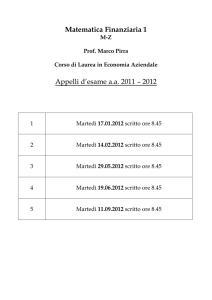
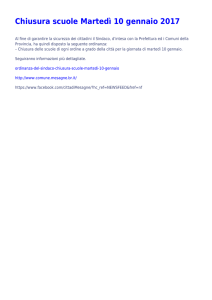
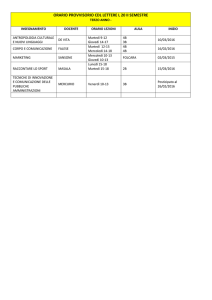
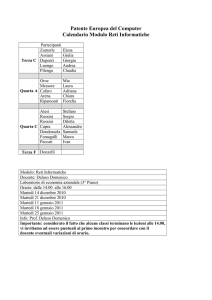
Basi di dati biologiche
Seminario per il corso di Basi di Dati II
Luana Rinaldi
[email protected]
martedì 11 maggio 2010
AGENDA:
•
Introduzione alla bioinformatica;
•
Concetti Biologici;
•
Banche dati biologiche;
•
Collaborazioni tra banche dati;
•
Ricerca in banche dati biologiche;
martedì 11 maggio 2010
Introduzione alla bioinformatica
Bioinformatics is “the study of the information content and information flow in biological systems and
processes”.
[Michael Liebman in “Bioinformatics: An Editorial Perspective” ]
(http://www.netsci.org/Science/Bioinform/feature01.html)
martedì 11 maggio 2010
Nascita della bioinformatica
fine anni ’80 [Hwa Lim (http://www.dtrends.com/HAL.html)]:
“Bioinformatics”
Applicazione di tecniche informatiche nel dominio applicativo delle scienze della vita
Definizione: “Studio del contenuto informativo e del flusso di informazione nei sistemi e nei
processi correlati alla biologia”
[Micheal Liebman in Bioinformatics: An Editoria Perspective” (http://www.netsci.org/Science/Bioinform/feature01.html)]
martedì 11 maggio 2010
Bioinformatica: definizione
“Bioinformatics”
La bioinformatica è il campo della scienza in cui la biologia e l’informatica si
fondono in un’unica disciplina per facilitare nuove scoperte biologiche e
determinare nuovi paradigmi computazionali sul modello dei sistemi viventi
[NCBI: National Center for Biotechnology Information --- www.ncbi.nih.gov/Education]
martedì 11 maggio 2010
Bioinformatica: ambiti applicativi
•
Sviluppo di regole e algoritmi per l’analisi delle sequenze di acidi nucleici e proteine;
•
Simulazione di processi biologici: dall’interazione tra coppie di proteine ai
pathways metabolici (biologia dei sistemi);
•
fornire modelli statistici validi per l'interpretazione dei dati provenienti da
esperimenti di biologia molecolare e biochimica al fine di identificare tendenze e leggi
numeriche;
•
generare nuovi modelli e strumenti matematici per l'analisi di sequenze di DNA,
RNA e proteine la fine di creare un corpus di conoscenze relative alla frequenza di
sequenze rilevanti;
•
organizzare le conoscenze acquisite a livello globale su genoma e proteoma in basi
di dati al fine di rendere tali dati accessibili a tutti, e ottimizzare gli algoritmi di ricerca
dei dati stessi per migliorarne l'accessibilità;
martedì 11 maggio 2010
Analisi di Sequenze
•
Sequenze --> proteine, geni, regioni regolative, rna, dna
•
1977: prima sequenza nucleotidica;
•
•
1983: 2000 sequenze in banca dati;
Strumenti e metodi per l’analisi delle sequenze sono alla base di tutta la
bioinformatica;
martedì 11 maggio 2010
Annotazione Funzionale
martedì 11 maggio 2010
•
Ricerca in banche dati;
•
Motivi funzionali;
•
Identificazione di domini;
Analisi filogenetiche
•
martedì 11 maggio 2010
Ricostruzione della storia
evolutiva di geni e organismi
basandosi sulle
caratteristiche osservate
sulle sequenze geniche e
proteiche;
Bioinformatica Strutturale
•
1958 - John Kendrew:
servendosi della
cristallografia a raggi X,
riesce a definire in modo
completo la struttura
atomica della Mioglobina di
Capodoglio, dimostrando
che la proteina presentava
una disposizione degli atomi
ben ordinata, necessaria a
definirne la sua funzione;
[Premio Nobel per la chimica (1962)]
martedì 11 maggio 2010
Predizione Strutturale
•
Ricostruzione della struttura 3D di una proteina a partire dalla sua sequenza
primaria;
martedì 11 maggio 2010
Simulazioni
prot 2
sol.1
prot 1
prot 2
prot 2
programma di
docking
prot 1
prot 1
prot 2
prot 1
.
.
.
martedì 11 maggio 2010
sol.2
sol.3
•
Drug Design;
•
Protein Design;
•
Docking;
Genomica
Studio del genoma degli organismi viventi. In particolare si occupa della struttura,
contenuto, funzione ed evoluzione del genoma.
martedì 11 maggio 2010
Genomica
•
Sequenziamento del
DNA
•
Assemblaggio:
Ricostruzione del genoma
da milioni di sequenze;
•
Annotazione
Genomica:
Identificazione di geni,
trascritti e regioni
reogolative;
martedì 11 maggio 2010
Genomica Comparata
Confronto tra i genomi di diversi organismi, nella loro organizzazione e sequenza.
martedì 11 maggio 2010
System Biology
martedì 11 maggio 2010
•
Studio dei processi
biologici, a livello cellulare e
molecolare, considerati
come sistemi composti da
molte parti interagenti;
•
Processo:
•
Raccolta dati;
•
Modello matematico;
•
Simulazione e
previsione;
•
Verifica sperimentale;
Analisi di testi
Estrazione automatica di informazione scientifica dalla letteratura esistente.
martedì 11 maggio 2010
Ontologie
Classificazione e ordinamento della conoscenza biologica.
martedì 11 maggio 2010
Cenni Biologici
martedì 11 maggio 2010
Genoma e DNA
Tutte le informazioni contenute nel DNA di un organismo vivente costituiscono il suo
Genoma, contenuto in ciascuna cellula dell’organismo stesso
•Il DNA è un polimero (catena) di 4 acidi
nucleici semplici, detti nucleotidi;
•Ciascun nucleotide è costituito di tre parti:
una molecola di base + uno zucchero + un
gruppo fosforico;
•Le basi sono 4:
•A = Adenina
•G = Guanina
•C = Citosina
•T = Timina
martedì 11 maggio 2010
Il DNA è costituito da due sequenze
nucleotidiche che assumono la
caratteristica forma a spirale, legate tra
loro da legami ad idrogeno
La lunghezza del DNA viene misurata in
termini di coppie di basi
[ Il DNA umano è lungo 3.3 miliardi di
coppie di basi]
DNA - Acido desossiribonucleico
martedì 11 maggio 2010
Tra le basi vale la legge di
complementarietà di Watson-Crick:
•Adenina si lega solo con Timina: A-T
•Guanina si lega solo con Citosina: C-G
Quindi una sequenza determina
completamente la sequenza
complementare: questo consente di
generare copie identiche dell’informazione
immagazzinata nel DNA;
La direzione di ciascuna sequenza è
convenzionalmente da 5’ a 3’: quindi le due
sequenze di DNA sono complementari e
antiparallele;
DNA - Acido desossiribonucleico
martedì 11 maggio 2010
Proteine
Le proteine sono le componenti primarie degli esseri viventi.
Tutte le proteine, nonostante le loro enormi
differenze, sono composte dagli stessi 20
componenti di base: gli amminoacidi.
Gli amminoacidi sono legati tra loro
attraverso il legame peptidico;
La sequenza amminoacidica è codificata
direttamente dal materiale genetico (DNA),
attraverso un processo detto sintesi
proteica;
martedì 11 maggio 2010
Proteine
La sequenza polipeptidica possiede diversi gruppi laterali che, interagendo tra loro o con
l’acqua circostante, provocano il ripiegamento (folding) della proteina stessa, generando
così la struttura secondaria e terziaria. A volte, la proteina può ripiegarsi ulteriormente,
generando la struttura quaternaria.
La struttura tridimensionale di
una proteina è una delle
principali aree di ricerca, in
quanto spesso la forma è
correlata alla funzione;
martedì 11 maggio 2010
Codice Genetico
Il codice genetico è lo schema attraverso cui la cellula traduce una sequenza di codoni (o
triplette di basi) di RNA in una sequenza di amminoacidi durante la sintesi proteica
martedì 11 maggio 2010
RNA - Acido Ribo-nucleico
L’RNA è un polimero simile al DNA, da cui però differisce per alcuni aspetti:
•
è costituito da un’unica catena
nucleotidica;
•
i suoi nucleotidi sono composti da uno
zucchero di tipo ribosio;
•
la base azotata uracile sostituisce la
timina, pur mantenendo valida la
complementarietà con l’adenina;
Coinvolto nei processi di traduzione e
trascrizione del DNA e nella successiva
sintesi proteica.
martedì 11 maggio 2010
Concetti biologici utili alla bioinformatica
•
La sequenza di DNA può essere trattata come una stringa sull’alfabeto {A,C,G,T};
•
La sequenza primaria di una proteina può essere trattata come una stringa
sull’alfabeto {A, R, D, N, C, E, Q, G, H, I, L, K, M, F, P, S, T, W, Y, V};
•
Il DNA è formato da:
•[Esoni] Regioni codificanti: ovvero contenenti geni, cioè istruzioni per creare
proteine;
•[Introni] Regioni non codificanti: ovvero senza una funzione conosciuta;
•
Due o più sequenze di DNA o proteine si dicono omologhe se provengono da un
antenato comune. L’omologia può anche indicare una funzione comune nelle
sequenze in esame;
martedì 11 maggio 2010
!"#$"%&"'%
Banche dati biologiche
martedì 11 maggio 2010
Nascita delle banche dati biologiche
•
Inizio anni 70: nasce la tecnologia del DNA ricombinante, che permette di
manipolare le sequenze nucleotidiche e di capire la struttura, la funzione e
l’organizzazione del DNA;
•
Fine anni 70: pubblicazione dei primi dati genomici, con le prime sequenze
nucleotidiche codificanti liberamente accessibili attraverso i rudimenti della rete
disponibili a quel tempo tra le varie università;
•
1965: Margareth Dayhoff compila un atlante di proteine omologhe, studiando le
relazioni tra le sequenze primarie; viene reso pubblico in versione elettronica nel
1970 nella banca dati NBRF (National Biomedical Research Foundation);
•
1981 [Kurt Stueber]: nasce nel Laboratorio Europeo di Biologia Molecolare (EMBL)
ad Heidelberg l’EMBL-datalibrary (519 entries con sequenze di DNA e RNA);
•
1982 [Walter Goad]: nasce una banca dati simile negli USA, che darà vita alla
GenBank;
•
1986: nel National Institute of Genetics in Mishima (Giappone) nasce un mirror della
GeneBank, la DDBJ;
•
2001: Il Consorzio Pubblico Internazionale e la Celera Genomics forniscono dati del
genoma umano completo, aprendo la strada ai progetti di sequenziamento a
tappeto;
martedì 11 maggio 2010
Organizzazione di un database biologico
- L’oggetto principale è la ENTRY, un’unità riconoscibile grazie ad un identificatore
univoco, che possiede una descrizione organizzata in campi standardizzati
riconoscibili grazie agli HEADERS univoci nella banca dati.
- Ogni banca dati presenta 2 versioni delle entries:
Flat File: un file di testo semplice, formattato, non interattivo;
HTML (o XML): interattivo, di facile consultazione;
- Ogni banca dati ha dei suoi codici univoci di identificazione e definisce le sue
entries secondo un rigido standard, imponendo a priori un certo numero di possibili
campi contrassegnati da tag specifici, che permettono l’utilizzo di questi file da parte
di programmi automatici per l’information retrieval.
- Sia i flat-file che le pagine XML sono ricchi di cross-references, ossia riferimenti
che rimandano ad altre banche dati generiche o specializzate. Si ottiene così una
serie di informazioni spesso ridondanti.
martedì 11 maggio 2010
Esempio di ENTRY (EMBL-data library) (1/4)
Ogni linea comincia con due caratteri che indicano il codice: questo codice è sempre
seguito da 3 spazi bianchi. Le informazioni cominciano quindi dal carattere in posizione
6.
•
ID: identificatore della entry; tipo di molecola; divisione tassonomica; lunghezza bp;
•
AC: accession number (identifica univocamente il record);
•
SV-DT: versione e data di creazione della entry;
•
DE: descrizione della entry;
•
OS-OC: nome della specie, classificazione tassonomica;
ID
XX
AC
XX
DT
DT
XX
DE
XX
KW
XX
OS
OC
OC
OC
XX
martedì 11 maggio 2010
AJ223854; SV 1; linear; mRNA; STD; MUS; 949 BP.
AJ223854;
02-MAY-1998 (Rel. 55, Created)
23-SEP-2008 (Rel. 97, Last updated, Version 3)
Mus musculus telethonin complete cDNA
telethonin.
Mus musculus (house mouse)
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia;
Eutheria; Euarchontoglires; Glires; Rodentia; Sciurognathi; Muroidea;
Muridae; Murinae; Mus.
Esempio di ENTRY (2/4)
•
RN
RP
RA
RT
RL
RL
RL
XX
RN
RX
RX
RA
RA
RT
RT
RL
XX
RN
RX
RX
RA
RA
RT
RL
XX
DR
DR
RN, RA, RT, RL: informazioni bibliografiche;
[1]
1-949
Ievolella
;
Submitted
Ievolella
G.Colombo
C.;
(10-FEB-1998) to the EMBL/GenBank/DDBJ databases.
C., CRIBI Biotechnology Centre, Universita' di Padova, viale
3, 35121, ITALY.
[2]
DOI; 10.1038/72822
PUBMED; 10655062.
Moreira E.S., Wiltshire T.J., Faulkner G., Nilforoushan A., Vainzof M.,
Suzuki O.T., Valle G., Reeves R., Zatz M., Passos-Bueno M.R., Jenne D.E.;
"Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene
encoding the sarcomeric protein telethonin";
Nat. Genet. 24(2):163-166(2000).
[3]
DOI; 10.1016/S0014-5793(97)01108-3
PUBMED; 9350988.
Valle G., Faulkner G.P., Deantoni A., Pacchioni B., Pallavicini A.,
Pandolfo D., Tiso N., Toppo S., Trevisan S., Lanfranchi G.;
"Telethonin, a novel sarcomeric protein of heart and skeletal muscle";
FEBS Lett. 415(2):163-168(1997).
Ensembl-Gn; ENSMUSG00000007877; Mus_musculus.
Ensembl-Tr; ENSMUST00000008021; Mus_musculus.
martedì 11 maggio 2010
Esempio di ENTRY (3/4)
•
FT (Feature Table): Regioni o siti della sequenza considerati interessanti ed eventuale
link (cross-referencing);
FH
FH
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
XX
Key
Location/Qualifiers
source
1..949
/organism="Mus musculus"
/mol_type="mRNA"
/tissue_lib="Stratagene cDNA library Uni-ZAP tm XR Vector"
/tissue_type="Diaphram muscle"
/db_xref="taxon:10090"
1..36
/experiment="experimental evidence, no additional details
recorded"
928
37..540
/codon_start=1
/product="telethonin"
/function="sarcomeric protein"
/db_xref="GOA:O70548"
/db_xref="InterPro:IPR015667"
/db_xref="MGI:1330233"
/db_xref="UniProtKB/Swiss-Prot:O70548"
/experiment="experimental evidence, no additional details
recorded"
/protein_id="CAA11585.1"
/translation="MATSELSCQVSEENQERREAFWAEWKDLTLSTRPEEGCSLHEEDT
QRHETYHRQGQCQAVVQRSPWLVMRLGILGRGLQEYQLPYQRVLPLPIFTPTKVGASKE
EREETPIQLRELLALETALGGQCVERQDVAEITKQLPPVVPVSKPGPLRRTLSRSMSQE
AQRG"
541..949
/experiment="experimental evidence, no additional details
recorded"
5'UTR
polyA_site
CDS
3'UTR
martedì 11 maggio 2010
Esempio di ENTRY (4/4)
•
SQ
SQ: sequenza nucleotidica
Sequence 949 BP; 215 A; 250 C; 331 G; 153 T; 0 other;
aggagcagga catagcagag ggagcaatca gaaatcatgg ccacttcaga
caagtgtctg aggagaacca ggaacgcagg gaagccttct gggctgagtg
actctgtcta cccggccgga agagggatgc tccttgcacg aggaggatac
gagacctacc accggcaggg acagtgtcag gcggtggtac agcgctcacc
atgcgcctgg gtatcctcgg ccgtgggcta caggaatacc agctgccgta
ctgcccctac ccatcttcac gcccaccaag gtgggggcct ccaaggagga
acccccatcc agcttcggga gctgctggcc ctggagacgg ccctgggcgg
gagcgccagg acgtggctga gatcacaaag cagcttcccc ctgtggtgcc
cccgggcccc tgcgccgtac cctgtctcga tccatgtctc aggaagctca
gatggactgt gtgactcaga ctccactgtg tctgtctcag gctaggcact
gacaatggag gagagctgct ggcagtggct gctttgtagt ttgcccagag
tgggaggagg gagcccgagg ccaggatgcc taggtgtcct gagtccccac
gcgaggatgg cgggcactag gagtggagag ctgagcaccc tcagccccag
aagagatcct ggtgagagga gaggcccctg ggaatggcct gctcgggaac
ggagaaggat gtgcaacgct ctggaaagga gggggatgtg aagagggtgg
gcccccagca ccctctggta gcactgcaat aaatgctcag ccatgttca
gctgagctgc
gaaagacctg
acagaggcat
atggctggtg
ccagcgggtg
gcgcgaggag
ccagtgcgtg
agtcagcaaa
gagaggctga
tcctggctag
gtgggagcta
agggaaggga
aagaagagac
agatggacta
aagtgggcag
//
[per la descizione completa delle etichette dei campi è possibile consultare:
http://www.ebi.ac.uk/embl/Documentation/User_manual/usrman.html#2]
martedì 11 maggio 2010
60
120
180
240
300
360
420
480
540
600
660
720
780
840
900
949
Tipi di banche dati di interesse biologico
•
Le banche dati possono essere suddivise
•
•
per tipo:
•
PRIMARIE;
•
DERIVATE;
per tipo di informazioni contenute;
•
sequenze nucleotidiche;
•
sequenze proteiche;
•
strutture;
•
letteratura;
•
....
martedì 11 maggio 2010
Banche dati primarie
Memorizzano essenzialmente le sequenze e poche altre informazioni generiche
correlate alla sequenza per identificarla dal punto di vista specie-funzione (es:
laboratorio dove è avvenuto il sequenziamento, data, specie, descrizione...).
Le banche dati primarie sono:
•
(1980) [EBI] EMBL datalibrary: Europa
•
(1982) [NCBI] GenBank: USA
•
(1986) DDBJ: Giappone
Le tre organizzazioni utilizzando DBMS e modalità di accesso diversi:
•
NCBI: DBMS personalizzato, accesso tramite Entrez;
•
EBI/DDBJ: DMBS SRS Oracle, accesso tramite SRS;
In tutti i casi, la struttura della base dati è nascosta agli utenti;
martedì 11 maggio 2010
Sistemi di interrogazione alle banche dati
Esistono dei sistemi integrati che permettono di interrogare, attraverso il Web, in
modo semplice ed intuitivo le banche dati biologiche. I tre sistemi principali sono:
Le banche dati primarie sono:
•
ENTREZ: Associato a GenBank;
•
SRS: Associato a EMBL;
•
DBGET: Associato a DDBJ; martedì 11 maggio 2010
EBI - European Bioinformatics Institute
Hinxton - Cambridge (UK) - http://www.ebi.ac.uk/embl/
ricerca con
parole-chiave
ricerca con
accession number
martedì 11 maggio 2010
EBI - European Bioinformatics Institute
Hinxton - Cambridge (UK) - http://www.ebi.ac.uk/embl/
•
European Molecular Biology Laboratory (EMBL) Nucleotide Sequence Database è una
completa collezione di sequenze nucleotidiche primarie, mantenuta all’European
Bioinformatics Institute (EBI).
•
I dati sono sottomessi da centri di ricerca genomica, ricercatori individuali o autori
attestati, e sono immediatamente disponibili alla comunità.
•
I database sono su base Oracle e l’interazione con essi è fornita via web tramite il
Sequence Retrieval System (SRS), motore di ricerca proprio dell’EBI per i database
biologici.
EMBL
162.000.000
sequenze
martedì 11 maggio 2010
SRS
•
è un sistema aperto, che può essere installato su calcolatori differenti (server) e può
integrare banche dati strutturate su altri server SRS o altre banche dati, previa
strutturazione o indicizzazione nel sistema SRS
martedì 11 maggio 2010
NCBI - National Center for Biotechnology Information
http://www.ncbi.nlm.nih.gov/genbank/index.html
M14752!
martedì 11 maggio 2010
NCBI - National Center for Biotechnology Information
http://www.ncbi.nlm.nih.gov/genbank/index.html
•
E' un database di sequenze genetiche dell'National Institute of Healt statunitense. E'
quindi una collezione annotata di tutte le sequenze di DNA disponibili pubblicamente;
•
Accesso ai dati attraverso ENTREZ: sistema di interrogazione delle diverse basi dati
gestite dall’NCBI che costituisce quindi un hub completo per la ricerca di informazioni.
•
Offre anche la possibilità di effettuare ricerche di tipo bibliografico e, soprattutto, di
avere un collegamento diretto tra i vari database (sequenza-struttura-mappa geneticaarticolo)
martedì 11 maggio 2010
ENTREZ
•
sistema disponibile via web per la ricerca e l’estrazione dei dati da banche dati di
sequenze nucleotidiche, proteiche, dalla banca dati bibliografica MEDLINE, dalla
banca dati delle malattie mendeliane OMIN, e da ogni banca dati sviluppata
dall’NCBI;
•
Sistema CHIUSO, e non è possibile ottenere il software che gestisce il sistema;
martedì 11 maggio 2010
DDBJ - DNA Data Bank of Japan
http://www.ddbj.nig.ac.jp/
martedì 11 maggio 2010
DDBJ - DNA Data Bank of Japan
http://www.ddbj.nig.ac.jp/
•
DNA Data Bank of Japan (DDBJ) ha iniziato la sua attività nel 1984.
•
E' utilizzata soprattutto dai ricercatori giapponesi, ma ovviamente è utilizzabile da
tutti attraverso internet.
martedì 11 maggio 2010
Banche dati proteiche
Un secondo grande aggregato di banche dati è quello per le sequenze proteiche, le
quali possono essere ottenute in seguito a:
•
Determinazione diretta della sequenza proteica;
•
Traduzione di sequenze nucleotidiche per le quali sia stata individuata o predetta la
funzione di gene codificante la proteina;
•
Studi di espressione genica;
•
Cristallografia e determinazione delle strutture secondarie e terziarie;
martedì 11 maggio 2010
Banche dati proteiche
•
(1986) SWISS-PROT (Protein
knoledgebase): banca dati di
riferimento sviluppata a Ginevra.
Contiene informazioni
accuratamente annotate, spesso
a mano.
•
(1996) TrEMBL (Translated
EMBL): risultato della traduzione
automatica in amminoacidi di
tutte le sequenze annotate nella
banca dati EMBL come
codificanti proteine; supplemento
a SWISS-PROT;
•
PIR (Protein Information
Resource): soprattutto
indirizzato a definire gli standard
di annotazione, con ridondanza
minima;
Insieme hanno formato il consorzio
UNIPROT, repository centralizzato
di tutte le sequenze proteiche
martedì 11 maggio 2010
Banche dati di strutture
•wwPDB (world wide Protein Data Bank): banca dati di riferimento per i dati strutturali
3D di proteine, comprendente le coordinate atomiche determinate attraverso analisi
cristallografiche ai raggi X, analisi NMR ed altre tecniche.
Comprende anche una sezione dedicata alle strutture delle proteine determinate tramite
metodi computazionali.
Creata dalla collaborazione di RCSB (USA), MSD-EBI (EBI), PDBj (Giappone)
•MMDB (Entrez’s Molecular Modelling Database):
•NDB: banca dati di strutture di acidi nucleidi, soli o assieme a proteine;
•CSD: banca dati di strutture di piccole molecole organiche e organometalliche
martedì 11 maggio 2010
Banche dati derivate
Le banche dati primarie contengono tutte le sequenze conosciute di tutti gli organismi,
genomiche di mRNA, etc...
Per rendere la ricerca di informazioni organizzata sono state costruite delle banche
dati derivate che raggruppano solo dati relativi a specifici argomenti.
Esempi:
•
Database di sequenze genomiche: GDB (uomo), MGI (topo), SGD (lievito);
•
Database di geni e trascritti: UniGene, LocusLink, dbEST, etc...
Esistono poi dei database integrati che raggruppano i dati provenienti da differenti
database fornendo informazioni particolareggiate di argomenti specifici.
martedì 11 maggio 2010
Database ‘non ridondanti’
Nei database primari sono inserite tutte le sequenze conosciute ottenute
sperimentalmente e/o ricostruite
La stessa regione genomica o lo stesso trascritto possono essere stati sequenziati più
volte
RIDONDANZA
Per evitare questo problema sono stati creati dei database ‘semplificati’ senza
ripetizione di informazioni. In particolare:
•
RefSeq: sequenze genomiche, mRNA, proteine;
•
UniGene: sequenze ottenute dal sequenziamento dei trascritti (mRNA)
•
Gene: (sottoinsieme di RefSeq) sequenze geniche;
martedì 11 maggio 2010
Domini Proteici
Molte proteine, specialmente quelle di grandi dimensioni, sono formate da più parti
funzionali organizzate in strutture tridimensionali distinte che vengono chiamate
domini proteici.
Esempio:
alcuni fattori di trascrizione del DNA hanno due domini, uno in grado di legarsi con
una particolare sequenza di DNA, l’altro in grado di attivare la trascrizione.
Proteine formate da più di un dominio si sono probabilmente evolute per fusione
di geni che contenevano tali domini.
martedì 11 maggio 2010
Banche dati di domini proteici
Database contenenti domini funzionali delle proteine:
•
PFAM: (http://pfam.sanger.ac.uk) Banca dati di famiglie di proteine accomunate da
elementi strutturali e funzionali;
•
PROSITE: (http://www.expasy.org/prosite) Annota patterns amminoacidici individuati
in un set di sequenze proteiche attraverso analisi in silicio e studi sperimentali
•
SMART: (http://smart.embl.de) Risorsa che raccoglie dati relativi a domini proteici e
consente la ricerca di domini in nuove sequenze proteiche
•
InterPro: (http://www.ebi.ac.uk/interpro) Raccoglie informazioni strutturali e funzionali
relativi ad una proteina o ad una famiglia di proteine. Comprende PROSITE e PFAM
martedì 11 maggio 2010
Sottomissione di sequenze
Esistono più di 20 differenti tipi di formati per la sottomissione di sequenze ad una
banca dati:
Esiste la necessità di avere quindi un sistema che possa effettuare la conversione da
un formato all’altro...
martedì 11 maggio 2010
ReadSeq: http://www.ebi.ac.uk/cgi-bin/
Software disponibile sul web che effettua la conversione di diversi formati di file
sequenze
martedì 11 maggio 2010
Il formato FASTA
Spesso i programmi che effettuano analisi bioinformatiche sulle sequenze richiedono
che esse vengano date come input in un formato particolare: FASTA Format;
FASTA è un formato per la descrizione di una sequenza ‘grezza’. Consiste
essenzialmente in una parte iniziale di intestazione, di solito limitata ad una linea di
testo, e da una o più linee che riportano una sequenza di DNA o di amminoacidi,
usando l’alfabeto standard.
martedì 11 maggio 2010
!"#$
%&'($
*#01"2'
-.$/'
789:;77;<#7'
8=>%0"7'
789:;77;<#7'
8=>%0"7'
!"#$%#&'
*+$,'
*$/'
789:;77;<#7'
8=>%0"7'
./$'
3"0"#014'
(($)'
!")$
Collaborazioni tra banche dati
martedì 11 maggio 2010
565'
The International Nucleotide Sequence Database
Collaboration
•
EMBL, GenBank e DDBJ collaborano dal 1982. Ogni database mantiene e
processa nuovi dati e sequenze e informazioni biologiche ad esse correlate,
sottomesse dagli scienziati e ricercatori delle loro regioni;
•
Questi tre database si sincronizzano automaticamente tra loro ogni 24 ore. Il
risultato di questa sincronizzazione è che ogni database contiene esattamente le
stesse informazioni, ad eccezione delle sequenze sottomesse nell’ultima giornata;
martedì 11 maggio 2010
The International Nucleotide Sequence Database
Collaboration
•
La sincronizzazione è organizzata secondo regole pubblicate e standardizzate
dall’International Advisory Board.
•
Le linee guida consistono nella definizione delle tabelle del database, che regolano
quindi il contenuto e la sintassi di ogni nuova entry.
•
Il formato delle linee guida è DDT.
•
La sintassi è chiamata INSDSeq, e consiste principalmente nello stabilire le lettere
accettate per la codifica delle sequenze nucleotidiche e amminoacidiche.
martedì 11 maggio 2010
Il problema della nomenclatura
•
Non esiste uno standard di assegnazione dei nomi ai geni; uno stesso gene può
avere diversi nomi, o uno stesso nome può individuare diversi geni;
•
I geni possono essere catalogati in base agli organismi a cui appartengono, alla
loro attivazione nel corso dello sviluppo di un organismo, alla funzione e alla
struttura delle proteine codificate;
•
Il problema della nomenclatura è stato risolto assegnando ad ogni nuova entry
nella basi di dati un numero di serie, in modo da poter identificare ogni sequenza in
modo univodo: ACCESSION NUMBER
martedì 11 maggio 2010
Ricerche in banche dati
martedì 11 maggio 2010
L’importanza della similarità
•
Due sequenze simili potrebbero derivare dalla stessa sequenza ancestrale, avere
quindi la stessa struttura, o una funzione biologica simile
martedì 11 maggio 2010
L’importanza della similarità
martedì 11 maggio 2010
Allineamento di sequenze
•
Il passo base per la ricerca di similarità è l’allineamento di due o più sequenze;
•
La similarità tra due o più sequenze si verifica effettuando prima un allineamento tra
le sequenze in esame, e poi decidendo se le eventuali parti comuni sono più
facilmente dovute al caso o ad una effettiva relazione tra loro;
•
Esistono due tipi di allineamento:
•
GLOBALE: si tenta di allineare il massimo numero di caratteri delle due sequenze,
incluse le parti finali. Candidate ideali sono le sequenze di lunghezza simile;
•
LOCALE: si tenta di allineare solo pezzi di sequenze molto simili. L’allineamento
termina quando termina l’isola di forte match. Candidate ideali sono sequenze con
lunghezze diverse, che presentano regioni fortemente conservate;
martedì 11 maggio 2010
Misure di similarità
Le mutazioni delle sequenze genetiche sono alla base dell’evoluzione. Esse sono
dovute principalmente a:
•
mutazioni in siti differenti di una sequenza occorrono in maniera indipendente;
•
la rilevazione di mutazioni conservative è più probabile quando le due sequenze sono
correlate e meno probabile quando l’allineamento è casuale;
•
la lunghezza di un GAP (spazi inseriti per mantenere l’allineamento) non è correlata
agli elementi allineati con il GAP stesso;
Il punteggio totale assegnato ad un allineamento è una somma di termini: un
termine per ciascuna coppia di residui allineati, più un termine per ciascun GAP.
martedì 11 maggio 2010
Matrici BLOSUM
[1992 da S. Henikoff e J.G. Henikoff]
Introdotte per attribuire un punteggio alle sostituzioni nei confronti tra sequenze
aminoacidiche.
martedì 11 maggio 2010
Algoritmi per l’allineamento
I principali metodi di allineamento a coppie sono:
•
•
Algoritmi di Programmazione Dinamica:
•
Needleman & Wunsh: (1970) allineamento globale
•
SMith & Watermann: (1981) allineamento locale
Tecniche euristiche:
•
FASTA
•
BLAST
martedì 11 maggio 2010
Algoritmi di Programmazione dinamica
- Algoritmo di Needlemann & Wunsch: è un algoritmo dinamico che permette di
trovare l’allineamento globale ottimo. Calcola ricorsivamente l’allineamento ottimo per
sottosequenze via via più lunghe.
Complessità computazionale:
Spazio: S(mn)
Tempo: O(mn)
-Algoritmo di Smith & Watermann: è una variante dell’algoritmo N-W che permette
di trovare l’allineamento locale ottimo. Non ci sono punteggi negativi. L’opzione zero
corrisponde all’iniziare un nuovo allineamento.
Complessità computazionale:
Spazio: S(mn)
Tempo: O(mn)
martedì 11 maggio 2010
Algoritmi euristici per l’allineamento
Gli algoritmi di programmazione dinamica trovano allineamenti ottimi, ma sono troppo
lenti nei casi pratici, come ad esempio una ricerca su una banca dati.
Si utilizzano allora degli algoritmi euristici che migliorano le prestazioni a scapito della
qualità della soluzione.
Due applicativi simili che usano queste tecniche sono FASTA e BLAST. Per entrambi
è difficile valutare in modo preciso sia l’efficienza che l’affidabilità.
martedì 11 maggio 2010
FAST-All (Lipman, Pearson 1985)
Consente di cercare una sequenza (detta query) in un database di sequenze (dette
subject). Prevede tre fasi:
1-indicizzazione: la query viene divisa in parole di lunghezza prefissata e si
memorizzano tutte le posizioni di inizio parola. Viene costruita una lookup-table.
2-ricerca: ogni volta che il programma trova una parola coincidente su entrambe le
sequenze, viene memorizzata nella lookup-table (indice). Una volta terminata la
lettura, vengono estratte le più lunghe e su di esse viene effettuato l’allineamento
locale. Alla fine della fase viene compilata una graduatoria di similarità su questi
allineamenti.
3-raffinamento: il programma tenta di migliorare l’allineamento congiungendo le best
initial region con gap. Sulle sequenze che hanno ottenuto i migliori punteggi viene
applicata una variante dell’algoritmo SW, che restringe l’analisi delle best initial region
congiunte.
martedì 11 maggio 2010
FAST-All (Lipman, Pearson 1985)
martedì 11 maggio 2010
BLAST (Altshul 1990)
Basic local alligment search tool: ottimizzato per trovare allineamenti locali privi di
gap. L’algoritmo prevede tre fasi:
1- leggendo la sequenza query viene formato un elenco di parole di lunghezza W. Per
ognuna viene creata una lista di parole affini (W-mers): vengono considerati tutti i Wmers che superano una soglia T quando viene allineato con la parola della query;
2-vengono esaminate tutte le sequenze subject, per cercare la presenza di tutti i Wmers dell’elenco. Ogni corrispondenza trovata viene considerata come parte di un
allineamento più esteso.
3- viene considerata la possibilità di estendere ogni hit in entrambe le direzioni, senza
l’aggiunta di gap. Si ottiene quindi un allineamento locale detto HSP (High Scoring
Segment Pair)
martedì 11 maggio 2010
BLAST (Altshul 1990)
martedì 11 maggio 2010
BLAST (Altshul 1990)
martedì 11 maggio 2010
Basi di dati biologiche
Seminario per il corso di Basi di Dati II
Luana Rinaldi
[email protected]
martedì 11 maggio 2010