L'interfase è il periodo tra
successive mitosi e consiste in
tre fasi: G1, S, e G2. Durante la
fase S (sintesi), le molecole di
DNA di ogni cromosoma
vengono replicate producendo un
identico paio di molecole di
DNA
chiamate
cromatidi
(talvolta cromatidi "fratelli").
Ogni
cromosoma
replicato
quindi entro in mitosi con due
molecole identiche di DNA.
Sottili filamenti di cromatina
appaiono comunemente come
materiale granulare amorfo nel
nucleo colorato di cellule in
interfase.
Prima e dopo la fase S vi sono due
periodi
di
intensa
attività
metabolica,
crescita
e
differenziamento, detti G1 (gap =
intervallo 1) G2 (gap 2). Durante la
fase G1 le cellule si preparano per la
sintesi di DNA (fase S) e durante la
G2 hanno luogo la crescita e
l'espansione cellulare. Le cellule
possono uscire dal ciclo cellulare in
fase G1 ed entrare in uno stato di
riposo, o G0. Le cellule in G0 non
proliferano ma sono vitali e
metabolicamente attive, e possono
rientrare nel ciclo attraverso la fase
G1. Una volta in G1 la cellula è
obbligata a completare il ciclo.
La fase M, o mitosi, comprende
quattro tappe principali considerate in
dettaglio più avanti: profase, metafase,
anafase e telofase. La mitosi è di solito
la fase più breve del ciclo cellulare,
occupando 1 h del tempo totale di 1824 h richiesto per il ciclo completo in
una cellula animale ideale. Il tempo
trascorso in ogni fase della mitosi
varia notevolmente. La profase
richiede molto più tempo delle altre; la
metafase è la più breve.
Cyclin/cdk complexes are required at
specific cell cycle transitions
G1 cyclins segnalano alla cellula di
preparare i cromosomi per la replicazione
S-phase promoting factor (SPF)
preparano la cellula a entrare in fase S e
duplicare il DNA
* durante la sintesi di DNA la ciclina E si
degrada e compaiono le cicline di fase M
(mitotiche)
M-phase promoting factor (MPF) inizia:
- L’assemblaggio del fuso mitotico
- Dissolvimento dell’involucro nucleare
- Condensazionedei cromosomi
Inizia la metafase
A questo punto MPF attiva il copmesso
APC (Anaphase Promoting Complex)che
permette:
E
A
B
1) la separazione dei cromatidi fratelli e la migrazione ai poli
2) la degradazione delle cicline mitotiche (M-phase)
3) avvia la sintesi delle cicline della fase G1per il ciclo successivo
4) degradazione della geminina, una proteina che blocca la replicazione pre-mitotica del DNA
sintetizzato in fase S. ,
Control of Cell Cycle
• The cell cycle has checkpoints where feedback
information from the cell can trigger the next phase
• Proteins are responsible for the control through bonding
to signal receptors.
• 3 checkpoints: G1 checkpoint- makes the decision as
to whether the cell will divide. If conditions are
favorable. Like the cell is healthy and large enough
proteins will stimulate the cell to start the S Phase. The
cell cycle can also be stopped or sent to a resting point,
Ex. Muscle and nerve cells, at this checkpoint.
• G2 checkpoint- DNA replication is checked hear by
DNA repair enzymes. If the DNA is copied correctly
with no errors proteins then signal mitosis phase.
• Mitosis Checkpoint- Triggers the exit from mitosis
phase.
G1 Checkpoint
• Cell Cycle Arrest
• DNA Repair
G1 Checkpoint
• Apoptosis
Danno prima dell’entrata in S
G1/S checkpoint
Mediato dalle chinasi Chk1 e Chk2
Arresto rapido della
replicazione, dovuto alla
degradazione della
fosfatasi Cdc25A
la cui normale funzione
e di defosforilare e così
attivare la chinasi Cdk2
del complesso SMP, che
a sua volta attiva la
proteina Cdc45, che è
essenaziale per
l’assemblaggio del
complesso di
replicazione sull’origine
di replicazione del DNA
Rapido e transiente
Mantenimento prolungato
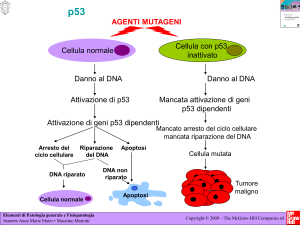
Il gene p53
Poco più del 50% dei tumori umani presenta mutazioni di questo gene. Nella
maggior parte dei casi, le mutazioni inattivanti che colpiscono entrambi gli
alleli p53 sono acquisite dalla cellula somatica. Meno frequentemente
alcuni soggetti ereditano un allele p53 mutato (sindrome Li-Fraumeni) e
possono sviluppare numerosi tipi di neoplasie quali:
1. Sarcomi;
2. Carcinomi della mammella;
3. Leucemie;
4. Tumori cerebrali;
5. Carcinomi della corticale del surrene.
Rispetto alle forme sporadiche, le forme ereditarie si presentano in soggetti più
giovani.
La proteina p53 è localizzata nel nucleo ed entra in gioco come freno
d’emergenza quando il DNA è danneggiato da radiazioni ionizzanti, raggi UV o
sostanze chimiche.
Attivazione di p53 in risposta al
danno del DNA
IR,UV, MMC.etposide,ROS
p21
p53, inattivo e stabile
Geni target on
Puma, Bax
Rilasco del citocromo c
apoptosi
p53 si lega al DNA e stimola la trascrizione di alcuni geni che mediano due funzioni
fondamentali: l’arresto del ciclo cellulare e l’apoptosi.
Sono stati identificati studiando malattie
rare come il retinoblastoma, che colpisce
1 bambino su 20.000.
40% = casi familiari con trasmissione
autosomica dominante, tumori multipli si
sviluppano nella retina di entrambi gli occhi
nella prima settimana dalla nascita.
60% = casi sporadici, un singolo
tumore si sviluppa in un occhio
nella prima infanzia.
Affinché il retinoblastoma si sviluppi è necessario che entrambi gli alleli siano inattivi.
Prodotti proteici degli oncosoppressori.
1. Molecole che regolano la trascrizione nucleare ed il ciclo cellulare.
Il gene Rb
Rb è stato il primo gene oncosoppressore scoperto. Il suo prodotto è una fosfoproteina
nucleare, pRb, che nel suo stato attivo (IPOfosforilato) costituisce un freno al passaggio dalla
fase G1 alla fase S del ciclo cellulare.
La forma attiva di pRb complessata ai fattori di trascrizione
E2F si lega al DNA ed inibisce la trascrizione dei geni i cui
prodotti sono necessari alla fase S del ciclo cellulare.
Quando i complessi ciclina D/CDK4,6 e ciclina E/CDK2 la
inattivano IPERfosforilandola, rilascia i fattori E2F.
I fattori E2F formano eterodimeri con la famiglia di proteine DP
ed attivano la trascrizione di diversi geni bersaglio
Quindi, se la proteina pRb è assente o alterata da mutazioni, la cellula progredisce
verso la fase S senza alcun freno.
REGOLAZIONE DEL CICLO CELLULARE
DA PARTE
DEI NUTRIENTI
Vitamina A
AF1
LBD - AF2
DBD
A/B
C
Helix 12
D
E/F
Nuclear Hormone Receptor
Superfamily
Type I family
Type II family
Steroid family
Non-steroid family
GR
PR
AR
MR
ER a, b
TR a, b
RAR a, b, g
RXR a, b, g
VDR
PPAR a, g, d
CAR, SXR/PXR
LXR a, b, FXR
Conserved domains of transcription factors in nuclear-hormone
receptor superfamily
A/B
AF-1 domain
C
Two non-repeating
C4 Zn finger motif
E
AF-2 domain
Fig 11.41 Lodish et al. Molecular Cell Biology
Retinoid X Receptor (RXR)
E’ espresso in tutte le cellule
Regola numerosi processi eterodimerizzando con altri recettori
nucleari (PPAR, VDR, LXR, FXR…….)
Il suo ligando naturale e’ l’acido 9 cis retinoico, …..altri ?
Diverse Structure of Ligands for Nuclear Receptors
Ligand Induces a Conformational Change in
the LBD that Repositions helix 12
No Ligand
Agonist
NR Antagonists Alter the Position of Helix 12
No Ligand
Agonist
(ER)
Antagonist
(ER)
Consensus sequences of DNA response elements for different
nuclear hormone receptors
The glucocorticoid receptor and oestrogen
receptor bind to their respective response
elements as homodimers. The response
element is an inverted repeat
The vitamin D receptor, the thyroid hormone
receptor and the retinoic acid receptor bind to
their respective response elements as
heterodimers (with RXR). The response
element is an direct repeat. The spacing between
these repeats determines the specificity of
the interaction.
Fig 11.42 Lodish et al. Molecular Cell Biology
RAR
A
A/B
C
D
E
F
B
C
D
E
F
dominio di transattivazione INDIPENDENTE dal ligando
dominio che lega il DNA
cerniera
dominio che lega il ligando e dimerizzazione
funzione non conosciuta
RARE
RAR-RXR
AGGTCAnnnnnAGGTCA
(DR5)
RXRE
RXR-RXR
AGGTCAnAGGTCA
(DR1)
Nuclear Hormone ReceptorLigand Complex Action 2
Recruitment of Coactivators is Necessary
They are an Obligate ‘Bridge’ to Pol II complex*
NH – receptor L
Ligand
Bound
NH – receptor
L
Ligand Bound
DNA Response Element
RNA polymerase II
or pol II complex
actually transcribes,
starting at TATA box
DNA site
TATA Box
* Receptor-ligand can bind DNA and NOT recruit pol II = Antagonist
In tumori sensibili l’acido retinoico causa un arresto della
crescita cellulare
cellule normali
Trattamento
con
+
a.retinoico
Trasfezione
Con
RARb
crescita
inibita
tumori sensibili
t. resistenti
+
-
crescita
inibita
crescita
inibita
RARb ha 2 sottotipi: RARb2 e RARb4
RARb2 funziona come un gene soppressore di tumore
RARb4 come un dominante negativo
Topi transgenici con poco RARb2 o con molto RARb4
sviluppano tumori.
Quindi l’acido retinoico agisce come soppressore della crescita
cellulare legandosi a RARb2, mentre elevati livelli di RARb4
sopprimono questo effetto (tumori resistenti all’acido retinoico)
RETINOIDI e NEUROBLASTOMA
Retinoidi inducono differenziamento in cellule nervose
Nei neuroblastomi inducono apoptosi
DNA Microarray hanno dimostrato l’induzione di geni
Pro-apoptotici da parte di retinoidi
Estrazione
dell’mRNA dai
2 campioni di
cellule che si
vogliono
confrontare
Conversione in
cDNA
Marcatura con
2 fluorocromi
diversi
Confronto dei profili di espressione genica in
due campioni cellulari diversi
(1)
(2)
(3)
(6)
Immagine a colori
raffigurante il
microarray
Eccitazione della
fluorescenza
tramite laser
Riconoscimento (4)
tra i cDNA
provenienti dai 2 campioni e
quelli già presenti sul microarray
(5)
L’Acido retinoico induce arresto della crescita cellulare oppure
proliferazione a seconda del tipo cellulare (es. epidermide o
epitelio tracheale).
Topi privati di vitamina A sviluppano molti tumori specialmente da
epitelio tracheale.
Trattamento con acido retinoico le blocca in G1:
Ac. Retinoico ciclinaD1
mRNA
+
Acido
retinoico
Induzione dei geni
Addetti alla Ubiquitinazione
Ub
Ub
Ub
Ub
Ciclina D1
Ciclina D1
Degradazione e arresto in G1
EFFETTO ANTIPROLIFERATIVO
DELL’ACIDO RETINOICO SU
VARI TUMORI
Tumore
ciclina repressa
Trachea
Neuroblastoma
Mieloide (U937)
Epatoma
D1
D3
A, B, D2, D3, E
D1
Vitamina D
Produzione della Vitamina D da
UVB
• UVB (290-315 nm) converte 7-deidrocolesterolo
in pre-vitamina D
• Previtamina D3 subisce una isomerizzazione che
risulta nella formazione della vitamina D3 (25
idrossivitamina D (25(OH)D))
• Questa e’ convertita in 1,25-diidrossivitamina D3
(1,25(OH)2D3) nel fegato e nei reni
AF1
LBD - AF2
DBD
A/B
C
Helix 12
D
E/F
Nuclear Hormone Receptor
Superfamily
Type I family
Type II family
Steroid family
Non-steroid family
GR
PR
AR
MR
ER a, b
TR a, b
RAR a, b, g
RXR a, b, g
VDR
PPAR a, g, d
CAR, SXR/PXR
LXR a, b, FXR
Diverse Structure of Ligands for Nuclear Receptors
Consensus sequences of DNA response elements for different
nuclear hormone receptors
The glucocorticoid receptor and oestrogen
receptor bind to their respective response
elements as homodimers. The response
element is an inverted repeat
The vitamin D receptor, the thyroid hormone
receptor and the retinoic acid receptor bind to
their respective response elements as
heterodimers (with RXR). The response
element is an direct repeat. The spacing between
these repeats determines the specificity of
the interaction.
Fig 11.42 Lodish et al. Molecular Cell Biology
VIT AMIN D (V) RECEPTOR (VDR)
ACT IVAT ION OF A VDR RESPONSIVE GENE
RNA Polymerase II
VDRE
Hormone Regulated Gene
+1
RXR VDR
V
NUCLEUS
Gene
T ranscription
RXR VDR
V
VDR
CYT OPLASM
V
V
La vitamina D causa un arresto della proliferazione
cellulare e differenziamento in
cheratinociti
osteoblasti
cellule ematopoietiche
VDR e’ specialmente espresso nelle sopracitate cellule
quando sono proliferanti
Crescita dopo trattamento con Vitamina D
Cellule di tumore prostatico
Alva-31(linea cellulare)
50%
0%
Arresto in G1
La mortalita’ del tumore alla prostata aumenta con la
diminuzione alla esposizione alla luce solare
I livelli di vitamina D circolanti diminuiscono con l’eta’
Alti livelli di sintesi di melanina correlano con un aumento
dell’incidenza di tumori alla prostata
Crescita dopo trattamento con Vitamina D
Alva-31(linea cellulare)
Alva-31(linea cellulare)
+ VDR antisenso
0%
100%
La forma attiva di pRb complessata ai fattori di trascrizione
E2F si lega al DNA ed inibisce la trascrizione dei geni i cui
prodotti sono necessari alla fase S del ciclo cellulare.
Quando i complessi ciclina D/CDK4,6 e ciclina E/CDK2 la
inattivano IPERfosforilandola, rilascia i fattori E2F.
I fattori E2F formano eterodimeri con la famiglia di proteine DP
ed attivano la trascrizione di diversi geni bersaglio
Quindi, se la proteina pRb è assente o alterata da mutazioni, la cellula progredisce
verso la fase S senza alcun freno.
Vitamina D
Induzione di p21
Rilascio di citocromo C
Repressione di geni
Antiapoptotici
X
Promotore di p27
Gene Luciferasi
Vitamina D
p27
(proteina: piu’ stabile
Arresto in G1
p27fosforilata
meno stabile)
Paracalcitolo (analogo della vitamina D)
HL60 (linea leucemica)
A concentrazione nanomolare riduce le cellule in fase S
Aumenta le cellule in fase G1
Ha effetto pro-differenziante
Aumenta il numero di cellule che vanno incontro ad apoptosi
Trattamento con Vit D3 aumenta l’espressione della Tuberina
(gene
Oncosoppressore)
or Vit D3
Attivita’ del promotore di
p21
TGF-b e Vit D3 aumentano l’espressione della Tuberina, di p21 e diminuiscono la ciclina A
Cellule ERa+
Il trattamento con vitamina D induce i geni per:
Molecole di adesione
p53
p21
Retinoblastoma
Varie caspasi
TGFb e il gene per il retinoblastoma sono indotti in tutti e due i tipi cellulari
Lo svantaggio maggiore
per un suo impiego come
farmaco antitumorale e’
la sua enorme influenza
sul metabolismo del
calcio e dell’osso.




![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)


![ESTRAZIONE DNA DI BANANA [modalità compatibilità]](http://s1.studylibit.com/store/data/004790261_1-44f24ac2746d75210371d06017fe0828-300x300.png)