln( N (
) )−
)2)−
ε)=
kT
2πkT
2 kT
2πkT
3
3
−
m 32 −1 − 2 3
−1 2
) V T ) − ) = N0 ln(V T ) + N0 ln N0 (
= N0 (ln N0 ((
= N 0 (ln( N (
2π k
2
m 32 3
) − N0 .
2π k
2
Infine:
3
3
−
m 32
3
S = −kN0 ln N0 (
) = −kN0 ln(V −1T 2 ) = kN0 ln(VT 2 ) = kN0 (lnV + ln T )
cui
2π ek
2
dobbiamo aggiungere una costante arbitraria. Otteniamo così, per ogni stato B:
∆S
3
T 2V
= nR ln( B B ) .
3
TA2VA
Come abbiamo visto, tale quantità ha la stessa espressione di
quella calcolata al paragrafo precedente a partire dal primo principio:
3
k ln(T 2V ) +costante.
S = N0
Data l’interpretazione della funzione S, possiamo
concludere che lo stato stabile, cioè di massima entropia, corrisponde allo stato
che può essere realizzato nel massimo numero di modi, ovvero lo stato più
“disordinato”27. Da notare che l’entropia risulta definita a meno di una costante.
Secondo il terzo principio della dinamica o teorema di Nerst, l’entropia di un
sistema allo zero assoluto è zero. Vogliamo notare, per inciso, che l’espressione
3
dell’entropia S = N0 k ln(T 2V ) diverge per T →0 . Questo fatto dipende dall’aver
3
preso CV = 2 R , cioè costante in funzione della temperatura, cosa vera solo nel
27
Si può notare che se l’universo fosse esistito da tempo infinito, allora avrebbe già
raggiunto il massimo dell’entropia o “morte termica”. Dunque deve esistere da un tempo
finito: ∼ 15 ⋅ 10 9 anni.
110
Cap. 5 – Termodinamica e calorimetria
limite di alta temperatura28. Come nel caso dei solidi, anche per i gas il calore
molare tende invece a 0 quando T →0 . Infine il terzo principio implica
dall’espressione: S = k ln H , che la quantità H sia uguale ad 1 allo zero
assoluto, ovvero che tutte le molecole si trovano nello stesso stato dinamico.
A titolo di esercizio, calcoliamo adesso l’entropia di un insieme statistico di
oscillatori lineari di cui abbiamo parlato precedentemente alla fine del par. 4.
Il numero di oscillatori nell’ennesima zona di area h è:
ni
n
e − β ihν
1
= 1 ) e l’entropia
wi = i =
= e − β ihν = (1 − e − β hν )e − β ihν ; (
− β ihν
N
N
e
Z
∑
∑
i
i
è:
∑ N ln( N N ) + N ∑ N + N ln N − N =
n
n
n
n
= −N
∑ N ln N N + N ln N = − N ∑ N (ln N + ln N ) + N ln N =
n
n
n
n
= −N
∑ N ln N − N ln N + N ln N = − N ∑ N ln N
n !...n k !
S
= − ln 1
= −N
k
N!
i
ni
ni
i
i
i
i
ni
i
i
i
i
i
i
i
i
i
28
E. Fermi riporta in Thermodynamics (pag. 148) la formula valida per un gas
monoatomico ad alta temperatura di Tetrode e Sakura, ottenuta dalla meccanica
3
5
3
(2π AR) 2 ω e 2
quantistica: S = R(ln V + ln T + ln
).
2
h3 N A4
111
Cap. 5 – Termodinamica e calorimetria
∑ (1 − e
= −N
∑ (1 − e
S
= −N
k
− β hν
)e − βihν ln((1 − e − βhν )e − βihν ) =
i
− βhν
)e − βihν (− βihν + ln(1 − e − βhν )) = Nβ ( Eν −
i
=N
− βhν
hν e
hν
− N ln(1 − e − βhν ) ⇒ S = N
kT 1 − e − βhν
T
e
−
hν
kT
hν
−
1 − e kT
1
hν ) − N ln(1 − e − βhν ) =
2
− Nk ln(1 − e
−
hν
kT
Nε
T
) = nR ln Z +
29
.
Da notare che l’entropia del sistema di oscillatori è definita in questo caso in
valore assoluto e tende a zero quando la temperatura va a zero, come vuole il
terzo principio della termodinamica o teorema di Nernst, il che è una buona
ragione per lasciare h non nullo ed avere una energia media come calcolata alla
fine del paragrafo 4.
Un ultimo punto: la funzione H = ∫ f ( v , t ) ln f ( v , t )dv ( f (v, t ) = P(v, t ) ) è nota
come funzione H di Boltzmann. Boltzmann dimostrò che negli stati di non
equilibrio, quando essa è funzione del tempo e f ( v , t ) è soluzione
dH
dell’equazione del trasporto, la funzione decresce sempre:
< 0 , se non ci
dt
sono forze esterne agenti sul sistema, mettendo così in evidenza il fatto che i
29
Fatta l’identificazione della entropia con
ln( n i ) , l’espressione n i = e −α e − β , per ottenere:
S
=α
k
∑i ni ln(ni ) + N ln N , sostituiamo in
S
=−
k
∑i ni (−α − βεi ) + N ln N cioè:
S
=−
k
∑i ni + β ∑i εi + N ln N = α ∑i ni + βU + N ln N . Differenziando rispetto a U
a
dS
) = k β . Come abbiamo visto nel par. precedente:
dU V
1
1
dS
1
o: β =
. Questa è una dimostrazione generale del
(
)V = , dunque: kβ =
T
kT
dU
T
valore di β . kT = 4,14 ⋅ 10 −21 J = 0, 026 eV ; β = 2, 41 ⋅ 10 20 J − 1 = 38, 46 eV − 1 a
volume costante: (
T = 300K .
112
Cap. 5 – Termodinamica e calorimetria
processi spontanei (non soggetti cioè ad azioni esterne) sono sempre irreversibili.
Come abbiamo visto H = −S .
dH
df ( v , t )
Calcoliamo la derivata di H :
dv . La derivata
= ∫ (1 + ln f ( v , t ))
dt
dt
df (v , t )
è stata calcolata nell’ottenere l’equazione del trasporto. Ponendo
dt
df
= v − w ( f ( w) f (v ) − f ( w) f (v ))σ dwx dw y dwz si ha:
dt
dH
= ∫ (1 + ln f ( v )) v − w ( f ( w ) f ( v ) − f ( w ) f ( v ))σ dw x dw y dw z dv
dt
Come si è detto Boltzmann dimostra che questa quantità è sempre minore di zero
e quando raggiunge il suo massimo valore si deve avere
f ( w ) f ( v ) − f ( w ) f ( v ) = 0 da cui egli deduce la forma della distribuzione
f = f ( v ) che risulta quella già calcolata da Maxwell.
∫
13. Relazione tra entropia e funzione di partizione.
La relazione trovata per l’entropia è generale. Infatti, detta funzione di partizione
e − βε i
N − βε i
− βε
la Z = i e i ; abbiamo: n i = N
;
=
e
− βε i
Z
∑i e
∑
e −α =
N
N
⇒ −α = ln .
Z
Z
S
N!
= ln
= N ln N − N −
k
n1!...n k !
= N ln N − N
e −βε i
∑ ∑e
i
− βε i
∑ n ln n + ∑ n = N ln N − ∑ n ln n =
i
i
i
i
i
(−α − βε i ) = N ln N − N ln
i
i
i
N
+
Z
i
+ βN
ε i e − βε i
∑ ∑e
i
− βε i
= N ln Z + Nβε = N ln Z + βU ⇒ S = nR ln Z +
U
T
i
In cui U è l’energia totale del sistema. Si vede che l’entropia totale va a zero con
la temperatura. Infatti Z =1, perché esiste un solo stato possibile a T = 0 cioè
quello
a
εi = 0
e dunque ln Z = 0 . Evidentemente anche l’energia interna
diventa zero. Notiamo tuttavia che perché
U
= CV vada a zero, deve avvenire
T
113
Cap. 5 – Termodinamica e calorimetria
che CV → 0 , ovvero che i calori specifici tendano a zero al tendere di T →0
Possiamo mostrare che ciò è vero sia per il sistema di oscillatori armonici che per
il sistema delle molecole. Per gli oscillatori armonici:
−
hν
−
hν
hν
e kT
dU
(hν ) 2
e kT
+ Nhν
⇒
=
=
U=N
C
N
V
hν
hν
−
−
2
dT
kT 2
1 − e kT
(1 − e kT ) 2
Per le molecole:
U=N
ε i e− βε
∑∑e
i
i
i
− βε i
dU
N
⇒ CV =
= 2
dT kT
∑ ε e (∑ e
(∑ e
i
2 − βε i
i
− βε i
i
i
)−(
− βε i 2
∑εe
i
i
− βε i 2
)
)
Si vede che il calore molare tende a zero per la temperatura che tende a zero in
entrambi i casi.
In conclusione: l’entropia, l’energia interna e il calore molare a volume costante
tendono tutti e tre a zero per T → 0 sia per gli oscillatori armonici sia per le
molecole. Nel caso di un gas perfetto tuttavia non vanno a zero né l’entropia (
3
S = k lnVT 2 ) che anzi diverge, né il calore molare C V = 3 R che è
2
indipendente dalla temperatura, ovvero le formule trovate sono valide solo ad alta
temperatura. Dovremo capire come mai. Occorrerà, infine, studiare lo stesso
problema per il calore specifico dei solidi (vedi note di Fisica Generale 2).
14. I potenziali termodinamici
Le variabili di stato che abbiamo incontrato fin ora sono: p,V , T , S ,U . Oltre a
queste se ne possono definire altre (potenziali termodinamici):
H = U + pV (entalpia)
F = U −TS (energia libera di Helmoltz)
G = H −TS (energia libera di Gibbs)
Differenziando:
dU = δ Q − pdV = TdS − pdV
dH = dU + pdV + Vdp = δ Q + Vdp = TdS + Vdp
dF = dU − TdS − SdT = − pdV − SdT
dG = dH − TdS − SdT = Vdp − SdT
114
Cap. 5 – Termodinamica e calorimetria
Il differenziale di H è zero se l’entropia e il volume rimangono costanti. La
variazione di entalpia nella combustione completa di un massa M di
combustibile bruciato a volume costante V (come accade in un motore) è:
∆H = Q + V ( p f − pi ) , in cui Q è il calore rilasciato e ( p f − p i ) è la variazione
di pressione.
Il differenziale dell’energia libera F è zero, se temperatura e volume sono
costanti. Se la sola temperatura è costante, allora la variazione di F è uguale al
lavoro fatto cambiato di segno. Questo fatto possiamo interpretarlo dicendo che
F rappresenta la massima quantità di lavoro ricavabile dal sistema e, poiché
F = U −TS , se ne deduce che dell’energia interna U solo la parte F sia
trasformabile in lavoro, mentre la quantità TS sia inutilizzabile ovvero vincolata e
rappresenti calore scambiato a temperatura costante. Dall’espressione del
∂F
F , si ricava che
= −S
e
dunque:
differenziale
di
∂T
F = U − TS = U + T
∂F
∂F
⇒ F −U = T
(equazione di Gibbs-Helmoltz), in cui si
∂T
∂T
rivede che l’energia libera e l’energia interna sono identiche allo zero assoluto,
altrimenti sono diverse per un termine. Si noti che possiamo scrivere
dU
dF dU
dS
dS
=
−T
− S = C −T
− S in cui C =
è il calore specifico. Quando
dT
dT dT
dT
dT
T →0 , il calore specifico tende a zero. Infatti, se l’entropia si mantiene limitata
con la temperatura che va a zero, allora il calore specifico deve andare a zero; in
T
C (T )
C dT
effetti, se S (T ) = ∫ V
, allora perché S sia finita, l’integrando V
deve
T
T
0
dS
tende anch’esso a zero e
dT
dF
dF
= − S . Nernst ipotizzò (1906) che anche
= 0 allo zero assoluto e
si ha:
dT
dT
rimanere finito, ovvero: limCV (T ) = 0 30. Il termine T
perciò che l’entropia andasse anche essa a zero allo zero assoluto (terzo principio
dF dU
=
=0 e
della termodinamica). In conclusione, a T = 0 abbiamo: F = U ,
dT dT
S = 0 . Poiché si trova che, per un centinaio di gradi a partire dallo zero assoluto,
30
Colgo l’occasione per puntualizzare che , se questo è vero, allora c’è qualcosa di
sbagliato con il teorema di equipartizione dell’energia che prevede per i gas dei calori
molari proporzionali alla costante R indipendentemente dalla temperatura.
115
Cap. 5 – Termodinamica e calorimetria
dF dU
=
avremo che i processi si svolgeranno senza aumento di entropia, come è
dT dT
dF dU
dS
=
−T
−S .
chiaro dalla relazione:
dT dT
dT
dF
= 0 è utile per risolvere problemi di equilibrio chimico.
L’ipotesi
dT
Come si vede, il differenziale di G è nullo per una trasformazione in cui la
pressione e la temperatura rimangono costanti, tipicamente si tratta di un
passaggio di stato; ne vedremo un’applicazione fra poche righe.
Dalle relazioni precedenti otteniamo le derivate prime e seconde:
(
∂U
∂U
∂ 2U
∂T
∂ 2U
∂p
)V = T ;(
)S = − p ⇒
=(
= − ( )V
)S =
∂S
∂V
∂S ∂ V
∂V
∂ V ∂S
∂S
(
∂H
∂H
∂2 H
∂T
∂2H
∂V
) p = T;(
)S = V ⇒ (
)S = (
)S = (
)p = (
)p
∂S
∂p
∂S ∂ p
∂p
∂p ∂S
∂S
(
∂F
∂F
∂2 F
∂S
∂2 F
∂p
)V = − S ;( )T = − p ⇒
= −(
)T = (
)T = − ( )V
∂T
∂V
∂T ∂V
∂V
∂V ∂T
∂T
(
∂G
∂G
∂ 2G
∂S
∂ 2G
∂V
) p = −S; (
)T = V ⇒
= − ( )T =
=(
)p
∂T
∂p
∂T ∂ p
∂p
∂p ∂T
∂T
E dunque le relazioni:
∂T
∂p
) S = −( )V
∂V
∂S
∂T
∂V
( )S = ( ) p
∂p
∂S
∂S
∂p
( )T = ( )V
∂V
∂T
∂S
∂V
( )T = −( ) p
∂p
∂T
(
Sono chiamate relazioni (o equazioni)
di Maxwell e vengono usate in molti
problemi termodinamici.
Un esempio di applicazione dei
potenziali termodinamici si ha con
l’equazione
di
Clapeyron:
dp
λ
. Questa equazione si
=
dT T (V A − VB )
riferisce alla transizione di fase tra lo stato A e lo stato B, per esempio, la
transizione tra fase liquida e fase gassosa (vapore) e lega la pressione e la
temperatura del passaggio di stato e può essere usata per ottenere la dipendenza
funzionale della pressione dalla temperatura. Poiché il passaggio di stato è una
transizione a temperatura e pressione costante, l’energia libera di Gibbs rimane
116
Cap. 5 – Termodinamica e calorimetria
costante
(vedi
differenziale
della
definizione)
e
si
ha:
dG = 0 ⇒ G A = G B ⇒ dG A = dG B .
Facciamo variare l’energia libera ( G = U + pV − TS ) facendo variare la pressione (e
la temperatura), si ha:
dG = dU + pdV + Vdp − TdS − SdT = δ Q + Vdp − δ Q − SdT = Vdp − SdT
Le variazioni di G della sostanza nelle due fasi devono essere uguali perché si
conservi l’eguaglianza della G al passaggio di stato:
dGA = dGB ⇒ (V f − Vi )dp − dT (S f − Si ) = 0 ⇒
il
calore
latente
f
f
S f − Si =
δQ
∫T
i
=
di
transizione,
m
dp S f − Si
λm
=
=
in cui λ è
dT V f − Vi T (V f − Vi )
è
la
massa
di
sostanza
e
1
λ
δQ = .
T
T
∫
i
Per la transizione da liquido a vapore (ebollizione), si ha: Vi << V f = R m T e,
A p
λA
sostituendo, si ha: dp = Aλ2 dT che integrata dà: log p = −
, nel caso in cui λ
RT
p
RT
sia costante. Altrimenti possiamo espandere λ e ottenere: λ = a + bT + cT 2 + ... e
sostituendo, avere:
λ A λA
λA
log p = −
a+
b log T +
cBT + ... Questa si chiama legge di Antoine.
RT
R
R
Possiamo calcolare la temperatura di ebollizione in funzione della pressione. Una
misura della temperatura di ebollizione dell’acqua darà dunque la pressione e,
potenzialmente, l’altitudine del luogo.
−
λ1
La figura che segue dà l’andamento nel piano p-T della funzione p = e R T per
l’acqua. In realtà, se la temperatura oltrepassa la temperatura critica TC , allora la
sostanza esiste solo nello stato di gas e la curva dell’equazione di Clapeyron non
vale più. Inoltre a temperatura bassa (sotto il punto triplo) eventualmente il vapore
diventa solido direttamente piuttosto che fare prima una transizione al liquido.
117
Cap. 5 – Termodinamica e calorimetria
F1
F2
1 ,4
O r i g in P r o
8
E v a l u a t i o n
O r i g in P r o
8
E v a l u a t i o n
O r i g i n P r o
8
E v a lu a t io n
O r i g i n P r o
8
E v a lu a t io n
1 ,2
L iq u id o
TC
p (atm)
1 ,0
O r i g in P r o
8
E v a l u a t i o n
O r i g i n P r o
8
E v a l u aV
t ia
o np o re
O r i g in P r o
8
E v a l u a t i o n
O r i g i n P r o
8
E v a lu a t io n
O r i g in P r o
8
E v a l u a t i o n
O r i g i n P r o
8
E v a lu a t io n
O r i g in P r o
8
E v a l u a t i o n
O r i g i n P r o
8
E v a lu a t io n
O r i g in P r o
8
E v a l u a t i o n
O r i g i n P r o
8
E v a lu a t io n
0 ,8
0 ,6
0 ,4
S o lid o
P u n to trip lo
H 2O , T c= 6 3 8 K
0 ,2
0 ,0
100
200
300
400
500
600
T (K )
Un’altra applicazione che possiamo discutere è la dipendenza dal volume
dell’energia interna. Per un gas perfetto, sappiamo che l’energia interna dipende
solo dalla temperatura, ma un gas reale risente delle forze intermolecolari come
abbiamo visto nella discussione dell’equazione di van der Waals. Aumentando il
volume si riduce l’energia cinetica delle molecole che viene usata contro le forze
di
coesione
per
allontanare
le
molecole.
∂S
∂S
δQ
∂S
dU = TdS − pdV = T
dT + ∂V dV − pdV = dT dT + (T ∂V − p )dV =
T
V
T
∂T V
∂S
∂U
∂U
= nCV dT + (T
− p )dV = ∂T dT + ∂V dV
∂V T
V
T
Da cui si deduce che: ∂ U = nCV e ∂ U = (T ∂ S − p ) . Questa seconda
∂ T V
∂ V T
∂V T
relazione può essere riscritta, usando (
∂S
∂p
)T = ( )V , ∂U = (T ( ∂p )V − p ) . In
∂V
∂T
∂T
∂ V T
effetti si vede che ∂U = (T ( ∂p )V − p ) = 0 per un gas perfetto perché:
∂T
∂ V T
∂p
nR
)V − p) = (T
− p) = 0 . Per un gas di van der Waals,
∂T
V
∂p
nR
nRT
n2
( )V =
e
,
sostituendo
si
ha
p=
−a 2
∂T
V −b
V −b
V
(T (
nR
nR
n2
n2
∂U
=
(
T
−
T
+
a
)
=
a
∂V
V −b
V −b
V2
V2
T
.
Infine,
possiamo
invece:
infine:
scrivere:
118
Cap. 5 – Termodinamica e calorimetria
dU = nCV dT + a
n2
V2
dV . Per un gas perfetto
a= 0 e ritorniamo alla vecchia
formula.
Possiamo adesso calcolare la differenza tra calori specifici per un gas reale
Possiamo scrivere le due relazioni (TDS):
∂S
∂S
∂P
TdS = T
dT + T ∂V dV = nCV dT + T ∂T dV .
∂T V
T
V
∂S
∂S
∂S
∂V
TdS = T
dT + ∂ P dV = nC P dT + T ∂ P dP = nC P dT − T ∂ T dP
∂
T
P
P
T
T
Sottraendo
membro
a
membro
la
seconda
dalla
prima:
∂S
∂S
∂S
∂S
0 = n(C P − CV )dT − T
dV + T
dP ⇒ n(CP − CV )dT = T
dV − T
dP =
∂V T
∂P T
∂V T
∂P T
∂P
∂V
= T
dV +
dP
∂T P
∂T V
∂P
∂V
T dV +
dP
∂T P ∂T
∂T
∂T V
=
dV + dP che
Da cui otteniamo: dT =
n(CP − CV )
∂V P
∂P V
∂P
∂V
T
T
∂
T
∂T
∂T
V
∂T P
fornisce:
e
, che infine danno:
=
=
∂P
∂V P n (C P − CV )
V n (C P − CV )
∂ P ∂ V . Relazione che può essere applicata ad un
n ( C P − CV ) = T
∂ T V ∂ T P
qualunque gas. D’altra parte, per un gas che obbedisce all’equazione dei gas
perfetti, abbiamo di nuovo:
n(CP − CV ) = T
nR nR
= nR ⇒ CP − CV = R , come sapevamo già. Notare che le
V P
derivate che sono a destra devono essere entrambe positive: sia il volume che la
pressione crescono con la temperatura. Dunque è sempre vero che: CP − CV > 0 .
Allo zero assoluto i due calori specifici sono uguali. Sono uguali anche ad ogni
temperatura per cui ∂ V = 0 .
∂T P
Ciò accade , per esempio, all’acqua alla temperatura di 4°C.
E’ anche possibile calcolare il rapporto tra i due calori specifici. Si trova, tenendo
l’entropia costante:
119
Cap. 5 – Termodinamica e calorimetria
∂V
nC P dT = T
dP ;
∂T P
∂V
C P ∂T P
=
CV
∂P
∂T
V
∂P
nCV dT = −T
dV ,
∂ T V
∂V
∂P
∂P
∂V = ∂V
S
∂P
da
cui:
T
= γ . Poiché le due derivate sono collegate coi
S
coefficienti di compressibilità: κ S = − 1 ∂ V (adiabatico) e κ T = − 1 ∂ V , il
V ∂P S
V ∂ P T
γ può essere calcolato a partire da questi due ultimi coefficienti.
1
Per un gas perfetto − 1 ∂V = 1 nRT
e usando
=
V ∂ P T V P 2
P
1
PγV = C
, abbiamo:
1
− −1
−
1 ∂V
CP γ
=
V ∂P S γ V
∂V
C P ∂P
=
CV
∂V
∂P
. Facendo il rapporto, abbiamo:
1
1
γ VP γ
p
T
=
=
= γ , come sapevamo.
1
C
− −1
C P γ
S
γ V
Calcoliamo allora C P − CV = T ∂P ∂ V (posto n=1 ) per un gas di Van der
∂T V ∂ T P
P=
Waals:
(
RT
1
R
∂P
,
−a 2 ⇒
=
V −b
V
∂ T V V − b
)
1
∂T
− 2 a (V − b ) + PV 3 + aV .
∂V =
3
RV
P
CP − CV = T
Da
e:
cui:
R
RV 3
. Si vede subito che questa espressione si
V − b −2a(V − b) + PV 3 + aV
riduce a R , se a = b = 0 , cioè per un gas perfetto.
Possiamo infine, dimostrare col calcolo esplicito, cioè a prescindere dal teorema
δQ
generale, che anche per un gas reale la quantità
è un differenziale esatto.
T
Infatti:
δQ
dU pdV
dT
n2
nRT
n 2 dV
+
= nCV
+a
dV
+
(
−
a
)
=
T
T
T
T
V −b
TV 2
V2 T
3
dT
nR
= nCV
+
dV = nCV d ln(T ) + nRd ln(V − b ) = nRd ln (T 2 )(V − b )
T
V −b
=
120
Cap. 5 – Termodinamica e calorimetria
(abbiamo preso il gas monoatomico), questa espressione si riduce a quella già
calcolata per i gas perfetti, se b = 0 .
15. Il campo elettrostatico e il campo magnetostatico nella materia
Consideriamo un’altra applicazione della statistica di Boltzmann. Un campo
elettrico (magnetico) costante diretto lungo l’asse Z viene applicato ad un pezzo
di materiale dielettrico (gas, liquido o solido isotropo e omogeneo). I dipoli
elettrici (magnetici) atomici si allineano lungo l’asse del campo, ma a causa
dell’agitazione termica non potranno essere tutti allineati lungo questa direzione.
Ci si domanda quale è il momento elettrico di dipolo medio lungo Z.
L’energia di un dipolo è data da: U = − p ⋅ E = − pEcosθ ( U = − d ⋅ B ), così
l’energia di una molecola sarà: U = ε − pE cos θ , dove ε è l’energia cinetica. Il
numero di molecole con energia tra U e U+dU è:
e
dn
= +∞
N
−
∫e
=
(ε − pE cosθ )
kT
dxdydzdpx dp y dp z d (cosθ )
−
(ε − pE cosθ )
kT
dxdydzdpx dp y dp z d (cosθ )
−∞
ε
−
kT
e dxdydzdp
+∞
∫
e
−
x dp y dpz
pE cosθ
e kT d (cos θ )
+1
ε
kT dxdydzdp
∫
x dp y dpz e
−∞
=
pE cosθ
kT d (cos θ )
−1
Integrando sullo spazio delle fasi, risulta che la frazione sul totale delle molecole
con coseno dell’angolo tra cos θ e cos θ + d (cos θ ) , è:
pE cosθ
dn
e kT d (cosθ )
= +1 pE cos
. Per ottenere il valore medio di
θ
N
e kT d (cos θ )
cos θ , occorre
∫
−1
moltiplicare per cos θ e integrare:
+1
+1
cos θ =
∫
−1
dn
cos θ =
N
∫
−1
cos θ e
+1
∫e
pE cosθ
kT
d (cos θ )
pE cos θ
kT d (cos θ )
=
−1
121
Cap. 5 – Termodinamica e calorimetria
=
d
da
=−
+1
d
a
−e −a + ea
ln e ax dx =
ln
=
−a
da
a
−e + ea
−1
∫
−e −a + ea e −a + ea
+
−
a
a2
=
1 e −a + ea
1
+
= − + coth( a ) (Funzione di Langevin), avendo posto
a ea − e −a
a
x = cos θ
pE
. Si vede che per E → +∞ , la funzione di Langevin tende
kT
asintotticamente ad 1 (saturazione). Per a << 1 , sviluppando in serie di
1
a
1 a a3
McLauren il coth(a ) = + − + ... si ha31: cos θ = − + coth(a ) ≈ , da cui,
a
3
a 3 45
e a =
infine, si ricava che il momento di dipolo medio lungo Z è: pz = p
, e la polarizzazione è: P = N 0
p2 E
in cui
3kT
pE p 2 E
=
3kT 3kT
N0 = numero di dipoli (molecole) per
unità di volume.
Sottoponendo un dielettrico ad un campo elettrico esterno, si forma dunque una
polarizzazione proporzionale al campo elettrico. Tutto ciò crea un momento di
dipolo complessivo nel materiale dato da: d =
∫ PdV = ∫ ρrdV . Si può poi
V
dimostrare
∇⋅P + ρ = 0
31
∫
∫
V
V
d = PdV = ρr dV
che
∫
= − r ∇ ⋅ PdV ,
V
da
cui
segue
che
V
. In effetti:
Il momento di dipolo della molecola d’acqua è: p = 1, 8 5 ⋅ 1 0 − 30 C ⋅ m = 1, 8 5 D eb ye e
kT = 4, 14 ⋅ 10 − 21 J a
T = 300K . Ne segue che a << 1 per campi elettrici fino a valori
9
dell’ordine di ∼ 10 V / m .
122
Cap. 5 – Termodinamica e calorimetria
F1
1,0
Funzione di Langevin
0,8
0,6
0,4
0,2
0,0
2
4
6
8
10
a
∂Px
∫ r ∇ ⋅ PdV = ∫ x( ∂x
V
+
V
∫
+ x(
V
=(
∂Py
∂y
+
∂Pz
∂z
∂Py
∂y
+
∂Pz
)dxdydzi + ... =
∂z
)dxdydzi + ... =(
∂Px
∫ x ∂x dxdydzi +
V
∂
∂
∫ ∂x ( xP )dxdydz − ∫ P dxdydz)i + ∫ ∂y ( xP )dxdydzi + ...
x
x
V
∂
y
V
∂
V
∂
∫ ∂x ( xP )dxdydz + ∫ ∂y ( xP )dxdydz + ∫ ∂z ( xP )dxdydz )i + ... − ∫ P dVi − ∫ P dV j − ∫ P dVk =
x
y
V
z
V
x
V
V
∫
∫
∫
∫
V
V
V
V
y
z
V
V
= ∇ ⋅ ( xP )dxdydz + ∇ ⋅ ( yP )dxdydz + ∇ ⋅ ( zP )dxdydz − PdV
I tre integrali
∫ ∇ ⋅ ( xP )dxdydz = ∫ ( xP ) ⋅ ndS
V
e simili sono nulli, perché la
S
polarizzazione è nulla sulla superficie, prendendo una superficie adeguata.
Pensiamo ad un blocchetto di dielettrico soggetto ad un campo esterno, la
superficie S può essere scelta come una superficie che contiene l’intero
blocchetto, appena fuori del blocchetto. Scegliendo in tal modo la superficie si
modifica anche il volume d’integrazione, ma evidentemente gli integrali sul
volume non cambiano, mentre quello di superficie si, almeno che non sia nullo.
Da cui:
∫ ρr dV = − ∫ r ∇ ⋅ PdV ⇒ ∇ ⋅ P + ρ = 0 , dove la densità di carica va
V
V
intesa come densità di carica di polarizzazione ρ P . Il potenziale generato da un
123
Cap. 5 – Termodinamica e calorimetria
dipolo in un punto a distanza R = ( x − ξ )2 + ( y − η )2 + ( z − ζ )2
(vedere capitolo 3): ϕ =
momento
1 p⋅R
4πε 0 R 3
. Possiamo riscrivere questa formula per un
di
dϕ ( x, y, z) =
(dal dipolo) è
PdV = Pd ξ dη d ζ
dipolo
1 P⋅R
1
1
1
1
P
dV =
( P ⋅ ∇ ( )) dV =
( ∇ ( ) − ∇ ⋅ P ) dV
4πε 0 R 3
4πε 0
R
4πε 0
R
R
:
32
Che, integrando su tutto il dielettrico, dà:
ϕ ( x, y , z ) =
1
4πε 0
P
1
1
∫ (∇( R ) − R ∇ ⋅ P )dV = 4πε (− ∫
0
V
V
∇⋅ P
dV +
R
P
∫ R ⋅ ndS )
formula che
S
possiamo interpretare dicendo che il potenziale è dato dal contributo di una
densità di carica superficiale σ P = P ⋅ n e dal contributo di una densità di carica di
σ
ρP
1
( P dS +
dV ) . Essendo poi il dielettrico
volume ρ P = − ∇ ⋅ P : ϕ =
4πε 0
R
R
∫
∫
S
V
neutro, dovrà essere: 0 = ∫ σ P dS + ∫ ρ P dV .
S
V
ρ + ρP
D’altra parte, abbiamo: ∇ ⋅ E =
, in cui abbiamo designato con ρ la
ε0
densità
di
carica
“vera”.
∇ ⋅ (ε 0 E ) = ρ + ρ P = ρ − ∇ ⋅ P ⇒ ∇ ⋅ (ε 0 E + P ) = ρ .
troviamo infine:
Posto allora
Troviamo:
D = ε0E + P ,
∇⋅ D = ρ .
2
Poiché abbiamo dimostrato che P = ε 0 N 0 p E = ε 0 χ E , possiamo dire che:
3 kT ε 0
D = ε 0 E + P = ε 0 (1 + χ ) E = ε 0 ε r E = ε E
, con:
ε r = 1 + χ ( ε r = costante dielettrica relativa, ε = ε 0ε r = costante dielettrica e χ =
suscettività). Come si vede, la costante dielettrica relativa è maggiore di 1.
32
Osserviamo che R è la differenza tra i vettori di posizione del momento di dipolo (
ξ ,η, ζ ) e del punto d’osservazione ( x, y, z ): R = ( x − ξ )2 + ( y − η )2 + ( z − ζ )2 . La
derivazione di 1/ R va fatta rispetto alle variabili greche non a quelle latine. In tal caso:
R
1
= ∇( ) , mentre la derivazione rispetto alle latine dà la stessa cosa, ma con un segno
3
R
R
meno davanti.
124
Cap. 5 – Termodinamica e calorimetria
Se oltre al momento di dipolo o al posto del dipolo permanente della molecola, si
deve considerare un momento indotto dal campo elettrico esterno, allora si pone:
p
pz = ( p
+ α p ) E , con α P la “polarizzabilità” del mezzo e si trova:
3kT
2
p2
)E , da cui33: ε r = 1 + N 0 1 ( p + α p ) .
P = N0 (α P +
3kT
ε 0 3kT
Prendiamo adesso un condensatore piano tra le cui armature possiamo inserire un
dielettrico di costante dielettrica relativa ε r . Chiamiamo E0 ed E
rispettivamente i campi prima e dopo l’inserzione del dielettrico. La differenza di
potenziale prima e dopo l’inserzione saranno:
distanza fra le armature. Abbiamo:
∇⋅ E =
∆V0 = E0l
e ∆V = El , con l
ρ + ρP
⇒ ∇ ⋅ (ε 0 E + P) = ∇ ⋅ D = ∇ ⋅ (ε 0ε r E ) = ρ = ∇ ⋅ (ε 0 E0 ) , da
ε0
cui: E = E0 / ε r e per conseguenza: ∆V0 = ε r ∆V > ∆V , cioè con l’inserzione
del dielettrico la differenza di potenziale si abbassa proprio di ε r .
Per il campo magnetico si procede nello stesso modo.
1
r ×∇× M può essere dimostrata con un calcolo
La eguaglianza: MdV =
2
diretto34.
33
∫
∫
V
V
∇ × B = µ 0 ( j + jM ) ⇒ ∇ × ( B − µ 0 M ) = µ 0 j
Per l’acqua per cui ε r = 80 (statica) si trova:
.
α p = 20,9 ⋅ 10−39 C ⋅ m2 / V . Ad alta
frequenza la polarizzabilità diventa nulla e ε r scende a circa 2, come si avrebbe se la
molecola fosse non polarizzabile. L’indice di rifrazione
n = εr diviene allora 1,33. Per
l’elio, che non ha un momento di dipolo intrinseco, la polarizzabilità è:
αp
ε0
= 2, 5 ⋅ 10 − 30 m 3 , da cui si ha: ε r = 1.0000678 ( n =
N0 =
34
6.02 ⋅ 10 23 m ol
22, 4 ⋅ 10 − 3 m 3
εr = 1,33 ). Abbiamo preso:
= 0, 27 ⋅ 10 26 m ol / m 3 .
A. S: Kompaneyets – Theoretical Physics – Dover p.146/147
125
Cap. 5 – Termodinamica e calorimetria
Ponendo: H =
B
H =
=
µ0
1
µ0µr
B
µ0
− M , si ha:
∇×H =
j . Se M = N0
− M ⇒ B = µ 0 (H + M ) = µ 0 (H + N 0
d 2B
, possiamo sostituire:
3kT
1
d2
d2
(1 − N 0 µ 0
)=
B) ⇒ B
3kT
3kT
µ0
B=H
µr =
1
e
µ = µ0 µr
(permeabilità
magnetica).
Anche
d2
(1 − N 0 µ 0
)
3kT
d2
µr ≅ 1 + N 0 µ0
= 1 + χ m è maggiore di 1. I materiali per cui tutto ciò è vero si
3kT
chiamano “paramagnetici” e µ è poco ( 1 / 10 5 ) maggiore dall’unità. Esistono
sostanze “diamagnetiche”, per cui µ differisce poco ( 1 / 10 5 ) dall’unità, ma è
inferiore a 1. Evidentemente nelle sostanze diamagnetiche la suscettività è
negativa. Possiamo capire questo osservando che, come effetto della legge di
Lenz (vedi Fisica Generale 2), la molecola diventa sede di una corrente che si
oppone alla variazione del campo magnetico esterno all’atto della sua accensione,
cioè acquisisce un momento magnetico opposto a quello del campo esterno,
ovvero una suscettività magnetica χm negativa. Dovrebbe risultare chiaro che
tale effetto di diamagnetismo si deve avere per ogni materiale. Tuttavia, se la
molecola non ha inizialmente momento magnetico essa non può che risultare
diamagnetica, se, viceversa, essa ha già un momento magnetico iniziale, il
paramagnetismo può risultare l’effetto dominante. Immaginiamo un’orbita
elettronica
intorno
ad un
nucleo.
L’equazione
del
moto
è:
m
v2
1 Ze 2
= mω 2r =
r
4 πε 0 r 2
. La velocità angolare è dunque: ω0 = ±
1 Ze 2
,
4πε 0 m r 3
1
dove il doppio segno si riferisce ai due versi di percorrenza dell’orbita. Se
applichiamo un campo magnetico lungo un asse perpendicolare al piano
dell’orbita, si ha: m ω 2 r =
1
Ze 2
4 πε 0 r 2
+ e ω rB
, che dà come soluzioni per la nuova
∫ r × jdV viene dall’espansione in multipoli del potenziale
∫ j (r ⋅ r ' )dV ' = − r × ∫ r '× jdV ' = m × r .
L’altra eguaglianza m =
1
2
V
vettore: A =
µ0 1
4π r 3
µ0 1
4π r 3
V'
µ0 1
4π r 3
1
2
V'
126
Cap. 5 – Termodinamica e calorimetria
eB
(frequenza di Larmor), nell’ipotesi
2m
che: ω L << ω 0 . In altre parole la velocità angolare aumenta. Pertanto varia la
corrente
e
il
momento
magnetico
dell’atomo
diminuisce
dω
d µ = Sdik = −e
Sk = −eSωL k , con S = area dell’orbita e k il versore nella
2π
direzione del campo magnetico esterno. Se l’atomo non aveva momento
magnetico, ne ha ora acquistato uno nella direzione del campo magnetico, ma con
verso opposto a questo.
Il momento della quantità di moto dell’elettrone cambia allora di dL = m r 2 ω l che
va moltiplicato per il rapporto giromagnetico e per il numero di elettroni per unità
velocità angolare: ω = ω L ± ω 0 , con ω L =
di volume per darci la suscettività: χ = − N 0 Z µ0
e2r 2
.
4m
Nella teoria precedente, abbiamo usato il fatto che la relazione tra induzione
magnetica e campo magnetico è lineare: B = µH , anche se essa tende a saturare
per campi molto alti (vedere la funzione di Langevin). Per certi materiali, detti
“ferromagnetici”, la saturazione è raggiunta invece rapidamente (almeno al di
sotto di una temperatura che dipende dal materiale, detta temperatura di Curie, al
di sopra della quale il materiale è paramagnetico). La ragione per questo
comportamento sta nell’interazione tra dipoli, che non è stata presa qui in
considerazione. Si ha inoltre un effetto di isteresi. Ciò vuol dire che, all’aumentare
di H , B raggiunge velocemente un massimo e poi non cresce più (saturazione)
ovvero riportando il valore di H a zero, B non si annulla più (isteresi), come
nello sketch sottostante. Usando i valori dei vari parametri dell’acqua e come
dipolo magnetico 1 magnetone di Bohr ( µ B =
eℏ
= 9, 274 ⋅ 10 − 24 A ⋅ m 2 35)
2m e
otteniamo:
χ dia = 8 , 18 ⋅ 1 0 − 6
−6
e χ para = 286 ⋅ 10 .
35
Il magnetone di Bohr è il momento magnetico di un elettrone che orbita intorno al
protone in un atomo d’idrogeno nell’orbita fondamentale.
127
Cap. 5 – Termodinamica e calorimetria
H
(solo qualitativo)
Vedremo in Fisica Generale 2 che la formula ∇ × H = j va modificata per tenere
conto del fatto che la divergenza della corrente non è nulla, in caso ci siano
dipendenze dal tempo. Poiché la divergenza del rotore è nulla e quella di j M non
∂
P . Ripetendo il
può esserlo, si dovrà aggiungere alla jM il termine
∂t
ragionamento di otterrà: ∇ × H = j +
∂D
.
∂t
Il corpo nero: introduzione
Un interessante problema, poi rivelatosi addirittura cruciale nello sviluppo della
fisica, è quello del corpo nero. Si tratta, secondo una definizione generica, di
studiare le caratteristiche dell’emissione dei corpi in funzione della loro
temperatura.
Il problema nasce in un contesto storico in Germania, in cui il problema
dell’illuminazione era diventato importante nello sviluppo industriale del paese.
I nomi principali sono: Max Planck. A. Einstein, W. Wien, Rayleigh, Jeans,
Paschen, Lummer, Pringsheim.
Planck, il cui conservatorismo scientifico è sottolineato costantemente dai suoi
biografi, si trovò a fare il rivoluzionario. Tuttavia propose sempre le soluzioni che
meno entravano in collisione con la fisica precedente. Einstein fu il fisico che più
spinse invece per una interpretazione rivoluzionaria di ciò che fu scoperto da
128
Cap. 5 – Termodinamica e calorimetria
Planck e da altri e portò la fisica verso la meccanica quantistica, della quale poi
non fu mai convinto.
Diamo ora una definizione precisa del problema del corpo nero.
2. Definizione del problema
Si definisce “emissività” di un corpo ε la quantità d’energia emessa dal corpo per
dE
unità di tempo ed unità di superficie: ε = emessa .
dSdt
dε
Chiamiamo “emissività specifica” invece la quantità: ελ =
o la quantità
dλ
dε
d ε d ε dν
c dε
εν =
. Le due quantità sono legate dalla relazione
e
=
=
dν
d λ dν d λ λ 2 dν
dunque la teoria può essere formulata in termini dell’una o dell’altra quantità,
+∞
secondo convenienza. Ovviamente è: ε =
∫
+∞
ελdλ =
0
∫ εν dν .
0
Il “potere d’assorbimento” a di un corpo, è la frazione dell’energia incidente su di
esso che il corpo assorbe: a =
aλ =
E ass
; il ”potere specifico d’assorbimento”
E inc
è
dE ass ( λ )
dE (ν )
( aν = ass ) ovvero la frazione dell’energia incidente su di esso
dE inc ( λ )
dEinc (ν )
con lunghezza d’onda λ (con frequenza ν ) che il corpo è capace di assorbire.
Nel seguito ammetteremo sempre che ε , ε λ , ε ν , a , a λ , aν siano costanti sulle
superfici del corpo nero.
Orbene, fu dimostrato da Kirchhoff che il rapporto
Kλ = Kλ (λ,T) indipendente dalla natura del corpo.
ελ
aλ
è una funzione universale
La legge di Kirchhoff si può dimostrare col seguente ragionamento. Prendiamo
una cavità con le pareti a temperatura T. La cavità abbia pareti perfettamente
riflettenti. Mettiamo nella cavità un corpo di forma e composizione qualsiasi. Il
corpo assorbirà la radiazione eventualmente già presente nella cavità e riemetterà
della radiazione su lunghezze d’onda in generale diverse da quelle di
assorbimento. Sia K l’intensità della radiazione (uguale ovunque, all’interno
della cavità perché supponiamo di essere in condizioni d’isotropia), cioè la
quantità di energia che incide sull’unità di superficie nell’unità di tempo e sia
dK
Kλ =
l’intensità della radiazione in un intervallo infinitesimo di lunghezze
dλ
129
Cap. 5 – Termodinamica e calorimetria
d’onda. Il meccanismo di assorbimento e di riemissione del corpo genererà col
tempo una distribuzione spettrale di energia Kλ stabile. In questa situazione
d’equilibrio, il corpo emetterà una quantità d’energia, per unità di tempo,
d’intervallo di lunghezze d’onda e di superficie, pari a ε λ e dovrà per forza
assorbirne una quantità uguale. Dunque: ε λ = a λ K λ . Peraltro K λ è una funzione
universale non dipendente dalla forma e costruzione della cavità, ma solo dalla
sua temperatura. Infatti se K λ non fosse universale, potremmo mettere in
contatto due cavità (1 e 2) diverse con la differenza K λ 1 − K λ 2 finita in qualche
intervallo di lunghezze d’onda alla stessa temperatura, usando un filtro che lasci
passare solo radiazione a lunghezza d’onda λ. Abbassando di una quantità
infinitesima la temperatura della cavità 1, potremo ancora fare in modo che
K λ 1 > K λ 2 , malgrado sia ora T1 < T2 . In questo caso, energia si trasferirà
spontaneamente da un corpo a temperatura inferiore ad un corpo a temperatura
maggiore, violando il secondo principio della termodinamica. Spesso si afferma
che un corpo emette onde elettromagnetiche alle stesse frequenze alle quali le
assorbe, poiché se a λ = 0 = ε λ K λ allora anche ε λ = 0 .
Se il corpo è capace di assorbire tutta l’energia che lo investe a qualunque
frequenza ( a λ = 1 ), allora esso ha un’emissività specifica che è la funzione
universale Kλ (λ, T ) . Un corpo nero, cioè, ad esempio, una superficie coperta di
nerofumo,
ha approssimativamente la proprietà di assorbire qualunque
radiazione, quasi completamente: definiamo quindi “corpo nero”, quello per cui è
rigorosamente vero che a λ = 1 . Dunque, calcolare l’emissività ε λ del corpo nero
equivale a trovare la funzione universale K λ ( λ , T ) .
Invece di una cavità a pareti perfettamente riflettenti con un pezzo di carbone
dentro, d’ora in poi considereremo una cavità a pareti nere (nel senso definito di
a λ = 1). nel cui interno si formerà una radiazione con densità spettrale K λ ( λ , T ) ,
per l’azione delle pareti, piuttosto che per l’azione di un corpo inseritovi dentro.
La radiazione è in effetti assorbita e riemessa dalle stesse pareti della cavità.
Poiché la
Kλ (λ,T )
rappresenta la quantità di energia che raggiunge una
superficie dS nel tempuscolo dt interna alla cavità, possiamo calcolare la
quantità d’energia che cade in particolare sulla superficie interna della parete della
cavità. Possiamo, in effetti, immaginare di aprire un foro nella parete di superficie
dS e misurare la K λ ( λ , T ) dall’esterno. Possiamo anche equiparare la superficie
del foro alla superficie di un corpo nero in quanto la radiazione che penetrasse
nella cavità dall’esterno sarebbe completamente assorbita. In tal caso la emissività
130
Cap. 5 – Termodinamica e calorimetria
della superficie del foro è proprio la funzione universale K λ ( λ , T ) . Dal punto di
vista sperimentale, l’esistenza di un foro che metta in comunicazione l’interno e
l’esterno della cavità, consente la misura dell’emissività del foro.
La misura
viene fatta con un bolometro, apparecchio inventato da S. P. Langley nel 1881 in
cui una sottile lamina di platino, coperta di nerofumo, assorbe la radiazione
incidente su di essa e di cui si può misurare la variazione di temperatura attraverso
una variazione di resistenza elettrica. Naturalmente ammetteremo sempre che il
foro sia sufficientemente piccolo da non disturbare l’equilibrio della radiazione
all’interno della cavità. Un esempio al quale si può pensare è quello di un forno
del quale si vuole misurare la temperatura T, proprio osservando le caratteristiche
della radiazione emessa (cioè con un “pirometro”) attraverso la sua bocca.
Il problema del corpo nero è dunque quello del calcolo di K λ ( λ , T ) , ma possiamo
ridurre tale calcolo a quello di un’altra quantità: la densità di energia specifica
presente nella cavità in un intervallo infinitesimo di lunghezze d’onda o di
dE
dE
o uν =
. In queste formule E è l’energia totale nella
frequenze uλ =
dVd λ
dVd λ
V il
cavità
e
suo
volume
interno.
Naturalmente
porremo:
u=
E
=
V
+∞
∫
0
+∞
uλ d λ =
∫ uν dν .
0
Radiazione
entrante
Fig. 1: Corpo nero.
c
u λ d λ . In conclusione, ciò che
4
dovremo calcolare è la densità specifica di energia nella cavità.
Possiamo infatti dimostrare che K λ d λ = ε λ d λ =
131
Cap. 5 – Termodinamica e calorimetria
c
u λ d λ . Si
4
tratta, in effetti, di vedere quale è la
radiazione emessa attraverso il foro di
superficie dS nella parete sommando i
contributi provenienti, con eguale
Dimostriamo che: ε λ d λ =
dS’
θ
Radiazione
uscente
Fig. 2: Radiazione uscente dal corpo nero.
probabilità, da ogni direzione dP =
dΩ
.
4π
L’energia uscente dal foro è quella
contenuta con densità u in un cilindro di
base dS e di altezza c o, nel caso
d’incidenza non normale, di altezza
c cos θ :
d ( − cos θ ) dφ
dE
dΩ
dΩ
=u
c ⋅ n dS = u
c cos θdS = u
c cos θdS
dt
4π
4π
4π
π
2
Integrando su tutte le direzioni, si ha: ε = dE = cu ∫ cos θd ( − cos θ ) = cu .
dSdt
2
4
0
c
Derivando la relazione K = ε = u rispetto a λ, si ottiene:
4
Kλ = ελ = c uλ .
4
3. La legge di Stefan-Boltzmann
Boltzmann dimostrò teoricamente che il potere emissivo del corpo nero obbediva
∞
alla legge ε = ∫ ε λ d λ = σT 4 , Stefan confermò sperimentalmente il risultato di
0
Boltzmann, trovando: σ
= 5, 67 ⋅ 10 − 8 W m − 2 / K
(costante di Stefan-Boltzmann).
Dimostriamo adesso la legge di Stefan-Boltzmann. Calcoliamo dapprima la
pressione esercitata dalla radiazione sulle pareti della cavità, considerate
perfettamente riflettenti, tenendo conto del fatto che l’impulso trasportato da
un’onda è p =
E
. La densità di impulso è data
c
da u / c . Conseguentemente
132
Cap. 5 – Termodinamica e calorimetria
u
dΩ
c ⋅ ndS
da
c
4π
una direzione θ . Il contributo all’impulso ceduto alla parete per unità di tempo da questa
dp
dF
u
dΩ
=
= dP = c ⋅ n 2cosθ
= u cos2 θ d (− cosθ ) . Integrando
radiazione sarà allora:
dSdt dS
c
4π
l’impulso che arriva nell’unità di tempo su una superficie infinitesima è
π /2
su tutto l’angolo solido si ha: P =
u 36
∫0 u cos θ d (− cosθ ) = 3
2
. La pressione può essere
ottenuta in modo simile a quella delle molecole in un gas perfetto, immaginando
che
la
radiazione
sia
un
gas
di
fotoni:
dF N
2πd (− cosθ )
dP =
= f (v)dv
2mvcosθv cosθ , che va modificata, facendo le
dS V
4π
E hν
h
sostituzioni: a p = mv →
=
= , Nf ( v )dv → N * (tutte le onde hanno
c
c
λ
v→
c
velocità c ) e
. Otteniamo:
dF N 2π d ( − cos θ ) E
NE
2 cos θ c cos θ =
d ( − cos θ ) cos 2 θ =
=
.
dS V
4π
c
V
2
= ud ( − cos θ )cos θ
dP =
Integrando su θ per 0 ≤ θ ≤
π
, otteniamo: P =
u
.
3
2
Prendiamo adesso una cavità piena di radiazione in equilibrio termico con le
pareti a temperatura T 37. Come si è visto la pressione esercitata dalla radiazione
1
3
sulle pareti è data da P = u , con
u
densità di energia del campo em nella cavità.
Immaginiamo di fare un’espansione isoterma
36
della cavità proprio come in
Nel caso si abbiano delle pareti perfettamente assorbenti, il fattore 2 va eliminato. Se si
ha radiazione perpendicolare alla parete, il fattore un terzo va eliminato. L’esistenza di
una pressione di radiazione è stata predetta anche da Adolfo Bartoli (Firenze 1851-Pavia
1896) sulla base del secondo principio della termodinamica ed è stata verificata
sperimentalmente da Lebedev, Nichols e Hull (1901, 1903) che usarono una bilancia di
torsione con appesi al braccio orizzontale due dischi riflettenti da una parte e anneriti
dall’altra. Il lato annerito fu usato per misurare, attraverso l’aumento di temperatura, la
quantità di luce che cadeva sul disco. La parte riflettente per misurare la pressione. Punto
essenziale era l’altissimo vuoto raggiunto nella campana di vetro in cui la bilancia era
contenuta. L’altissimo vuoto era necessario per evitare l’effetto che fa ruotare il mulinello
dei radiometri di Crookes, secondo la spiegazione del radiometro data dallo stesso Bartoli.
37
Dimostrazione a pag. 61 di The theory of heat radiation – M. Planck
133
Cap. 5 – Termodinamica e calorimetria
termodinamica facciamo espandere un gas contro un pistone che scivola a tenuta
in un cilindro. Dal primo principio della Termodinamica, abbiamo:
1
1
4
δQ = TdS = pdV + dU = udV + d (uV ) = udV + Vdu + udV = udV + Vdu
3
3
3
da cui:
4
V
∂S
4
e
dS =
u
udV + du ⇔
=
3T
T
∂V 3T
∂S V du
∂2S
1 du
∂2S
4
4 du
1 du
4
=
⇔
=
=
=− 2 u+
⇔
=
u
∂T T dT
∂V∂T T dT ∂T∂V
3T dT
3T dT 3T 2
3T
du
dT
⇔
=4
⇔ ln(u ) = ln(T 4 ) + costante
u
T
ovvero: u ≈ T 4 38e poiché il flusso d’energia uscente per unità di tempo e
superficie ε dal corpo nero è proporzionale a u, abbiamo: ε = σ T 4 . Troviamo
pure che l’entropia è: S ≈ VT 3 .
4. La legge di Wien
Un’altra legge fondamentale fu scoperta da W. Wien (legge dello spostamento)
che asserisce che la emissività del corpo nero deve avere la forma:
ελ =
1
λ5
f (λ ⋅ T ) con
f
funzione del prodotto delle due variabili temperatura
assoluta e lunghezza d’onda. La legge di Wien può anche essere riscritta nella
T5
f (λ ⋅ T ) = T 5 g(λT ) . L’interesse di tale legge risiede,
forma: ε λ =
5
(λT )
naturalmente, nel fatto che, nota la funzione g = g ( x ) con x = λT , conosciamo
l’emissività del corpo nero a qualunque temperatura.
Spesso la legge dello spostamento viene riportata nella forma del prodotto tra la
lunghezza d’onda a cui occorre il massimo della curva d’emissione per la
temperatura a cui si considera tale curva: λ max T = cost . Questa seconda forma
della legge si ottiene dalla prima ponendo a zero la derivata dell’emissività
dε λ
= 0 per trovare il massimo della curva a T fisso:
dλ
38
Sulla legge di S.B. sono basati i comuni termometri a infrarossi per la misura della
temperatura corporea.
134
Cap. 5 – Termodinamica e calorimetria
dε λ
= −5λ −6 f (λ ⋅ T ) + λ −5 f '(λ ⋅ T )T = 0 ⇒ −5λ −1 f (λ ⋅ T ) + f '(λ ⋅ T )T = 0 . Detto C il
dλ
valore del prodotto λ ⋅ T per cui questa equazione è verificata, si ha: λmax ⋅ T = C .
Questa soluzione C è valida a qualunque temperatura, naturalmente, e pertanto la
λmax cambierà al cambiare della temperatura. Dunque tale legge fornisce la
lunghezza d’onda λmax al massimo dell’emissione in funzione della temperatura
del corpo nero. Inoltre il valore di ε λ per λ = λmax dipende dalla quinta potenza
della temperatura: ε λ ( λ max ) ≅ T 5 .
Si noti che dalla legge dello spostamento segue la legge di Stefan-Boltzmann.
∞
∫
Infatti: ε = ε λ dλ =
0
+∞
1
∫λ
5
0
f (λ ⋅ T ) dλ = T 4
+∞
∫
0
f ( x)
x
5
dx = σ T 4 , con
x = λ ⋅T .
Dimostriamo adesso la legge di Wien.
Consideriamo una cavità formata da un cilindro a pareti perfettamente riflettenti.
La cavità è chiusa da un pistone anche esso perfettamente riflettente. La cavità è
inizialmente piena di radiazione di corpo nero la cui distribuzione viene
modificata a causa del fatto che il pistone si muove verso l’esterno, con velocità v
, aumentando il volume V della cavità. Le modifiche alla distribuzione della
radiazione tra le varie frequenze è dovuta a due fattori: l’effetto Doppler sulla
superficie S del pistone in moto e il lavoro fatto dalla radiazione sul pistone per
muoverlo. Il primo dei due effetti cambia la frequenza da ν ' a ν in accordo con
la formula: ν = ν '(1 − 2 β cos θ ) . Il secondo riduce l’energia che ritorna dopo la
riflessione diminuita di un fattore: (1 − 2β cos θ ) 39. Naturalmente tutta l’energia
presente nell’intervallo tra dν salta in un nuovo intervallo di frequenze, mentre
l’energia nell’intervallo dν ' finisce nell’intervallo dν , se ν ' = ν (1 + 2 β cos θ ) .
Calcoliamo adesso l’energia che incide sul pistone riflettente di area S a
dΩ
cdt cos θ S questa viene inviata in un altro intervallo
frequenza ν : dEν = uν dν
4π
dΩ
. La pressione
4π
2
dΩ
2
2I
)cos θ = dI cos θ ⇒ P =
cos θ . Il lavoro
infinitesima su dS è dP = (uc cos θ
c
4π
c
c
2I
cos θ dSvdt = 2β dE cosθ . Pertanto: dE − dE ' = 2β dE cos θ ⇒
fatto sarà: dL =
c
dE ' = dE(1 − 2β cos θ ) .
39
Vediamo perché. L’energia che incide su dS in dt è: dI = uc cos θ
135
Cap. 5 – Termodinamica e calorimetria
di frequenze, svuotando l’intervallo dν , mentre l’energia riflessa dalla frequenza
ν ' = ν (1 + 2 β cos θ ) riempirà dν . Tale energia è quella incidente abbassata del
fattore (1 − 2β cos θ )
dΩ
dE 'ν ' = uν (ν ')dν '
cdt cos θ S(1 − 2β cos θ ) =
4π
dΩ
cdt cos θ S(1 − 2β cos θ )
= uν (ν + 2νβ cos θ )dν (1 + 2β cos θ )
4π
I due fattori, (1 + 2β cos θ ) e (1 − 2β cos θ ) dovuti al cambio di frequenza e alla
riduzione di energia discussi prima, danno un termine correttivo in β 2 che si può
trascurare. Dunque, abbiamo:
2π d ( − cos θ )
d ( − cos θ )
c
dE 'ν ' = uν (ν + 2νβ cos θ )dν
cdt cos θ S = uν (ν + 2νβ cos θ )dν
cos θ dV
4π
2
v
dV = Svdt . Facciamo adesso una espansione in serie di
Con
∂u (ν )
uν (ν + 2νβ cos θ ) = uν (ν ) + ν
2νβ cos θ + ... e, sostituendo otteniamo:
∂ν
∂u
∂u
1
d ( − cos θ )
dE 'ν ' = uν (ν ) + 2νβ ν cos θ
cos θ dV = dEν + ν ν cos 2 θ d ( − cos θ )dV .
2
∂ν
β
∂ν
∂uν
cos2 θ d ( − cos θ )dV è il cambiamento d’energia nell’intervallo
∂ν
1 ∂u
dν . Integrando sull’angolo, si ha: ν ν dV . Possiamo allora scrivere che la
3 ∂ν
1 ∂u
variazione d’energia totale nel volume V è: d (Vuν ) = ν ν dV . Ovvero:
3 ∂ν
∂uν
1 ∂uν
V= ν
− uν . Se troviamo una soluzione di questa equazione
3 ∂ν
∂V
differenziale, abbiamo la soluzione del nostro problema. Si può verificare che la
soluzione generale è della forma: uν = ν 3 f (ν 3V ) . Questo è quello che possiamo
dire, dunque non otteniamo la soluzione completa del problema, ma solo una
forma generale. Possiamo assumere che la espansione fosse adiabatica e porre la
variazione dell’entropia uguale a zero che sappiamo significare: VT 3 = costante
La quantità: ν
(vedi anche par. 8) e sostituire, ottenendo così:
ν
uν = ν 3 f ( )
T
ovvero:
1
f (λT ) . Quest’ultima è la forma in cui si trova espressa di solito la legge
λ5
di Wien. Possiamo ricordare che W. Wien aveva proposto una legge del corpo
uλ =
136
Cap. 5 – Termodinamica e calorimetria
nero nella forma: uν = αν 3 e
−β ν
T
−β ν
o ( u λ = α c5 e T ) ; in altre parole, aveva
λ
ipotizzato che la funzione f = f (λT ) della sua legge fosse un esponenziale.
Dal punto di vista sperimentale, si conosceva l’andamento di ελ . Si veda uno dei
risultati ottenuti da Lummer e Pringsheim nel 1897 nella figura che segue.
5. Il calcolo classico dell’emissività del corpo nero
Passiamo adesso al calcolo di uν (ν , T )
La densità media d’energia nella cavità è il prodotto del numero di frequenze
dn ν (per unità di volume) contenute in un intervallo di frequenze tra ν e ν + dν
moltiplicato per l’energia media della radiazione a queste frequenze. Il calcolo
delle frequenze possibili in una cavità è in effetti il calcolo delle frequenze
stazionarie nella cavità e può essere facilmente eseguito.
Possiamo riformulare il problema in un modo leggermente diverso. Il sistema
delle onde stazionarie presenti nella cavità possiede un numero di gradi di libertà
infinito: esistono cioè un infinito numero di frequenze stazionarie possibili.
Tuttavia tra due frequenze infinitamente vicine, ν e ν + dν , abbiamo un numero
finito di frequenze, cioè un numero finito di gradi di libertà. Il sistema delle onde
viene considerato in equilibrio con le molecole delle pareti, in altre parole alla
stessa temperatura. Ne segue che ogni grado di libertà (frequenza) avrà la stessa
1
energia media delle molecole delle pareti cioè kT . Trattandosi di oscillazioni
2
con metà della propria energia cinetica e metà potenziale, useremo in effetti il
valore kT , come avremmo fatto nel caso di oscillatori armonici. Dovrebbe
apparire così evidente che per calcolare l’energia media stiamo utilizzando il
teorema di equipartizione di Maxwell. Notare che usando il teorema di
equipartizione dell’energia, possiamo svolgere l’intera teoria senza affrontare i
dettagli del problema dell’interazione molecola-campo. In altre parole Rayleigh
cerca ancora di risolvere il problema usando solo la termodinamica mettendo da
parte il problema dell’interazione della radiazione con la materia.
Calcoliamo dapprima il numero di frequenze tra ν e ν + dν delle oscillazioni
elettromagnetiche possibili in una cavità a cubo di spigolo l . Naturalmente
prenderemo il cubo con tre pareti giacenti sui piani coordinati.
5a. La densità di frequenze possibili
137
Cap. 5 – Termodinamica e calorimetria
I campi devono soddisfare le equazioni di Maxwell40:
Risultati di Lummer e
Pringsheim (da E. Perucca,
Fisica Generale e Sperimentale)
Risultati di Lummer e
Pringsheim (da M. Born,
Atomic Physics)
Da questi grafici, determinando la posizione dei
massimi, si può verificare la legge di Wien e
trovare la costante C = λmax ⋅ T . Moltiplicando i
punti di ciascun grafico per λ 5 e mettendo in un
grafico i dati in funzione di λT , si ottiene la
funzione universale f (λT ) .
40
Rayleigh considerò invero, il caso di oscillazioni meccaniche stazionarie non onde em. Il
risultato è lo stesso.
138
Cap. 5 – Termodinamica e calorimetria
1 ∂2E
=0
∆E − 2
c ∂t 2
∇ ⋅ E = 0
∂B
∇ × E = − ∂t
∇ ⋅ B = 0
Prendiamo la soluzione nella forma: E = E ( x , y , z )e iω t e sostituendo otteniamo:
∆E + k 2 E = 0 .
E ( x, y, z ) = X ( x)Y ( y ) Z ( z ) ,
Se poniamo
otteniamo tre equazioni:
d2X
dx 2
+ k x2 X = 0 ;
d 2Y
dy 2
k2 =
+ k x2Y = 0 :
ω2
c2
d 2Z
dz 2
= k x2 + k y2 + k z2 ,
+ k x2 Z = 0 , che
ammettono come soluzioni delle funzioni trigonometriche.
Con la condizione al contorno che le componenti del campo elettrico tangenti ad
E y = E z = 0 per x = 0, l
una parete siano nulle sulle pareti: E x = E z = 0 per y = 0, l ,
E = E = 0 per z = 0, l
y
x
E x = E x 0 cos k x xsenk y ysenk z ze iωt
le soluzioni possono scriversi nella forma: E y = E y 0 senk x x cos k y ysenk z ze iωt
iωt
E z = E z 0 senk x xsenk y y cos k z ze
che, a causa della condizione sulla divergenza, sono soggette al vincolo:
E x 0 k x + E y 0 k y + E z 0 k z = 0 . Inoltre per soddisfare le condizioni al contorno deve
k x l = n x π
essere: k y l = n y π .
k z l = n z π
Sono soluzioni della equazione delle onde, quelle funzioni per cui:
k x2 + k y2 + k z2 =
n x2 + n 2y + n z2 =
ω2
2
c
4l 2
λ2
⇒π (
ny
nx 2
n
2π
, ovvero per cui:
) + ( )2 + ( z )2 =
l
l
l
λ
. Notiamo che la condizione E x 0 k x + E y 0 k y + E z 0 k z = 0 , ci
dice che, fissate le tre componenti del vettore d’onda, esistono due ampiezze
139
Cap. 5 – Termodinamica e calorimetria
indipendenti per il campo elettrico, la terza essendo fissata automaticamente da
questa relazione. Esistono dunque due gradi di libertà per il campo elettrico.
Passiamo al campo magnetico adesso.
Calcoliamo il rotore del campo elettrico:
(∇ × E ) x = (k y E z 0 − k z E yo ) senk x x cos k y y cos k z ze iωt
(∇ × E ) y = (k z E x 0 − k x E zo ) cos k x xsenk y y cos k z ze iωt
(∇ × E ) z = (k x E y 0 − k y E xo ) cos k x x cos k y ysenk z ze iωt
Ne segue che il campo magnetico è ( − iω B = ∇ × E ):
i
Bx =
ω
i
By =
Bz =
ω
i
ω
( k y E z 0 − k z E yo ) senk x x cos k y y cos k z ze iω t
( k z E x 0 − k x E zo )cos k x xsenk y y cos k z ze iω t
( k x E y 0 − k y E xo )cos k x x cos k y ysenk z ze iω t
per cui è soddisfatta automaticamente anche la condizione ∇ ⋅ B = 0 .
Notare che i due campi sono perpendicolari l’uno all’altro: E ⋅ B = 0 . In complesso
esistono due ampiezze arbitrarie per il campo elettrico, mentre le ampiezze del
campo magnetico sono fissate in funzione delle ampiezze del campo elettrico.
Calcoliamo adesso il numero di possibili oscillazioni con lunghezza d’onda
compresa tra λ e λ+dλ.
Sia k = ( k 1 , k 2 , k 3 ) il vettore d’onda. Perché esso corrisponda ad un’onda
stazionaria
deve
essere:
kili = niπ
k = k12 + k 22 + k 32 =
per
i = 1,2,3 .
Sarà:
n3
n
n
n
= π ( 1 )2 + ( 2 )2 + ( 3 )2 ⇒
l1
l2
l3
1
λ
=
ν =
n
n
k
1 n1 2
=
( ) + ( 2 )2 + ( 3 )2
2π 2 l1
l2
l3
c
λ
=
n2
n
n
c n1 2
( ) + ( 2 )2 + ( 3 )2
2 l1
l2
l3
In particolare per un cubo per cui
l1 = l2 = l3 = l , si ha:
n1 Fig. 5: Spazio con assi n1, n2, n3.
140
Cap. 5 – Termodinamica e calorimetria
n1 + n2 + n3 =
2
2
2
4l 2 2 4l 2
ν = 2 .
c2
λ
Nello spazio n1, n2, n3 (fig. 23), questa relazione è l’equazione di una sfera di
2lν
raggio R =
. Da questa espressione si ricava che nello spazio delle frequenze
c
si ha un punto rappresentativo per ogni cubetto di lato unitario, ovvero una densità
di punti rappresentativi unitaria. Dunque, per calcolare il numero di punti, ovvero
il numero di possibili oscillazioni tra le frequenze 0 e ν , occorre calcolare il
2lν
volume della sfera di raggio R =
(e moltiplicare per la densità che è uno).
c
In verità, la frequenza minima non è zero, ma piuttosto
ν min =
3c 41
, come si
2l
ricava ponendo n1 = n2 = n3 = 1 nella relazione precedente. In conclusione, il
numero di oscillazioni possibili tra ν min ≈ 0 e ν è:
nν = 2
1 4
8π l 3ν 3
8π l 3
, dove il fattore 2 è dovuto ai due stati di
( π R3 ) =
=
8 3
3c 3
3λ 3
polarizzazione e il fattore 1/8 deriva dal fatto che i tre numeri n1, n2, n3 devono
essere positivi e, dunque, si è interessati solo ad un ottante della sfera
Dividendo per il volume ( V = l 3 ) e differenziando, si ottiene infine il risultato
8π
8 2
cercato: dnν = πν3 dν e dnλ = 4 d λ .
λ
c
41
Evidentemente, si può dire anche che esiste una
λmax = l / 2 . Questo in effetti implica
che il numero di stati tra λ e λ + dλ dovrà diminuire all’aumentare di λ. E’ in effetti
questo l’aspetto chiaro della formula di Rayleigh-Jeans. La diminuzione a piccole λ
risulta invece incomprensibile, perché non esiste un meccanismo classico che limita lo
scambio energetico tra oscillatori nelle pareti e radiazione.
141
Cap. 5 – Termodinamica e calorimetria
F1
6
2,0x10
6
1,8x10
6
1,6x10
6
1,4x10
6
dnν/dν
1,2x10
6
1,0x10
5
8,0x10
5
6,0x10
5
4,0x10
visibile
5
2,0x10
0,0
0,00E+000
2,00E+014
4,00E+014
6,00E+014
8,00E+014
1,00E+015
ν(Hz)
5b. L’energia media
Per il calcolo dell’energia media pertinente a queste frequenze, Reyleigh decise di
usare il teorema di equipartizione dell’energia già visto nella teoria di Maxwell,
che per il caso di un oscillatore armonico lineare dà kT (metà per l’energia
cinetica e metà per quella potenziale). In effetti le onde stazionarie hanno
un’energia proporzionale al quadrato dell’ampiezza. Esistono però due ampiezze
arbitrarie, come si è visto, per il campo elettrico. Poniamo ε = α x 2 , dove x è una
delle quattro ampiezze, e assumiamo che la probabilità di avere un’ampiezza x
+∞
sia: dP =
dx
e
+∞
∫
0
−
∫x
2
ax
kT
ax 2
−
e kT
allora l’energia media sarà in effetti: ε = α
2
e
−
ax 2
kT
0
+∞
∫
ax 2
−
e kT
dx
=
kT
2
0
che moltiplicata per le due ampiezze dà appunto kT . In effetti, questa formula
implica una sostanziale equivalenza tra un sistema di oscillatori armonici e il
nostro sistema di onde stazionarie. Un altro modo di giustificare la scelta
dell’energia media è il seguente. Ricordiamo che la densità d’energia del campo
142
Cap. 5 – Termodinamica e calorimetria
elettromagnetico è data da ε 0 E 2 +
B2
tutto il volume V , H = (ε 0 E 2 +
B2
∫
V
µ0
µ0
. L’energia totale si otterrà integrando su
)dV .
Torniamo alle componenti del campo elettrico:
E x = E x 0 cos k x xsenk y ysenk z zeiωt
iω t
E y = E y 0 senk x x cos k y ysenk z ze
iωt
E z = E z 0 senk x xsenk y y cos k z ze
e calcoliamo il quadrato del modulo del campo elettrico integrato su tutto lo
spazio interno alla cavità. Partiamo da una delle componenti:
Ex2 = Ex20 (cos k x xsenk y ysenk z z )2 e2iωt . Poniamo F (t ) =
eiωt
⇒ eiωt = Fɺ ( x) e
iω
riscriviamo, integrando:
l
l
l
l3 2 ɺ 2
V
2
ɺ
E = E ∫ cos k x xdx ∫ sen k y ydy ∫ sen k z zdzF (t ) = Ex 0 F (t ) = Ex20 Fɺ 2 (t )
8
8
0
0
0
Sommando le tre componenti e moltiplicando per ε 0 otteniamo per l’energia del
V
2
2
2
2
campo elettrico: H E = ε 0 E02 Fɺ (t ) , con E0 = Ex 0 + E y 0 + Ez 0 . Andiamo adesso
8
2
x
2
x0
2
2
2
al campo magnetico:
Bx =
(k × E0 ) x
senk x x cos k y y cos k z zeiωt = (k × E0 ) x senk x x cos k y y cos k z zF (t )
iω
e simili per le altre due componenti. Quadriamo e integriamo sul volume:
l
B = (k × E )
2
x
2
0 x
l
∫ sen k xdx ∫ cos
2
x
0
0
l
2
k y ydy ∫ cos 2 k z zdzF 2 (t ) =
0
V
(k × E0 ) 2x F 2 (t ) .
8
Sommando sulle tre componenti e dividendo per µ0 , otteniamo per l’energia del
campo magnetico: H B =
V 2 2 2
E0 k F (t ) . Sommando i due contributi all’energia,
8
abbiamo:
143
Cap. 5 – Termodinamica e calorimetria
H=
V
1 2 2 2 V
1 2 2
2 2
2
2
k F (t ) =
ε 0 E0 Fɺ (t ) + k E0 F (t ) = ε 0 E0 Fɺ (t ) +
8
µ0
µ 0ε 0
8
(
)
(
)
V
V
ε 0 E02 Fɺ 2 (t ) + c 2 k 2 F 2 (t ) = ε 0 E02 Fɺ 2 (t ) + ω 2 F 2 (t )
8
8
1
Poniamo adesso: x =
ε 0V E0 F (t ) e sostituendo si ottiene:
2
1
1
H = xɺ 2 + ω 2 x 2 che rappresenta appunto l’energia di un oscillatore lineare di
2
2
=
pulsazione ω e di massa unitaria. Il valore medio dell’energia di un sistema di
tali oscillatori è appunto kT .
8 2
Dunque, sostituendo nella formula precedente, si otterrà: uν = πν3 kT
uλ =
8π
λ4
o
c
kT .
L’ultima formula è detta formula di Rayleigh-Jeans (1900-1905)42. Essa
rappresenta un risultato catastrofico, in quanto per λ →0 , ε λ → ∞ (fig. 21): ciò
significa che l’energia totale emessa dal corpo nero è infinita!
Il comportamento di questa funzione, a grandi λ, descrive, invece, abbastanza
bene i risultati sperimentali: il problema a piccole λ sorge perché l’energia media
è indipendente da λ e dunque non offre un andamento accettabile per λ →0
(catastrofe ultravioletta). Si noti anche che questa formula soddisfa la legge dello
spostamento di Wien, potendosi scrivere: u λ =
8π
λ5
k ⋅ ( λ T ) . Possiamo ricordare che
W. Wien aveva proposto una legge del corpo nero nella forma: uν = αν 3 e
uλ = α
c
λ5
−β ν
e T
−β ν
T
o(
) ; in altre parole, aveva ipotizzato che la funzione f = f (λT )
della sua legge fosse un esponenziale. Questa formula si adatta bene ai dati a
grande frequenza ovvero piccola λ. Ciò fa pensare che la legge di Rayleigh-Jeans
e quella proposta da Wien siano i limiti a cui tende uλ a grande e a piccola λ
42
Anche H.A. Lorentz ottenne questa nel caso di un metallo contenente un gas di elettroni
liberi, modello già usato per calcolare la conduttività e poi il coefficiente di assorbimento,
Calcolando l’energia media irraggiata dagli elettroni durante le collisioni col reticolo
cristallino, si ottiene l’emissività. Dividendo infine per il coefficiente di assorbimento si
otteneva la funzione universale di Kirchhoff. Dai dettagli del calcolo era chiaro che la
formula valeva solo per grandi lunghezze d’onda (1902).
144
Cap. 5 – Termodinamica e calorimetria
3,0x10
10
2,5x10
10
2,0x10
10
1,5x10
10
1,0x10
10
Wien
Planck
R.-J.
3
ελ (J/(sm )
rispettivamente. Nella figura precedente sono riportate le curve di Wien, la curva
di Rayleigh-Jeans e la curva finale del corpo nero (linea tratteggiata) calcolata nei
prossimi paragrafi. Tutte queste curve sono calcolate a T = 10 3 K . Vogliamo anche
menzionare che, qualche anno più tardi, Einstein giudicava che la formula di
Rayleigh-Jeans fosse l’unica compatibile con la fisica classica.
Secondo Rayleigh l’origine della difficoltà stava nella non applicabilità del
principio di equipartizione dell’energia, peraltro mai dimostrato in generale. Per
inciso, Jeans riteneva invece che gli spettri sperimentali erano spettri non
d’equilibrio e che la distribuzione d’energia d’equilibrio si poteva raggiungere solo
in tempi lunghissimi. Questo tuttavia avrebbe reso inaccettabili le dimostrazioni
delle leggi di Stefan-Boltzmann e di Wien in cui abbiamo applicato la
termodinamica a stati d’equilibrio.
3
T=10 K
9
5,0x10
0,0
0,0
-6
5,0x10
-5
1,0x10
1,5x10
-5
-5
2,0x10
λ(m)
Si può ottenere la relazione tra la densità di energia della radiazione e quella
media degli oscillatori immaginando di introdurre il meccanismo di interazione
radiazione-materia.
Il modello di cavità che usiamo è quello di una cavità le cui pareti siano formate
da oscillatori unidimensionali (atomi). Gli oscillatori assorbono ed emettono la
radiazione e sono in equilibrio con il campo (tanta energia emessa tanta energia
assorbita). E’ vero che gli atomi non sono degli oscillatori, ma la cosa non è
rilevante: il sistema fisico immaginato è costruito secondo le leggi della fisica e
deve comunque condurre a risultati sensati.
145
Cap. 5 – Termodinamica e calorimetria
L’energia emessa per unità di tempo da un oscillatore armonico con frequenza
propria ω 2 =
k
m
è data dalla formula di Larmor: P =
2e 2
1
4πε 0 3c 3
a2.
Per un
x (t ) = A cos(ωt + ϕ )
oscillatore armonico (libero) abbiamo: xɺ (t ) = −ωAsen (ωt + ϕ ) .
2
ɺxɺ(t ) = −ω A cos(ω t + ϕ )
La potenza media irraggiata in un periodo dall’oscillatore armonico è:
T
T
2e2 1 2
1 2e2 1 2 4
1 e2 2 4
P=
a dt =
A ω cos2 (ωt + φ )dt =
Aω .
4πε 0 3c3 T
4πε 0 3c3 T
4πε 0 3c3
0
0
1
∫
∫
Mediando sulle ampiezze, si ha: P =
1
e2
4πε 0 3 c 3
A 2ω 4 .
Possiamo riscrivere questa
relazione notando che l’energia media di un oscillatore è:
1
1 2
mA 2ω 2
. Mediando sulle ampiezze A degli oscillatori:
kx ) =
2
2
2
2
mA2ω 2
, si ha infine: P = 1 2 e ε (2πν ) 2 . Questa è l’energia ceduta in
ε =
2
4πε 0 3 mc 3
ε = ( mxɺ 2 +
media dagli oscillatori di frequenza ν e ampiezza arbitraria al campo em
(calcolata per primo da H. Hertz).
Si può anche calcolare l’energia ceduta dal campo em agli oscillatori di frequenza
ν
2
per unità di tempo che risulta essere: P = 1 e uν .Il calcolo dell’energia
4ε 0 3m
ceduta agli oscillatori viene fatto nel modo seguente.
Poniamo Ex (t ) =
1
+∞
iωt
g (ω)e
2π ∫
−∞
dω , in cui g(ω) =
1
2π
trasformata di Fourier del campo elettrico e inoltre g *(ω) =
densità d’energia è (indicando con
+∞
−iωt
∫0 Ex (t)e
1
2π
+∞
dt è la
−iωt
∫0 Ex (t)e
dt . La
T →∞ un lungo intervallo di tempo):
146
Cap. 5 – Termodinamica e calorimetria
+∞
u=
∫
uν dν = ε 0 E 2 = 3ε 0 E x2 = 3ε 0
1
T
T
∫
0
= 3ε 0
E x2 (t ) dt = 3ε 0
0
1
T
+∞
∫
+∞
1
g (ω ) d ω
2π
−∞
∫
E x (t ) eiω t dt = 3ε 0
0
1
T
T
∫
E x (t )
0
1
T
2π
+∞
∫
g (ω ) g * (ω ) d ω = 3ε 0
−∞
+∞
1
∫ g (ω )e
iω t
d ω dt =
−∞
2
2π
T
+∞
∫
2
g (ν ) dν
0
1 uν T
si trova così: g (ν ) =
.
2π 3ε 0 2
2
ɺɺ + ω02 x =
L’equazione dell’oscillatore armonico è: x
e
g(ω )e iωt . Una soluzione della
m
quale, nulla assieme alla velocità all’istante zero, è:
e − iω0 t
ω e iω0t
ω
x( t ) = f (ω )e iω t − f (ω )
1 −
+
1 +
2
ω0
2 ω0
la cui derivata prima è:
e − iω0 t
ω e iω0t
ω
xɺ (t ) = iω f (ω )e iω t − iω0 f (ω ) −
1 −
+
1 +
e la seconda è:
2 ω0
2 ω0
e − iω0 t
ω e iω0t
ω
ɺɺ
x(t ) = −ω 2 f (ω )e iω t + ω02 f (ω )
1 −
+
1 +
. Si vede
2
ω0
2 ω0
immediatamente che la soluzione ha x( t ) = 0 e xɺ ( t ) = 0 . Che si tratti di una soluzione si
vede rimpiazzando nell’equazione. Si ottiene:
e − iω0 t
ω e iω0 t
ω
ɺɺ
1
x ( t ) = −ω 2 f (ω )e iω t + ω02 f (ω )
−
+
1 +
+
2
ω0
ω0
2
e − iω0 t
+ω02 f (ω )e iω t − ω02 f (ω )
2
ω
1 −
ω
0
e iω0 t
+
2
(
ω
1 +
ω
0
2
2
iω t
= ω0 − ω f (ω )e
(
)
2
2
iωt
Perché questa sia una soluzione basta che: ω0 − ω f (ω )e =
f (ω ) =
m
(
e
ω02
− ω2
)
scrive dunque: x ( t ) =
)
e
g(ω )e iωt , ovvero:
m
g (ω ) . La soluzione generale sommata su tutte le frequenze si
1
2π
+∞
eg (ω )
∫ m (ω
−∞
2
0
−ω2
)
i t e − iω 0 t
ω e iω 0 t
ω .
ω
1 −
+
1 +
e −
2
ω0
ω 0
2
Il lavoro fatto dal campo sull’oscillatore nell’unità di tempo sarà:
147
Cap. 5 – Termodinamica e calorimetria
1
T
δW =
T
∫ F (t ) xɺ (t )dt =
0
T −∞
e2
=
2π mT
∫∫
g(ω )e iω t
1
(ω
2
0
0 −∞
− ω '2
e − iω0t
ω ' e iω0t ω '
iω ' t
− iω0 g(ω ') −
1 −
+
1 +
d ω d ω ' dt
iω ' g(ω ')e
2 ω0
2
ω0
)
Per convenienza, scriviamo l’integrale come tre integrali e invertiamo l’ordine
dell’integrazione, integrando prima sul tempo, abbiamo:
δ W1 =
e2
2π mT
T −∞
∫∫
g (ω ) i ω '
0 −∞
T
g (ω ')
ω02 − ω '2
∫
d ω d ω ' e i (ω + ω ') t dt , ma e i (ω + ω ') t dt = 2πδ (ω + ω ')
0
, per cui:
δ W1 =
e2
mT
−∞
∫
g (ω ) i ω '
−∞
g (ω ')
ω 02 − ω 2
δ (ω + ω ')d ω d ω ' = −
e2
i
mT
. Ricordare che g ( −ω ) = g * (ω ) .
Questo integrale si risolve con i residui e si ha:
−∞
g (ω )
∫ω
−∞
2
0
2
−ω
2
ω d ω = 2π i lim(ω 0 − ω )
conclusione: δ W1 =
δ W2 =
=
e2
2π mT
e2
mT
= −i
ω = 2π i
g (ω 0 )
2
ω0 + ω0
ω0 =
ω 02 − ω 2
dω = −
e2
i
mT
1
2
2π i g (ω 0 ) . In
2
e
2
2π g (ω0 ) .
2mT
T −∞
∫ ∫ g(ω )e
i (ω −ω0 ) t
iω0 g(ω ') ω0 − ω '
(
2 ω02 − ω '2
0 −∞
)
ω0
d ω d ω ' dt =
ig(ω ')
d ω dω ' =
0 + ω ')
0
−∞
e
ig (ω0 )
=
mT
=
−ω
2
−∞
g ( −ω )
∫ g(ω )δ (ω − ω ) 2 (ω
2
δ W3 =
ω 02
2
∫
g (ω )ω
2
2 −∞
e
mT
g (ω )
−∞
−∞
∫
−∞
e2
2π mT
g (ω ')
e2
e2
2
dω ' = −
2π g (ω0 ) g ( −ω0 ) = −2π
g (ω 0 )
2mT
2mT
2 ( ω0 + ω ' )
T −∞
∫ ∫ g(ω )e
0 −∞
−∞
∫ g(ω )δ (ω
0
−∞
e2
g (ω 0 )
mT
−∞
∫
−∞
+ ω)
i (ω0 + ω ) t
− iω0 g(ω ') ω0 + ω '
(
2 ω02 − ω '2
)
ω0
d ω d ω ' dt =
− ig(ω ')
d ω dω ' =
2 ( ω0 − ω ' )
g (ω ')
e2
2
d ω d ω ' = 2π
g (ω 0 ) .
2 mT
2 (ω0 − ω ')
148
−∞
g (ω )
∫ω
−∞
2
0
2
−ω2
ω dω
Cap. 5 – Termodinamica e calorimetria
In conclusione:
δ W = δ W 1 + δ W 2 + δ W 3 = 2π
e2
e 2 1 uν T
e 2 uν .
2
g ( ω 0 ) = 2π
=
2m T
2 m T 2π 3 ε 0 2
3 m 4ε 0
Poiché, all’equilibrio, l’energia ceduta dagli oscillatori al campo e quella ceduta
dal campo agli oscillatori devono essere eguali, abbiamo:
1
2e 2
4πε 0 3 m c
3
ε (2πν ) 2 =
1
4ε 0
e2
8π
u ⇔ uν = 3 ν 2 ε
3m ν
c
che è esattamente la formula
trovata precedentemente. La differenza tra la trattazione di Rayleigh e questa
appena presentata sta nel fatto che il simbolo ε rappresenta l’energia media delle
oscillazioni em nel primo caso e quello degli oscillatori nel secondo caso. Nel
secondo caso l’energia media è senza dubbio kT , almeno che la statistica di
Boltzmann non risulti inapplicabile in questo caso per ragioni ignote. Ne segue, tra l’altro,
che l’energia media delle oscillazioni (onde) e quella degli oscillatori sono uguali come
era in effetti stato assunto nella trattazione di Rayleigh43.
6. La teoria del corpo nero secondo Planck
La strada verso la soluzione del problema la indicò Max Planck (1900). Il
modello usato è quello della cavità piena di oscillatori armonici (risuonatori). Egli
fece notare che il terzo principio della termodinamica (teorema di Nernst)
prevedeva che l’entropia di un sistema e, dunque anche di un sistema di oscillatori
armonici, deve essere nulla allo zero assoluto: l’entropia di un sistema
termodinamico, come è stata definita da Boltzmann, non è tuttavia zero, essendo
definita a meno di una costante. La ragione per cui la costante additiva rimane
arbitraria viene identificata nel fatto che le cellette dello spazio delle fase hanno
una dimensione comunque piccola. Secondo Planck, se si assume che le cellette
hanno una dimensione piccola, ma non infinitamente piccola, allora si può
ottenere un’entropia nulla allo zero assoluto. Si introduce così una costante hche
ha le dimensioni del prodotto di una quantità di moto per una lunghezza (o, se si
preferisce di un’azione: un’energia per un tempo). Ciascuna celletta dello spazio
delle fasi avrà così un volume pari a h 3 . All’interno di ciascuna celletta l’energia
è la stessa per ogni oscillatore. Il
procedimento di Planck è detto
“quantizzazione” dello spazio delle fasi, ma esso quantizza anche le possibili
energie di oscillazione del campo e.m. La ragione che induce Planck a suggerire
una quantizzazione dello spazio delle fasi è la seguente.
Boltzmann ha calcolato l’entropia di un gas come il logaritmo della probabilità
W di quello stato, lasciando indeterminata una costante di proporzionalità che per
43
Che le due energie medie debbano essere uguali è, secondo Poincaré conseguenza del
secondo principio della termodinamica. Vedi: H. Poincaré, L’ipotesi dei quanti in
Geometria e caso Bollati – Boringhieri. Pag. 183.
149
Cap. 5 – Termodinamica e calorimetria
le proprietà del logaritmo si traduce in una costante additiva incognita sul valore
dell’entropia. Vediamo perché. Se mettiamo insieme due sistemi le cui probabilità
siano W1 e W2 , la probabilità di quel sistema è il prodotto delle due probabilità
W = W 1W 2 mentre l’entropia deve essere additiva. Posta dunque l’entropia come
una
funzione
della
probabilità
S = f (W ) ,
abbiamo:
f (W ) = f (W1W 2 ) = f (W1 ) + f (W 2 ) . Derivando rispetto a W1 , abbiamo:
fɺ (W )W 2 = fɺ (W 1 ) , derivando nuovamente, ma questa volta rispetto a W2
abbiamo: fɺ (W ) + W ɺɺf (W ) = 0 , la cui soluzione è f (W ) = k ln(W ) + costante .
Questo giustifica la scelta della funzione logaritmo che risulta più che una
semplice convenienza, ma ci dà anche una costante additiva incognita. La costante
moltiplicativa viene identificata con la costante di Boltzmann. Planck obietta alla
costante additiva e dichiara di volere assegnare un valore assoluto all’entropia,
perciò la propone come una costante moltiplicativa nel logaritmo. Sceglie poi di
n
riscrivere l’entropia usando le probabilità matematiche wi = i (invece di n i ) di
N
44
occupazione di ogni cella dello spazio delle fasi e ottiene : S = − kN wi ln( wi ) .
∑
i
Eventualmente,
S = kN
l’entropia
deve
∫ w ln wdxdydzdp dp dp .
x
y
z
essere
calcolata
usando
un
integrale:
Tuttavia così facendo, abbiamo cambiato le
Vsf
dimensioni dell’espressione a destra. Per risolvere il problema possiamo dividere
per una costante h 3 con le dimensioni dello spazio delle fasi. Planck postula
allora l’esistenza di una dimensione naturale per le cellette. Egli scrive: “That
such a definite quantity (cioè h) really exists is a characteristic feature of the
theory we are developing, as contrasted with that due to Boltzmann and forms the
content of the so-called hypothesis of quanta”45. In conclusione, le cellette hanno
una dimensione finita, come indicato dall’esistenza della costante h. All’interno
di ogni celletta è impossibile definire le variabili dinamiche eccetto che come
medie. Da questo nasce la quantizzazione dell’energia che useremo sugli
oscillatori armonici. Vediamo cosa succede.
n1
44
−
∑ n ln w
i
i
45
n
n1
n
n
( ) 1 ...
( ) 1 ...
n ! n !...
n 1
ln W = − ln 1 2
= − ln eN N = − ln Ne n1 +n2 ... = − ln 1 ... = − ln( w1n1 ...) =
(e)
(e)
N!
N
i
= −N
∑ w ln w
i
i
i
M. Planck - The theory of heat radiation. Dover, pag. 125
150
Cap. 5 – Termodinamica e calorimetria
Consideriamo lo spazio delle fasi degli oscillatori armonici. L’energia di un oscillatore è:
1
1
1 2 1
p2 +
mω 2 x 2 = 1
E=
p + mω 2 x 2 , che, riscritta nella forma:
2mE
2E
2m
2
rappresenta un’ellisse nello spazio delle fasi di semiassi: a = 1 2 E e b = 2 mE , la cui
ω m
E
area è: S = pdx = πab = 2πE / ω = . Consideriamo adesso una sequenza di queste
ν
ellissi di area h, 2h, 3h… in modo che l’intercapedine tra due ellissi di contorno sia
E
sempre h, avremo46: S n = pdx = nh = n ⇒ E n = nhν per i valori dell’energia
ν
corrispondenti alle varie ellissi.
La condizione posta da Planck è che la statistica degli oscillatori a frequenza ν conduca ad
un valore nullo dell’entropia allo zero assoluto, come richiesto dal principio di Nernst (o
terzo principio della termodinamica). Possiamo calcolare l’energia media nel modo
seguente. L’equazione parametrica del moto di un oscillatore armonico è: x (t ) = A sin ϕ ,
con ϕ = ωt +α . La quantità di moto è: p x = Amω cos ϕ . Da queste si ricava:
∫
∫
2
2
x px
A + Amω = 1. In parole: la traiettoria nello spazio delle fasi di un oscillatore
armonico è un’ellisse, la cui area è: S = π A 2 mω . Poniamo adesso le aree di queste ellissi
uguali a: 0, h,..., nh . In questo modo l’area compresa tra due ellissi in sequenza è h
L’ellisse che limita l’ennesima è caratterizzata da un’ampiezza d’oscillazione:
nh
An2 =
. Supponiamo che all’interno di ogni area limitata da due ellissi sequenziali,
π mω
la distribuzione di punti rappresentativi sia uniforme, allora l’energia in ogni area è:
Nν ,n 1 2
Nν ,n
2
Eν ,n =
kA dxdpx , in cui
è la densità dei punti e 12 kA è l’energia
2
h
h
∫
C
dell’oscillatore. Cambiamo variabili ed usiamo piuttosto ϕ , A al posto di x, px . Abbiamo:
46
Notare l’identità di questa condizione con quella di Bohr per l’atomo d’idrogeno. Bohr
estende in effetti questa condizione ad un sistema periodico con due gradi di libertà
151
Cap. 5 – Termodinamica e calorimetria
Eν ,n =
Nν ,n
h
∫
1 mω 2 A3 mω dAd ϕ
2
C
Eν ,n =
Nν ,n mω 3
h
Nν ,n m ν
∫
π A dA =
3
2 3
=2
h
Nν ,n mω3
C
otteniamo:
π4
h
2
π m (2πν )2
2 2
h
A4
π
4
An
=
n = 1,... + ∞
An −1
(
)
(n2 − (n − 1)2 ) = Nν ,n hν n − 12 ,
Ovvero l’energia media in ciascuna areola tra due ellissi sequenziali d’ordine
Eν , n =
ϕ,
, in cui mωA è lo jacobiano. Integrando su
Eν , n
Nν , n
(
= hν n −
1
2
n
e n−1 è:
).
p
E3
E2
E1
x
h
h
h
Planck calcola poi l’entropia, la sua relazione con l’energia totale Eν degli oscillatori a
frequenza ν e quale frazione del loro numero totale Nν ( wn ) si trovi in ogni areola di
superficie h , in modo da calcolarne l’energia media da cui poi ottenere uν .
Abbiamo come espressione dell’entropia:
Nν !
, come per Botzmann. Planck riscrive questa espressione in una
S = k ln
n1 ! n2 !...
maniera diversa da Boltzmann, facendo apparire esplicitamente le probabilità di
n
occupazione di un’areola di superficie h, w i = i ( ( w1 + w 2 + ...) = 1 ) .
Nν
152
Cap. 5 – Termodinamica e calorimetria
S = − k ln
n1 ! n2 !...
= − k ( − Nν ln Nν − Nν + n1 ln n1 − n1 + n2 ln n2 − n2 ...) =
Nν !
= − k ( − Nν ln Nν + n1 ln n1 ...) = − kNν ( −( w1 + w2 + ...) ln Nν + w1 ln n1 + ...) =
= − kNν ( w1 ln
n1
+ ...) = − kNν
Nν
∑ w ln w
i
i
i
Troviamo adesso lo stato d’equilibrio col solito metodo dei moltiplicatori arbitrari, a
energia totale fissa e numero totale di oscillatori fisso.
∞
∑
∞
wn = 1 ⇒
n =1
=0
n
n =1
Eν = Nν hν
S = −kNν
∑
n =1
∑δ w
δE
(n − )wn ⇒ ν = ∑(n − )hνδ wn = ∑nhνδ wn = 0
∑
2
2
Nν
n=1
n=1
n=1
1
1
δS
wn ln wn ⇒
= 0 = ∑( ln wn +1) δ wn = ∑ln wnδ wn
∑
kNν
n=1
n=1
n=1
(ln wn + β nhν + α )δ wn = 0 ⇒ ln wn + β nhν + a = 0 ⇒ wn = e − a e − β nhν =
Poniamo adesso:
e − β nhν
e − β nhν
∑
n =1
.
wn = aγ n , con γ = e−βhν e
1
1− γ
=
1
γ
e
γ −1
−1
γ
1
−
n =1
n =0
Da questa espressione ricaviamo l’energia media degli oscillatori a frequenza ν
E
1
1− γ
1
1 1− γ γ
1− γ
Eν = ν = hν
hν
+
hνγ 2
nγ n −1 =
(n − ) wn =
(n + )γ n +1 = hν
γ
γ 1− γ
γ
Nν
2
2
2
a=
∑
1
− β nhν
=
∑
1
=
n
∑
∑
n =1
1
1− γ
d
= hν +
hνγ 2
2
dγ
γ
∑
n=0
∑
n =0
1
1− γ
d
1
hνγ 2
(
)=
γ n = hν +
2
dγ 1 − γ
γ
n=0
1
1 − γ 2 hν
1
hνγ
1
hν e β hν
1
hν
hν +
= hν +
= hν +
= hν +
γ
2
h
−
β
ν
β
2
2
1− γ 2
2
γ
(1 − γ )
1− e
e hν − 1
Per quanto riguarda l’entropia abbiamo:
∞
S
−
=
( wn ln wn ) =
(aγ n ln aγ n ) = a
γ n (ln a + n ln γ ) =
kNν
n =1
n =1
n=1
∞
∞
∞
∞
γ n + ln γ
= a ln a
nγ n = a ln a( γ n − 1) + ln γ
nγ n =
n =1
n =1
n =0
n =1
=
∑
∑
∑
∑
∑
∑
∑
153
Cap. 5 – Termodinamica e calorimetria
1
= a ln a (
− 1) + γ ln γ
1− γ
1− γ
il
γ
γ
+ γ ln γ
nγ n −1 = a ln a
1− γ
n =1
∑
∞
∑ nγ
n −1
n =1
=
1− γ 1− γ
1
d 1
γ ln γ
γ ln γ
ln
=
+
+
=
= ln
γ
γ 1− γ
γ
γ
γ
dγ 1 − γ
(1 − γ ) 2
1− γ
1
1
γ
= ln
+ ln γ
= − ln γ + ln(1 − γ ) +
ln γ =
ln γ + ln(1 − γ ) . Identificando β
γ
1− γ
1− γ
1− γ
con
1− γ
∞
1− γ
1
,
kT
solito
otteniamo
hν
−
hν
1
S = kNν
− ln 1 − e kT
hν
kT
e kT − 1
Eν =
infine:
1
hν +
2
hν
hν
e kT
e
−1
Il cui valore è zero allo zero assoluto come
volevamo in osservanza del teorema di Nernst. Si noti pure che se h→ 0 , S → ∞ .
1
β=
L’identificazione
si
fa
con
la
relazione:
TdS = dEν
kT
T
dEν
dS
kT
1
=
( − β hν ) =
=
hν .
2
Nν
Nν
(1 − γ )
(1 − γ ) 2
L’energia media della radiazione sarà la stessa degli oscillatori a parte il valore a T=0:
Eν , rad = hν
e
−
hν
kT
hν
−
kT
1− e
=
hν
hν
kT
e
.
−1
Che ci dà il risultato cercato sull’energia media degli oscillatori a frequenza ν , risultato
che sostituisce quello del teorema di equipartizione. Si noti anche che se h → 0 ,
Eν → kT . Questo porta ad una conclusione interamente nuova, perché, usando questa
2
funzione al posto di kT , si ha: uν = 8π v
3
c
hν
hν
e kT
=
8π h
−1
ν3
hν
e kT
c3
−1
Si ha cioè, che Eν è una funzione della frequenza, tale che per
Eν ≈ ν e
−
hν
kT
.
ν → ∞
(ovvero
λ →0 )
→ 0 . La catastrofe ultravioletta è così scongiurata. In conclusione, si ha per
2
lo spettro del corpo nero: εν = c 8π v
3
4
c
hν
hν
e kT − 1
=
2π h
c2
ν3
hν
e kT − 1
154
Cap. 5 – Termodinamica e calorimetria
formula che descrive bene i dati sperimentali. Si può evidentemente anche scrivere:
1
2π hc 2
1
(che soddisfa la legge di Wien: ε λ = 5 f (λ T ) !). Si vede
ελ =
5
hc
λ
λ
λ kT
−1
e
comunque che, per energia del fotone piccola rispetto all’energia media a
temperatura T ( hv << kT ), cioè per ν → 0 o T →+∞, si ritorna al limite classico:
uν =
8π v 2
c3
kT . Si può anche verificare che ad alta frequenza la formula di Planck si
− hν
riduce a quella proposta da Wien: uν = 8π3h ν 3 e kT . Evidentemente è solo ad alta
c
frequenza o a bassa temperatura che la radiazione e.m. acquista la sua
caratteristica quantistica . Si poteva ottenere l’energia media degli oscillatori,
meno rigorosamente, usando il solito fattore di Boltzmann e − βε e sostituendo
all’integrale:
+∞
E=
∫ εe
0
+∞
− βε
dxdydzdp x dp y dpz
la sommatoria E ν
∫e
− βε
dxdydzdp x dp y dpz
∑ nhν e
=
∑e β
n =0
− β nhν
− nhν
+
hν
.
2
n =0
0
Calcolando infatti l’energia media di tutti gli oscillatori di frequenza ν , senza fare
il limite per h → 0 . Ci troviamo a calcolare la somma di una serie geometrica.
Sostituiamo all’integrale la somma sui valori medi appena calcolati:
nhν e − β nhν
hν
d
hν
d
hν
Eν = n
ln( e − β nhν ) +
ln( γ n ) +
+
=−
=−
=
− β nhν
2
d
2
d
2
β
β
e
n
n
∑
∑
∑
∑
n
d
1
hν
d
dx hν
1
hν hν e − β hν hν
ln(
)+
ln((1 − γ ))
( −hνγ ) +
=−
=
+
=−
=
+
dβ
1−γ
2 dx
dβ
2
1−γ
2 1 − e − β hν
2
con γ
= e − β hν
.
La questione che si pone è, però, relativa al senso fisico di questa operazione:
perché mai un’onda elettromagnetica dovrebbe avere delle energie quantizzate? È
questo un fenomeno relativo agli scambi (che sarebbero quantizzati) di energia tra
materia e radiazione o la quantizzazione è invece una caratteristica della
radiazione?
Come si vedrà successivamente, risulta che il modo corretto di interpretare il
postulato di Planck è di ricorrere nuovamente alle particelle di luce di Newton,
155
Cap. 5 – Termodinamica e calorimetria
nella forma dei moderni “fotoni”. Questo, tra l’altro, condurrà a comprendere
anche l’altra quantizzazione, quella operata da Bohr nel suo modello atomico47.
Per dare ulteriore credibilità al risultato di Planck, facciamo vedere che dalla
2
distribuzione ε λ = 2π hc
5
λ
1
è possibile ricavare la legge di Stefan-
hc
e λ kT
−1
∞
∞
ε = 2π hc
∫
ε = ε λ d λ . Cioè:
Boltzmann. Occorre calcolare
2
1
∫λ
0
0
5
dλ
hc
e λ kT
.
−1
L’integrale si può mettere nella forma:
∞
0
dλ
1
∫λ
5
hc
e λkT
0
=
−1 1
∫λ
λ
2
3
∞
−1
dλ
µ
eλ
=
−1
1
µ
ν
∞
xν −1dx
∫e
0
x
−1
=
1
µν
Γ(4)ς (4) ∗,
hc
µ
, x = e ν = 4. Sostituendo i valori numerici di Γ(4)ς (4) , si
kT
λ
avendo posto: µ =
ottiene infine
∞
dλ
1
∫λ
5
hc
e λkT − 1
0
=
1
µν
Γ(4)ς (4) = (
∞
∫
ε = ε λ d λ = 2π hc 2
0
kT 4 π 4 1 πkT 4
) 3!
= (
) . In conclusione:
hc
90 15 hc
1 π kT 4
2 5 k4
T 4 , da cui si deduce che la costante
(
) =
π c
3
15 hc
15
( hc )
4
di Stefan-Boltzmann è: σ = 2 π 5 c k 3 . Questo permette di assegnare un valore
15
( hc )
numerico alla costante di Planck h a partire dal valore empirico di
σ
. Risulta:
−34
h = 6,63⋅10 Js .
47
In realtà Planck propone un modello di interazione quantizzata, ma non una
quantizzazione del campo e/o delle energie degli oscillatori.
∗
Applichiamo qui la formula di Riemann: ς ( z ) =
1
Γ( z )
∞
xν −1
∫e
0
x
−1
, dove Γ = Γ(z ) è la
funzione gamma di Eulero e la funzione ς = ς (z ) è la funzione zeta di Riemann. Per z=4
Γ(4) = 3! e ς (4) =
π4
90
.
156
Cap. 5 – Termodinamica e calorimetria
Non è poi difficile ricavare la legge di Wien, nella forma più elementare,
calcolando la derivata di ε λ ed eguagliandola a zero, si ottiene:
hc
λmaxT =
= 2,89 ⋅ 10−3 m ⋅ K .
5k
157
Cap. 5 – Termodinamica e calorimetria
Spettro del corpo nero secondo
Planck.
Si noti che nel grafico della
emissività specifica ε λ ha il
massimo a λmax = 500nm , cui
corrisponde una frequenza:
c
ν0 =
= 6 ⋅ 1014 Hz . Questa
λmax
frequenza è diversa dalla
frequenza a cui la ε ν ha il
massimo che appare piuttosto
essere: ν m a x = 3 ⋅ 1 0 7 H z .
3
ελ (J/(sm )
F1
2,2x10
14
2,0x10
14
1,8x10
14
1,6x10
14
1,4x10
14
1,2x10
14
1,0x10
14
8,0x10
13
6,0x10
13
4,0x10
13
2,0x10
13
T=6000K
Ricordando che: ε λ =
visibile
0,0
-7
5,0x10
-6
1,0x10
-6
1,5x10
-6
2,0x10
2,5x10
-6
-6
3,0x10
λ(m)
3,00E-007
0
T=6000 K
2,50E-007
2,00E-007
2
εν(J/m )
λ2
εν e
derivando, si vede subito che i
due massimi non possono
essere in posizioni coincidenti.
0,0
1,50E-007
1,00E-007
5,00E-008
0,00E+000
0,00E+000
c
5,00E+014
1,00E+015
1,50E+015
2,00E+015
ν(Hz)
158
Cap. 5 – Termodinamica e calorimetria
10. La teoria del corpo nero secondo Einstein
Nel 1912, H. Poincarè dimostrò che, se lo spazio delle fasi è un continuo senza
zone proibite, allora il teorema dell’equipartizione dell’energia deve valere. In
altre parole, una teoria come quella di Planck, cioè una teoria che assumesse dei
salti quantici tra una zona dello spazio delle fasi ad un’altra era l’unico tipo di
teoria che poteva evitare la legge di Rayleigh-Jeans e giungere ad una energia
media a frequenza fissa come quella di Planck e non kT . E’ questo l’assunto
iniziale di un procedimento col quale Einstein calcolò nel 1917 lo spettro del corpo nero
senza utilizzare gli oscillatori armonici, ma riferendosi all’atomo di Bohr (191315). Supponiamo che l’atomo abbia solamente un livello fondamentale (1) e un
livello eccitato (2). All’equilibrio il campo cede agli atomi tanta energia quanta ne
assorbe. L’energia assorbita da un atomo dipende dalla densità d’energia presente
nella cavità, dal numero di atomi al livello fondamentale e da un coefficiente B12
che dipende dal tipo di atomo, mentre quella ceduta dipende da quanti atomi si
trovano nello stato eccitato e da un coefficiente A21 , che dipende dal tipo di atomi.
Tuttavia è necessario supporre anche che l’emissione di fotoni sia stimolata dalla
presenza della radiazione, per cui occorre considerare anche un termine
proporzionale alla densità di radiazione, al numero di atomi nello stato eccitato e
ad un coefficiente B21 . Se così non fosse all’equilibrio avremmo B12 uν n1 = A21 n2
che, per temperature alte cioè per alta uν , diverrebbe impossibile perché il
termine a sinistra aumenta, mentre quello a destra aumenta solo perché aumenta la
popolazione n 2 . Essendo n 2 limitato, eventualmente non ci potrebbe essere più
eguaglianza. Il termine di emissione indotta B21uν n2 deve essere aggiunto pena la
violazione della statistica di Boltzmann. All’equilibrio dunque abbiamo:
B12 uν n1 = B21uν n2 + A21n2 da cui ricaviamo:
uν = A21
1
n
B12 1 − B21
n2
hν
∆E
. Il rapporto n1 = e kT = e kT , come si evince dalla statistica di
n2
−E
Boltzmann, per cui: n = Ce
kT
1
, abbiamo così: uν = A21
B12
hν
kT
e
. Per T → ∞
− B21
(o per ν →0 ), questa formula si deve ridurre a quella di Rayleigh-Jeans:
uν =
8πν 2
c3
kT ,
per
cui
dobbiamo
avere:
159
Cap. 5 – Termodinamica e calorimetria
1
uν = A21
B12
hν
kT
e
≅ A21
− B21
1
B12 − B21 + B12
hν
kT
che ci dà la formula di Rayleigh-
3
Jeans, se B12 = B21 e A21 = 8πν h La teoria può essere generalizzata ad un atomo
3
B12
c
avente più di due livelli, sostituendo al pedice 12, il pedice ij e sommando. Infine notiamo
che queste formule costituiscono la base teorica dei laser.
11. La termodinamica della radiazione elettromagnetica
Facciamo ora una precisazione. Nella formula dell’energia media, appare una
temperatura assoluta: evidentemente si tratta della temperatura della radiazione,
ma cosa è la temperatura della radiazione? Per Planck, è la temperatura delle
pareti della cavità una volta che si sia stabilita una situazione di equilibrio
energetico tra pareti e radiazione.
4
Poiché risulta: u = 4 ε = 8 π 5 k 3 T 4 = bT 4 , si ha che l’energia interna del
c
15 c
( hc )
campo di radiazione è: U = Vu = VbT 4 . Possiamo anche calcolare l’entropia del
sistema d’onde, integrando:
4
V
4
V
4
d (T 3 )
4
dS =
udV + du =
bT 4 dV + 4 bT 3 dT = bT 3 dV + 4Vb
⇔ S = bVT 3 .
3T
T
3T
T
3
3
3
A partire dalle relazioni: P =
bT 4
3
e U = VbT 4 , si può dimostrare che, in
un’espansione adiabatica del corpo nero VT 3 = costante.
principio della Termodinamica, si ha:
Infatti, dal primo
dV
4
4
dT
dV
dT
+ udV + duV = udV + Vdu = udV + 4Vu
⇒
= −3
3
3
3
T
V
T
Integrando, si trova il risultato quotato VT 3 = costante. Possiamo anche scrivere
δ Q = 0 = pdV + dU = u
che: costante= VT / T = 3Vp/(bT) ⇒ pV = AT con A=costante.
Per completezza si deve aggiungere che la pressione esercitata dalla radiazione (o
dal gas di fotoni) sulle pareti della cavità può essere calcolata utilizzando la
formula già trovata per un gas di molecole (par. 3) e sostituendo all’energia della
molecola quella di un fotone:
N
1
1 nν
1
1
P = nm A v 2 ⇒ Pν =
h ν ⇒ P = u = bT 4 . Da notare che si tratta
3
V
3 V
3
3
di una pressione molto bassa a temperatura ambiente , ma a temperature del tipo
di quelle presenti al centro delle stelle tale pressione diviene enorme.
4
160
Cap. 5 – Termodinamica e calorimetria
Da notare anche i procedimenti usati per dimostrare, con tecniche tipicamente
termodinamiche, le leggi di Stefan-Boltzmann e di Wien.
C’è però da notare che i concetti di entropia e di temperatura sono stati qui
implicitamente estesi alla radiazione em. Si può fare? Se si può fare, la radiazione
è in definitiva trattata come un gas. Infatti la radiazione ha un volume, ha una
pressione (quella esercitata sulla parete) e una temperatura (quella della parete
della cavità con cui è in equilibrio termico), dunque ha i suoi bravi parametri di
stato come un gas. Inoltre ha un’energia interna, fa lavoro, ha un’entropia come
un qualsiasi sistema termodinamico. Secondo Planck, il sistema della radiazione
nella cavità è un sistema termodinamico con lo stesso buon diritto di un recipiente
pieno di gas. Nell’uno e nell’altro caso una conoscenza dettagliata (conoscere,
cioè, i campi ad ogni istante ed ogni punto) non ha senso, come non ha senso
tentare di conoscere posizione e velocità di ogni molecola di gas. Ciò che,
evidentemente, ci interessa è conoscere le funzioni globali che definiscono lo
stato macroscopico del sistema. Nel caso della nostra cavità, basta conoscere uν
(o Kν o εν ). Concettualmente, dunque, la quantizzazione, cioè l’introduzione del
quanto di luce o fotone è molto vicina. Introdotto il fotone, dovremo per forza
introdurre l’equivalente della distribuzione di Boltzmann, cioè una distribuzione
in energia che data la relazione tra frequenza ed energia risulta in effetti una
distribuzione di frequenza: quella del corpo nero! Dunque il corpo nero è un
contenitore pieno di un gas di fotoni cui si possono applicare tutti i concetti usati
per un gas ordinario.
13. Le statistiche quantistiche
Il problema del corpo nero può essere studiato considerando la cavità illustrata nel
capitolo precedente come un contenitore pieno di particelle: i fotoni appunto. Un
gas di fotoni però non obbedisce alla statistica di Bolzmann, ma ad una statistica
diversa, con leggi che sono state trovate dal fisico indiano Bose (1926). Per questa
ragione il fotone viene detto “bosone”. Il termine bosone, in effetti, sta ad indicare
tutte le particelle a spin intero che sono mediatrici di una forza. Così come il
fotone media l’interazione elettromagnetica, altre particelle mediano interazioni di
diverso tipo: le W ± e la Z 0 mediano l’interazione debole, il gravitone quella
gravitazionale, i gluoni mediano l’interazione forte. Tutte queste particelle (ed
altre) obbediscono alla statistica di Bose-Einstein.
13a. La statistica di Bose-Einstein
161
Cap. 5 – Termodinamica e calorimetria
Come si è visto, la statistica di Boltzmann fornisce la distribuzione in energia
delle particelle in un gas. Se si è in grado di definire la distribuzione energetica
dei fotoni nella cavità, questo equivale, grazie alla relazione E = hν , a conoscere
la loro distribuzione in frequenza e si può quindi ottenere lo spettro in frequenza
del campo dentro la cavità del corpo nero. Moltiplicando la frazione di fotoni con
frequenza tra ν e ν + dν per E = hν , si ottiene la densità di energia nella cavità
e moltiplicando per il solito fattore c/4, si otterrà l’emissività specifica del corpo
nero. Un risultato positivo, cioè identico a quello ottenuto a pag. 8, è una
conferma forte dell’interpretazione corpuscolare del campo elettromagnetico.
Ricalcando il percorso logico già fatto per la statistica di Boltzmann, cerchiamo in
quanti modi si può realizzare una configurazione dell’insieme dei fotoni W e
massimizziamo il lnW con la condizione che l’energia totale sia costante,
cerchiamo cioè la configurazione per cui S = k lnW è massima. Non possiamo
però richiedere la costanza del numero totale di fotoni, perché, a differenza di ciò
che accade in un gas, il numero di particelle non è, nel caso dei fotoni, fissato.
Cerchiamo dunque il massimo del logaritmo di W al variare del numero totale
dei fotoni Nν in ciascun intervallo infinitesimo di frequenza. Il massimo dovrà
essere ottenuto con la condizione che l’energia totale U =
hν Nν
∑
ν
del sistema sia
costante, secondo quanto detto precedentemente. Dovranno allora valere
simultaneamente le equazioni:
d ( k ln W ) = 0
dU =
hvdNν = 0
ν
∑
Utilizzando il metodo dei moltiplicatori arbitrari di Lagrange, si moltiplichi la
seconda equazione per β e si sottragga dalla prima: d ln W − β dU = 0 . A questo
punto, per poter procedere occorre conoscere l’espressione esatta di W .
Calcoliamola, tenendo presente altri due punti:
a.
Più fotoni possono trovarsi in uno stesso stato. Questa regola, come
vedremo, non si applica agli elettroni.
b. I fotoni non sono distinguibili.
.
Ricordiamo che il numero di stati con frequenza tra ν e ν + dν è:
dnν =
8πν 2
c3
dν = gν .
Per risolvere il nostro problema, oltre al numero di stati tra ν e ν + dν degli stati,
si deve calcolare anche la popolazione Nν in tali stati. Ci si chiede:
162
Cap. 5 – Termodinamica e calorimetria
1) In quanti modi si possono distribuire Nν fotoni in dnν =
8πν 2
c3
dν = gν stati con
frequenza (energia) compresa tra ν e ν + dν ?
2) In quanti modi si può realizzare una configurazione con Nν fotoni per ogni
stato caratterizzato da una frequenza ν ?
Per rispondere alla domanda 1), si indichi con una x un fotone (di frequenza ν ) e
con una barra il limite di uno stato del gruppo di stati gν . Per esempio, una
distribuzione di Nν = 12 fotoni su gν = 5 differenti stati sarà indicata con la
sequenza di simboli: xxx/xx/xxxx/x/xx, cioè tre fotoni nel primo stato, due nel
secondo, quattro nel terzo, uno nel quarto e due nel quinto. Allo stesso modo si
sarebbe potuta considerare la distribuzione: xx/xxxx/xx/x/xx, cioè due fotoni nel
primo stato, quattro nel secondo, ecc.: quanti modi si hanno dunque per distribuire
questi 12 fotoni nei 5 stati? Si devono permutare 12+5-1 oggetti (le dodici x e le
quattro barrette) e dunque calcolare il numero di permutazioni di 12+5-1 o, più in
generale, di Nν + gν − 1 oggetti: questo numero è pari a ( Nν + gν − 1)! Tuttavia, si
deve prestare attenzione al fatto che, permutando due fotoni tra di loro, non si
ottiene una distribuzione diversa (i fotoni sono indistinguibili secondo quanto
detto al punto b). Questo significa che le N ν ! permutazioni tra le ( Nν + gν − 1)! ,
che possono essere realizzate invertendo la posizione di 2 fotoni, non vanno in
realtà considerate perché non danno stati distinti. E neppure vanno considerate le
(gν −1)! permutazioni che si ottengono scambiando le barre. In conclusione si
avranno: Wν =
( N ν + g ν − 1)!
stati diversi. Questo risponde alla prima domanda.
N ν !( g ν − 1)!
La risposta alla domanda 2. è semplice: il numero di modi ( W ) in cui si può
realizzare una configurazione generale con Nν fotoni per ogni stato caratterizzato
da una energia hν è il prodotto del numero di modi ( Wν ) in cui si può realizzare
la distribuzione di Nν sui dn ν stati di frequenza tra ν e ν + dν :
( Nν + gν − 1)!
W=
Wν =
.
Nν !( gν − 1)!
∏
ν
∏
ν
Si calcoli adesso il differenziale di lnW nelle variabili Nν e si sostituisca.
Utilizzando anche la formula di Stirling ln( n!) = n ln n − n , si ottiene:
163
Cap. 5 – Termodinamica e calorimetria
( N ν + g ν − 1)!
)=
ν
ν − 1)!
∏ N !( g
dS
= d ln W = d ln(
k
ν
( N ν + gν − 1)!
)=
ν
ν − 1)!
∑ d (ln N !( g
ν
∑ d (ln( N + g −1)! − ln N !− ln( g − 1)!) =
= ∑ d (( N + g − 1) ln( N + g − 1) − ( N + g − 1) − N
=
ν
ν
ν
ν
ν
ν
ν
ν
ν
ν
ν
ν
ln N ν + N ν ) =
ν
=
∑ν dNν ln
( Nν + gν −1)
.
Nν
Sostituendo: ∑ dN ν (ln
ν
( Nν + gν − 1)
− β hν ) = 0 .
Nν
Perché questa relazione sia vera a prescindere dai dN ν , deve essere:
ln
( Nν + gν − 1)
− β hν = 0 per ogni ν .
Nν
Svolgendo questa relazione, si trova:
( Nν + gν − 1)
g −1
= e β hν ⇒ 1 + ν
= e β hν ⇒ gν = Nν ( e β hν − 1) ,
Nν
Nν
dove
abbiamo
8 2
trascurato 1 rispetto a g ν . Infine si può scrivere ( gν = πν3 dν ):
c
dnν =
gν
e β hν − 1
= 8π l 3
1
ν2
c 3 e β hν − 1
dν , considerando Nν un numero infinitesimo
d nν .
A questo punto, dividiamo per l 3 e moltiplichiamo per h ν per ottenere uν e,
3
c
h
ancora per
, per ottenere εν . In conclusione, si ottiene: εν = 2π 2 β νhν
.
c e
−1
4
1
Questa è la stessa espressione trovata a pag. 8, se si identifica β con
. Questa
kT
volta però, εν è stata calcolata sulla base dell’esistenza dei fotoni e della loro
statistica. Nel nostro gas di fotoni valgono le relazioni già trovate:
8
k4
VT 4 ,
π5
15 c
( hc ) 3
64 5 k 4
π
VT 3 ,
3
45c
U = uV =
S=
p=
(hc)
u
U
8
k4
T4
=
=
π5
3
3V
45 c
( hc ) 3
164
Cap. 5 – Termodinamica e calorimetria
.
Infine il numero totale di fotoni in una cavità a temperatura T è:
n=
8π
∞
∫
ν 2dν
hν
0 kT
e
∞
=
x 2dx
∫e
≈ Γ(3)ς (3) = 2.404 ⋅ 8π (
kT 3
) (= 2,02 ⋅ 1016 m−3a T = 103 K )
hc
−1
−1 0
Per Γ (3) = 2! e ς (3) ≈ 1, 202 .
Una domanda sorge spontanea: quale è la relazione tra la statistica di Bose e
quella di Boltzmann. La differenza sta nel considerare le molecole distinguibili e
nella condizione che il numero totale di molecole è costante. Rifacendo il calcolo
fatto per i fotoni, ma aggiungendo la condizione che il numero di molecole sia
c
3
x
4 p2
costante si ottiene (usiamo g = π 3 dp
48
, vedi oltre la discussione della statistica
h
di Fermi): dnν =
4π l 3
h
p2
3
e
α
dp =
ε
e kT
4π l 3
−1
h
2mε
3
e
α
ε
e kT
−1
m dε
4π l 3
=
2 ε
h3
2m
e
α
3
2
ε
ε
e kT
dε .
−1
Si vede che per e α molto più grande di uno si ottiene un andamento simile a quello della
statistica di Boltzmann:
dnν =
4π
h3
Ae
−
ε
kT
2m
3
2
−α
e d nν
ε d ε , con A = e
è stato scritto qui per unità di
ε
volume. Riprendiamo la formula di Boltzmann:
−
dn
1
=
e kT dxdydz 4πp 2 dp ,
N V ( 2πmkT ) 3
integriamola sul volume e otteniamo: dn = 4π
cui deve essere: A =
Nh
N
(2π m kT )
3
e
−
ε
kT
2 m 3 ε d ε , per
3
(2π mkT ) 3
per ottenere la normalizzazione corretta. Il parametro
A , è detto “parametro di degenerazione” e riotteniamo la statistica di Boltzmann, se A
è molto più piccolo di 1. Si noti che il parametro A diventa grande a bassa temperatura e
per numero di particelle/unità di volume (densità) N grandi. In tali condizioni la
statistica di Boltzmann non sarà più valida.
13b. La statistica di Fermi-Dirac
48
Manca un fattore 2 rispetto ai fotoni, perché per le molecole non c’è il doppio stato di
polarizzazione, come per i fotoni.
165
Cap. 5 – Termodinamica e calorimetria
La formula trovata è diversa per gli elettroni poiché, come già detto, per essi non
vale la condizione espressa precedentemente al punto a.: più elettroni non possono
occupare uno stesso stato, in accordo con il principio di esclusione di Pauli.
Tuttavia, è applicabile la condizione di conservazione del numero totale di
elettroni. Supponiamo di avere dunque un contenitore cubico in cui si trovano N
elettroni con un’energia totale E. Un tale contenitore è appunto un metallo in cui
oltre gli elettroni legati nel reticolo cristallino, un certo numero di elettroni si
muova liberamente (vedi per esempio la modellizzazione dell’effetto termoionico
al prossimo capitolo). Come nel caso dei fotoni, anche gli elettroni vanno
rappresentati come un’onda, pertanto il numero di stati con impulso tra p e p+dp,
8π
h
si ottiene come per il caso dei fotoni: g p = 3 p 2 dp . Basta porre p =
λ
h
dλ
nell’analoga espressione dei fotoni dnλ = 8π
(un fattore 2 per gli stati di spin
λ4
sostituisce il fattore 2 che nel caso dei fotoni è il numero di stati di
polarizzazione).
Il numero di modi per disporre N p elettroni su g p livelli è allora la combinazione
di
Np
oggetti su
gp
Pp =
elettroni :
posti diviso per il numero di modi di permutare gli
gp !
N p !( g p − N p )!
Np
. Rifacendo il calcolo precedente con la
condizione che il numero di elettroni si conservi dN =
∑ dN
p
= 0 , che si
k
conservi l’energia totale dE =
∑ε
p dN p
= 0 e usando il metodo dei moltiplicatori
p
arbitrari, abbiamo:
dS = 0 = d ln
∏N
k
=
∑ −d (ln N
gp !
p !( g p
p !+ ln( g p
− N p )!
=
− N p )!) = −
∑ d ln N
k
∑ dN
k
=−
p !( g p
p (ln
− N p )!
=
N p + 1 − 1 − ln( g p − N p ) − 1 + 1) =
k
∑ dN
p (ln
N p − ln( g p − N p )) ⇒
∑ dN
k
⇒ ln
dn =
gp !
p (ln
k
gp − N p
Np
8π
h
= α + βε p ⇒
2
p dp
3
e
α+
ε
kT
+1
gp − N p
Np
α + βε p
=e
gp − N p
Np
⇒ Np =
− α − βε p ) = 0 ⇒
gp
α+
e
. Infine abbiamo:
εp
kT
+1
. Si noti che abbiamo, senza tanti complimenti, posto: β =
1
. La
kT
166
Cap. 5 – Termodinamica e calorimetria
ragione è che esiste un teorema che ci consente in generale questa identificazione senza
passare per il calcolo dell’energia media come si è fatto nel caso della statistica di
εF
8π
p 2 dp . Esaminiamo
Boltzman. Poniamo adesso:
. Abbiamo:
α =−
kT
dn =
h3
ε −ε F
e
kT
+1
adesso il significato della costante ε F . Consideriamo dapprima il caso ε > ε F . In
questo caso, portando la temperatura allo zero l’esponenziale diverge e troviamo che i
dn
livelli non sono affatto occupati:
= 0 . Se, viceversa, ε < ε F , portando la temperatura
dp
a zero l’esponenziale va a zero e troviamo che ogni guscio sferico è tutto occupato. A più
1
alta temperatura, invece a ε = ε F . L’andamento di dn = ε − ε
è disegnato in
F
e
kT
+1
basso.
La statistica degli elettroni è detta “statistica di Fermi-Dirac” e le particelle che,
come gli elettroni, obbediscono al principio di Pauli, sono detti “fermioni”. Le
particelle che obbediscono al principio di Pauli sono caratterizzate del resto da
uno spin semintero: le particelle che costituiscono la materia cioè sono ben
distinte dalle particelle che mediano le interazioni. Da notare che, per
entrambe le statistiche si riducono a quella classica di Boltzmann.
ε k >> kT ,
167
Cap. 5 – Termodinamica e calorimetria
Possiamo adesso ottenere l’energia media e il numero di elettroni, calcolando gli
integrali (seguendo A. J. Dekker – Fisica dello stato solido, pag. 212 (1965)):
+∞
N =
∫
0
8π
h3
ε −ε F
e
kT
+∞
+∞
m
8π 2 m ε 2ε dε
8π m 3 / 2 2ε
=
=
dε e
ε −ε F
3
ε −ε F
h3
h
0
+1 0
e kT + 1
e kT + 1
p 2 dp
∫
∫
+∞
+∞
8π m 3 / 2 ε 2ε
1 8π ( m ε ) 3 / 2 2
=
d
ε
dε
ε −ε F
N h3
h 3 ε −kTε F
kT
0
0
e
+1
e
+1
Allo zero assoluto, il denominatore diviene infinito almeno che non sia ε < ε F ,
nel qual caso il denominatore vale 1, dunque gli integrali sono limitati
all’intervallo 0 - ε F :
1
ε =
N
∫
εF
0
N =
8π
∫h
3
∫
m 3 / 2 2ε dε =
8 2π
h3
m3/ 2
2 3/ 2
h 2 3N 2 / 3
(
)
ε F0 ⇒ ε F0 =
3
2 m 8π
0
ε0 =
1
N
ε F0
∫
0
8 2π
( m )3/ 2
3
8 2π
2 5/ 2 3
3/ 2
h
( mε ) d ε =
ε = ε F0
5
h3
8 2π 3/ 2 2 3/ 2 5 F0
m
εF
3
3 0
h
Per gli stessi integrali a temperatura diversa da zero, si ottiene:
5π 2 kT 2
ε = ε 0 1 +
12 ε F0
e per il “livello di Fermi”:
π 2 kT
ε F = ε F0 1 −
12 ε F0
2
.
Dunque il livello di Fermi è una funzione della temperatura, ma debole abbastanza da
essere considerata costante. Dalla precedente formula si deduce che il contributo al calore
2
3
5π 2 kT
dU
molare dato dagli elettroni è piccolo: U = N A ε F 1 +
⇒ CV =
=
dT
5 0
12 ε F0
= N Ak
π 2 kT
π 2 kT
=R
. L’energia di Fermi può essere anche dell’ordine di 10 eV,
2ε F0
2ε F0
mentre kT = 0, 025 eV a temperatura ambiente, per cui il rapporto
π 2kT
= 1, 2 ⋅10−2 ,
2ε F0
ovvero il contributo del calore specifico degli elettroni è dell’ordine di CV = 0, 01 ⋅ R ,
rispetto a quello del reticolo: CV = 3R .
168
Cap. 5 – Termodinamica e calorimetria
Utilizzando questo ultimo risultato possiamo trovare l’equazione di stato di un gas di
2
2
Fermi a temperatura prossima allo zero. Infatti la relazione PV = nε ⇒ P = Nε (con
3
3
ε = energia media delle “molecole”, N = numero di particelle per unità di volume e n
numero totale di particelle) è valida anche per un gas di Fermi, perché tutti gli argomenti
usati a suo tempo per trovarla sono validi anche qui. Tuttavia l’energia media è adesso
23
h2 3 2/ 3 5/ 3
data dalla formula precedente. In conclusione: P =
N ε F0 =
( ) N , che
35
5m 8π
come si vede è indipendente dalla temperatura. In realtà è indipendente dalla temperatura
solo nella gamma di temperature prossima allo zero per cui l’approssimazione fatta di una
distribuzione piatta è valida. La pressione dipende così dalla densità alla 5/3: N 5/3 .
Questa pressione indipendente dalla temperatura, ma solo dalla densità può nelle stelle di
neutroni compensare la pressione dovuta alla gravitazione conducendo ad una situazione
di equilibrio.
Anche la statistica di Fermi si riduce a quella di Boltzman per il valore del parametro di
degenerazione molto più piccolo di 1. Il parametro di degenerazione è lo stesso di quello
visto per i fotoni, a parte un fattore due: A =
Nh3
2 (2π mkT )3
dovuto al fatto che per gli
elettroni esiste un doppio stato di spin. Sostituendo nella formula i valori delle costanti e
della massa dell’elettrone, si ottiene che già a T = 300K , un gas di elettroni è
completamente degenere.
Secondo le moderne teorie sulle particelle elementari, il mondo è costituito da particelle
che obbediscono alla statistica di Fermi, dette “fermioni”. Sono particelle dotate di un
momento della quantità di moto intrinseco detto “spin” che, in certe unità, è dato da un
numero semi intero. Queste particelle sono le costituenti della materia, per esempio, gli
elettroni, i protoni ecc… I fermioni interagiscono fra loro attraverso lo scambio di
particelle obbedienti alla statistica di Bose e perciò chiamate “bosoni”. Nel caso delle
interazioni elettromagnetiche, il bosone che viene scambiato è il fotone. Nella teoria
elettrodebole unificata di Weinberg e Salam, in cui l’interazione elettromagnetica e quella
nucleare debole sono unificate nello stesso senso in cui abbiamo visto l’unificazione del
magnetismo e dell’elettricità, i bosoni scambiati sono quattro: il fotone (0), la 1 2 e le due
3 ± . Queste ultime tre particelle sono state scoperte al CERN di Ginevra negli anni ’70.
Nel caso delle interazioni nucleari forti, i fermioni sono i “quark” che interagiscono fra
loro (in particolare per formare i protoni, i neutroni, ecc…) e i bosoni scambiati sono
chiamati “gluoni”, dall’inglese “glue” (colla).
Per le interazioni gravitazionali si ipotizza un bosone chiamato “gravitone”.
Nel caso delle interazioni elettrodeboli, la teoria funziona solo se si ammette che le
particelle di materia e i bosoni abbiano massa nulla. In caso contrario, la teoria presenta
delle infinità che la rendono inutilizzabili. Tuttavia nel mondo reale le particelle hanno
massa. Per risolvere questo problema si è dovuto inventare una teoria che producesse le
masse attraverso l’interazione con un campo quantizzato, detto campo di Higgs. Di
169
Cap. 5 – Termodinamica e calorimetria
recente è stato annunziato al CERN la scoperta di una particella che è, verosimilmente, il
bosone di Higgs.
Conclusione
Come si è visto, la teoria cinetica dei gas dà una spiegazione completa e
comprensibile del comportamento dei gas. Le leggi dei gas sono state tutte
giustificate sulla base della teoria atomica. Al di là dunque dei singoli risultati che
hanno pure spesso un importante valore applicativo, la visione del mondo che
emerge è quella immaginata dagli antichi atomisti greci. Vogliamo aggiungere
che, oltre alla teoria dei gas, l’atomismo della materia implica che anche i
composti allo stato solido o liquido vanno studiati come aggregati di atomi. In
diversi casi è ciò che abbiamo appunto fatto, per esempio nel caso dello studio dei
campi elettrici e magnetici nei mezzi materiali: la teoria per quanto elementare si
applica infatti anche ai solidi e ai liquidi. Stessa cosa per la viscosità che è molto
importante per la meccanica e i problemi di lubrificazione. La teoria dello stato
solido può essere elaborata però solo con la meccanica quantistica e pertanto
rinunzieremo ad attaccare questo problema.
Su un punto, cui si è solo brevemente accennato, possiamo spendere un po’ più
tempo: il verso di scorrimento del tempo. Se si chiede a qualcuno in che verso un
pianeta, diciamo Marte, percorre la sua orbita, certamente la risposta sarà “non lo
so”. E’ una conoscenza per quanto semplice che normalmente solo quelli
interessati nel soggetto hanno. Proviamo a vedere se possiamo indovinare la
risposta. Il pianeta può girare in un verso o in un altro, ma non c’è verso di
pensare ad una ragione per cui dovrebbe girare in senso orario o antiorario:
entrambi i moti sembrano essere possibili. Se ci riflettiamo un po’ su possiamo
pervenire all’idea che i due moti sono l’uno il film all’inverso dell’altro. Se
facessimo il film del moto orbitale di Marte intorno al Sole e lo proiettassimo,
non troveremmo modo di dire quale delle due proiezioni diretta o inversa
corrisponda alla realtà. Possiamo concludere dunque che il moto diretto o
invertito nel tempo sono due fenomeni egualmente possibili e che la meccanica
non ci dice quale dei due moti, diretto o inverso, sia quello possibile. Proviamo
adesso a confrontare questo caso con il caso di un pendolo che oscilla. Sappiamo
che per effetto degli attriti il pendolo si fermerà. Facciamo anche qui il film e
proiettiamolo una volta in senso diretto e una volta in senso inverso. In un caso
vedremo il pendolo fermarsi, nell’altro vedremo il pendolo accelerare. Qualunque
osservatore cui venga chiesto quale dei due versi di proiezione sia quello giusto,
non avrebbe esitazione a scegliere quello in cui il pendolo si ferma. In altri termini
è possibile in questo caso scegliere un verso del tempo, perché mentre in un verso
osserviamo un fenomeno fisico con cui siamo familiari, nell’altro osserviamo
qualcosa che sappiamo non avvenire mai. La differenza tra i due fenomeni, il
170
Cap. 5 – Termodinamica e calorimetria
moto di Marte e quello del pendolo, è l’introduzione dell’attrito, non considerato
per ovvie ragioni nel caso del moto del pianeta. Possiamo fare un altro esempio:
un gas che fuoriesca da un recipiente che vada a riempire un secondo recipiente
inizialmente vuoto. Anche in questo caso c’è un verso di scorrimento del tempo
che restituisce un fenomeno non osservabile: il ritirarsi del gas che inizialmente
occupa i due recipienti, in uno solo di essi.
Possiamo concludere che nella meccanica il senso del tempo non è deciso da
nulla, ciò evidentemente perché la seconda legge della dinamica contiene una
derivata seconda rispetto al tempo: cambiando il segno del tempo la seconda
legge della dinamica rimane invariata. Considerazioni perfettamente analoghe si
possono fare per l’elettromagnetismo.
Se tuttavia introduciamo oltre al fenomeno puramente meccanico, anche il calore
generato dall’attrito o fenomeni termodinamici, la storia cambia: l’entropia può
solo aumentare. Un fenomeno può solo andare in un verso, ma non nell’altro,
quello cioè in cui l’entropia diminuisce, non viene osservato e spesso sembra
ridicolo. Il verso del tempo è dunque determinato dal verso di aumento
dell’entropia, non dai fenomeni meccanici o elettromagnetici elementari.
Appendice 1. La formula di Stirling
Dimostriamo la formula di Stirling. Abbiamo, calcolando l’integrale per parti che.
+∞
n! =
∫0 e
+∞
n! =
∫0 e
−t n
t dt . Facciamo la sostituzione: t = n (1 + x ) per ottenere:
−t n
t dt =
+∞
∫−1 e
−n(1+ x)
[ n(1 + x)]
n
può essere approssimato come:
−n n+1
+∞
ndx = e n
n!
e− n n n +1
+∞
=
∫e
−1
− nx
∫−1 e
−nx
[(1 + x)]n dx .
[ (1 + x)]
n
+∞
dx =
∫e
−∞
−n
L’integrale
x2
2 dx
1
2π 2
=
n
1
2
2π
1
1
⇒ ln n ! = −n + (n + 1) ln n + 2 ln 2π − 2 ln n ≃ n ln n − n , che
n
da cui: n ! = e− n n n+1
è la formula usata nel testo.
Appendice 2
4
La formula di Poiseuille φ = π R ∆ p che dà la portata di un tubo di raggio R e
8 ηl
di lunghezza l in cui scorre un fluido newtoniano di viscosità η dà la quantità di
171
Cap. 5 – Termodinamica e calorimetria
liquido che scorre nel tubo in funzione della differenza di pressione agli estremi
del tubo ∆p .
Consideriamo una corona cilindrica di raggio r < R , di spessore dr e lunghezza
l . Sul cilindro si esercitano due forze, una è la forza di viscosità che si esercita
sul mantello esterno e su quello interno e che rallenta il moto:
dv
dv d 2 v
dv
d 2v
d
dv
dF = η 2π rl
− η 2π ( r + dr )l ( + 2 dr ) = −η 2π l ( + r 2 )dr = −η 2π l ( r )dr
dr
dr dr
dr
dr dr
dr
(trascurando infinitesimi al quadrato) l’altra è la forza dovuta alla pressione:
dF = ∆p 2π rdr . In condizioni di equilibrio, queste due forze devono essere uguali
d dv
in
valore:
Da
cui
ricaviamo:
∆p 2π rdr = −η 2π l ( r )dr .
dr dr
dv
dv
dv
∆p r 2
∆p r
∆prdr = −η ld ( r ) ⇒ r
=−
⇒
=−
. Integrando di nuovo:
dr
dr
dr
ηl 2
ηl 2
v=−
r 2 ∆p
+ C . Per
4 lη
r =R, v= 0 cioè:
∆p 2
R 2 ∆p
(R − r2 ) .
= C e sostituendo: v ( r ) =
4lη
4lη
La quantità totale di fluido che passa attraverso una sezione del tubo per unità di
R
∆p
( R2 − r 2 )2π rdr =
tempo è pertanto: φ =
4lη
∫0
R
∆p
∆p 2 R2 R4
∆pR4
π
(R2 − r 2 )rdr = π
−
R
=π
.
2lη
2lη
2
4
8lη
0
∫
Forza di viscosità agente sul mantello
interno:
F = η 2π rl
dv
dr
e esterno:
dv d 2 v
dr )
+
dr dr 2
Forza agente sulla base della corona
cilindrica dF = 2π rdr ∆p
dF = −η 2π ( r + dr )l (
A titolo di completamento, troviamo adesso la relazione tra pressione, forza peso
e velocità per un liquido non viscoso. L’equazione che risolve questo problema è
172
Cap. 5 – Termodinamica e calorimetria
detta equazione di Bernoulli (Daniel). Immaginiamo di avere un tubo anche a
sezione variabile, non necessariamente orizzontale in cui scorre un fluido a bassa
viscosità. Prendiamo due sezioni del liquido successive l’una all’altra di
lunghezza dx e dx' e sezioni A e A' . I volumetti di questi liquidi sono dV = Adx
e dV ' = A' dx ' . Se li prendiamo contenenti la stessa massa, considerando che la
densità ρ è costante allora dV = dV ' .
Il lavoro fatto dalla pressione sui due volumetti è: PdV e P ' dV , mentre le
1
variazioni di energia potenziale e cinetica sono: ρ dVgz , ρ dVgz ' nonché ρdVv2 e
2
1
2
ρ dVv ' .
Per
la
conservazione
dell’energia,
abbiamo
allora:
2
1
1
PdV + ρ dVgz + ρ dVv2 = P ' dV + ρ dVgz '+ ρ dVv '2 eliminiamo il volumetto e
2
2
1 2
abbiamo: P + ρ gz + ρ v = costante, che costituisce appunto la equazione di
2
Bernoulli. Abbiamo visto che l’equazione di Bernoulli rappresenta la
conservazione dell’energia in un fluido.
Appendice 3
Viene spesso puntualizzato a proposito del teorema di equipartizione dell’energia
che l’energia cinetica di un sistema di particelle assume sempre la forma di una
somma di termini quadrati. Ne segue che, in assenza di potenziali come è il caso
di un gas perfetto, il teorema di equipartizione vale necessariamente. Supponiamo
che una molecola sia una combinazione di n sotto-componenti. Il sistema è
dunque un sistema di particelle dotate di energia cinetica complessiva pari a
2T =
∑ mv
i
2
i i
2
2
2 2
dove le vi = xɺi + yɺi + zɺi . Se passiamo dalle coordinate cartesiane
alle coordinate generalizzate q1 ,..., qk , abbiamo: xi = xi ( q1 ,..., qk ) e pertanto:
xɺi =
∑
j
∂ xi
qɺ j
∂q j
(e simili per le altre coordinate cartesiane). Sostituendo
nell’espressione
2T =
∑
i
mi (
∑
dell’energia
jk
( a ijk qɺ j qɺ k
+b ijk qɺ j qɺ k
+
c ijk qɺ j qɺ k
cinetica
otterremo:
) . Questa espressione può essere però
trasformata in una espressione che contenga solo quadrati, usando una
trasformazione lineare a nuove variabili η1 ,...,η k , con η j = η j ( q1 , ..., q k ) :
173
Cap. 5 – Termodinamica e calorimetria
2T =
∑ m∑
del tipo
i
i
k
( Aki ηk2 + Bki ηk2 + Cki ηk2 ) . Ne segue che ad ogni termine dell’energia
1 i 2
1
Akη k spetta un valore medio pari a kT .
2
2
Bibliografia
Per la teoria cinetica:
1. Loeb, Leonard B. The Kinetic Theory of Gases, McGraw-Hill (1934)
2. Jeans, James, An Introduction to the Theory of Gases, Cambridge
University Press, 1940
3. Jeans, James, The Dynamical Theory of Gases, Cambridge University
Press, 1904
4. Bloch, Eugene, The Kinetic Theory of Gases, Metheun & Co. Londra,
1924
5. Meyer, Oscar E., The Kinetic Theory of Gases, Bibliolife, versione
originale pubblicata da Longmans, Green & co, 1899
6. L. Boltzmann, Lectures on Gas Theory, Dover Publications Inc., New
York, 1992 (originale: Vorlesungen uber Gastheorie, 1896)
7. Knudsen Martin, Kinetic theory of gases, Meuthen’s monographs on
physical subjects, 1934
8. Kennard, Kinetic theory of gases, McGraw-Hill
9. Presenr, R. D. Kinetic theory of gases, McGraw-Hill Book Company, Inc.
1958
10. Perrin, Jean, Brownin movement and molecular reality, Taylor and
Francis London, 1910
11. Conduction of electricity through gases, Cambridge University Press
1928
Per la termodinamica:
1. Fermi, Enrico, Thermodynamics, Dover Publications (1936)
2. Schroedinger, Erwin, Statistical Thermodynamics, Dover Publications
(1944).
3. Planck, Max, Treatise on Thermodynamics, Dover publications (1926)
4. Zemansky, Mark W., Heat and thermodynamics, McGraw-Hill Book
Company (1957)
5. Boato, Giovanni, Lezioni di termodinamica, libreria universitaria Pacetti
Genova, (1967)
Per la teoria del corpo nero:
174
Cap. 5 – Termodinamica e calorimetria
1. Planck, Max, The theory of Heat Radiation, Dover Publications (1914).
2. Born, Max, Atomic Physiscs, Dover Publications Inc.
175