Che cos’è il clonaggio?
L’utilizzo della riproduzione asessuale per produrre organismi geneticamente identici tra loro e
alla «madre»:
nel giardinaggio: propagazione delle piante per talee (es. di cloni)
in batteriologia: colture di espansione da una singola colonia
Clonaggio applicato ai geni
Inserimento di un gene in una cellula qualsiasi (batterio, linea cellulare, cellula uovo):
Trasmissione del gene inserito attraverso la replicazione della cellula stessa (gene clonato)
Produzione di organismi geneticamente modificati (transgenici)
Storia del clonaggio molecolare:
le prime molecole di DNA ricombinante sono stati generati e studiati nel 1972
I microbiologi, cercando di comprendere i meccanismi molecolari attraverso i quali batteri
limitavano la crescita di un batteriofago:
o Isolamento di enzimi di restrizione
o hanno dimostrato che gli enzimi di restrizione tagliano le molecole di DNA cromosomico in
frammenti di lunghezza in posizioni specifiche
o La sezione della molecola di più grandi dimensioni potrebbe essere purificate mediante
separazione in base al peso molecolare e purificazione (da gel d’agarosio)
o Usando un secondo enzima, la DNA ligase, I frammenti generate per restrizione e purificati
possono essere uniti creando un DNA ricombinante.
o Ricombinando il frammenti d’interesse col DNA del vettore, ad esempio un batteriofago or
plasmidi, che si replicano naturalmente all’interno di batteri
o Grandi quantità di molecole di DNA ricombinante possono così essere prodotte in colture
batteriche
Step nel clonaggio molecolare
o Scelta dell’organismo ospite e del vettore per il
clonaggio
o Preparazione del DNA vettore
o Preparazione del DNA da clonare
o Creazione del DNA ricombinante con la ligasi
o Introduzione del DNA ricombinante nell’organismo
ospite
o Selezione degli organsmi contenente il vettore
ricombinante
o Screening dei cloni col DNA d’interesse e con le
proprietà biologiche da questo derivate
Scelta dell’organismo ospite e del vettore per il clonaggio
Vettori plasmidici (A) di E. coli. Sono di uso comune, perché sono tecnicamente abbastanza sofisticati, versatili, ampiamente disponibili, e
offrono rapida crescita di microrganismi ricombinanti con attrezzatura minima a disposizione.
Se il DNA da clonare è eccezionalmente grande (da centinaia di migliaia a milioni di coppie di basi), è spesso scelto un cromosoma artificiale
batterico (B) o di lievito come vettore (C).
Applicazioni specializzate possono richiedere sistemi ospite-vettore specializzati (D). Ad esempio, se gli sperimentatori intendono isolare una
proteina ricombinante da un particolare dall'organismo ospite, deve essere scelto un vettore di espressione contenente segnali appropriati
per trascrizione e traduzione nell'organismo ospite desiderato. Oppure, se la si vuole ottenere la replicazione del DNA ricombinante in
diverse specie (per esempio, in applicazioni che richiedono il trasferimento del DNA ricombinante da batteri alle piante), deve essere
selezionato un vettore con caratteristiche tali da crescere in una più ampia gamma di ospiti (vettore shuttle). Spesso, gli esperimenti di
clonaggio molecolare richiedono, infatti, un clonaggio in un plasmide batterico, seguito da un subclonaggio in un vettore specializzato.
(A)
(B)
(C)
(D)
Scelta dell’organismo ospite e del vettore per il clonaggio
4
4 segmenti DNA di importanza critica per funzioni ed
utilità sperimentale:
2
1. L’origine di replicazione nel vettore adatta per la
duplicazione nell’organismo ospite
2. Uno o più siti per enzimi di restrizione per introdurre
nel vettore il DNA esogeno (multiple cloning
site=MCS)
3. Un gene marker per la selezione genetica delle
cellule che hanno internalizzato il VETTORE
4. Un gene tag che può essere usato per individuare le
cellule con il vettore RICOMBINANTE
3
1
Uso di enzimi di restrizione
A. Taglio del plasmide
B. Taglio del DNA esogeno con gli stessi enzimi di
restrizione
C. Inserzione del DNA esogeno nel vettore
ricombinante
Uso di enzimi di restrizione
Clonaggio di DNA genomico
Per clonare un DNA genomico, questo deve
prima
essere
estratto
dalle
cellule
dall’organsmo d’interesse. Virtualmente ogni
tessuto può costituire una sorgente di materiale
(anche quelli da animali/piante estinti), basta non
sia troppo degradato. Il DNA genomico viene
purificato con metodi semplici di estrazione per
rimuovere la componente proteica.
Sticky-end cloning (clonaggio direzionale)
o
Per inserire un frammenti di DNA in un plasmide è
preferibile usare due diversi enzimi di restrizione per
digerire il DNA, generando due diverse estremità.
Questo per impedire il ri-annealing del vettore di DNA
su se stesso senza l’inserto, ciò permette, inoltre,
l’inserimento del frammento ricombinante in un’unica
direzione.
o
Se non è possibile usare due diversi siti per enzimi di
restrizione, allora il DNA del vettore deve per
necessariamente essere defosforilato per evitare
l’elevato grado di ricircolarizzazione del vettore senza
l’inserto. Il vettore tagliato col RE può poi essere
defosforilato usando la Fosfatasi Acalina: in questo
modo le molecole di vettore coi terminali
defosforilato non riescono a duplicarsi nelle cellule
dell’ospite, e la replicazione può avvenire solo per i
vettori con integrato l’inserto nel dito di taglio con l’RE.
o
fosfatasi alcalina del gamberetto (SAP) o fosfatasi
antartica (AP) sono un’adatta alternativa, in quanto
possono essere facilmente inattivate dopo la reazione.
Blunt-end cloning
o
La reazione di ligasi su terminali blunt-end non coinvolge
l’appaiamento di basi delle estremità di DNA digerito con RE
o
Il vantaggio maggiore del clonaggio blunt-end è che l’inserimento del
DNA esogeno non richiede che ci sia nel vettore un sito di
restrizione corrispondente, come quando dobbiamo clonare prodotti
di PCR.
o
La reazione di ligasi su terminali blunt-end è meno efficiente di
quella su sticky-end, tipicamente la reazione ha una resa 100X più
bassa, perchè dipende dall’incontro casuale tra I blunt-ends.
o
La concentrazione dell’enzima Ligase è usata più alta (10x o più) è
più lunghi sono anche I tempi di incubazione.
o
Il vettore necessita di essere defosforilato per limitare la selfligation.
o
Mentre l’inserto deve essere fosforilato. I prodotti di PCR bluntended normalmente mancano del 5'-fosfate, perchè i primers sono
sempre sintetizzato non fosforilato. La fosforilazione può essere
condotta con T4 polynucleotide kinase.
o
Usando la Taq polymerase per la PCR, questa può aggiungere un’extra
adenine (A) al 3' del prodotto PCR. In questo caso, i terminali blunt del
vettore devono essere adattati aggiungendo ai terminali una T. Ciò si
ottinene aggiungendo dideoxythymidine triphosphate (ddTTP) con la
Terminal Transferase al vettore.
Clonaggio di prodotti di PCR
La reazione di polimerizzazione a catena
(PCR) è spesso usata per produrre
frammenti di DNA o RNA (RT-PCR)
aventi sequenze specifiche e creare
porzioni di DNA esogeno per il clonaggio
molecolare.
Il DNA per il clonaggio può essere
ottenuto da RNA usando la reverse
transcriptase (complementary DNA or
cDNA cloning), o rappresentato da
frammenti
di
DNA
sintetizzato
artificialmente (artificial gene synthesis).
Uso di linkers attaccati alle molecole di DNA
Spesso il DNA è digerito con
enzimi di restrizione oppure,
ottenuto per PCR usando primer
contenente siti per RE
Questo per generare frammenti
con estremità in grado di essere
legate a quelle del vettore.
Se necessario, brevi frammenti di
dsDNA (linkers) conteneti i siti
per RE possono essere aggiunti
Per creare strutture terminali del
DNA
esogeno
che
siano
compatibili col vettore.
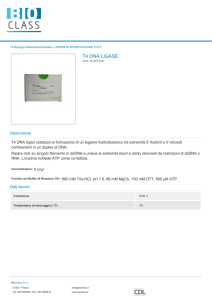
Reazione ligasica
o La reazione della Ligasi è l’unione, operata da
questo enzima, di due frammenti di acido nucleico.
É una procedura essenziale per il clonaggio e la
creazione di molecole di DNA ricombinante.
o La reazione prevede l’utilizzo di un co-fattore, in
genere ATP or NAD+.
o L’enzima più usato in laboratorio è la T4 DNA
ligase.
Fattori che influenzano la reazione ligasica
o
o
o
o
o
La concentrazione di enzima
• Alta concentrazione di DNA ligase concentration può essere
usata in presenza di PEG (polyethylene glycol) per una
reazione più rapida
La temperature di reazione ed il tempo d’incubazione
• L’optimum di temperature per l’attività della DNA ligase è
37°C, ma le estremità possono essere stabilmente legate
anche a temperature più basse. Molti protocolli preferiscono I
12-16°C, la temperature ambiente, o i 4°C.
La concentrazione di DNA
•
Una molecola di DNA può ricircolarizzare unendo le sue
estremità
•
Più bassa è la concentrazione di DNA, più bassa la
ricircolarizzazione intramolecolare
•
La concentrazione di DNA totale non deve essere meno di 10
ng/μl
•
La concentrazione dei frammenti di DNA, così come la loro
lunghezza, sono fattori che possono interferire nelle reazioni
ligasiche intermoleculari o intramoleculari.
La composizione del buffer di reazione
• Un eccesso di Na+ può irrigidire la strutture del DNA e
favorire le reazioni ligasiche intermolecolari. Tuttavia, alte
concentrazione di cationi monovalenti (>200 mM) possono
completamente inibire la reazione.
Il rapporto molare tra insert e vettore usualmente è circa 3:1
• Rapporti superiori possono causare inserzioni multiple. Il
rapport può essere variato in base alla lunghezza dell’inserto.
Altri metodi per legare il DNA nel vettore
Un elevato numero di kit commerciali per il clonaggio di DNA usa altri metodi e non DNA ligase.
Questi metodi servono per aumentare la rapidità di clonaggio. Tuttavia questi metodi richiedono
l’uso di vettori appositamente disegnati e specificamente progettati, e mancare per questo di
flessibilità.
o Topoisomerase-mediated ligation
La topoisomerase può essere usata invece della ligase, il clonaggio può essere fatto più
rapidamente senza bisogno di RE per digerire il vettore e l’inserto. Col TOPO cloning
method un vettore linearizzato è attivato dall’attacco con la topoisomerase I ai suoi terminali,
tale "TOPO-activated" vector può poi ricevere l’inserzione di prodotti PCR product
rilasciando un vettore circolare ricombinante.
o Homologous recombination
Un altro metodo di clonaggio senza l’uso della ligase, che si basa sul fenomeno della
ricombinazione, usando ad esempio un Gateway cloning system. Con questo Sistema un
gene, una volta inserito in un vettore (chiamato clone d’entrata), poi può essere introdotto in
una varietà di altri vettori per l’espressione in sistemi specifici.
Topoisomerase-mediated ligation in TOPO cloning
o Tecnica
La TA TOPO cloning technique si basa sulla capacità di creare nel vettore una (T) che può complementarizzare con una
una (A) presente su un differente frammento di DNA da inserire. In questo caso l’inserto DNA di è creato per PCR: infatti, la
Taq polymerase aggiunge un extra "A" come nucleotide al terminale 3‘ durante la reazione di polimerizzazione, producendo
una 3'-adenine protrundente.
La DNA topoisomerase I (Vaccinia virus topoisomerase I) riconosce specifiche sequenze sul DNA, come 5´-(C/T)CCTT-3'.
Quest’enzima può essere usato per tagliare in particolare il DNA superavvolto in corrispondenza di queste sequenze
appositamente inserite in questi vettori per creare I terminali 3' fosfato di (T) che fungono da accettore ed ibrizizzano con la
(A) del prodotto PCR.
Plasmid
5’-AGTGATTCAACATATTCCGTTCCCTTATTCCGATACTATG-3’
3’-TCACTAAGTTGTATAAGGCAAGGGAATAAGGCTATGATAC-5’
Plasmid/Topoisomerase I
complex
TOPO
5’-AGTGATTCAACATATTCCGTTCCCTT-3’
3’-TCACTAAGTTGTATAAGGCAAGGGA
3’-A acceptor DNA PCR
products
5’- - GGATTGACAACATATTCCGA-3’
3’-ACCTAACTGTTGTATAAGGC- - 5’
Topoisomerase-mediated ligation in TOPO cloning
o TOPO vectors
I TOPO vectors contengno la sequenza specifica riconosciuta dalla topoisomerasi 5´-(C/T)CCTT-3' ai due terminali
linearizzabili. Sono plasmidi ingegnerizzati per essere linearizzato con una 3‘-deossitimidina (T) sporgente e legata
covalentemente dalla topoisomerase I.
Il 3‘-A sporgente di un prodotto di PCR è complementare al 3‘-T sporgente del vettore e viene legato in presenza di of the
topoisomerase I.
Topoisomerase-mediated ligation in TOPO cloning
Homologous recombination in molecular cloning
Il gene d’interesse è facilmente
veicolato in un plasmide come
vettore di destinazione.
Questa
reazione
è
mediata
da mix enzima Invitrogen™ LR Clon
ase™, che contiene le proteine
necessarie per asportare il gene di
interesse dal clone di entrata
e
integrarlo
nel
vettore
destinazione,
che
poi
diventa
il
vostro
clone
di espressione.
Questa reazione è semplice:
richiede
una
reazione BP (ricombinazione tra i
siti attB e attP) utilizzando una mix
di
enzima Invitrogen™ BP Clonase™.
Vi è una vasta scelta di vettori d’espressione disponibili
per applicazioni con Gateway cloning technology, come
per l’espressione di proteine in E. coli, lievito, insetti, o
cellule di mammiferi per studi di RNAi, o per
caratterizzazione cristallografiche di proteine, oppure
studi d’interazione proteina–proteina, c’è un vettore di
Destinazione adatto per ogni applicazione specializzato
e customizzato.
https://www.thermofisher.com/it/en/home/life-science/cloning/gateway-cloning.html?gclid=COLht6-xNICFRSeGwodj7kA8w&s_kwcid=AL!3652!3!78852797961!p!!g!!gateway%20cloning&ef_id=WNnprgAA
BQ4vba5S:20170328044222:s
Homologous recombination in molecular cloning
To popularize this method, we tested critical parameters
influencing the efficiency of PCR fragments cloning into
PCR-amplified vectors by homologous recombination in
the widely used E. coli strain DH5α.
These results support the idea that homologous recombination
in E. coli might be one of the most effective methods for
cloning one or two PCR fragments. For its simplicity and high
efficiency, we believe that recombinational cloning in E. coli
has a great potential to become a routine procedure in most
molecular biology-oriented laboratories.
Optimal Cloning of PCR Fragments by Homologous Recombination in Escherichia coli
Ana Paula Jacobus, Jeferson Gross. PLOS ONE | DOI:10.1371/journal.pone.0119221
March 16, 2015
Cloning using homopolymer tailing reaction catalyzed by Terminal
Deoxynucleotidyl Transferase
La DNA polimerasi gioca un ruolo essenziale nei
processi di replicazione, riparo, e ricombinazione di
acidi nucleici. Durante ciascuno di questi processi
biologici, la polimerasi estende un filamento di DNA a
partire da un primer su uno stampo di DNA (o cDNA).
Tuttavia non tutti gli enzimi necessitano di un primer
per la reazione di polimerizzazione, ne di un templato.
La terminal deoxynucleotidyl transferase (TdT),
possiede l’INUSUALE capacità di incorporare
nucleotide in modo indipendente dalla presenza di
un templato usando solo sDNA (single-stranded
DNA) come substrato (esempio B).
Cloning using homopolymer tailing reaction catalyzed by Terminal
Deoxynucleotidyl Transferase
PCR product
homopolymeric tail
at 3’-OH end
homopolymeric tail
at 3’-OH end
The gap is
not a
problem
when the tail
is >20nt
homopolymeric
tail prevents selfligation of the
vector
It’s possible recover the
insert of DNA using
restriction enzymes of
the MCS
Multiple cloning site (MCS): the pUC18 and pUC19 polylinker
A multiple cloning site (MCS), also called a polylinker, is a short segment of DNA which contains many (up to ~20)
restriction sites - a standard feature of engineered plasmids. Restriction sites within an MCS are typically unique,
occurring only once within a given plasmid. MCSs are commonly used during procedures involving molecular cloning
or subcloning. Extremely useful in biotechnology, bioengineering, and molecular genetics, MCSs let a molecular biologist
insert a piece of DNA or several pieces of DNA into the region of the MCS. This can be used to create transgenic
organisms, also known as genetically modified organisms (GMOs).
Replicazione plasmidi - ori
o
o
o
o
o
Sono presenti in copie multiple nella cellula batterica (cellula ospite)
Possono essere trasferiti alla progenie quando la cellula batterica si divide
Sono in grado di replicarsi all’interno dei batteri
Utilizzano per lo più enzimi batterici per la replicazione
Contengono solo poche (a volte una proteina) necessarie alla sua replicazione, poste vicino
all’ori, il resto può essere «tolto» (non necessario)
o ori, origine di replicazione sul plasmide, rappresentata da un singolo nucleotide
Replicazione plasmidi - regolato da un RNA antisenso
o
RNA antisenso (RNAII di 555 bp) un primer d’innesco della
replicazione
o
Lega l’ori formando un ibrido RNA/DNA
o
L’RNasi H taglia l’ibrido esponendo un 3’-OH per l’inizio della
polimerizzazione
o
L’inibizione della replicazione è invece determinata dall’RNA I (108
bp) complementare all’RNA II, che può con questo formare un
ibrido RNA/RNA non tagliabile dall’RNasi H.
o
Altro regolatore negativo è la proteina plasmidica Rop, che
aumenta la formazione dell’ibrido RNA I/RNA II
o
Nei plasmidi ad ampio spettro vicino all’ori c’è una sequenza detta
iterone (17-22 bp) legata dalla proteina repA (unica proteina
coinvolta nella replicazione plasmidica il cui gene è situato sul
plasmide). E un regolatore trascrizionale: lega il suo stesso
promotore e in presenta di numerose copie di plasmide lo inibisce,
riattivandolo subito dopo la divisione cellulare quando la sua
concentrazione cala.
Replicazione plasmidi - ori
Plasmidi ingegnerizzati: molte centinaia di copie nella
cellula batterica (solo 15 copie per i wild-type):
o Produce un’elevata espressione anche del gene
clonato
o Potrebbe generare instabilità e rallentare la crescita
batterica
o Effetti tossici sulla cellula ospite
o Alcuni plasmidi non possono coesiste insieme nella
stessa cellula (incompatibilità)
o Alcuni plasmidi si replicano in ampia gamma di specie
batteriche, ma quelli impiegati nel clonaggio hanno uno
spettro di selettività per l’ospite più ristretto (per evitare
rischi per ambiente/salute)
o COSTRUZIONE di plasmidi con un’ori funzionale per la
specie da usare come ospite; se necessario replicare il
plasmide in un ospite e studiare il comportamento del
gene clonato in un altro: costruire plasmidi shuttle,
con due ori specifiche per le diverse specie.
Instabilità segregazionale
o Specifica funzione: par, equa ripartizione delle copie
ad ogni evento cellulare. Nel plasmide pBR322, la
regione genica par è rimossa: in carenza di nutrienti o
fattori di stress si possono formare sottopopolazioni
batteriche prive di plasmide
o Superavvolgimento del DNA: ruolo nella ripartizione
plasmidica: assenza di par, inferiore superavvolgimento
o TopA, (DNA girasi) stabilizza il superavvolgimento
negativo dei mutati par, migliorando la ripartizione
durante la divisione cellulare
o Formazione di multimeri: ogni plasmide ha un’origine
di replicazione, plasmidi multimerici hanno più ori, ma
non hanno un numero equivalente di molecole
plasmidiche, ciò causa instabilità.
o La risoluzione dei multimeri in monomeri, può avvenire
quando sul plasmide è presente una sequenza cer,
riconosciuto dalla proteina Xer sintetizzata dall’ospite.
Marcatori di selezione – Resistenza agli antibiotici
o Necessità dipendente dall’inefficienza della
reazione di ligasi e di trasformazione batterica
(max efficienza in E. coli 1%)
o Prevenire la crescita delle non trasformate
(senza vettore)
o Geni che contribuiscono alla resistenza ad
antibiotici (β-lattamasi, idrolizzante antibiotici
β-lattamici, come la penicillina in acido
penicillico, AMPR)
Non discrimina le cellule batteriche trasformate col vettore plasmidico
da quelle col vettore contenente l’inserto genico clonato
Inattivazione inserzionale
I moderni vettori di clonaggio (ad esempio pUC19 e
derivati successivi, tra cui i vettori pGEM) utilizzano il
sistema di screening blu-bianco per distinguere le colonie
(cloni) delle cellule transgeniche da quelle che contengono
il vettore senza sequenza ricombinante inserita.
In questi vettori, DNA estraneo viene inserito in una
sequenza che codifica una parte essenziale della βgalattosidasi, un enzima che produce una colonia di colore
blu nel terreno di coltura.
L’inserimento del DNA estraneo nel sequenza codificante βgalattosidasi inattiva l'enzima, in modo che le colonie
contenenti DNA trasformato rimangono incolore (bianco) e
facilitando l’identificare dei cloni batterici contenenti il
plasmide ricombinante.
Inattivazione inserzionale
Il primo gene in E. coli lac operone è lacZ (β-galattosidasi,
β-gal).
La forma attiva di β-gal è un tetramero e idrolizza il lattosio
in glucosio e galattosio.
Eliminazione aminoacidi 11-41 della β-gal produce un
enzima
FORMANTE
UN
TETRAMERO
NON
FUNZIONANTE (processo α-complementazione) potrebbe
essere utilizzato per lo screening in colonie di E. coli della
presenza di inserti nel sito di clonaggio.
Nel bel mezzo della regione codificante l’α-peptide in un
plasmide pUC è introdotto un sito di clonazione multipla
(MCS). Quando un frammento di DNA viene inserito nel
MCS, interrompe l'α-peptide, rendendo la β-gal non
funzionale.
vettori per lo screening bianco/blu:
pGEM-T, pBluescript, pUC18 e pUC19
Inattivazione inserzionale
5-bromo-4-cloro-indolil-β-D-galattopiranoside (X-gal) è un analogo incolore di lattosio.
Quando la β-galattosidasi idrolizza X-Gal, si crea un prodotto blu (5,5'-dibromo-4,4dicloro-indaco).
In questo sistema di screening, un ceppo di E. coli è trasformato con una reazione
ligasica, poi viene fatto crescere su piastre di agar contenenti X-Gal. Una colonia di
colore blu indica che l'α-peptide nel plasmide è intatto (senza inserto), mentre una
colonia bianca indica che l'α-peptide è interrotta (presenza dell’inserto clonato).
Vettori con la regione codificante α-peptide e il MCS: pGEM-T, pBluescript, pUC18 e
pUC19.
X-gal può essere acquistato disciolto o in polvere. Esso può essere sciolto in DMSO o
DMF ad una concentrazione di 20 mg/ml. X-gal deve essere conservato a -20 °C e al
riparo dalla luce.
Isopropilico β-D-1-thiogalactopyranoside (IPTG) 10 mm è necessario per indurre
l'espressione β-gal in E. coli.
Preparazione delle piastre per la coltura delle colonie batteriche per lo screening:
Preparare alcune piastre di agar LB contenenti l'antibiotico appropriato per selezionare
per il plasmide prescelto. Su ogni piastra spargere
• 100 µl di 20 mg/ml x-Gal
• 100 µl di 10 mM IPTG
Stendere la reazione di trasformazione su una piastra contenete IPTG e X-Gal.
Incubare la piastra per una notte a 37 ° C.
Estrazione DNA plasmidico – Lisi alcalina
Coltura
batterica liquida
o
Rimuovere il terreno di coltura e pellettare le cellule batteriche
mediante centrifugata (per 1 ml di coltura)
o
A volte le cellule batteriche possono essere trattate col lisozima, per
rompere la parete batterica
o
Risospendere i batteri in una soluzione contenente (300 µl):
EDTA, che sottrae Mg inibendo le nucleasi che potrebbero degradare il
DNA; e RNasi, che alla rottura delle cellule frammenterà l’RNA; Tris
pH=8 manterrà invece in soluzione il DNA
o
Aggiungere una soluzione contenente (300 µl):
NaOH e SDS. Le membrane si denaturano per l’SDS causando la
lisi delle cellule e il DNA si denatura per il pH alcalino della soda,
ma essendo il plasmide superavvolto non si denatura
completamente
Birnboim and Doly 1979
Estrazione DNA plasmidico – Lisi alcalina
Coltura
batterica liquida
o Aggiungere una soluzione contenente (300 µl):
Acetato di potassio pH=5.1, per neutralizzare il pH e il DNA plasmidico tende ad acquistare
la conformazione originale, mentre il DNA cromosomico si ingarbuglia
o L’elevata concentrazione salina fa precipitare l’SDS insieme a proteine e detriti cellulari e
legato alla membrana anche il DNA cromosomale
o Il DNA plasmidico resta in soluzione, sarà recuperato col supernatante limpido, dopo
centrifugazione
o Aggiungere 0.9 volumi di Isopropanolo (720 µl su 800 µl di soluzione), per sottrarre
molecole di acqua al DNA e facendolo così precipitare, centrifugare e gettare il
surnatante
o Aggiungere 500 µl di etanolo 70% al pellet di DNA, per eliminare i sali, esiccare all’aria
o Risospendere il DNA in acqua o TE (Tris pH=8 ed EDTA)
Estrazione DNA plasmidico – Lisi alcalina
Estrazione DNA plasmidico –
Purificazione mediante estrazione fenolo/cloroformio
o Dopo la precipitazione con Acetato di potassio pH=5.1 e la centrifugazione per eliminare
membrane e DNA cromosomico, il supernatante potrebbe essere ulteriormente purificato
mediante estrazione con fenolo/cloroformio.
Estrazione DNA plasmidico –
mediante ultracentrifugazione in gradient di densità EtBr-CsCl
o EtBr riduce il superavvolgimento del DNA
plasmidico
o Rendendolo
più
cromosomico
denso
del
DNA
o Favorendo la sua separazione in
gradiente
di
CsCl
mediante
ultracentrifugazione
Radloff et all. 1967
Estrazione DNA plasmidico –
mediante ultracentrifugazione in gradient di densità EtBr-CsCl
1.DNA circolare plasmidico+
2.DNA lineare plasmidico
3.DNA plasmidico superavvolto
Il DNA aggiunto ad una provetta contenente una soluzione concentrata di Cloruro di Cesio (CsCl 6M) viene sottoposto a centrifugazione. In
queste condizioni nella provetta si forma un gradiente di densità, dal momento che il CsCl tende a concentrarsi verso il fondo della stessa.
Una volta che si è formato il gradiente di densità, il DNA (ma in generale qualunque molecola nella provetta) migra per fermarsi nella
regione della soluzione che ha densità uguale alla sua.
Il DNA può essere messo in evidenza in seguito all'introduzione di particolari sostanze che si legano ad esso e divengono visibili se illuminate
da luce ultravioletta (EtBr).
Estrazione DNA plasmidico –
mediante ultracentrifugazione in gradient di densità EtBr-CsCl
RCF (Relative Centrifugal Force)
Rappresenta
la
forza
di
sedimentazione
sviluppata
artificialmente dalla centrifuga viene
chiamata ed è indicata con un
numero che rappresenta un multiplo
della forza di gravità terrestre "x g".
rpm (revolutions per minute)
sono un'unità di misura della velocità di
rotazione, pari al numero di giri o cicli
compiuti in un minuto da un oggetto o
dagli organi rotanti di una macchina.
RCF = (1.12 · 10-5) . r. (rpm)2
Centrifughe a bassa velocià
(MAX 3.000-6.000 g) si usano per raccogliere
organelli di grandi dimensioni e precipitati
grossolani.
La velocità massima applicabile dipende
anche dal materiale dei porta-campioni (es. i
tubi Falcon da 50 ml si deformano con RCF
superiori a 4000 g)
Microcentrifughe
(10.000 g) sono capaci di accelerazioni rapide.
Le utracentrifughe, che sviluppano RCF
anche superiori a 100.000 g, sono dotate di
un sistema di aspirazione che crea il vuoto
nella camera di centrifugazione, e di un
sistema di refrigerazione che limita il
surriscaldamento per attrito del rotore e dei
campioni.
Ultracentrifughe
(MAX 600.000 g) possiedono sistemi di vuoto
e refrigerazione sofisticati e vengono usate
per isolare piccoli organelli come i ribosomi e
le vescicole di membrana.
Estrazione di DNA plasmidico - con il kit Miniprep QIAGEN
PROTOCOLLO:
•Centrifugare per 5 minuti a 4000 rpm in centrifuga da banco
•Eliminare il surnatente
•Risospendere il pellet in 500ul d'H20 e trasferire la sospensione in una provetta Eppendorf d 1.5ml
•Centrifugare per 2 minuti alla massima velocità in una microcentrifuga.
•Eliminare il surnatante.
•Aggiungere 250ul di tampone P1 [50mM Tris-HCl pH 8.0, 10mM EDTA, 100 ug/ml RNasiA
•Risospendere le cellule.
•Aggiungere 250 ul di tampone P2 [Tampone di lisi alcalina: 200mM NaOH,1%SDS(p/v)] e miscelare capovolgendo per 3-4 volte la provetta (non
usare vortex per evitare di frammentare il DNA cromosomale - circa 400Kb - e non superare i 5 minuti a questo stadio). Questo passaggio
provoca la denaturazione di DNA e proteine e la solubilizazione delle membrane.
•Aggiungere350 ul di tampone N3 [Tampone di neutralizazzione: contenente potassio acetato, acido acetico,pH4.8], miscelare come al punto
precedente. Questo passaggio contente la rinaturazione selettiva del solo DNA plasmidico, infatti quello cromosomale, si riassocia male,
ingarbugliandosi.
•Centrifugre alla massima velocita (13000 rpm) in una microcentrifuga per 10 minuti. Il DNA plasmidico rimane in soluzione.
•Aspirare il surnatante (volume di circa 950ul) e trasferirlo in una nuova provetta Eppendorf. Il sedimento contenente pareti, complessi di DNA
cromosomale e proteine denaturate viene scartato.
•Trasferire il contenuto della provetta su una microcolonna QIA-Prep.
•Centrifugare per 1 minuto in microcentrifuga.
•Eliminare il liquido che è filtrato attraverso la microcolonna.
•Aggiungere alla microcolonna 0.75 ml di tampone PE [Tampone di lavaggio] e centrifugare per 1 minuto.
•Scartare il liquido filtrato e centrifugare per un minuto per eliminare il liquido residuo.
•Trasferire la microcolonna su una provetta Eppendorf da 1.5 ml.
•Per eluire il DNA aggiungere al centro della microcolonna 50 ul di tampone EB [Tampone di eluizione: 10mM Tris-Hl, pH 8.5]
•Lasciare assorbire per un minuto e poi centrifugare per 1 minuto.
•Tenere l'eluato ed eliminare la microcolonna.
•Allestire i campioni per l'elettroforesi, e farla partire.
Analysis of plasmid DNA in agarose gel
The movement of the DNA may be affected by the conformation of the
DNA molecule, for example, supercoiled DNA usually moves faster than
relaxed DNA because it is tightly coiled and hence more compact. In a
normal plasmid DNA preparation, multiple forms of DNA may be present.
Gel electrophoresis of the plasmids would normally show the negatively
supercoiled form as the main band, while nicked DNA (open circular
form) and the relaxed closed circular form appears as minor bands. The
rate at which the various forms move however can change using
different electrophoresis conditions, and the mobility of larger circular
DNA may be more strongly affected than linear DNA by the pore size of
the gel.
Ethidium bromide which intercalates into circular DNA can change
the charge, length, as well as the superhelicity of the DNA
molecule, therefore its presence in gel during electrophoresis can
affect its movement.
Agarose gel electrophoresis can be used to resolve circular DNA with
different supercoiling topology.
*Note however that the size of a circular DNA like plasmids cannot be
accurately gauged using standard markers unless it has been linearized
by restriction digest, alternatively a supercoiled DNA marker may be
used.


![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)
![(Microsoft PowerPoint - PCR.ppt [modalit\340 compatibilit\340])](http://s1.studylibit.com/store/data/001402582_1-53c8daabdc15032b8943ee23f0a14a13-300x300.png)