Corso di Laurea in Biologia Sanitaria
Universita' di Padova
C.I. DI METODI STATISTICI PER LA BIOLOGIA,
INFORMATICA E LABORATORIO DI INFORMATICA
(MOD. B)
8 + 32 ore
Docente:
Dr. Stefania Bortoluzzi
Dipartimento di Biologia
Universita' di Padova
viale G. Colombo 3, 35131, Padova
Tel. 0039 049 8276214
Email: [email protected]
IV LEZIONE
Uso di Genome Browser per l'annotazione di
sequenze genomiche.
Allineamento di sequenze trascritte con
sequenze genomiche: BLAT.
PROGETTO GENOMA UMANO
Milestones:
• 1990: Inizio (U.S. Department of Energy and the
National Institutes of Health)
• Giugno 2000: Completamento della sequenza
“working draft” dell’intero genoma umano
• Febbraio 2001: Pubblicazione prime analisi sul
genoma completo
• Aprile 2003: Completamento della sequenza
•
Una sequenza viene detta “finita” quando presenta un livello di
errore inferiore a 1/10000 basi e non ha gaps.
Il Progetto Genoma Umano era complesso dal punto di vista
tecnico ma anche dal punto di vista computazionale.
L’output di una singola reazione di sequenza (read) = 500-800
bp Tutti i singoli frammenti dovevano essere assemblati in
una singola stringa lineare.
NCBI fornisce ora l’assembly di riferimento per i 3 principali
“portali genomici”:
• MapView
• Ensembl
• Genome Browser
La sequenza primaria del genoma non è sufficiente…
Annotazione del genoma
• E’ necessario riportare sull’assembly le informazioni e i dati sperimentali
già ottenuti.
• Riconciliare e integrare l’assembly con le mappe fisiche, genetiche e
citogenetiche
• Gli STS sono mappati sulla sequenza usando e-PCR
• La corrispondenza con la mappa citogenetica utilizzando FISH
sistematica di BAC.
L’annotazione dei geni è attuata con metodi leggermente diversi dai 3
“genome browser”
– L’NCBI allinea mRNA di RefSeq, mRNA di GenBank utilizzando
MegaBlast.
– Ensembl allinea tutte le proteine umane note di SP/Trembl
utilizzando un suo algoritmo
– UCSC allinea mRNA di Refseq e GenBank e dalle ultime release
SP/Trembl con BLAT
Annotazione dei geni
• ab initio, in base a “sensori”, funzioni che tentano di dedurre la presenza
di una caratteristica genica in base a motivi o proprietà statistiche del DNA.
– Sensori per TSS (G+C)
– Sensori per siti splicing (AG-GT)
– Sensori che misurano la composizione in basi di esoni putativi
L’output dei vari sensori è combinato per generare un “modello genico”
• metodi basati sulla similarità: l’allineamento di una regione
genomica con un cDNA o un EST sono una buona evidenza.
Lo splicing alternativo complica l’interpretazione degli allineamenti tra DNA
genomico, cDNA e ESTs
I dati di similarità sono incompleti: trascritti poco espressi o espressi
transientemente sono assenti…
I programmi di ultima generazione come Grail/Exp, Genie EST,
GenomeScan combinano predizioni ab inizio con dati di similarità ottenendo
risultati migliori
Viral Genomes
3 milioni di basi in formato testo = nessuna utilita’
Servono:
•Annotazione dell’informazione sulla sequenza
•Possibilita’ di recuperare velocemente la sequenza di
regioni specifiche del genoma in base a criteri di
• Contenuto di informazione
• Caratteristiche di sequenza
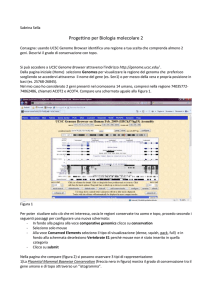
UCSC Genome Browser
Sistema per la “navigazione” della sequenza e
dell’annotazione di genomi, che permette la
visualizzazione dell’informazione a “diverso
ingrandimento” ed il recupero di porzioni di sequenza
con associate le informazioni di annotazione, come:
Geni noti e geni predetti
ESTs, mRNAs
Isole CpG
assembly gaps e coverage, bande cromosomiche
Omologia con altri genomi
…
Genomi
disponibili
Human
Homo sapiens
assembly
• 99% delle regioni
contenenti geni
• accuratezza 99.99%
• 2.84 Gb finite “highly
contiguous”
Species
A. gambiae
A. mellifera
C. briggsae
C. elegans
C. intestinalis
Chicken
Chimp
Cow
D. ananassae
D. erecta
D. grimshawi
D. melanogaster
D. mojavensis
D. persimilis
D. pseudoobscura
D. sechellia
D. simulans
D. virilis
D. yakuba
Dog
Fugu
Human
Mouse
Opossum
Rat
Rhesus
S. purpuratus
SARS
Tetraodon
X. tropicalis
Yeast
Zebrafish
UCSC Genome Browser
Molte possibilita’ per la ricerca di una regione specifica:
• chr7
un cromosoma intero
• 20p13
una regione (banda p13 del cr. 20)
• chr3:1-1000000
il primo milione di basi del cr. 3 dal ptel
• D16S3046
regione intorno al marcatore (100,000 basi per lato)
• RH18061;RH80175 regione tra i due marcatori
• AA205474
regione genomica che si allinea con la sequenza con
questo GB accession number
• PRNP
regione del genoma che comprende il gene PRNP
• NM_017414
• NP_059110
• 11274 (LLID)
Oppure di liste di regioni:
• pseudogene mRNA Lists transcribed pseudogenes, but not cDNAs
• homeobox caudal
Lists mRNAs for caudal homeobox genes
• zinc finger
Lists many zinc finger mRNAs
• huntington
Lists candidate genes associated with Huntington's
disease
Genome Browser
Categorie di annotazione:
• Mapping and
Sequencing Tracks
• Genes and Gene
Prediction Tracks
• mRNA and EST Tracks
• Expression and
Regulation
• Comparative Genomics
• ENCODE Tracks
• Variation and Repeats
Genome Browser: display mode
Known Genes
Hide
Genome Browser: display mode
Known Genes
Dense
Genome Browser: display mode
Known Genes
Squish
Genome Browser: display mode
Known Genes
Pack
Genome Browser: display mode
Known Genes
Full
Genome Browser
Categorie di annotazione: Expression and Regulation
CpG Islands, ISOLE CpG
Regioni in cui seqenze CpG sono significativamente piu’ frequenti che nel resto del
genoma. Associate ai geni, soprattutto agli housekeeping. Di solito si trovano vicino ai TSS,
associate ai promotori. Nei vertebrati le sequenze CpG sono rare, e le C nelle isole CpG
tendono ad essere metilate e, nel tempo, le C metilate tendono a mutare a T per
deaminazione spontanea. Percio’ sono rare e di solito “vengono mantenute solo per ragioni
regolative”.
Metodo: Finestra a scorrimento per dinucleotidi (punteggio +17 per CG, -1 tutti gli altri) e
successiva identificazione dei segmenti con punteggi massimali. Poi, per ogni segmento,
calcolo contenuto in GC (>= 50%), lunghezza (> 200), e rapporto tra frazione di
dinucleotide CG osservato e atteso in base al contenuto in GC del segmento (> 0.6).
GNF Gene Expression Atlas Ratios Using Affymetrix GeneChips
Dati d’espressione ottenuti dall’analisi di Affymetrix GeneChips GNF (The Genomics
Institute of the Novartis Research Foundation).
I colori mostrano il segnale medio tra diversi esperimenti su scala logaritmica: colore scuro
segnale forte.
FirstEF: First-Exon and Promoter Prediction
Predizioni del programma First Exon Finder: primi esoni, promotori e isole CpG.
Due predizioni consecutive a distanza inferiore a 1000 bp sono rappresentate come primi
esoni alternativi.
Metodo: FirstEF is a 5' terminal exon and promoter prediction program. It consists of
different discriminant functions structured as a decision tree. The probabilistic models are
optimized to find potential first donor sites and CpG-related and non-CpG-related promoter
regions based on discriminant analysis.
Genome Browser
5-Way Regulatory Potential
Traccia che mostra il punteggio RP lungo la sequenza in esame,
computato in base all’analisi dell’allineamento multiplo di regioni ortologhe
dei genomi considerati
Description
This track displays regulatory potential (RP) score, computed from
alignments of human (hg17), chimpanzee (panTro1), mouse (mm5), rat
(rn3), and dog (canFam1).
Genome Browser
Categorie di annotazione: Comparative Genomics
Genome Browser
Mouse Net
Mouse Net è la traccia che mostra
l’insieme degli allineamenti ottimali tra il
genoma umano e quello di topo,
ottenuti attraverso BLASTZ.
BOX = ungapped alignments
LINEE = gaps
Genome Browser
Mouse Net
Genome Browser
Categorie di annotazione: Variation and Repeats
1.
2.
3.
4.
5.
6.
7.
8.
9.
Variation and Repeats
SNPs
Overlap SNPs
Random SNPs
Affy 120K SNPs
Affy 10K SNPs
RepeatMasker
Simple Repeats
Self Chain
Genome Browser
SNPs, Simple Nucleotide Polymorphisms
Traccia che mostra
• diversi tipi di polimorfismi: SNPs propri, inserzioni e delezioni, duplicazioni
• polimorfismi osservati in base alla comparazione di cloni, polimorfismi
inclusi nei kit per il Genotyping dell’Affimetrix (10K e 120K).