PEDIATRIC HEPATOLOGY
a cura di
Francesco Cirillo
I difetti di sintesi degli acidi biliari:
una diagnosi che non dovrebbe mai
essere omessa
Bile acid synthesis defect:
a diagnosis that should never be missed
Mara Cananzi1
Giuseppe Giordano2
Gastroenterologia, Endoscopia
digestiva, Epatologia e Cura
del bambino con trapianto di
fegato, UOC Clinica Pediatrica,
Dipartimento di Salute della
Donna e del Bambino, Azienda
Ospedaliera Università degli
Studi di Padova; 2 Laboratorio
Spettrometria di Massa,
Dipartimento di Salute della
Donna e del Bambino, Azienda
Ospedaliera Università degli Studi di Padova
1
Key words
*OCPSOFSSPSTPGCJMFBDJENFUBCPMJTNt%FGFDUT
PGQSJNBSZCJMFBDJETZOUIFTJTt$IPMJDBDJEt
Chenodeoxycholic acid
Abstract
Inborn errors of bile acid synthesis
are rare genetic disorders that cause
chronic liver disease, fat malabsorption and fat-soluble vitamin deficiency
in childhood. The diagnosis is made by
liquid chromatography-tandem mass
spectrometry. If the disorder remains
untreated end-stage liver disease may
develop, while bile acid replacement
therapy allows resolution of the hepatic disorder with excellent prognosis.
Indirizzo per la corrispondenza
Mara Cananzi
via Giustiniani 3, 35100 Padova
E-mail: [email protected]
14
Introduzione
I difetti di sintesi degli acidi biliari (DSAB) sono
un gruppo di alterazioni del metabolismo degli
steroli causati da alterazioni, geneticamente
determinate, del processo di sintesi degli acidi biliari 1. Da un punto di vista clinico i DSAB
possono esordire a tutte le età: in età pediatrica
si manifestano prevalentemente con quadri di
epatopatia e con i segni del malassorbimento
delle vitamine liposolubili mentre in età adulta
si presentano con sintomi neurologici 2. L’esatta epidemiologia dei DSAB non è nota; si stima
che la prevalenza di questi disordini in Europa
sia pari a 1-9 soggetti/1.000.000 persone. Si
ritiene inoltre che i DSAB siano responsabili di
circa l’1-2% di tutte le epatopatie croniche in
età pediatrica 2.
L’obiettivo di questa revisione è quello di fornire indicazioni utili per sospettare, diagnosticare e
trattare le epatopatie secondarie a DSAB in età
pediatrica.
Acidi biliari:
basi biochimiche
e fisiologiche
Gli acidi biliari sono un gruppo eterogeneo di steroli acidici (molecole anfipatiche costituite da un
nucleo sterolico idrofobico e da un gruppo acido
carbossilico idrofilico) sintetizzati tramite multiple reazioni enzimatiche a partire dal colesterolo (Fig. 1). Il processo di sintesi degli acidi biliari
avviene esclusivamente negli epatociti e consiste
nella produzione degli acidi biliari primari (acido
colico [cholic acid, CA] e acido chenodesossicolico [chenodeoxycholic acid, CDCA]) e dei loro
coniugati (Fig. 2). Gli acidi biliari secondari sono
prodotti nel lume intestinale tramite processi di
deidrossilazione e deconiugazione operati dalla
Giorn Gastr Epatol Nutr Ped 2015;VII:14-20
PEDIATRIC HEPATOLOGY
I difetti di sintesi degli acidi biliari
Figura 1.
Classificazione degli acidi biliari e loro strutture molecolari.
flora batterica; la loro sintesi
non è pertanto rilevante ai fini
dei DSAB 3.
Gli acidi biliari possiedono
numerose funzioni: stimolano la produzione biliare; consentono l’escrezione di colesterolo e di tossici esogeni
ed endogeni con la bile; permettono di assorbire i grassi
e le vitamine liposolubili; facilitano l’assorbimento intestinale di calcio; prevengono
l’eccessiva crescita batterica
intestinale. In condizioni di
colestasi, tuttavia, l’aumento
della concentrazione intraepatocitaria degli acidi biliari
produce effetti epatotossici.
A causa del loro elevato potere detergente, in concentrazioni troppo elevate, gli
acidi biliari innescano meccanismi di stress ossidativo
e generano effetti citotossici
e pro-apoptotici 3.
DSAB:
classificazione
e aspetti
fisiopatologici
I DSAB sono classificati in “primitivi” e “secondari” 2. I DSAB
primitivi sono causati da deficit congeniti di specifici enzimi
epatici coinvolti nel processo
di sintesi degli acidi biliari. I
DSAB secondari si realizzano
nell’ambito delle malattie perossisomiali (es. sindrome di
Zellweger, adrenoleucodistrofia neonatale) in cui l’alterato
assemblaggio dei perossisomi
impedisce lo svolgimento delle reazioni enzimatiche a sede
perossisomiale implicate nella
biosintesi degli acidi biliari. In
questo articolo sono trattati
soltanto i DSAB primitivi.
Dal 1974 fino ad oggi sono stati descritti 8 difetti primitivi di
sintesi degli acidi biliari (Fig. 2,
Tab. I), tutti a trasmissione au-
tosomica recessiva; fra questi
i più frequenti sono costituiti
dal deficit di 3-β-idrossi-C27steroido deidrogenasi e dal
deficit di δ4-3-ossisteroide-5-β
reduttasi, seguiti dalla xantomatosi cerebrotendinea e dai
difetti di coniugazione 1. Nonostante le loro singole peculiarità, i DSAB condividono i medesimi principi fisiopatologici:
il deficit di un enzima causa, “a
monte” del blocco enzimatico,
un accumulo di intermedi tossici e, “a valle” del blocco enzimatico, una ridotta produzione
biliare.
In condizioni fisiologiche gli acidi biliari primari regolano la loro
sintesi tramite un meccanismo
di feedback negativo modulato
dal recettore nucleare X Farnesoide (FXR). Nei DSAB l’assenza degli acidi biliari primari impedisce l’attivazione di questo
meccanismo di auto-regolazio-
15
M. Cananzi, G. Giordano
Figura 2.
Sintesi degli acidi biliari all’interno degli epatociti. La principale via biosintetica (in arancione) è costituita
dalla via classica, responsabile della formazione del 90% degli acidi biliari primari (CA e CDCA). La via
alternativa di sintesi degli acidi biliari (in rosa) è responsabile della formazione di una quantità di CDCA
pari a circa il 10% di tutto il pool di acidi biliari primari. La terza via di sintesi (in azzurro) è costituita dal
percorso di 25-idrossilazione che porta alla formazione di un’ulteriore piccola quota di CA. Dopo la sintesi,
gli acidi biliari primari vengono coniugati. Il processo di coniugazione con glicina o taurina, è indispensabile per la loro escrezione nella bile poiché ne garantisce la solubilità in ambiente acquoso a diversi
pH (ovvero nella bile e nel liquido enterico); per tale ragione, sia in condizioni fisiologiche che in corso di
colestasi, gli acidi biliari presenti nella bile sono quasi esclusivamente costituiti da acidi biliari coniugati.
In giallo e blu sono rappresentati gli enzimi i cui deficit sono responsabili dei DSAB attualmente noti (da
Heubi et al., 2007 2, mod.).
ne per cui gli epatociti, nel tentativo di ripristinare un normale
pool di acidi biliari, continuano
a metabolizzare il colesterolo sintetizzando un accumulo
inarrestabile di metaboliti epato- e/o neuro-tossici 4.
16
In condizioni normali, gli acidi
biliari rappresentano i principali componenti organici
della bile e, una volta secreti
nelle vie biliari, sono i maggiori determinanti del gradiente
osmotico responsabile dell’e-
screzione biliare. Nei DSAB
l’assenza degli acidi biliari
determina una riduzione della
produzione di bile con conseguente
malassorbimento
intestinale di lipidi e vitamine
liposolubili (Fig. 3) 4.
PEDIATRIC HEPATOLOGY
I difetti di sintesi degli acidi biliari
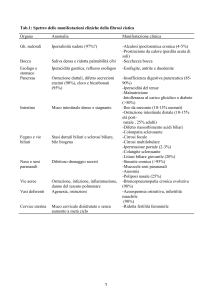
Tabella I.
Principali caratteristiche dei DSAB attualmente noti.
#OMIM
Gene/i
Sede
intracellulare
della reazione
biochimica
Deficit di colesterolo
7α-idrossilasi
118455
CYP7A1
microsomi
A
IperC, LB
Deficit di ossisterolo
7α-idrossilasi
(DSAB3) *
613812
CYP7B1
microsomi
I
E
Deficit di 3-β-idrossi-C27-steroido
deidrogenasi (DSAB1)
607765
HSD3B7
microsomi
I
E, M
Deficit di δ4-3-oxosteroide-5-β
reduttasi (DSAB2)
235555
AKR1D1
citosol
I
E
Deficit di sterolo 27 idrossilasi
(xantomatosi cerebrotendinea)
213700
CYP27A1
mitocondri
I, A
E, N, X,
C, IpoC
Deficit di 25-idrossilasi **
604551
CH25H
microsomi
I
E
Deficit di 2-metilacil-CoA racemasi
(DSAB4)
214950
AMACR
perossisomi
I, A
E, M, N,
IpoC
Deficit di CoA ligasi e difetti
dell’amidazione (Ipercolanemia
familiare)
607748
EPHX1,
TJP2,
BAAT
perossisomi
I
E, M
Reazione
biochimica
Deficit enzimatico
(DSAB)
Modificazioni
dell’anello
sterolico
Modificazioni
delle catene
laterali
dell’anello
sterolico
Coniugazione
Età di
esordio
Quadro
clinico
Abbreviazioni. E = epatopatia, M = segni malassorbimento dei grassi e delle vitamine liposolubili, N = neuropatia, IpoC = ipocolesterolemia,
IperC = ipercolesterolemia, LB = litiasi biliare, X = xantomi, C = cataratta, I = età pediatrica, A = età adulta.
* Il deficit di ossisterolo 7α-idrossilasi è stato descritto in un unico paziente: lattante di 10 settimane affetto da una grave epatopatia colestatica
a GGT bassa e con assenti acidi biliari plasmatici. L’epatopatia, a causa dell’importante tossicità dei precursori monoidrossilati, non ha risposto
alla terapia con CA ed il bambino ha necessitato di un trapianto di fegato.
** Il deficit di 25 idrossilasi è stato descritto in un unico paziente: lattante di 9 settimane affetto da epatite colestatica a GGT bassa associata a
ridotti livelli plasmatici di acidi biliari. L’epatopatia è stata curata con CA + CDCA.
DSAB:
quadri clinici e
bioumorali
I DSAB possono manifestarsi in
modo eterogeneo sia per età di
insorgenza dei sintomi (pediatrica o adulta) che per manifestazioni cliniche; queste possono essere costituite da quadri
di epatopatia cronica, di malassorbimento intestinale e di disturbi neurologici progressivi. Il
principale fattore che determina
la variabilità clinica dei DSAB è
costituito dal difetto enzimatico. In generale, i DSAB causati da deficit di enzimi implicati
nelle reazioni di modificazione
del nucleo sterolico (con l’eccezione del deficit di colesterolo
7α-idrossilasi) sono più precoci e più severi ed esordiscono
nell’infanzia con quadri di epatopatia associati a segni di malassorbimento delle vitamine liposolubili. Nei DSAB causati da
deficit di enzimi implicati nelle
reazioni di modificazione delle
catene laterali dell’anello sterolico e nei processi di coniugazione degli acidi biliari primari
sono invece prevalenti i sintomi
extra-epatici 2.
I DSAB determinati da deficit
enzimatici di enzimi implicati
nelle reazioni di modificazione
del nucleo sterolico rappresentano i DSAB più frequenti e di
maggiore interesse per l’epatologo pediatra. Essi sono principalmente costituiti dal deficit di 3-β-idrossi-C27-steroido
deidrogenasi e dal deficit di
δ4-3-ossisteroide-5-β reduttasi. Da un punto di vista clinico
si manifestano in età pediatrica
con quadri variabili di epatopatia (epatite neonatale, epatite
cronica, cirrosi) e con segni
di malassorbimento delle vitamine liposolubili talora con
17
M. Cananzi, G. Giordano
Figura 3.
A. In condizioni fisiologiche gli acidi biliari primari auto-regolano la loro sintesi tramite un meccanismo
di feedback negativo modulato da FXR. In presenza di sufficienti quantità di CA e CDCA, FXR riduce la
sintesi di nuovi acidi biliari inibendo la trascrizione di 3 enzimi: colesterolo 7 α-idrossilasi (via classica),
sterolo-27 idrossilasi (via alternativa), 12α-idrossilasi (via classica e pathway di 25-idrossilazione).
B. Nei DSAB l’assenza degli acidi biliari primari impedisce l’attivazione di FXR con conseguente accumulo
di metaboliti tossici a monte del difetto enzimatico e ridotta escrezione biliare nel lume intestinale.
franca steatorrea. L’età media
alla diagnosi è di circa 1,3 anni
(range: 4 settimane-11 anni di
vita). I segni clinici di esordio
sono principalmente costituiti
da: ittero, epatomegalia, steatorrea, scarsa crescita, rachitismo. Il prurito è di regola
assente anche se è stato segnalato in rari casi 5. Il deficit
di δ4-3-ossisteroide-5-β reduttasi tende a manifestarsi più
precocemente e ad avere un
decorso più rapidamente evolutivo verso la cirrosi e l’insufficienza epatica rispetto al deficit di 3-β-idrossi-C27-steroido
deidrogenasi. I test epatici dimostrano valori bassi di GGT
(mediana 20U/L, range 11-53)
e valori variabili di transaminasi (ALT mediana 157U/L, range
55-600) e bilirubina (mediana
110umol/L, range 40-350). Da
un punto di vista bioumorale
questi disordini sono contraddistinti da valori bassi di acidi
18
biliari plasmatici e da abnormi
livelli di precursori anomali degli acidi biliari nel plasma e nelle urine 2, 4, 5-8.
I DSAB determinati da deficit
degli enzimi deputati a catalizzare le reazioni di modificazione delle catene laterali
dell’anello sterolico del colesterolo sono principalmente
costituiti dalla xantomatosi cerebrotendinea e dal deficit di
2-metilacil-CoA racemasi. Si
manifestano principalmente in
età adulta con disordini neurologici degenerativi e sintomi
quali atassia, paralisi pseudobulbare, demenza precoce. A
questi si associano segni di
malassorbimento delle vitamine liposolubili talora con franca
steatorrea. Il quadro epatologico è variabile ma nella maggior
parte dei casi consiste in un
quadro di epatite colestatica
neonatale a GGT bassa, con
acidi biliari plasmatici bassi e a
risoluzione spontanea nel primo anno di vita. Da un punto
di vista bioumorale questi disordini sono contraddistinti da
ridotti valori ematici di colesterolo e acidi biliari, e da abnormi livelli di precursori anomali
degli acidi biliari nel plasma e
nelle urine 2.
I DSAB determinati da deficit
di enzimi deputati alla coniugazione degli acidi biliari primari sono raggruppati sotto il
termine di Ipercolanemia Familiare. Da un punto di vista
bioumorale sono contraddistinti dalla completa assenza
di acidi biliari coniugati nella
bile e da elevati livelli plasmatici e urinari di CA. Da un punto
di vista clinico sono principalmente caratterizzati dai segni
del malassorbimento delle vitamine liposolubili in assenza
di evidente steatorrea. A causa
degli elevati livelli plasmatici di
CA, molti soggetti lamentano
PEDIATRIC HEPATOLOGY
I difetti di sintesi degli acidi biliari
prurito pur in assenza di una
vera e propria epatopatia. Solo
nel corso dei primi mesi di vita
una quota di pazienti si presenta con un quadro di epatite
colestatica neonatale a GGT
bassa usualmente a risoluzione spontanea ma, in rari casi,
potenzialmente severa 9.
DSAB: diagnosi
In età pediatrica i DSAB devono essere sospettati in presenza di:
rVOFQBUPQBUJBDPMFTUBUJDBDSPnica a GGT bassa, associata
a normali livelli degli acidi biliari plasmatici, soprattutto se
in assenza di prurito;
rVOFQBUPQBUJBDSPOJDBDSJQUPgenetica;
rTFHOJ EJ NBMBTTPSCJNFOUP
intestinale di lipidi e vitamine
liposolubili (es. scarsa crescita, steatorrea, rachitismo,
ariflessia, discoagulopatia) 1.
La biopsia epatica può mostrare quadri istologici con gradi
variabili di necrosi epatocitaria,
fibrosi, colestasi canalicolare
ed epatocellulare, sempre in
assenza di reazione duttulare;
l’esame istologico pertanto, è
utile nella diagnosi differenziale dell’epatopatia ma non permette di formulare una diagnosi di certezza 5.
La diagnosi si basa sull’utilizzo della spettrometria di massa che non solo consente di
diagnosticare la presenza di
DSAB ma anche di riconoscere il deficit enzimatico presente. In condizioni normali l’escrezione urinaria degli acidi
biliari è irrilevante; in caso di
colestasi, invece, l’escrezione
urinaria degli acidi biliari aumenta in modo direttamente
proporzionale alla severità della colestasi. Analogamente, nei
DSAB, i metaboliti “atipici” degli acidi biliari prodotti a monte
del blocco enzimatico vengono eliminati con l’urina dove
possono essere identificati e
quantificati tramite l’indagine
spettrometrica. Per ciascun
DSAB, quindi, la spettrometria
di massa permette di riconoscere “l’impronta metabolica”
dei singoli difetti in modo non
invasivo (Fig. 4) 1, 2.
L’analisi genetica è necessaria
per stabilire il difetto molecolare alla base del deficit funzionale enzimatico e per effettuare una consulenza genetica
alla famiglia.
Trattamento
La terapia dei difetti di sintesi
degli acidi biliari primari si basa
sulla somministrazione per via
orale di CA e/o CDCA con lo
scopo di ricostituire un normale pool di acidi biliari 1. Ciò
permette da un lato di inibire,
tramite l’attivazione di FXR, il
Figura 4.
Profili degli acidi biliari urinari ottenuti tramite spettrometria di massa rispettivamente da un soggetto
sano (A) e da un paziente affetto da deficit di 3-β-idrossi-C27-steroido deidrogenasi (B). IS = standard
interni marcati. A, B, C, D = metaboliti biliari atipici presenti nel DSAB e non presenti in condizioni normali
19
M. Cananzi, G. Giordano
processo endogeno di biosintesi di precursori “tossici”
degli acidi biliari e, dall’altro,
di ripristinare la produzione di
bile acido biliare-dipendente 2.
CA e CDCA si sono dimostrati
efficaci nel trattamento di tutti i DSAB (i.e. scomparsa dei
precursori tossici degli acidi
biliari nell’urina, guarigione
dell’epatopatia) fatta eccezione per il deficit di ossisterolo
7α-idrossilasi (Tab. I). CA e
CDCA possono essere utilizzati singolarmente e in combinazione; le dosi di partenza
sono di 5-15 mg/kg/die per
CA o CDCA in monoterapia 4-8,
e di 7 + 7 mg/kg/die per l’associazione di CA + CDCA 5. In
corso di trattamento la spettrometria di massa permette
di stabilire l’adeguatezza della
dose monitorando la quantità dei metaboliti degli acidi
biliari nell’urina 7. La terapia
con acidi biliari primari è priva
di effetti collaterali e sicura a
lungo termine. Sono stati segnalati casi di transitoria epatotossicità (ipertransaminase-
mia, colestasi a GGT elevata,
prurito, diarrea) in caso di assunzione accidentale di dosi
elevate.
Nei pazienti con difetti di coniugazione degli acidi biliari
primari si è recentemente dimostrata efficace la terapia
con acido glicocolico (15 mg/
kg/die) 10.
Bibliografia
1
Setchell KD, Heubi JE. Defects
in bile acid biosynthesis-diagnosis and treatment. J Pediatr Gastroenterol Nutr 2006;43(Suppl
1):S17-22.
2
Heubi JE, Setchell KD, Bove
KE. Inborn errors of bile acid
metabolism. Semin Liver Dis
2007;27:282-94.
3
Monte MJ, Marin JJ, Antelo A, et
al. Bile acids: chemistry, physiology, and pathophysiology. World
J Gastroenterol 2009;15:804-16.
4
5
Gonzales E, Gerhardt MF, Fabre
M, et al. Oral cholic acid for hereditary defects of primary bile acid
synthesis: a safe and effective
long-term therapy. Gastroenterology 2009;137:1310-20.
Subramaniam P, Clayton PT, Port-
mann BC, et al. Variable clinical
spectrum of the most common
inborn error of bile acid metabolism-3beta-hydroxy-Delta 5-C27steroid dehydrogenase deficiency.
J Pediatr Gastroenterol Nutr
2010;50:61-6.
6
Mizuochi T, Kimura A, Ueki I, et
al. Molecular genetic and bile
acid profiles in two Japanese patients with 3beta-hydroxy-DELTA5-C27-steroid dehydrogenase/
isomerase deficiency. Pediatr Res
2010;68:258-63.
7
Riello L, D’Antiga L, Guido M, et al.
Titration of bile acid supplements
in 3beta-hydroxy-Delta 5-C27steroid dehydrogenase/isomerase
deficiency. J Pediatr Gastroenterol Nutr 2010;50:655-60.
8
Seki Y, Mizuochi T, Kimura A, et al.
Two neonatal cholestasis patients
with mutations in the SRD5B1
(AKR1D1) gene: diagnosis and bile
acid profiles during chenodeoxycholic acid treatment. J Inherit
Metab Dis 2013;36:565-73.
9
Setchell KD, Heubi JE, Shah S,
et al. Genetic defects in bile acid
conjugation cause fat-soluble vitamin deficiency. Gastroenterology
2013;144:945-55.
10
Heubi JE, Setchell KD, Jha P, et al.
Treatment of bile acid amidation
defects with glycocholic acid. Hepatology 2015;61:268-74.
I difetti di sintesi degli acidi biliari:
t sono rari disordini epatici causati da deficit geneticamente determinati degli enzimi coinvolti nel metabolismo degli
acidi biliari;
t nel bambino si manifestano tipicamente con quadri di colestasi a GGT bassa e con i segni del malassorbimento dei
grassi e delle vitamine liposolubili;
t sono diagnosticati tramite l’identificazione di metaboliti “atipici” degli acidi biliari nei liquidi biologici (siero, urine)
mediante spettrometria di massa;
t se non trattati possono evolvere verso la cirrosi e l’insufficienza epatica;
t rispondono prontamente al trattamento con acidi biliari che, nella maggior parte dei casi, determina la completa
regressione dell’epatopatia.
20