Inquadrare il fenomeno della carnificazione polmonare. Pg 500 mottura
La c.p. fa parte dei fenomeni evolutivi anomali dei processi polmonitici, tra cui vi sono anche
-polmonite in organizzazione con bronchiolite obliterante
-polmoniti croniche
-necrosi, suppurazione e gangrena
-infine, la carnificazione.
Può inoltre verificarsi nei quadri di congestione cronica polmonare (notizia trovata on line ma non
confermata dal mottura...Per gli esiti della congestione cronica (pg 467 mottura): non causa
carnificazione, ma indurimento bruno!)
(-Sviluppare evoluzione normale delle polmoniti)
Eziopatogenesi. Sono chiamati in causa diversi fattori...
Per gli esiti polmonitici:
-l' introduzione nella pratica clinica di nuovi e più efficaci farmaci, soprattutto gli antibiotici, ha
causato la riduzione degli esiti mortali post-polmonitici, determinando l'aumento dei casi in
questione;
-la sterilizzazione precoce dell'essudato grazie ai farmaci che da un lato arriva tardi per impedire
completamente il processo infettivo, dall'altro riduce drasticamente gli stimoli chemiotattici nei
confronti dei granulociti.
Quindi abbiamo un essudato fibrinoso abbondante, sterile, non accompagnato da essudazione
granulocitaria. Ci sarà perciò una drastica riduzione dei fenomeni di digestione (fermentativa della
quota proteica) della fibrina, a causa della riduzione degli enzimi litici granulocitari. Questo da il
via libera ai fenomeni della fibrosi e della carnificazione.
Meccanismi: gli alveoli sono pieni di fibrina che permane e non viene riassorbita, dopo un paio di
settimane compaiono fibroblasti e fibre collagene che trasformano la fibrina in tessuto connettivo
neo formato; compare anche neoangiogenesi. I tralci di fibrina ormai trasformati in connettivo
lasciano dei piccoli spazi vuoti che vengono rivestiti di epitelio polmonare, con pneumociti di tipo 2
(principalmente).
Nb: fenomeni quali la necrosi estesa e la gangrena sono favoriti da meccanismi simili, nel senso che
processi infettivi che un tempo avrebbero causato la morte del pz, ora si curano coi farmaci
risultando però in un prolungamento del processo infettivo e di situazioni ischemiche con comparsa
di estese aree di necrosi.
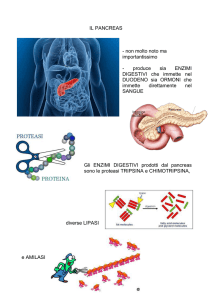
Discutere il quadro morfologico descritto nel pancreas diabetico
Diabete mellito: alterazione cronica del metabolismo dei carboidrati, caratterizzata da iperglicemia,
glicosuria, iperlipemia, acidosi chetonica e bilancio dell'azoto negativo.
Frequenza 2-3% pop italiana;
complicanze frequenti: aterosclerosi; micorangiopatia; nefropatia; neuropatia;
è tra le prime 10 cause di morte;in USA 7° causa di morte
Il quadro descritto nel caso, dominato da estesi fenomeni autolitici è di riscontro comune in quanto
il pancreas è ricco di cellule contenenti enzimi litici già attivi (bloccati da inattivatori) a ph cellulare
in grado di digerire proteine, carboidrati e lipidi, cioè tutte le strutture cellulari.
Affinchè si sviluppi diabete del pancreas endocrino, esso deve perdere il 90% (prove sperimentali)
della sua funzionalità.
Forme secondarie:
pancreatiti acute > emorragiche e croniche ad evoluzione sclerosante
neoplasie maligne voluminose
emocromatosi
acromegalia per eccesso di GH (forse perchè aumenta la produzione surrenalica di
glucocotricoidi ad azione antinsulinica)
sindrome di cushing (i glucocorticoidi causano gluconeogenesi e inibiscono la captazione
periferica di insulina = effetto antinsulinico)
infezioni, rosolia congenita, citomegalovirus
farmaci: coricosteroidi, pentamidina, vacor
Sindrome di Down
DM gestazionale
Il danno coinvolge prima la componente esocrina, poi le isole e si deve necessariamente spingere
fino alla coda (condizione necessaria ma non sufficiente), cioè al settore dove la loro
concentrazione è maggiore.
Nella pancreatite acuta emorragica le estese aree di necrosi possono portare allo sviluppo del
diabete.
Nelle pancreatiti croniche invece si manifesta di rado, solo in forme molto avanzate dove la sclerosi
si è piano piano diffusa sino alle isole.
I traumi contusivi possono comportare estesi fenomeni necrotici con comparsa di DM.
In caso di Litiasi, la sclerosi che ne consegue porta a DM solo se viene complicata da processi
flogistici. (NB: l'occlusione del dotto di Wirsung non causa mai la distruzione delle isole, per
esempio ciò avviene nella fibrosi cistica (vedi)).
Nell' emocromatosi si instaura il Diabete Bronzino, a causa dell'accumulo di granuli di emosiderina
nel citoplasma delle cellule B, che hanno effetto tossico.
Forme primitive:
tipo 1 giovanile: esordio brusco, rapida evoluzione, prognosi peggiore;
tipo 2 dell'adulto: comparsa 4°/6° decennio, prognosi migliore, decorso lento;
incidenza 3% pop mondiale
Alterazioni morfologiche del diabete primario (pg 906 mottura):
Macro: sono aspecifiche e difficilmente distinguibili da pancreas di soggetti non affetti da DM e di
pari età;
Micro:
-tipo I:
-deficienza numero e volume isole B (Langherans), difficili da trovare nella sezione istologica
(ciò indica la riduzione del numero delle isole);
-infiltrazione linfocitaria (insulite)soprattutto nei casi gravi e di breve durata (pare che concorrano
nella distruzione delle cell B): linfociti T CD8+ >>, CD4+ e macrofagi <; pochissimi eosinofili;
Questo infiltrato a dato il via a ipotesi patogenetiche di natura virale (coxakie) e autoimmunitaria
(correlata anche alla presenza di anticorpi anti-insulari nel 50% dei giovani pz, e nel 10% di questi
ci sono anche ab-anti mucosa gastriva, tiroide e surrene).
PATOGENESI DM1: sembra essere causato da una infezione virale e ritardato dal lungo periodo di
latenza necessario perchè si realizzi la perdita immunomediata di cell B, dipendente inoltre dal
background genetico del pz. Cause:
suscettibilità genetica (> frequenza nord europa; concordanza gemelli omozigoti al 70% indica
penetranza incompleta; HLA-DR3 e/o DR4 nel 95% dei malati mentre nella pop normale siamo al
45%, con probabili alterazioni del riconoscimento dei linfociti T degli autoantigeni)
autoimmunità (insulite con aumento espressione MHCI e MHC2 aberranti (causata da INFgamma
dei linfociti T); l'impianto di CD4+ di animali malati in sani causa la comparsa di malattia; presenza
di autoanticorpi GAD (decarbossilasi acido glutammico), IA-2 (autoantigene insulare tipo 2),
insulina, gangliosidi; NB la presenza simultanea di anti GAD, IA-2, insulina, provoca la comparsa
di DM clinicamente evidente entro 5 anni; presenza di altre pato autoimmuni come M. Graves,
Addison, tiroidite Hashimoto)
fattori ambientali (virus, le nuove dx di DM hanno andamento stagionale correlato alle infezioni
virali di Coxsakie B, morbillo, parotite, citomegalo, rosolia, mononucleosi; riscontro di genoma
retrovirale nelle cellule retrovirali che può comportarsi come superantigene)
-tipo II: lieve riduzione di volume, lieve fibrosi del pancreas esocrino, lesioni delle B cell:
-amiloidosi o degenerazione ialina (pare sia causa e non conseguenza, è un reperto anche degli
insulinomi e in soggetti non affetti da DM anziani, potrebbe essere legato alla sofferenza delle B
cell): colpisce il 51-72% pz, non associata ad amiloidosi sistemica; correlata all'età dei pz;
riscontrata nel 2% dei pz non diabetici, per questo non viene considerata strettamente specifica.
Consiste nella progressiva deposizione di amiloide (sostanza omogenea, eosinofila, ialina, fibrillare
al microscopio elettronico) che in realtà è falsa amiloide perchè costituita da fibrille di amilina negli
spazi interstiziali tra capillari ed endotelio e provoca col tempo collasso dei capillari e dall'altro
atrofia e necrosi delle cellule endocrine. In fase avanzata le isole diventano un cumulo di zone
ialine, a volte confluenti, frammiste a cellule con nucleo picnotico. Da notare che il pancreas
esocrino non viene coinvolto nel processo!
L' amiloide in teoria impedisce gli scambi nutritivi e respiratori delle cellule e anche le funzioni
paracrine.
-degenerazione idropica insulare: i granuli citoplasmatici delle B cell scompaiono e lasciano al
loro posto un unico vacuolo che provoca nel tempo atrofia cellulare e necrosi; in realtà nelle cellule
si accumula glicogeno e alcuni autori sottolineano come il nome di tale lesione sia sbagliato e la
chiamano “glicogenosi”; è più frequente nel DM giovanile a rapido decorso; i granuli si esauriscono
per eccessiva stimolazione, mentre il glicogeno si accumula probabilmente per effetto secondario
dell'insufficienza insulare.
-in tutti i casi in cui le alterazioni sopraesposte sono assenti, la degranulazione è invece sempre
presente.
PATOGENESI DM2: disordine complesso e multifattoriale, coinvolgente sia l'alterato rilascio di
insulina con relativo deficit, che l'insensibilità dei tessuti periferici. Cause:
disregolazione della secrezione di insulina (la normale secrezione pulsatile di insulina viene persa,
manca il picco rapido iniziale; non evidenza di danno insulare immunomediato o virusmediato)
resistenza insulinica (obesità e gravidanza la causano anche senza DM; riduzione recettori GLUT4 cioè periferici, presenza di difetto postrecettoriale)
obesità (80% pz con DM2 è obeso; obesità di tipo centrale; la riduzione di peso precoce causa
regressione dell'alterata tolleranza al glucosio)
amilina (peptide normalmente prodotto dalle cell B e cosecreto con l'insulina, provoca una
pseudoamiloidosi)
Discutere il problema del riconoscimento morfologico delle isole e della loro composizione
Le isole pancreatiche sono circa 1-2 milioni, hanno un diametro di 100-200 micron, sono sparse nel
pancreas esocrino e sono più numerose verso la coda, peso totale 1-2 gr.. Il limite anatomico con il
p. esocrino è netto e ben visibile nella sezione istologica di un organo ben conservato, le isole hanno
uno stroma fibrillare percorso da numerosi capillari che scorrono tra travate e file singole di cellule,
(situazione che aumenta la superficie di contatto col sistema ematico).
Troviamo 4 tipi cellulari maggiori:
1. A glucagone: periferiche, 20-30%, + ab-antiglucagone; + colorazione Grimelius (argirofilia
+); rapporto cell A/B=1:3;
2. B insulina: centrali,60-80%, poligonali, citopl chiaro, granuli + all'aldeide fucsica, +
all'immunoistochimica per ab anti-insulina e anti peptide-C;(vedi pg 917 mottura per M.E.);
3. D somatostatina: 5-10%, forma allungata con prolungamenti citopl, + al metodo argentico di
Davenport, NB: la somatostatina inibisce il rilascio sia di glucagone che di insulina.
4. PP polipeptide pancreatico, stimola secrezione gastrica e enzimatica intestinale, inibisce la
motilità gastro; queste cellule sono presenti anche nel pancreas esocrino;
2 tipi cellulari minori:
1. D1 peptide intestinale vasoattivo (VIP), induce glicogenolisi, iperglicemia e stimola la
secrezione gastroenterica causando diarrea secretoria;
2. ENTEROCROMAFFINI, serotonina; con colorazione di Gomori (Argento nitrato e
Metenamina borato), colora le strutture argentaffini (cioè fissano i sali d'argento e lo
riducono ad argento metallico, l'argentofilia invece consiste solo nel legarli)
Il riconoscimento: colorazione con blu di toluidina dopo metilazione e successiva demetilazione
allo studio delle isole pancreatiche di cavia. Tre tipi cellulari sono chiaramente distinguibili con tale
metodo, e cioè: le cellule A, deputate alla produzione del glucagone colorate in blu, le B insulinosecernenti, praticamente incolori ed un terzo tipo di cellula, identificabile con quella argentofila,
colorata metacromaticamente in rosso. Correva l'anno 1965
NB: la dimostrazione istochimica della presenza di G6PDH nelle cellule pancreatiche è ingrado di
discriminare tra pancreatite cronica (+) e carcinoma (-).
Insufficineza cardiaca sinistra = ↓gittata (ins, anterograda)
Vsx: in genere ipertrofico e dilatato, con dilatazione Asx, fibrillazione e stasi ematica che favorisce
la comparsa di trombi; (rischio emboli cerebrali e riduzione flusso encefalo e reni);
Polmoni: aumento pressione vene polmonari e capillari con formazione di trasudato perivascolare e
interstiziale; ampliamento edematoso dei setti alveolari; accumulo liquidi negli alveoli (edema
polmonare); emoglobina e transferrina fagocitate dai macrofagi alveolari causano la comparsa dei
granuli di emosiderina; CLINICA: dispnea, ortopnea, dispnea parossistica notturna, tosse;
Reni: ↓ gittata con ipoperfusione, attivazione sistema renina-angiotensina-aldosterone, ritenzione di
acqua e sali e aumento della volemia e dei liquidi extraematici; il rilascio di peptide natriuretico
atriale bilancia il riassorbimento dei sali riducendo il volume ematico eccesivo; se il deficit di
perfusione è severo aumentano le scorie azotate con iperazotemia prerenale;
Encefalo: encefalopatia ipossica negli stadi finali fino a perdita della coscienza coma e morte;
Insufficienza cardiaca destra = stasi venosa (ins. Retrograda)
Fegato: epatopatia congestizia con aumento di peso e volume, con congestione cronica passiva: aree
centrolobulari rossastre e congeste, circondate da zone pallide; se coesiste ins. Sx l'ipossia severa
provoca necrosi ischemica centrolobulare intorno ai sinusoidi congesti; se il quadro è più severo la
combinazione di congestione e ipoperfusione causano necrosi emorragica centrolobulare dando al
fegato l' aspetto a NOCE MOSCATA; raramente, col tempo queste aree diventano fibrotiche
portando alla “cirrosi cardiogena” (non è una vera cirrosi perchè la fibrosi è solo centrolobulare);
Sistema portale: splenomegalia congestizia (milza dilatata fino a >300gr (normale 150), con
dalatazione sinusoidale e aree di recente emorragia; solo nelle congestioni di lunga durata,
tipicamente dovute a cirrosi con marcata fibrosi (e non per stasi venosa sistemica) , la milza può
arrivare a superare i 4/5 kg, avere la capsula ispessita e fibrosa e la consistenza dura, a livello
siunsoidale aumenta il collagene della membrana basale che si dilatano a causa della rigidità della
parete, il sangue staziona più tempo nei cordoni e le cellule ematiche vanno incontro a distruzione
eccesiva (ipersplenismo), possono esserci focolai emorragici più o meno recenti con emosiderina
negli istiociti. Questi focolai possono organizzarsi con fibrosi e deposito di sali di calcio e ferro sui
tessuti connetivi e le fibre elastiche (noduli di Gandy-Gamna)
Intestino: può comparire edema cronico della parete che interferisce con l'assorbimento; Ascite,
dovuta ad accumulo abnorme di trasudato nella cavità peritoneale;
Reni: congestione marcata (più che nell'ins. sx) maggiore ritenzione liquidi, edema periferico,
azotemia prerenale;
Encefalo: congestione venosa e ipossia (come sopra);
Cavità pleuriche e pericardiche possibili versamenti, > pleura dx; eventuale atelettasia polmonare
per versamenti importanti;
Tessuti sottocutanei: edema periferico nelle sedi declivi, in particolare edema malleolare e pretibiale
possono esser considerati segno distintivo di ins. Dx; nei casi molto gravi anasarca (presenza
abnorme di trasudati nei tessuti e nelle cavit&agrave; sierose (pleura, pericardio ecc.) di tutto il
corpo, in occasione di grave insufficienza renale, di scompenso cardiocircolatorio, di cirrosi epatica
e di stati cachettici.