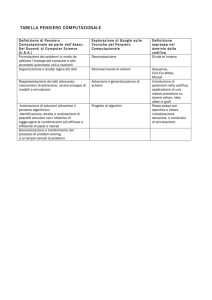
Genomics Session
Lezione 4
Identificazione di segnali
nel genoma
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Segnali nel genoma
Segnale:
Una sottosequenza (sottostringa), con un pattern particolare, nel DNA o
RNA, che riveste un ruolo biologico. Generalmente, è riconosciuto da una
proteina o da un'altra molecola e si ritrova in varie locazioni nel genoma.
Esempi:
- Sequenze riconosciute da endonucleasi di restrizione (es. GAATTC per
EcoRI);
- Sequenze nei promotori;
- Sequenze segnale dei geni;
- Elementi dell'origine di replicazione e terminatori.
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Segnali nel genoma
Il segnale può essere rappresentato da particolari sequenze di nucleotidi, ma
queste sono solo approssimazioni di ciò che è effettivamente riconosciuto
dall'interattore.
Ad esempio, una proteina riconoscerà particolari gruppi chimici del DNA in una
particolare conformazione nello spazio. Il riconoscimento è sempre dettato da
leggi fisiche.
Alcune di queste interazioni sono transienti, altre possono essere più durature.
L'interazione non è quasi mai covalente.
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Interazione DNA-proteine
Come le proteine riconoscono il DNA:
Le proteine riconoscono DNA o RNA per
mezzo di legami idrogeno ed interazioni
elettrostatiche principalmente con la
catena proncipale dll'acido nucleico.
Il riconoscimento specifico dei siti di
interazione avviene mediante legami con
i bordi degli anelli della base che
sporgono nel solco maggiore o minore
della doppia elica.
Ci possono essere anche interazioni
idrofobiche mediate da catene laterali
che si intercalano fra due coppie di basi.
[Sun et al., 2004]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Interazione DNA-proteine
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Interazione DNA-proteine
Come le proteine riconoscono il DNA:
I siti di interazione possono avere strutture
semplici o complesse.
Ci può essere simmetria interna (ad es.,
ripetizioni invertite di motivi, sequenze
palindromiche).
Questo è dovuto al fatto che le proteine
leganti DNA sono spesso oligomeri (dimeri, o
tetrameri).
Se l'oligomero è ad esempio un dimero con
struttura “testa-a-testa“, la posizione relativa
die due siti di legame del dimero si rifletterà
sulla sequenza riconsciuta nel genoma.
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Interazione DNA-proteine
Gal4: attivatore di geni relativi al metabolismo del galattosio
L'unità biologica funzionale è un omodimero
5'-NNNTCGGNNNACTNTNNNCCGANNN-3'
3'-NNNAGCCNNNTGANANNNGGCTNNN-5'
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Segnali nel genoma
- Segnali nel DNA possono essere identificati con tecniche sperimentali;
- Analisi genetica classica
- Sfruttando le modificazioni delle proprietà del DNA duplex quando
legato all'interattore;
- gel-shift assay
- Sfruttando la “protezione“ che l'interattore offre al sito
- chromatin immunoprecipitation
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Chromatin immunoprecipitation (ChIP)
Immunoprecipitazione (IP): tecnica per
isolare un antigene (es. una proteina)
mediante il legame di un anticorpo che ci si
leghi specificamente.
Chromatin immunoprecipitation (ChIP):
tecnica per identificare dove una data
proteina si lega su una sequenza genomica
in vivo.
Procedura:
1. Si induce la formazione di legami covalenti
fra proteine e DNA (usando formaldeide o
DTBP);
2. Si lisano le cellule e si frammenta il DNA;
3. Con un anticorpo specifico per la proteina
di interesse, si isolano i frammenti di DNA
legati alla proteina stessa.
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Chromatin immunoprecipitation (ChIP)
4. Il legame fra DNA e agente di cross-linking
è rotto (ad es. usando alte temperature) e il
DNA purificato;
5. La sequenza di frammenti di DNA è
determinata;
6. I frammenti sono mappati sul genoma;
7. Si può una stima quantitativa di quante
sequenze sono trovate per ogni sito.
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Regolazione della trascrizione
Lo studio ed identificazione di segnali nel DNA è spesso
applicato per l'analisi dell'espressione genica.
●
Cosa attiva o disattiva un gene?
●
Quando un gene è attivato o disattivato?
●
Dove (in quali cellule, tessuti, organi) un gene è attivo?
●
Lezione 4
Quanto è attivo un gene (quante copie del suo prodotto
sono sintetizzate)?
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Regolazione della trascrizione
Restrizione spaziale e temporale dell’espressione genica:
Geni housekeeping
Geni con espressione ristretta nello spazio
• Espressione in piu’ organi/tessuti diversi
Stesso ruolo in piu’ tessuti
Il gene codifica per diverse isoforme (promotori alternativi e/o
splicing alternativo tessuto
specifico)
• Espressione specifica per tessuto, linea o tipo cellulare
• Espressione solo in singole cellule
• Distribuzione intracellulare o extracellulare
•
Geni con espressione ristretta nel tempo
Stadio di sviluppo
Stadio di differenziamento
Momento del ciclo cellulare
Espressione inducibile da parte di fattori ambientali o extracellulari
[source: Stefania Bortoluzzi 2003]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Controllo dell'espressione genica
L'espressione di un gene è regolata a vari livelli, con meccanismi
diversi;
La maggior parte di questi controlli è mediata da motivi di
sequenza, sul DNA, RNA o sulla proteina.
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Regolazione della trascrizione
●
●
●
●
Lezione 4
La trascrizione di ogni gene è controllata da una regione
regolatoria intorno al sito di inizio della trascrizione (transcription
start site, TSS);
Regioni che esercitano influenza sulla trascrizione possono però
essere anche molto lontane dal TSS;
La regolazione è dovuta a due tipi di componenti:
●
Sequenze (corte) di DNA regolatorio;
●
Proteine che vi si legano specificamente;
La combinazione di diversi componenti determina l'effetto
complessivo di regolazione;
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Regolazione della trascrizione
[Wasserman, Nature Rev. 2004]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Regolazione della trascrizione
POL II PROMOTER ELEMENTS
●
●
●
CORE PROMOTER ELEMENTS
● TATA box
● Initiator
● Downstream promoter element
TRANSCRIPTION FACTORS (TF) BINDING SITES
● CAAT box
● GC box
● Sp-1 sites
● GAGA boxes
ENHANCER(S) SITES
[source: Stefania Bortoluzzi 2003]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Regolazione della trascrizione
PROMOTORE CORE
→
PROM. PROSSIMALE
→
PROMOTORE DISTALE
→
regione sufficiente a deteminare
il TSS esatto
200-300 bp upstream al TSS,
responsabile, almeno in parte,
della modulazione
dell’espressione
100 bp – 2 Mb
[source: Stefania Bortoluzzi 2003]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Regolazione della trascrizione
[source: Stefania Bortoluzzi 2003]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Regolazione della trascrizione
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Regolazione della trascrizione
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Motivi di sequenza
●
Segnali regolatori della trascrizione:
●
Riconosciuti da proteine (fattori di trascrizione, TF);
●
Sequenze corte (5-25 bp);
●
A corta o lunga distanza dal TSS, fino a 1000 bp (o più);
●
Lezione 4
Sequenza, distanza dal TSS, e orientamento di un
motivo riconosciuto dall stesso TF sono variabili.
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Banche dati di TF e motivi di sequenza
TRANSFAC: http://www.gene-regulation.com/pub/databases.html#transfac
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Banche dati di TF e motivi di sequenza
TRANSFAC: http://www.gene-regulation.com/pub/databases.html#transfac
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Banche dati di TF e motivi di sequenza
TRANSFAC: http://www.gene-regulation.com/pub/databases.html#transfac
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Banche dati di TF e motivi di sequenza
TRANSFAC: http://www.gene-regulation.com/pub/databases.html#transfac
TRANSFAC matrix entry: TATA box
Campi:
Accession
ID
Descrizione
TF associati a questa entry
Matrice pesata
Numero di siti usati
Altre info
Referenze
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Banche dati di TF e motivi di sequenza
Banche dati specie-specifiche:
SCPD (yeast) http://rulai.cshl.edu/SCPD/
DPInteract (e. coli) http://arep.med.harvard.edu/dpinteract/
Drosophila DNase I Footprint Database (v2.0) http://www.flyreg.org/
Motivi di sequenza
Come si rappresenta un motivo
●
●
●
●
●
Lezione 4
Sequenza consenso: stringa con la sequenza più frequente
posizione per posizione (+/- wildcards);
Espressione regolare;
Profilo;
Modello statistico;
Logo.
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Matrici pesate
[Wasserman, Nature Rev. 2004]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Matrici pesate
[Wasserman, Nature Rev. 2004]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Matrici pesate
Scanning a sequence against a PWM
Sp1
ACCCTCCCCAGGGGCGGGGGGCGGTGGCCAGGACGGTAGCTCC
A
C
G
T
[-0.2284 0.4368
[-0.2284 -0.2284
[ 1.2348 1.2348
[ 0.4368 -0.2284
-1.5
-1.5
2.1222
-1.5
-1.5
-1.5
-1.5 1.5128
2.1222 0.4368
-1.5 -0.2284
0.4368
-1.5
-1.5 -0.2284
1.2348 1.5128
0.4368 0.4368
-1.5 -0.2284
-1.5 -0.2284
1.7457 1.7457
0.4368
-1.5
0.4368
-1.5
-1.5
1.7457
]
]
]
]
Abs_score = 13.4 (sum of column scores)
Relative score
A
C
G
T
[-0.2284 0.4368
[-0.2284 -0.2284
[ 1.2348 1.2348
[ 0.4368 -0.2284
-1.5
-1.5
2.1222
-1.5
-1.5
-1.5
-1.5 1.5128
2.1222 0.4368
-1.5 -0.2284
0.4368
-1.5
-1.5 -0.2284
1.2348 1.5128
0.4368 0.4368
-1.5 -0.2284
-1.5 -0.2284
1.7457 1.7457
0.4368
-1.5
0.4368
-1.5
-1.5
1.7457
]
]
]
]
0.4368
-1.5
-1.5
1.7457
]
]
]
]
Max_score = 15.2 (sum of highest column scores)
A
C
G
T
[-0.2284
-0.2284 0.4368
[-0.2284 -0.2284
[ 1.2348 1.2348
[ 0.4368 -0.2284
-1.5
-1.5
2.1222
-1.5
-1.5
-1.5
-1.5 1.5128
2.1222 0.4368
-1.5 -0.2284
0.4368
-1.5
-1.5 -0.2284
1.2348 1.5128
0.4368 0.4368
-1.5 -0.2284
-1.5 -0.2284
1.7457 1.7457
0.4368
-1.5
Min_score = -11.0 (sum of lowest column scores)
Abs_score - Min_score
⋅ 100 %
Max_score - Min_score
13.4 - (-11.0)
=
⋅ 100% = 93%
15.2 − (− 11.0)
Rel_score =
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Logo della sequenza
●
●
●
●
●
L'altezza relativa di ogni lettera riflette la sua abbondanza nell'allineamento
multiplo; l'altezza della pila di lettere è una basata su una misura della
conservazione basata sull'entropia;
Entropia(i) = -SUM { p(base, i)* ln[p(base, i)] }
Conservazione(i) = 2- Entropia(i)
Si esprime in bits di informazione
Posizione molto conservata -> bassa entropia -> pila alta
Posizione poco conservata -> alta entropia -> pila bassa
[Wasserman, Nature Rev. 2004]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Identificazione di motivi di sequenza
Scanning a sequence against a PWM
Sp1
ACCCTCCCCAGGGGCGGGGGGCGGTGGCCAGGACGGTAGCTCC
A
C
G
T
[-0.2284 0.4368
[-0.2284 -0.2284
[ 1.2348 1.2348
[ 0.4368 -0.2284
-1.5
-1.5
2.1222
-1.5
-1.5
-1.5
-1.5 1.5128
2.1222 0.4368
-1.5 -0.2284
0.4368
-1.5
-1.5 -0.2284
1.2348 1.5128
0.4368 0.4368
-1.5 -0.2284
-1.5 -0.2284
1.7457 1.7457
0.4368
-1.5
0.4368
-1.5
-1.5
1.7457
]
]
]
]
Abs_score = 13.4 (sum of column scores)
Problema: enorme numero di predizioni positive
Quante sono false predizioni?
Livello basale di trascrizione e binding dei TF
Come ridurre il numero di false predizioni?
Analizzare il contesto in cui il sito si viene a trovare (isole CpG)
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Identificazione di motivi di sequenza
Se la specificità di un TF è nota, si tratta di analizzare sequenze
genomiche con il modello di specificità.
Se la specificità del TF non è nota, o se si vogliono identificare
nuovi motivi di interazione, il problema diventa più difficile
Alcuni fattori possono facilitare la loro identificazione:
- over-rappresentati in prossimità di TSS;
- conservazione evolutiva;
- caratteristiche strutturali.
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Identificazione di motivi di sequenza
Motivi over-rappresentati in prossimità di TSS
daf-19 Binding Sites in C. elegans
-150
GTTGTCATGGTGAC
GTTTCCATGGAAAC
GCTACCATGGCAAC
GTTACCATAGTAAC
GTTTCCATGGTAAC
-1
che-2
daf-19
osm-1
osm-6
F02D8.3
[source: Peter Swoboda]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Analisi del promotore di geni co-regolati
●
●
●
Geni espressi in maniera simile e coordinata possono essere
identificati sperimentalmente (microarrays, RT-PCR, etc.);
Supponiamo di avere a disposizione la regione del promotore di n
geni G1, G2, … Gn;
Problema: trovare siti di legame di TF senza necessariamente
conoscerne la specificità. Due approcci:
●
Ricerca esaustiva di parole;
●
Lezione 4
Modelli probabilistici;
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Consensus, Wconsensus (Hertz & Stormo, 1999)
Si cerca il motivo che conduce alla
matrice pesata con più altro
contenuto di informazione
1. Si parte da un dataset di
promotori G1, G2, … Gn
2. Si seleziona un k-mero da un
promotore Gi
3. Si costruisce una matrice dal kmero
4. Per ogni altra sequenza Gj, si
usa ogni k-mero per aggiornare la
matrice
5. Si sceglie la matrice a maggiore
contenuto di informazione
6. Si riparte dal punto 4 fino ad
esaurimento delle sequenze
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
MEME (Bailey & Elkan, 1995)
Algoritmo di EM: Expectation-Maximization
Un algoritmo EM è un metodo per trovare la stima della massima verosimiglianza
dei parametri di un modello statistico. EM funziona iterativamente, alternando una
fase E (Expectation), in cui la verosmiglianza è calcolata stanti i parametri correnti,
e una fase M (Maximization) dove nuovi parametri sono calcolati cercando di
massimizzare la funzione usata in E.
Funziona iterativamente:
1. Generazione di un modello del motivo di sequenza
a. Inizia con un motivo (k-mero) da una posizione casuale o specificata
b. Costruisce una matrice incorporando frequenze stimate di
background
2. Identifica esempi del motivo usando il modello
a. Per ogni k-mero nel set di geni, calcola la probabilità dato il modello
b. Seleziona i k-meri a più alta probabilità
3. Revisione del modello
a. Costruisce una nuova matrice, basata sulle frequenze pesate di tutti i
k-meri pescati nel dataset
4. Continua fino a convergenza
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
MEME (Bailey & Elkan, 1995)
EM algorithm
●
Iterative EM algorithm
●
●
●
E-step
●
●
Lezione 4
Compute expected posterior probability of binding
locations, based on current preferences
M-step
●
●
Exact binding locations for all protein-DNA pairs
recognition preferences: Pp(N|A)
Update DNA-recognition preferences to maximize the
likelihood of current binding locations based on the
distribution of possible binding locations in previous Estep
Local optima
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
MEME (Bailey & Elkan, 1995)
1. Si parte da una serie di k-meri e si calcola la matrice pesata
[source: Benos 2007]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
MEME (Bailey & Elkan, 1995)
2. Si identificano nuovi k-meri (blu) e si ri-calcola la matrice pesata
[source: Benos 2007]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
MEME (Bailey & Elkan, 1995)
3. Usando la nuova matrice pesata, alcuni dei k-meri di partenza (verdi) saranno
non più sufficientemente simili
4. Si continua finchè non si aggiungono/scartano più k-meri e i punteggi
rimangono simili
[source: Benos 2007]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
MEME (Bailey & Elkan, 1995)
5. Una volta trovato un motivo, si cancella dalle sequenze del dataset e si
ricomincia
6. Si finisce una volta che i motivi trovati scendono sotto una soglia di punteggio,
e si ripete per tutti i valori di k
[source: Benos 2007]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
MEME (Bailey & Elkan, 1995)
http://meme.sdsc.edu/meme/website/meme.html
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
MEME (Bailey & Elkan, 1995)
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Gibbs samplers
Altra strategia per esplorare ampi spazi conformazionali
1. Dati una serie di promotori G1, G2, … Gn, seleziona un k-mero
s1,s2, ... ,sn da ciascuno di essi
[source: Benos 2007]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Gibbs samplers
2. Scarta un k-mero si a caso dall'insieme s1,s2, ... ,sn
3. Calcola una matrice pesata dai rimanenti n-1
4. Usa la matrice per analizzare un altro k-mero si' dalla sequenza i da
cui era stato pescato si
5. Se lo score di si' è migliore di quello di si, prendi si', altrimenti prendi si'
con probabilità score(si')/score(si), e ricomincia
[source: Benos 2007]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi basati su conservazione evolutiva
Approccio: usare la conservazione fra specie per migliorare la predizione di
segnali
Si basa su alcune ipotesi:
1. La funzione e la specificità di legame dei TF è ben conservata in specie
diverse sufficientemente vicine;
2. Siti funzionali nel DNA (non codificanti) sono sottoposti a una diversa
pressione selettiva rispetto alle sequenze che li circondano;
3. Geni ortologhi di specie diverse hanno espressione simile, e
necessariamente sono controllati da un simile set di TF
I metodi più usati appartengono alla classe dei metodi per phylogenetic
footprinting
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi basati su conservazione evolutiva
Omologhi: sequenze che hanno origine comune, posono avere o no una funzione simile;
Ortologhi: omologhi prodotti da speciazione. Sono geni derivati da un ancestore comune la cui
divergenza deriva dalla divergenza delle due specie in cui si trovano. Tendono ad avere funzioni simili;
Paraloghi: omologhi prodotti da duplicazione genica. Sono geni derivati da un ancestore comune che si
è duplicato. Le due copie accumulano mutazioni e non sono soggette alla stessa pressione evolutiva.
Tendono ad avere funzioni diverse (poco o anche molto).
[Baxevanis and Ouellette, 2001]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi basati su conservazione evolutiva
[Baxevanis & Ouellette 2005]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi basati su conservazione evolutiva
Quali specie scegliere?
Bilanciare fra:
- specie abbastanza vicine tali che:
1) Ci sia sufficiente similarità da garantire un buon
allineamento di sequenze
2) Si possa assumere conservazione funzionale e regolativa
degli ortologhi
- specie abbastanza lontane tali che:
1) le sequenze non funzionali abbiano avuto tempo di
divergere
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi basati su conservazione evolutiva
- Si parte da motivi corti perfettamente conservati (ad es. Triplette, o coppie di
triplette separate da uno spaziatore);
- Si estendono questi seeds consentendo minore conservazione;
- Si combinano motivi simili identificati in tutto il genoma e si costruiscono le
matrici pesate.
[Kellis et al. 2003]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi basati su alberi filogenetici
●
Dati:
●
●
●
●
●
Una lunghezza stipulata del motivo k
Il punteggio massimo consentito di parsimonia d
Problema
●
Lezione 4
n sequenze ortologhe S1, S2, S3, …, Sn
un albero filogenetico T che mette in relazione
queste sequenze
Trovare il set di k-meri con score di parsimonia <= d
in T
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi basati su alberi filogenetici
AGTCGTACGTGAC... (Human)
AGTAGACGTGCCG... (Chimp)
ACGTGAGATACGT... (Rabbit)
GAACGGAGTACGT... (Mouse)
TCGTGACGGTGAT... (Rat)
k=4
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi basati su alberi filogenetici
ACGT
AGTCGTACGTGAC...
AGTAGACGTGCCG...
ACGT
ACGTGAGATACGT...
GAACGGAGTACGT...
ACGT
ACGG
TCGTGACGGTGAT...
Parsimony score: 1 mutazione
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi basati su conservazione evolutiva
Problemi:
- Bisogna scegliere le specie giuste;
- Genomi diversi evolvono a tassi diversi;
- Regioni diverse nello stesso genoma evolvono a tassi diversi;
- L'allineamento di regioni con scarsa pressione selettiva può essere
problematico;
- Ancora pochi genomi.
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Metodi ibridi
- Metodi sia basati su modelli statistici del motivo, sia basati su conservazione
evolutiva, soffrono di un elevato numero di predizioni false
- Metodi ibridi combinano i due approcci per ridurre il numero di falsi positivi
- Questo a discapito della sensibilità della precisione (molti veri positivi sono persi)
Alcuni lavori:
- Sinha et al. 2004 “PhyME: A probabalistic algorithm for finding motifs in
sets of orthologous sequences”
- Moses et al. 2004 “Monkey: identification of transcription factor binding sites
in multiple alignments using a binding site-specific evolutionary model
- Siddharthan et al. 2005 “PhyloGibbs: A Gibbs sampling motif finder that
incorporates phylogeny.”
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
ConSite (Adams, 2000)
[Wasserman, Nature Rev. 2004]
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Predizione usando informazioni strutturali (Kaplan, 2005)
Approccio: sfruttare la conoscenza sulla modalità di legame del TF per
predizione di siti di legame;
Applicazione: Cys2His2 Zinc Finger
- Famiglia più numerosa di proteine leganti il DNA;
- Solitamente monomerica, ma contenente diverse ripetizione del ”dito“
- Il dito interagisce con il solco maggiore del DNA
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Predizione usando informazioni strutturali (Kaplan, 2005)
Assunzioni:
- proteine della stessa famiglia hanno la stessa modalità di riconoscimento;
- la specificità dipende da quali aminoacidi si trovano nelle posizioni di contatto e dal
loro contesto;
- si possono estrarre regole dai membri della famiglia già ben caratterizzati, ed
estrapolarle a membri meno noti per determinarne la specificità.
Modo di legame canonico del dito di zinco sul DNA
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Predizione usando informazioni strutturali (Kaplan, 2005)
- Per molti zinc-fingers, sono noti solo i
promotori che essi regolano, ma non gli
esatti siti di legame
- Si identificano le matrici di specificità di
questi zinc-fingers con algoritmi EM
- Le specificità di ogni zinc-finger sono
combinate in una matrice che riporta
posizione per posizione le preferenze (cioè
quale nucleotide è trovato più
frequentemente quando in quella posizione
c'è uno specifico aminoacido)
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Predizione usando informazioni strutturali (Kaplan, 2005)
Dall'allineamento di tutti gli zinc-finger
in Transfac si determinano le
frequenze di riconoscimento
posizione-specifiche
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Predizione usando informazioni strutturali (Kaplan, 2005)
Dato un nuovo zinc-finger a specificità ignota, si identificano le posizioni di contatto, e si
usa la matrice di interazione per stimarne la specificità.
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010
Predizione usando informazioni strutturali (Kaplan, 2005)
- In Drosophila sono stati trovati 29 zinc-fingers con struttura canonica ma specificità
sconosciuta;
- Per ciascuno di essi, se ne è stimata la specificità e si e' usata la matrice ottenuta per
analizzare il promotore di tutti i geni noti di Drosophila;
- Di tutti i geni aventi un sito putativo di legame per un dato zinc-finger, si sono valutate
caratteristiche comuni.
Celle blu: arricchimento significativo di termini GO
Lezione 4
Genomica Computazionale, Laurea Magistrale A.A. 2009/2010