La corrente tardiva di sodio:
fisiopatologia e farmacologia
di un nuovo bersaglio terapeutico
Antonio Zaza
Dipartimento di Biotecnologie e Bioscienze, Università degli Studi di Milano-Bicocca, Milano
The sodium current (INa) plays a pivotal role in the propagation of electrical activity in the heart. However, a
large body of evidence indicates that the “late” component of INa (INaL) may be enhanced in diseased myocardium. INaL enhancement has consequences on the electrical stability, contractile function and metabolism
of cardiac myocytes, which may significantly contribute to disease progression. The possibility of blocking INaL
selectively, that is to say without affecting the INa component involved in electrical propagation, has recently
emerged. INaL has hence become a “therapeutic target”, thus far clinically validated in the treatment of angina and arrhythmias but, as suggested by bench evidence, potentially relevant to a wider range of cardiac disorders. Such multiplicity of effects originates from the complex network of cell functions affected by INaL enhancement; acquaintance with such a network is useful in fully exploiting the therapeutic potential of selective INaL inhibition. This review deals with the pathophysiology of INaL enhancement and with the basic principles underlying its selective inhibition. Clinical evidence of antianginal and antiarrhythmic efficacy of INaL inhibition is available, but its discussion goes beyond the scope of this review.
Key words. Arrhythmias; Late sodium current; Myocardial ischemia; Pathophysiology; Pharmacology;
Ranolazine.
G Ital Cardiol 2011;12(2 Suppl 10):3S-11S
L’attuale approccio farmacologico alla cardiopatia ischemica
cronica si basa principalmente sulla riduzione del lavoro meccanico del cuore, compendiato dal prodotto fra pressione arteriosa e frequenza cardiaca (doppio prodotto). Anche gli interventi mirati primariamente ad aumentare il flusso coronarico agiscono sul tono della muscolatura vascolare e sono quindi destinati ad influenzare anche le resistenze sistemiche. Pur
avendo indubitabili meriti, questo approccio è concettualmente limitato dal fatto che implica una riduzione della performance cardiaca, intesa come capacità di sostenere lavoro emodinamico. Sarebbe teoricamente possibile ridurre il consumo di
ossigeno senza ridurre la performance cardiaca? La risposta è
affermativa perché, a parità di lavoro emodinamico, il consumo
di ossigeno dipende dall’“efficienza” del ciclo cardiaco. Inoltre,
il flusso coronarico può essere modulato anche agendo sul rilassamento diastolico e, quindi, senza interferire sul controllo
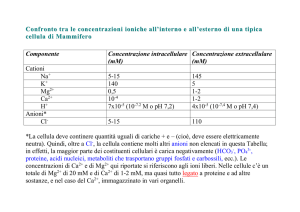
della pressione arteriosa. La possibilità di agire in modo positivo su efficienza e rilassamento diastolico è emersa dalla recente individuazione del ruolo patogenetico di una anomalia della corrente di sodio (INa) miocardica (trasportata dai canali voltaggio-dipendenti), consistente nell’aumento della sua componente persistente (late sodium current, INaL) (Figura 1)1, e dal© 2011 Il Pensiero Scientifico Editore
L’autore dichiara di avere rapporti di consulenza con Menarini (Firenze)
e di finanziamento della ricerca con Gilead (Palo Alto, CA).
Per la corrispondenza:
Prof. Antonio Zaza Dipartimento di Biotecnologie e Bioscienze,
Università degli Studi di Milano-Bicocca, Piazza della Scienza 2,
20126 Milano
e-mail: [email protected]
la disponibilità di un farmaco in grado di contrastarla selettivamente.
I prossimi due paragrafi illustrano rispettivamente il meccanismo e le conseguenze funzionali dell’aumento di INaL; l’ultimo paragrafo discute il meccanismo con cui è possibile inibire
INaL selettivamente. Le evidenze cliniche dell’efficacia terapeutica del blocco di INaL, seppur disponibili, non vengono discusse
nella presente rassegna.
PATOGENESI DELL’AUMENTO DI INaL
INa è condotta da una proteina canale la cui conformazione è
sensibile al potenziale esistente a cavallo della membrana cellulare. Sulla scorta delle evidenze sperimentali, le variazioni della conformazione del canale, che ne modulano la conduttanza
durante le vari fasi del ciclo elettrico (Fasi 0-4), sono schematizzabili come illustrato nella Figura 22.
A potenziale diastolico (Fase 4) il poro è chiuso dalla giustapposizione dei quattro domini della proteina (D1-D4) che
formano il canale. La depolarizzazione oltre la “soglia di attivazione” (circa -70 mV) trascina verso l’esterno i domini in modo sequenziale. La deformazione della proteina che segue allo
spostamento dei primi tre domini (D1-D3) apre il poro, permettendo il passaggio della corrente (Fase 0, attivazione). Lo
spostamento dell’ultimo dominio (D4) segue con qualche millisecondo di ritardo e completa l’attivazione, ma permette contemporaneamente alla “particella di inattivazione” (un segmento citoplasmatico della proteina) di legarsi alla bocca intracellulare del canale, causandone inattivazione (Fase 1).
G ITAL CARDIOL | VOL 12 | SUPPL 2 AL N 10 2011
3S
LA CORRENTE TARDIVA DI SODIO
L’inattivazione è quindi secondaria all’attivazione e limita la
durata di INa a pochi millisecondi, corrispondenti alla fase di depolarizzazione rapida del potenziale d’azione. Lo stato inattivato persiste durante la fase di plateau (Fase 2) e viene rimosso (recupero dall’inattivazione) solo dal ritorno del dominio D4
alla posizione iniziale, causato dalla ripolarizzazione (Fase 3).
In condizioni normali, lo stato inattivato del canale è stabile e la probabilità che esso ritorni aperto durante la fase di plateau (Fase 2), seppur non nulla, è molto piccola (INaL quasi nulla). Tuttavia, in condizioni patologiche, tale probabilità aumenta e permette ad una frazione più significativa di INa di persistere durante la fase di plateau (Fase 2 patologica in Figura 2).
L’aumento patologico di INaL è quindi interpretabile come destabilizzazione dello stato inattivato dello stesso canale che conduce la INa fisiologica.
Aumento di INaL si osserva in risposta a diversi stimoli patogenetici (ipossia, esposizione a superossidi, metaboliti ischemici, ecc.)3,4 ed è sostanziale nell’insufficienza cardiaca di varia
eziologia5,6. Esso è quindi verosimilmente una conseguenza relativamente aspecifica di sofferenza cellulare. Osservazioni recenti suggeriscono che i vari stimoli che inducono INaL riconoscano un mediatore ultimo comune nell’eccessiva fosforilazione del canale da parte della Ca2+-calmodulina-kinasi (CaMKIIδ)7.
Quest’ultima è attivata dal calcio (Ca2+) citosolico e dai radicali liberi dell’ossigeno, le cui concentrazioni aumentano precocemente nella disfunzione cellulare. Uno spiccato aumento di
INaL consegue inoltre a mutazioni del canale del sodio, che alterano la stabilità del meccanismo di inattivazione, e spiega le
manifestazioni cliniche della sindrome di prolungamento della
ripolarizzazione denominata LQT38.
CONSEGUENZE DELL’AUMENTO DI INaL
L’aumento di INaL ha due effetti diretti: 1) rallenta la velocità di
ripolarizzazione; 2) cambia il bilancio dei flussi da cui dipende
la concentrazione intracellulare dello ione sodio (Na+). Questi
effetti determinano una cascata di alterazioni secondarie, spesso legate da feedback positivo, di significato sia per la funzione elettrica che per quella contrattile e metabolica.
Il rallentamento della ripolarizzazione implica prolungamento del potenziale d’azione (rilevabile all’ECG come aumento del QTc) e tende ad aumentarne la variabilità temporale e spaziale9,10. La stabilità della ripolarizzazione è infatti condizionata dalla velocità di variazione del potenziale di membrana11, la cui riduzione facilita fenomeni a chiaro significato
aritmogeno, quali i post-potenziali precoci12. Non sorprende,
quindi, che l’aumento di INaL causi direttamente tali fenomeni e
ne faciliti l’induzione da parte di altri farmaci13,14.
In condizioni normali, INaL ha ampiezza molto minore della
componente transiente di INa (INaT) (dell’ordine dell’1%); tuttavia la durata di INaL eccede di circa 100 volte quella di INaT. Se ne
conclude che le due componenti di INa hanno più o meno lo
stesso peso nel mediare l’ingresso di Na+ nella cellula15. Un aumento patologico di INaL può quindi perturbare in modo assai significativo l’omeostasi ionica intracellulare. L’eccessivo ingresso di Na+ causa in primis corto-circuito della pompa Na+/K+; infine, se la capacità di trasporto della pompa viene ecceduta, la
concentrazione intracellulare di Na+ aumenta, una condizione
che caratterizza l’insufficienza contrattile16. Le conseguenze secondarie a tale perturbazione appaiono subito chiare se si considera che la gran parte dei trasporti attivi di sostanze attraver-
so le membrane cellulari di tutto l’organismo è alimentato dal
gradiente di concentrazione del Na+.
La Figura 3 confronta i movimenti di Na+ e Ca2+ durante l’accoppiamento eccitazione-contrazione normale (pannello A) e in
presenza di aumento patologico di INaL (pannello B). Ad ogni ciclo, una quota del Ca2+ necessario ad innescare la contrazione
entra nella cellula attraverso i canali del Ca2+ voltaggio-dipendenti del sarcolemma e, per evitare accumulo, deve esserne
espulso. La maggior parte del flusso di espulsione del Ca2+ (contro gradiente) è sostenuta dallo scambio con il Na+ (gradiente a
favore), mediato dalla proteina di trasporto NCX. Un aumento
della concentrazione di Na+ nel citoplasma riduce l’espulsione
del Ca2+, che viene captato dalla pompa SERCA nel reticolo sarcoplasmico17. L’apertura dei canali RyR, normalmente confinata
alla sistole elettrica, viene facilitata dal sovraccarico di Ca2+ 18;
ne consegue una elevata probabilità che il Ca2+ venga liberato
dal reticolo sarcoplasmico in modo spontaneo durante la diastole. Le conseguenze immediate di tale situazione vanno dalla
compromissione del rilassamento dei sarcomeri (funzione diastolica) alla genesi di fenomeni elettrici a significato aritmogeno
(delayed afterdepolarization)17. Più a lungo termine, il Ca2+ svolge nella cellula svariate funzioni, che includono la modulazione
di numerose attività enzimatiche, della trascrizione genica e del
bilancio fra crescita e morte cellulare19,20. Di fatto, vie di segnalazione importanti per il “rimodellamento” cellulare e tissutale
sono attivate dal Ca2+ 21,22; verosimilmente per questo, il Ca2+
cellulare viene attivamente confinato in specifici compartimenti dai sistemi di trasporto. È quindi ipotizzabile che l’aumento di
INaL, compromettendo tale compartimentazione, abbia un ruolo
attivo nell’evoluzione cronica del danno miocardico.
L’aumento del Ca2+ cellulare che deriva da INaL ha, nell’immediato, un effetto inotropo positivo, che potrebbe essere significativo nel sostenere l’attività meccanica del miocardio con
insufficienza avanzata23. Quindi, l’aumento di INaL nel miocardio ipertrofico/insufficiente potrebbe rappresentare, nell’immediato, un meccanismo adattativo al deficit contrattile. Tuttavia, analogamente a quanto accade per l’attivazione neuroumorale, l’aumento di INaL ha, a lungo termine, significato verosimilmente maladattativo, cioè tale da contribuire alla progressione del deficit contrattile24. Ciò nondimeno, in presenza
di insufficienza cardiaca avanzata, un blocco di INaL può teoricamente comportare transitoria diminuzione della funzione
contrattile e ciò dovrebbe essere tenuto in considerazione nell’applicazione terapeutica.
Equivalenti acidi (H+) vengono continuamente prodotti dalla componente anaerobica del metabolismo energetico.
L’espulsione attiva di H+ dalla cellula è largamente affidata ad
uno scambiatore Na+/H+ (proteina NHE in Figura 3) e viene quindi rallentata dall’eccesso di Na+ intracellulare25. Tale difetto nell’espulsione di H+, conseguente alla dissipazione del gradiente
di Na+, acquisisce particolare significato durante ischemia, in
cui la produzione di ATP dipende maggiormente dal metabolismo anaerobico (lattacido).
Un aumento di INaL ha potenzialmente conseguenze dirette
sull’efficienza meccanica del miocita, quantizzabile come rapporto fra lavoro emodinamico e consumo di ossigeno. Esso rappresenta infatti un meccanismo di “corto-circuito” del potenziale chimico generato, con dispendio di ATP, dalla pompa
Na+/K+ (proteina NaK in Figura 3) e, secondariamente, di Ca2+
(SERCA). Inoltre, la persistenza di una contrazione attiva dei
sarcomeri durante la diastole consuma energia senza contribuire alla gittata.
G ITAL CARDIOL | VOL 12 | SUPPL 2 AL N 10 2011
5S
A ZAZA
Figura 3. Relazione fra i movimenti di Na+, Ca2+ e H+ durante un ciclo di accoppiamento eccitazione-contrazione normale (A) e in presenza di aumento patologico della corrente tardiva di sodio (INaL) (B). Nel pannello A, i riquadri numerati descrivono la sequenza di eventi nel ciclo; gli eventi che si riferiscono a sistole e diastole sono illustrati rispettivamente nella parte sinistra e destra della figura a scopo di schematizzazione. Nel pannello B, i riquadri descrivono le anomalie degli eventi del ciclo, derivanti dall’eccesso di INaL. La bibliografia a supporto del contenuto della figura
è citata nel testo.
FFA, acidi grassi liberi; RS, reticolo sarcoplasmatico.
6S
G ITAL CARDIOL | VOL 12 | SUPPL 2 AL N 10 2011
LA CORRENTE TARDIVA DI SODIO
Questi effetti si traducono in un incremento della componente del consumo energetico che non si estrinseca in lavoro meccanico e, quindi, a diminuzione dell’efficienza della pompa (aumento del consumo di ossigeno per ogni livello di lavoro emodinamico). È stato inoltre osservato che aumenti contemporanei delle concentrazioni citosoliche di Na+
e di Ca2+ conducono a caduta del potenziale chimico (rapporto NADH/NAD+) che il mitocondrio utilizza per produrre
ATP26.
In conclusione, le potenziali conseguenze di un aumento
patologico di INaL sull’omeostasi ionica cellulare sono di ampio
spettro, comprendendo aspetti “acuti” (aritmie, disfunzione
diastolica, alterazioni del controllo del pH intracellulare, aumento del consumo di ossigeno) e cronici (rimodellamento miocardico). È quindi ipotizzabile che il blocco di INaL possa costituire
un intervento terapeutico a significato “pleiotropico”; tuttavia,
la sua efficacia clinica è stata per ora valutata solo in termini di
effetto antischemico ed esistono solo dati preliminari circa l’effetto antiaritmico. Ci soffermeremo qui sui meccanismi relativi
a questi effetti.
INaL e ischemia miocardica
La registrazione di INa richiede l’isolamento dei miociti, condizione in cui l’ischemia non può essere integralmente riprodotta. Tuttavia, l’esposizione dei miociti a componenti del
quadro ischemico, quali ipossia, metaboliti ischemici, specie
reattive dell’ossigeno, è in grado di indurre aumento patologico di INaL (per una rassegna della letteratura vedi Zaza et
al.27).
I meccanismi con cui un aumento di INaL può condurre ad alterazione del rapporto fra flusso coronarico e consumo di ossigeno (CF/VO2) sono conseguenza diretta delle alterazioni descritte nel paragrafo precedente. La compromissione del rilassamento diastolico28,29 limita il flusso coronarico per compressione estrinseca del circolo intramurale30. Tale meccanismo è limitato al circolo coronarico; quindi, la sua eliminazione non interessa necessariamente le resistenze di altri circoli. Il ruolo dell’eccesso di INaL nel limitare il flusso coronarico è indirettamente dimostrato dall’aumento della perfusione miocardica che deriva dal suo blocco selettivo31.
La diminuzione dell’efficienza contrattile dei miociti implica aumento del consumo di ossigeno a parità di doppio
prodotto (frequenza cardiaca x pressione arteriosa, espressione del lavoro emodinamico). Sia la dinamica del Ca2+ citosolico, che la risposta dei sarcomeri al Ca2+, vengono depressi
dall’acidosi intracellulare25,32; quindi, la diminuita capacità di
eliminazione dei H+ comporta verosimilmente una sensibilizzazione della funzione contrattile all’ischemia. Se ne deduce
che un blocco di INaL può migliorare il rapporto CF/VO2 e la
tolleranza del miocardio all’ischemia con meccanismi largamente indipendenti da variazioni della frequenza cardiaca o
della pressione arteriosa e, pertanto, complementari a quelli
caratteristici della terapia antischemica tradizionale.
INaL e aritmie
La destabilizzazione della ripolarizzazione e il sovraccarico intracellulare di Ca2+, conseguenti ad aumenti patologici di INaL,
hanno chiaro significato aritmogeno. L’osservazione di un effetto antiaritmico del blocco selettivo di INaL, ampiamente dimostrato sperimentalmente33-35, non trova quindi difficoltà in-
terpretative. Tale effetto dovrebbe tuttavia essere limitato alle
condizioni in cui INaL è effettivamente aumentata, forse più probabili nel ventricolo che nell’atrio. Si è invece osservato un considerevole effetto della ranolazina, un bloccante che a livello
ventricolare mostra elevata selettività per INaL27,33, anche sulle
aritmie atriali. Benché la rilevanza di INaL sia stata recentemente estesa alle aritmie atriali36, la spiegazione di questa osservazione può risiedere anche nella peculiarità di INa espressa nel
muscolo atriale (vedi sotto)37,38.
MECCANISMO DEL BLOCCO SELETTIVO DI INaL
Il farmaco che dimostra maggior selettività per INaL (rispetto a
INaT) è la ranolazina27,33,39-41. Il meccanismo di tale selettività, illustrato nella Figura 4, può essere interpretato sulla scorta dei
seguenti dati.
La ranolazina si lega ad una porzione del canale, localizzata sul dominio D4 della proteina, con cui interagiscono tutti i
generici bloccanti di INa (lidocaina, chinidina, ecc.), definito per
questo “recettore degli anestetici locali”42. Il comportamento
del blocco di INa durante depolarizzazioni cicliche suggerisce
che la ranolazina si leghi al canale solo quando esso è nel suo
stato inattivato37,43. Come rappresentato in modo schematico
nella Figura 4, ciò è interpretabile assumendo che la conformazione assunta dalla proteina nello stato inattivato renda accessibile il sito di legame al farmaco (Fasi 1-2) e il recupero dall’inattivazione (Fase 3) determini quindi la dissociazione del farmaco44. Tuttavia, essendo il sito di legame comune ad altri farmaci, questo meccanismo potrebbe non essere sufficiente a
spiegare la peculiare selettività per INaL dimostrata dalla ranolazina. È intuibile come un requisito per la mancanza di effetto
su INaT sia la completa rimozione del blocco durante la diastole, prima della successiva eccitazione (Fase 4). Perché ciò avvenga, quando lo stato inattivato viene rimosso dalla ripolarizzazione della membrana, il farmaco deve rapidamente dissociare dal canale, e non può farlo attraverso l’ambiente idrofilico del poro, che rimane inaccessibile in questa fase. Un fattore che può favorire tale condizione è quindi un’elevata solubilità (mobilità) del farmaco nell’ambiente lipidico (idrofobico)
della membrana, a cui contribuisce l’assenza di carica45. La Figura 5 confronta la frazione di farmaco presente in forma neutra a pH fisiologico (pannello A) per i farmaci la cui selettività
per INaL (INaL/INaT block ratio) è stata misurata (pannello B)39-41. La
non perfetta correlazione fra selettività e neutralità suggerisce
l’influenza di fattori aggiuntivi; tuttavia, nel suo insieme, questa analisi supporta l’ipotesi che la ranolazina dissoci dal canale attraverso l’ambiente lipidico della membrana (Figura 4) e,
più in generale, che il grado di ionizzazione contribuisca alla selettività per INaL.
Diversamente da quanto accade nel ventricolo, nell’atrio
una significativa frazione dei canali del Na+ permane nello stato inattivato durante tutta la diastole37. Ciò spiega perché nell’atrio la ranolazina perda parzialmente la sua selettività per
INaL e alteri velocità di conduzione e refrattarietà più o meno
come fa la chinidina (classe IA dei farmaci antiaritmici)37, farmaco di notevole efficacia sulle aritmie atriali. Dato che il rischio proaritmico dei farmaci come la chinidina dipende dalla loro azione sul ventricolo, l’atrio-selettività rappresenta un
potenziale vantaggio nella terapia delle aritmie sopraventricolari.
G ITAL CARDIOL | VOL 12 | SUPPL 2 AL N 10 2011
7S
A ZAZA
RIASSUNTO
La corrente di sodio (INa) ha un ruolo fondamentale nella propagazione dell’attività elettrica nel muscolo cardiaco. Tuttavia, recenti
evidenze indicano che la sua componente tardiva (late sodium current, INaL) sia aumentata nel miocardio patologico. Tale aumento ha
importanti conseguenze sulla stabilità elettrica, sulla funzione contrattile e sul metabolismo cellulare, che contribuiscono a condizionare l’evoluzione della malattia. È recentemente emersa la possibilità di bloccare INaL selettivamente, cioè senza influenzare la
componente di INa coinvolta nella propagazione. INaL è quindi diventata un “bersaglio terapeutico” la cui validità, per ora dimostrata clinicamente nella terapia della sindrome anginosa e delle
aritmie, potrebbe teoricamente estendersi ad altre patologie. Tale
pleiotropismo di effetti è interpretabile sulla base della rete di
funzioni cellulari con cui l’aumento di INaL interferisce. La cono-
scenza degli effetti dell’aumento di INaL è utile per sfruttare al meglio le potenzialità offerte dal suo blocco farmacologico. Questa
rassegna illustra la fisiopatologia dell’aumento di INaL e i principi di
base con cui è possibile contrastarlo in modo selettivo. Non vengono invece discusse le evidenze cliniche dell’efficacia del blocco
di INaL nella terapia antianginosa, per cui si rimanda il lettore ad altra fonte.
Parole chiave. Aritmie; Corrente tardiva di sodio; Farmacologia; Fisiopatologia; Ischemia miocardica; Ranolazina.
RINGRAZIAMENTI
Si ringrazia Giunti O.S. Organizzazioni Speciali per la gentile collaborazione offerta nella realizzazione iconografica di questo documento.
BIBLIOGRAFIA
1. Berecki G, Zegers JG, Bhuiyan ZA, Verkerk AO, Wilders R, van Ginneken AC. LongQT syndrome-related sodium channel mutations probed by the dynamic action
potential clamp technique. J Physiol 2006;
570(Pt 2):237-50.
2. Armstrong CM. Na channel inactivation from open and closed states. Proc Natl
Acad Sci U S A 2006;103:17991-6.
3. Fearon IM, Brown ST. Acute and chronic
hypoxic regulation of recombinant hNav1.5
alpha subunits. Biochem Biophys Res Commun 2004;324:1289-95.
4. Gautier M, Zhang H, Fearon IM. Peroxynitrite formation mediates LPC-induced
augmentation of cardiac late sodium currents. J Mol Cell Cardiol 2008;44:241-51.
5. Valdivia CR, Chu WW, Pu J, et al. Increased late sodium current in myocytes
from a canine heart failure model and from
failing human heart. J Mol Cell Cardiol
2005;38:475-83.
6. Huang B, El-Sherif T, Gidh-Jain M, Qin
D, El-Sherif N. Alterations of sodium channel kinetics and gene expression in the
postinfarction remodeled myocardium. J
Cardiovasc Electrophysiol 2001;12:218-25.
7. Wagner S, Dybkova N, Rasenack EC, et
al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin
Invest 2006;116:3127-38.
8. Wang Q, Shen J, Li Z, et al. Cardiac
sodium channel mutations in patients with
long QT syndrome, an inherited cardiac
arrhythmia. Hum Mol Genet 1995;4:16037.
9. Shimizu W, Antzelevitch C. Cellular basis for long QT, transmural dispersion of repolarization, and torsade de pointes in the
long QT syndrome. J Electrocardiol 1999;
32(Suppl):177-84.
10. Maltsev VA, Silverman N, Sabbah HN,
Undrovinas AI. Chronic heart failure slows
late sodium current in human and canine
ventricular myocytes: implications for repolarization variability. Eur J Heart Fail 2007;9:
219-27.
10S
11. Zaza A. Control of the cardiac action
potential: the role of repolarization dynamics. J Mol Cell Cardiol 2010;48:106-11.
12. January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and
block. A role for L-type Ca2+ current. Circ Res
1989;64:977-90.
13. Wu L, Shryock JC, Song Y, Belardinelli L.
An increase in late sodium current potentiates the proarrhythmic activities of low-risk
QT-prolonging drugs in female rabbit hearts.
J Pharmacol Exp Ther 2006;316:718-26.
14. Wu L, Rajamani S, Shryock JC, et al.
Augmentation of late sodium current unmasks the proarrhythmic effects of amiodarone. Cardiovasc Res 2008;77:481-8.
15. Makielski JC, Farley AL. Na+ current in
human ventricle: implications for sodium
loading and homeostasis. J Cardiovasc Electrophysiol 2006;17(Suppl 1):S15-S20.
16. Pieske B, Houser SR. [Na+]i handling in
the failing human heart. Cardiovasc Res
2003;57:874-86.
17. Bers DM, Despa S, Bossuyt J. Regulation
of Ca2+ and Na+ in normal and failing cardiac
myocytes. Ann N Y Acad Sci 2006;1080:
165-77.
18. Gyorke S, Gyorke I, Lukyanenko V, Terentyev D, Viatchenko-Karpinski S, Wiesner
TF. Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac
muscle. Front Biosci 2002;7:d1454-d1463.
19. Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res
Commun 2004;322:1178-91.
20. Bers DM, Guo T. Calcium signaling in
cardiac ventricular myocytes. Ann N Y Acad
Sci 2005;1047:86-98.
21. Heineke J, Molkentin JD. Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol
2006;7:589-600.
22. Anderson ME, Brown JH, Bers DM.
CaMKII in myocardial hypertrophy and heart
failure. J Mol Cell Cardiol 2011 Jan 27 [Epub
ahead of print].
G ITAL CARDIOL | VOL 12 | SUPPL 2 AL N 10 2011
23. Schillinger W, Teucher N, Christians C,
et al. High intracellular Na+ preserves myocardial function at low heart rates in isolated myocardium from failing hearts. Eur J
Heart Fail 2006;8:673-80.
24. Maltsev VA, Undrovinas A. Late sodium
current in failing heart: friend or foe? Prog
Biophys Mol Biol 2008;96:421-51.
25. Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart.
J Mol Cell Cardiol 2009;46:318-31.
26. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake
during excitation-contraction coupling and
impairs energetic adaptation in cardiac myocytes. Circ Res 2006;99:172-82.
27. Zaza A, Belardinelli L, Shryock JC.
Pathophysiology and pharmacology of the
cardiac “late sodium current“. Pharmacol
Ther 2008;119:326-39.
28. Fraser H, Belardinelli L, Wang L, Light
PE, McVeigh JJ, Clanachan AS. Ranolazine
decreases diastolic calcium accumulation
caused by ATX-II or ischemia in rat hearts. J
Mol Cell Cardiol 2006;41:1031-8.
29. Moss AJ, Zareba W, Schwarz KQ,
Rosero S, McNitt S, Robinson JL. Ranolazine
shortens repolarization in patients with sustained inward sodium current due to type3 long-QT syndrome. J Cardiovasc Electrophysiol 2008;19:1289-93.
30. Duncker DJ, Bache RJ. Regulation of
coronary blood flow during exercise. Physiol
Rev 2008;88:1009-86.
31. Venkataraman R, Belardinelli L, Blackburn B, Heo J, Iskandrian AE. A study of the
effects of ranolazine using automated quantitative analysis of serial myocardial perfusion images. JACC Cardiovasc Imaging
2009;2:1301-9.
32. Blanchard EM, Solaro RJ. Inhibition of
the activation and troponin calcium binding
of dog cardiac myofibrils by acidic pH. Circ
Res 1984;55:382-91.
33. Antzelevitch C, Belardinelli L, Zygmunt
AC, et al. Electrophysiological effects of ra-
LA CORRENTE TARDIVA DI SODIO
nolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 2004;
110:904-10.
34. Song Y, Shryock JC, Wagner S, Maier
LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther 2006;318:
214-22.
35. Song Y, Shryock JC, Wu L, Belardinelli L.
Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in
guinea pig ventricular myocytes. J Cardiovasc Pharmacol 2004;44:192-9.
36. Sossalla S, Kallmeyer B, Wagner S, et al.
Altered Na+ currents in atrial fibrillation: effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am
Coll Cardiol 2010;55:2330-42.
37. Burashnikov A, Di Diego JM, Zygmunt
AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy
for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 2007;116:1449-57.
38. Antzelevitch C, Burashnikov A. Atrialselective sodium channel block as a novel
strategy for the management of atrial fibrillation. J Electrocardiol 2009;42:543-8.
39. Maltsev VA, Sabbah HN, Undrovinas AI.
Late sodium current is a novel target for
amiodarone: studies in failing human myocardium. J Mol Cell Cardiol 2001;33:92332.
40. Undrovinas AI, Belardinelli L, Undrovinas
NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left
ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol 2006;17(Suppl 1):
S169-S177.
41. Persson F, Andersson B, Duker G, Jacobson I, Carlsson L. Functional effects of
the late sodium current inhibition by
AZD7009 and lidocaine in rabbit isolated
atrial and ventricular tissue and Purkinje fibre. Eur J Pharmacol 2007;558:133-43.
42. Fredj S, Sampson KJ, Liu H, Kass RS.
Molecular basis of ranolazine block of LQT-3
mutant sodium channels: evidence for site
of action. Br J Pharmacol 2006;148:1624.
43. Rajamani S, El-Bizri N, Shryock JC,
Makielski JC, Belardinelli L. Use-dependent block of cardiac late Na+ current by
ranolazine. Heart Rhythm 2009;6:162531.
44. Starmer CF. Theoretical characterization
of ion channel blockade: ligand binding to
periodically accessible receptors. J Theor Biol
1986;119:235-49.
45. Hille B. Mechanisms of block. In: Hille B,
ed. Ionic channels of excitable membranes.
Sunderland, MA: Sinauer Associates, 1984:
272-302.
G ITAL CARDIOL | VOL 12 | SUPPL 2 AL N 10 2011
11S