B. MARINO - O. MILANESI - R. FORMIGARI - G. SANTORO
CARDIOLOGIA
PEDIATRICA
EDIZIONI MINERVA MEDICA
MARINO cardiologia pediatrica.indd 1
17/09/15 07:54
ISBN: 978-88-7711-849-3
© 2016 – EDIZIONI MINERVA MEDICA S.p.A. – Corso Bramante 83/85 – 10126 Torino
Sito Internet: www.minervamedica.it / e-mail: [email protected]
I diritti di traduzione, memorizzazione elettronica, riproduzione e adattamento totale o parziale, con qualsiasi mezzo (compresi
microfilm e copie fotostatiche), sono riservati per tutti i Paesi.
MARINO cardiologia pediatrica.indd 2
17/09/15 07:54
Vogliamo dedicare questo libro alle nostre famiglie
che ci hanno supportato e sopportato in questi anni
e alla generazione precedente alla nostra in particolare al
Dott. Luigi Ballerini, al Prof. Raffaele Calabrò,
al Prof. Fernando Picchio, al Prof. Gaetano Thiene
e al Prof. Franco Zacchello
che ci sono stati maestri e mentori
MARINO cardiologia pediatrica.indd 3
17/09/15 07:54
INTRODUZIONE
Consultare questo gran bel libro di cardiologia pediatrica è un autentico piacere e uno stimolo culturale
prezioso. E non è un piacere di nicchia poiché coinvolge – e stimola l’interesse – non soltanto dei cardiologi, ma specificamente dei pediatri ad ogni livello, universitario, ospedaliero, di territorio, in formazione.
L’impostazione editoriale e la partitura dei singoli capitoli permettono di seguire il bambino con problemi
cardiaci a partire dal concepimento, dall’epoca fetale e dalla nascita attraverso le età successive – attività
scolare e sportiva incluse – fino allo sviluppo adolescenziale verso l’età adulta: ogni età comporta diversi e
importanti rischi di malattia e al contempo specifiche opportunità di salute. Lungo questo percorso, il volume propone problemi e quesiti offrendo al lettore risposte di altissimo livello qualitativo. Perché i problemi
sono quelli che i pediatri generalisti e specialisti incontrano nella propria vita professionale e che devono
saper gestire con competenza e autorevolezza.
Mancava un manuale che con sapiente maestria spiegasse e motivasse quali e quante nuove acquisizioni abbiano mutato le prospettive del paziente con cardiopatia congenita e della sua famiglia e quali
nuove responsabilità, di conseguenza, siano oggi affidate ai medici che ne devono guidare e condividere
le scelte.
Ognuna di queste piccole, grandi rivoluzioni, dalla genetica molecolare alle straordinarie tecniche di
imaging che consentono diagnosi e interventi all’interno del cuore, come in una sorta di realtà virtuale, viene affrontata e orientata al sapere, al saper fare e, non da ultimo, al saper comunicare.
Conoscere le basi genetiche delle cardiopatie congenite è indispensabile per orientare la famiglia ad
affrontare future gravidanze al meglio delle attuali possibilità diagnostiche e terapeutiche; le tecniche interventistiche consentono oggi di correggere alcune malformazioni già alla nascita o addirittura in utero: il trasferimento del paziente è quindi programmabile quando è ancora in utero, verso centri nascita che abbiano
competenze e strutture idonee. Ma non vengono trascurati anche altri aspetti apparentemente più semplici
come il dolore toracico, i disturbi del ritmo o i limiti della pratica sportiva, aspetti che quotidianamente il
pediatra deve affrontare e orientare come patient manager, interfacciandosi con i centri cardiologici di eccellenza.
Ci troviamo sempre più spesso a trattare bambini con problemi cronici complessi. I cosiddetti children
with special health care needs (CSHCN), bambini con bisogni assistenziali speciali, e i più gravemente
compromessi children with medical complexity (CMC), sono nel nostro Paese poco meno di un milione e
mezzo. Sono affetti molto spesso da sindromi genetiche complesse di cui fanno parte cardiopatie congenite dalle più alle meno gravi. I progressi davvero impressionanti della cardiologia, della cardiochirurgia
e dell’interventistica permettono oggi di sopravvivere e spesso di diventare adulti a un gran numero di
bambini che un tempo morivano alla nascita o sopravvivevano per poche settimane o per pochi mesi.
Anche in questa chiave di lettura, il volume è uno strumento prezioso per il pediatra perché molto spesso
le malattie croniche complesse sono malattie rare che il singolo medico avrà l’opportunità di incontrare
una, due volte nel corso di tutta la sua vita professionale. L’abbondanza di illustrazioni ed esemplificazioni iconografiche, con tabelle riassuntive e flow chart esplicative, si rivela preziosa anche per la consultazione.
V
MARINO cardiologia pediatrica.indd 5
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
Gli editor hanno saputo trasmettere a questo testo la propria egregia competenza accademica unita a
un’altrettanta pregevole esperienza professionale in cardiologia pediatrica. Hanno saputo raccogliere intorno a sé numerosi esperti, tra i più autorevoli, della pediatria, della genetica clinica e della cardiologia che
hanno contribuito fattivamente all’eccellenza di questo libro. Ne hanno saputo coagulare saperi e competenze professionali mettendoli a disposizione dei pediatri e dei cardiologi per soddisfarne appieno, armonicamente, la domanda di conoscenza e di cultura e le esigenze dell’aggiornamento e dell’approfondimento
clinico-professionale.
Prof. Alberto G. Ugazio
Direttore Dipartimento di Pediatria
Ospedale Pediatrico Bambino Gesù IRCCS, Roma
VI
MARINO cardiologia pediatrica.indd 6
17/09/15 07:54
PRESENTAZIONE
Mi è stato gentilmente chiesto di scrivere una presentazione per questo trattato di cardiologia pediatrica,
una presentazione vista con gli occhi di un cardiologo tradizionalmente “adulto” quale io mi considero. Ai
tempi della mia iniziale formazione cardiologica i casi di cardiopatia congenita che capitavano all’osservazione clinica non erano rari. Poi, gradualmente, l’espandersi esplosivo delle conoscenze fisiopatologiche,
delle tecnologie diagnostiche e delle opzioni terapeutiche ha portato al progressivo sviluppo di competenze
specifiche e alla loro concentrazione in centri super specialistici nelle mani di esperti del settore.
Questa evoluzione ha portato a una progressiva ottimizzazione e standardizzazione delle procedure
diagnostiche e terapeutiche che rappresenta ora la base di partenza per il trattamento delle cardiopatie
congenite e della loro evoluzione nel tempo. Questo trattato fa il punto sullo stato dell’arte in questo campo
in maniera comprensiva e ammirevole. Tuttavia ritengo sia giunto il momento di prepararsi a un ulteriore
passo avanti, prendendo proprio come punto di partenza questo trattato. Infatti, dopo la correzione dei loro
difetti, molti pazienti hanno potenzialmente davanti a loro molti anni di vita normale, se seguiti nel tempo
secondo le direttive specifiche. Queste direttive possono essere implementate direttamente anche dai cardiologi adulti e dai medici di medicina generale ora che hanno a disposizione il razionale e le indicazioni
generali che sono dettagliate in questo trattato, per indirizzarli quando fosse opportuno, agli esperti del
settore e ai centri super specialistici.
Prof. Attilio Maseri
Presidente Fondazione “Per il Tuo Cuore” Onlus
VII
MARINO cardiologia pediatrica.indd 7
17/09/15 07:54
Prefazione
Cardiologia pediatrica: quo vadis?
Conoscere un argomento anche a fondo non costituisce affatto una garanzia di saperne anticipare gli
sviluppi, poiché “quando parliamo del domani spesso pensiamo alle nostre idee di oggi” (Tim Harford,
divulgatore economico inglese).
Prevedere quindi quale possa essere l’evoluzione tecnica e di pensiero che la cardiologia pediatrica è
destinata a percorrere nel corso dei prossimi anni rappresenta un difficile obiettivo, soprattutto qualora si
considerino le profonde mutazioni che il nostro sistema sanitario, e sociale più in generale, si appresta a
sperimentare nel futuro più vicino.
La ristrutturazione del modello assistenziale, che potrebbe prevedere il progetto di una progressiva
scomparsa delle enclavi specialistiche negli ospedali e negli istituti di cura, impone un cambiamento non
solo strutturale, ma anche concettuale, della figura e dell’identità del “cardiologo pediatra” e del modo con
il quale questo interagisce con le altre figure professionali di riferimento, che siano mediche, infermieristiche o tecniche.
Il quesito quindi andrebbe forse così riformulato:
Cardiologo pediatra, quis es?
La definizione di una figura professionale di questo tipo, sempre sospesa fra la cardiologia e la pediatria,
è complessa e risulterebbe scarsamente comprensibile qualora non si faccia riferimento alla memoria storica, l’epopea di quasi un secolo, costruita da donne e uomini dei quali la passione e la generosità hanno
permesso di alimentare la speranza per la sopravvivenza e per una vita migliore a un numero sconfinato di
persone cardiopatiche.
Fino ai primi decenni del Novecento, il cardiologo pediatra ovviamente non esisteva e le malformazioni
cardiache venivano viste come rari, se pur interessanti, scherzi della natura per i quali non vi era rimedio. È
per opera di un chirurgo, Robert E. Gross al Children’s Hospital di Boston, che nel 1938 viene tradizionalmente fatta iniziare l’era della diagnosi e della terapia delle cardiopatie congenite 1. Poi, nel 1944 a Baltimora fu eseguito il primo intervento palliativo per la tetralogia di Fallot: l’anastomosi succlavio-polmonare
secondo Blalock e Taussig e a Minneapolis nel 1954 Walton Lillehei eseguì il primo intervento di cardiochirurgia a “cuore aperto”. La via era aperta, e dato che finalmente si cominciava a poter curare le cardiopatie
congenite, si impose la necessità di eseguire diagnosi sempre più accurate 1.
Dalla metà degli anni Sessanta del Novecento, la cardiologia pediatrica uscì dalla fase pionieristica e si
affermò come una sottospecialità della pediatria e della cardiologia; la diagnostica strumentale si sviluppò
e gli interventi cardiochirurgici furono coronati da sempre maggiore successo. Ma è negli anni Settanta che
si realizzarono alcune condizioni che rivoluzionarono la conoscenza, la diagnosi e la cura delle cardiopatie
congenite. Lo studio dettagliato dei reperti anatomici, il raffronto con la clinica e l’esperienza chirurgica
permisero a un gruppo di medici dedicati, come Richard Van Praagh, Jesse E. Edwards, Maurice Lev e
Maria Victoria De la Cruz, per citarne solo alcuni, di gettare una prima concreta luce sull’apparente caos
delle malformazioni congenite del cuore, compiendo un incredibile sforzo di classificazione e ricognizione
nosologica, migliorandone sostanzialmente la comprensione anatomica e chirurgica 1.
IX
MARINO cardiologia pediatrica.indd 9
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
Gli studi di fisiopatologia neonatale, di emodinamica e di angiocardiografia chiarirono gli aspetti funzionali e resero possibile la diagnosi morfologica in vita. Nei primi anni Ottanta irruppe, poi, l’ecocardiografia bidimensionale quale metodica di visualizzazione del cuore, rendendo precise e veloci la diagnosi
e l’indicazione non invasiva al trattamento chirurgico delle cardiopatie congenite. Sempre in quegli anni,
poi, l’uso delle prostaglandine in epoca neonatale permise, riaprendo il dotto arterioso, la sopravvivenza
di alcune gravi malattie dotto-dipendenti che, quindi, potevano essere diagnosticate e operate precocemente 1.
La cardiochirurgia ovviamente continuò ad essere protagonista di quel periodo, con una tendenza alla
correzione delle cardiopatie in epoca sempre più precoce, realizzando soluzioni chirurgiche palliative o
correttive fino ad allora ritenute impossibili, mentre le nuove acquisizioni nel campo dell’immunologia
migliorarono grandemente la sopravvivenza dei pazienti sottoposti a trapianto cardiaco. Le innovazioni
culturali, tecniche e di mentalità degli anni Settanta e Ottanta del secolo scorso cambiarono radicalmente
la cardiologia pediatrica ponendo le basi per tutti gli sviluppi futuri. Negli anni Novanta, le tecniche di
emodinamica interventistica raggiunsero inoltre la piena maturità, consentendo il trattamento palliativo o
correttivo non chirurgico di un numero sempre maggiore di difetti cardiaci 1.
Infine, più di recente, risultati straordinari nella diagnosi e nella terapia interventistica e chirurgica delle
cardiopatie congenite ottenuti nell’ultimo decennio del secolo scorso hanno ridotto drasticamente, nei paesi sviluppati, la mortalità neonatale e infantile di queste malformazioni.
La cardiochirurgia pediatrica si presenta oggi come un campo nel quale i grandi progressi del passato
sono stati oramai confermati e consolidati, lasciando intravedere, almeno per il prossimo futuro, margini di
miglioramento sostenuti soprattutto dall’avanzamento delle tecniche di sostegno circolatorio extracorporeo
e dalla riduzione delle sequele perioperatorie. I contributi più nuovi nel campo delle cardiopatie congenite
stanno arrivando (e sono da attendersi sempre di più nel prossimo futuro) da due settori: il primo precede la
nascita e include la genetica, la morfogenesi e la vita prenatale; il secondo segue il trattamento chirurgico e
riguarda quindi il follow-up in età adolescenziale e adulta.
Con l’approssimarsi del nuovo millennio, la crescente specializzazione delle metodiche diagnostiche
e terapeutiche ha portato il cardiologo pediatra a differenziarsi in maniera sempre maggiore rispetto alle
discipline di origine, indipendentemente dalla sua formazione in pediatria o cardiologia, e il suo ambito di
azione è diventato sostanzialmente ben delimitato e delineato.
Tuttavia, è proprio nei recenti anni che il mondo apparentemente stabile della cardiologia pediatrica è
stato di nuovo rimesso in discussione da fattori che, se pur ampiamente previsti in passato, iniziano ora a
pesare in maniera più forte. Il primo è la riduzione alla nascita del numero delle cardiopatie congenite, in
particolare di quelle complesse, dovuta all’ampia diffusione dell’ecocardiografia fetale con la conseguente
e frequente scelta abortiva 2. Questo fenomeno è in parte bilanciato però, in alcune nazioni sviluppate,
dall’afflusso di bambini con cardiopatia congenita provenienti da Paesi con alti tassi di mortalità e con
scarse disponibilità di tecnologie mediche. Esportare, quindi, nei Paesi del terzo mondo le conoscenze, le
professionalità e le strumentazioni necessarie alla diagnosi e alla cura delle cardiopatie in età pediatrica è
una grande sfida per il presente e per il futuro della nostra specialità.
Contemporaneamente, nei Paesi altamente sviluppati vi è stato un crescente aumento del numero dei
pazienti adulti portatori di cardiopatia congenita, conseguenza diretta del continuo progresso che induce
la lunga sopravvivenza di pazienti pediatrici con cardiopatie anche gravi. Recenti dati epidemiologici dimostrano, infatti, che il numero dei pazienti adulti con cardiopatia congenita ha superato di molto quello
dei pazienti in età pediatrica e rappresenta attualmente circa i due terzi della popolazione di pazienti con
cardiopatia congenita 3; i sistemi sanitari si presentano ancora impreparati a ciò 3. Il cardiologo dell’adulto,
infatti, non ha la preparazione culturale per affrontare una cardiopatia congenita e, d’altra parte, il pediatra
cardiologo non ha la competenza e gli strumenti tecnici per affrontare le patologie correlate all’invecchiamento (aterosclerosi, malattie degenerative ecc.), con conseguenze importanti sia in termini economici che
di aumento di rischio per i pazienti 4. L’adulto portatore di cardiopatia congenita (il cosiddetto GUCH, da
grown up congenital heart patient) si presenta quindi come molto complesso e ben poche strutture sono
preparate e disposte a farsi carico di pazienti con criticità multiple. Le degenze, in questi casi, sono spesso
prolungate, con particolari difficoltà quando nel reparto “pediatrico” si debbano assistere anche pazienti di
età relativamente avanzata.
X
MARINO cardiologia pediatrica.indd 10
17/09/15 07:54
Prefazione
L’identità del cardiologo pediatra, consolidata alla fine del secolo scorso torna, dunque, a vacillare davanti a nuove sfide che, oltre alle problematiche di stretta pertinenza clinica, sono rappresentate da temi in
passato scarsamente considerati, quali l’abilità al lavoro, l’idoneità assicurativa, la gravidanza, lo sport. Non
si tratta più soltanto di conoscere l’anatomia, la clinica e la terapia delle cardiopatie congenite, ma viene
richiesto di rispondere alle esigenze di una popolazione adulta in continua espansione e con necessità e
problemi diversi da quelli per i quali la cardiologia pediatrica tradizionale, nella sua la storia ultradecennale, si era preparata.
La formazione specialistica del cardiologo pediatra, ritenuta ottimale negli anni Novanta, risulta ora
insufficiente davanti alle nuove sfide, soprattutto in Italia dove per lungo tempo si è rimasti fermi alla
didattica istituita dalla tabella XVIII del DPR 27/7/1987 e all’istituzione dei Master di II livello. Nel resto
del mondo, Gran Bretagna e Canada storicamente per prime, le strutture sanitarie nazionali e le società
scientifiche già si erano strutturate ad affrontare questa realtà emergente e hanno contribuito alla realizzazione di centri appositamente dedicati all’assistenza del cardiopatico congenito adulto con specifici
indirizzi educativi per il personale medico e infermieristico. L’organizzazione e l’attività di questi centri
specializzati per gli adulti con cardiopatie congenite si sono dimostrate efficaci per ridurre la mortalità e
la morbilità in questo crescente gruppo di pazienti 3, 4. La necessità di dover seguire pazienti sottoposti
a palliazioni permanenti o con sequele dopo correzioni chirurgiche, a torto ritenute “complete”, impone
ora al cardiologo pediatra non solo l’acquisizione di ulteriori nuove competenze tecniche, ma anche di
uscire dall’isolamento culturale e di interagire in maniera sempre maggiore con la cardiologia e la medicina interna dell’adulto.
L’altro problema emergente è quello dei costi. Il paziente cardiopatico congenito necessita di assistenza
dedicata per una durata che spesso è quella di tutta la sua vita, con ripetute procedure diagnostiche e terapeutiche di elevato costo. Anche sul piano della terapia medica, i nuovi farmaci, come ad esempio quelli
per il trattamento dell’ipertensione polmonare, presentano aspetti di rapporto costo/beneficio molto complessi che, nell’attuale scenario economico mondiale, non possono essere trascurati.
Appare quindi evidente che è necessario evitare quanto più possibile la “deregulation” dell’assistenza al
cardiopatico congenito, con relativi rischi oggettivi per il paziente e rischi economici per la sanità, concentrando esperienza, competenza e risorse in centri appositamente dedicati 5, 6. Allo stato attuale, almeno a
livello nazionale italiano, tale azione appare ancora lontana, al contrario di quanto ha fatto la Svezia già dai
primi anni Novanta, riducendo la mortalità operatoria dal 9,5 all’1,9% in breve tempo 7.
L’affacciarsi di ambiti tecnici sempre più nuovi mette dunque alla prova il concetto “tradizionale” che abbiamo del cardiologo pediatra, coinvolto sempre di più in ambiti un tempo preclusi, come la sala operatoria
a fianco del cardiochirurgo per le procedure cosiddette “ibride”, o la sala di risonanza magnetica nucleare,
insieme al radiologo, per la valutazione anatomica e funzionale di cardiopatie complesse.
Stiamo entrando in un’era di progressi tecnologici sempre più entusiasmanti e il ruolo del cardiologo
(“pediatra”?) dovrà adattarsi alle nuove sfide. La terapia genica, l’interventistica prenatale, la realizzazione
di grandi banche dati e studi epidemiologici su larga scala, la prevenzione primaria delle patologie cardiovascolari dell’età adulta, sono solo alcuni degli ambiti sui quali sarà necessario confrontarsi e la cui strada è
stata già indicata dalle grandi società scientifiche 8, 9. Dobbiamo sempre ricordare, però, che in tutti i campi
della medicina, e ancora di più in cardiologia pediatrica, la tecnologia non potrà sostituire la cultura, la
robotica non potrà sostituire l’accuratezza e la computeristica non potrà sostituire l’umanità del medico e il
suo amore per il malato.
Possiamo forse prevedere che, accanto al cardiologo pediatra classico si affermerà la figura di un cardiologo che, avendo perso il connotato (o almeno l’esclusività) di “pediatra”, ha acquisito la caratteristica
di “cardiologo del congenito”. Questa operazione, lessicalmente facile per gli anglosassoni (the congenital
cardiologist) risulta decisamente più faticosa per noi italiani, motivo per cui gli autori di questo manuale
hanno deciso di intitolarlo e dedicarlo, ancora, alla cardiologia pediatrica.
In questo nostro libro abbiamo voluto coinvolgere i migliori specialisti italiani nel settore per fornire un
panorama, crediamo piuttosto completo e approfondito, che riguarda tutti quei problemi di gestione pratica del cardiopatico congenito in età pediatrica e adulta, che raramente vengono affrontati nei classici libri
dedicati all’argomento. Non vi è una trattazione sistematica degli aspetti diagnostici e chirurgici di tutte le
cardiopatie congenite, per la quale rimandiamo agli ottimi testi che sono già a disposizione. Troverete inve-
XI
MARINO cardiologia pediatrica.indd 11
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
ce una trattazione aggiornata di vari argomenti di carattere pratico che possono interessare ed essere utili
certo ai cardiologi pediatri, ma anche agli specialisti pediatri, neonatologi, cardiologi, anestesisti e cardiochirurghi che sono in qualsivoglia modo coinvolti nella diagnosi e nella cura dei bambini con cardiopatia
(congenita o acquisita) e degli adolescenti e adulti con cardiopatia congenita.
Se questi argomenti e la loro trattazione otterranno la vostra attenzione, il nostro lavoro avrà avuto un
significato.
Gli autori
BIBLIOGRAFIA
1. Marino B. Cardiopatia congenita. Dai pionieri alle nuove frontiere. In: Burgio GR, Notarangelo LD (editors).
Malattie maestre. Milano: UTET; 2002. p. 159-67.
2. Egbe A, Uppu S, Lee S et al. Changing prevalence of severe congenital heart disease: a population-based study.
Pediatr Cardiol 2014;35:1232-8.
3. Marelli AJ, Ionescu-Ittu R, Mackie AS et al. lifetime prevalence of congenital heart disease in the general population from 2000 to 2010. Circulation 2014;130:749-56.
4. Mylotte D, Pilote L, Ionescu-Ittu R et al. Specialized adult congenital heart disease care: the impact of policy on
mortality. Circulation 2014;129:1804-12.
5. Kim YY, Gauvreau K, Bacha EA et al. Resource use among adult congenital heart surgery admissions in pediatric hospitals: risk factors for high resource utilization and association with inpatient death. Circ Cardiovasc
Qual Outcomes 2011;4:634-9.
6. Pasquali SK, Gaies MG, Jacobs JP et al. Center variation in cost and outcomes for congenital heart surgery. Cardiol Young 2012;22:1010-7.
7. Lundström NR, Berggren H, Björkhem G et al. Centralization of pediatric heart surgery in Sweden. Pediatr
Cardiol 2000;21:353-7.
8. Lenfant C. Report of the Task Force on Research in Pediatric Cardiovascular Disease. Circulation 2002;106:
1037-42.
9. Kaltman JR, Schramm C, Pearson CD. The National Heart, Lung, and Blood Institute bench to bassinet Program: a new paradigm for translational research. J Am Coll Cardiol 2010;55:1262-5.
XII
MARINO cardiologia pediatrica.indd 12
17/09/15 07:54
AUTORI
Anwar Baban
Bruno Dallapiccola
Marco Bonvicini
Fabio Dardi
Genetica Medica e Cardiologia Pediatrica, Ospedale
Pediatrico Bambino Gesù, Roma
Cardiologia Pediatrica e dell’Età Evolutiva, Policlinico
“S.Orsola-Malpighi”, Università degli Studi di
Bologna, Bologna
Giovanna Bosco
Dipartimento di Pediatria, Policlinico Umberto I,
“Sapienza” Università di Roma, Roma
Lorenzo D. Botto
Genetica Medica, Ospedale Pediatrico Bambino
Gesù, Roma
AOU di Bologna, Dipartimento di Medicina
Specialistica, Diagnostica e Sperimentale, Università
degli Studi di Bologna, Bologna
Giangiacomo Di Nardo
UOSD di Cardiopatie Congenite dell’Adulto (G.U.C.H.
Unit), A.O.R.N “Ospedali dei Colli”, II Università di
Napoli, Napoli
Divisione di Genetica Medica, Dipartimento di
Pediatria, Università dello Utah, Salt Lake City, UT,
USA
Giovanni Di Salvo
Gianfranco Butera
Maria Cristina Digilio
Cardiopatie Congenite del bambino e dell’adulto,
IRCCS Policlinico San Donato, San Donato Milanese
(MI)
Raffaele Calabrò
UOC di Cardiologia, A.O.R.N. “Ospedale dei Colli”,
II Università di Napoli, Napoli
Alessandro Capestro
Cardiologia e Cardiochirurgia Pediatrica e
Congenita, Dipartimento di Malattie Cardiovascolari,
AOU Ospedali Riuniti, Ancona
UOSD di Cardiologia Pediatrica, A.O.R.N. “Ospedale
dei Colli”, II Università di Napoli, Napoli
Genetica Medica, Ospedale Pediatrico Bambino
Gesù, Roma
Andrea Donti
Cardiologia Pediatrica e dell’Età Evolutiva, Policlinico
“S.Orsola-Malpighi”, Bologna
Fabrizio Drago
Aritmologia, Dipartimento Medico Chirurgico di
Cardiologia Pediatrica, Ospedale Bambino Gesù,
Palidoro, Roma
Rossella Capolino
Ilaria Ernesti
Genetica Medica, Ospedale Pediatrico Bambino
Gesù, Roma
Dipartimento di Pediatria, Policlinico Umberto I,
“Sapienza” Università di Roma, Roma
Mario Carminati
Silvia Favilli
Cardiopatie Congenite del bambino e dell’adulto,
IRCCS Policlinico San Donato, San Donato Milanese
(MI)
Adriano Carotti
Cardiochirurgia Pediatrica, Ospedale Pediatrico
Bambino Gesù, Roma
Biagio Castaldi
UOSD di Cardiologia Pediatrica, A.O.R.N. “Ospedale
dei Colli”, II Università di Napoli, Napoli
Enrico Chiappa
Cardiologia Pediatrica, AOU A. Meyer, Firenze
Roberto Formigari
Cardiologia Pediatrica e dell’Età Evolutiva, Policlinico
“S.Orsola-Malpighi”, Bologna
Eliana Franchi
Cardiologia e Cardiochirurgia Pediatrica e
Congenita. Dipartimento di Malattie Cardiovascolari,
AOU Ospedali Riuniti, Ancona
Nazzareno Galiè
Cardiologia Pediatrica, AOU A. Meyer, Firenze
Dipartimento di Medicina Specialistica, Diagnostica e
Sperimentale, Università degli Studi di Bologna, Bologna
Chiara Colantoni
Giuseppe Limongelli
Dipartimento di Pediatria, “Sapienza” Università di
Roma, Policlinico Umberto I, Roma
Pier Luigi Colonna
Cardiologia e Cardiochirurgia Pediatrica e
Congenita, Dipartimento di Malattie Cardiovascolari,
AOU Ospedali Riuniti, Ancona
U.O.S.D. Cardiologia Riabilitativa, Intensiva e
Scompenso Cardiaco, A.O.R.N. “Ospedali dei Colli”,
II Università di Napoli, Napoli
Maristella Lombardi
UOC Cardiologia Pediatrica, Azienda Policlinico,
Ospedale Pediatrico Giovanni XXIII, Bari
XIII
MARINO cardiologia pediatrica.indd 13
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
Alessandra Manes
Maria Giovanna Russo
Sergio Manieri
Sabrina Salvadori
Dipartimento di Medicina Specialistica, Diagnostica e
Sperimentale, Università degli Studi di Bologna, Bologna
UOC Pediatria, AO San Carlo, Potenza
Maurizio Marasini
UO di Cardiologia, Dipartimento di Cardiologia e
Chirurgia Cardiovascolare, Istituto Giannina Gaslini
IRCS, Genova
MARCO VALERIO MARIANI
Dipartimento di Pediatria “Sapienza” Università di
Roma, Policlinico Umberto I, Roma
Bruno Marino
Dipartimento di Pediatria “Sapienza” Università di
Roma, Policlinico Umberto I, Roma
Elisabetta M. Mariucci
Cardiologia Pediatrica e dell’Età Evolutiva, Policlinico
“S.Orsola-Malpighi”, Bologna
Francesco Martino
Dipartimento di Pediatria, “Sapienza” Università di
Roma, Policlinico Umberto I, Roma
Eliana Martino
Dipartimento di Pediatria, “Sapienza” Università di
Roma, Policlinico Umberto I, Roma
Vanessa Martucci
Dipartimento di Pediatria, Policlinico Umberto I,
“Sapienza” Università di Roma, Roma
Daniele Masarone
UOSD di Cardiologia Pediatrica, A.O.R.N. “Ospedale
dei Colli”, II Università di Napoli, Napoli
UOC Patologia Neonatale, Dipartimento di Salute
della Donna e del Bambino, Università degli Studi di
Padova, Padova
Tiberio Santoro
Dipartimento di Cardiologia, Casa Sollievo della
Sofferenza, San Giovanni Rotondo (FG)
Giuseppe Santoro
UOSD di Cardiologia Pediatrica, A.O.R.N. “Ospedale
dei Colli”, II Università di Napoli, Napoli
Berardo Sarubbi
UOSD di Cardiopatie Congenite dell’Adulto (G.U.C.H.
Unit), A.O.R.N “Ospedali dei Colli”, II Università di
Napoli, Napoli
Giancarlo Scognamiglio
UOSD di Cardiopatie Congenite dell’Adulto (G.U.C.H.
Unit), A.O.R.N “Ospedali dei Colli”, II Università di
Napoli, Napoli
Lucia Martina Silvestri
Dipartimento di Pediatria, Policlinico Umberto I,
“Sapienza” Università di Roma, Roma
Giorgio Svaluto Moreolo
U.O.S.D. Cardiologia Riabilitativa, Intensiva e
Scompenso Cardiaco, A.O.R.N. “Ospedali dei Colli”,
II Università di Napoli, Napoli
UOC Pediatria, Ospedale Santa Maria della
Misericordia, Rovigo
Pierpaolo Mastroiacovo
Dipartimento di Pediatria, Policlinico Umberto I,
“Sapienza” Università di Roma, Roma
ICBD - Alessandra Lisi International Centre on Birth
Defects and Prematurity, Roma
Giancarlo Tancredi
Ornella Milanesi
Giulia Tuo
UOC Cardiologia Pediatrica, Dipartimento di Salute
della Donna e del Bambino, Università degli Studi di
Padova, Padova
UO di Cardiologia, Dipartimento di Cardiologia e
Chirurgia Cardiovascolare, Istituto Giannina Gaslini
IRCS, Genova
Giuseppe Pacileo
Ugo Vairo
U.O.S.D. Cardiologia Riabilitativa, Intensiva e
Scompenso Cardiaco, A.O.R.N. “Ospedali dei Colli”,
II Università di Napoli, Napoli
UOC Cardiologia Pediatrica, Azienda Policlinico,
Ospedale Pediatrico, Giovanni XXIII, Bari
Massimiliano Palazzini
Flavia Ventriglia
Dipartimento di Medicina Specialistica, Diagnostica e
Sperimentale, Università degli Studi di Bologna, Bologna
Dipartimento di Pediatria, Policlinico Umberto I,
“Sapienza” Università di Roma, Roma
Giovanna Passarella
Paolo Versacci
UOC Pediatria, Ospedale Santa Maria della
Misericordia, Rovigo
Iva Pollini
Cardiologia Pediatrica, AOU A. Meyer, Firenze
Luca Ragni
Cardiologia Pediatrica e dell’Età Evolutiva, Policlinico
“S.Orsola-Malpighi”, Bologna
Alessandra Rea
U.O.S.D. Cardiologia Riabilitativa, Intensiva e
Scompenso Cardiaco, A.O.R.N. “Ospedali dei Colli”,
II Università di Napoli, Napoli
Dipartimento di Pediatria, Policlinico Umberto I,
“Sapienza” Università di Roma, Roma
Cristina Zanoni
Dipartimento di Pediatria, “Sapienza” Università
di Roma, Policlinico Umberto I, Roma
Francesco Zulian
Centro Regionale Specializzato di Reumatologia
Pediatrica, Dipartimento A.I.S. per la Salute della
Donna e del Bambino, Università degli Studi di
Padova, Padova
XIV
MARINO cardiologia pediatrica.indd 14
17/09/15 07:54
INDICE
Introduzione ................................................................................................................................................. V
Presentazione ........................................................................................................................................... VII
Prefazione ...................................................................................................................................................... IX
Autori ............................................................................................................................................................. XIII
1 CARDIOLOGIA FETALE ................................................................................................................. 1
G. Tuo, M. Marasini
2 NEONATO CON SOSPETTA CARDIOPATIA CONGENITA ............................... 27
S. Salvadori, G. Passarella, O. Milanesi
3 TRATTAMENTO INTERVENTISTICO
DELLE CARDIOPATIE CONGENITE ..................................................................................... 59
G. Butera, T. Santoro, M. Carminati
4 TRATTAMENTO CHIRURGICO DELLE CARDIOPATIE CONGENITE ............ 74
A. Carotti
5FOLLOW-UP E TRANSIZIONE ALL’ETÀ ADULTA
DEL CARDIOPATICO CONGENITO ................................................................................... 88
E.M. Mariucci, R. Formigari, M. Bonvicini
6 CARDIOPATIE CONGENITE NELL’ETÀ ADULTA:
PROBLEMATICHE CLINICHE ................................................................................................. 106
B. Sarubbi, G. Scognamiglio, G. Di Nardo
7 Cardiopatie congenite a esordio tardivo
e nell’età adulta .................................................................................................................... 119
A. Donti, E.M. Mariucci, R. Formigari
8 Consulenza genetica .................................................................................................... 134
M.C. Digilio, L.M. Silvestri, A. Baban, R. Capolino, M.V. Mariani, B. Marino, B. Dallapiccola
XV
MARINO cardiologia pediatrica.indd 15
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
9 Prevenzione primaria delle cardiopatie congenite .................. 146
P. Mastroiacovo, L.D. Botto
10 Sincope e morte improvvisa .................................................................................... 163
G. Svaluto Moreolo
11 Aritmie in età pediatrica .............................................................................................. 186
F. Drago
12 Dolore toracico .................................................................................................................. 215
P. Versacci, G. Tancredi, V. Martucci, I. Ernesti, L.M. Silvestri, F. Ventriglia, G. Bosco
13 Ipertensione polmonare in età pediatrica ........................................... 227
N. Galiè, F. Dardi, M. Palazzini, A. Manes
14 Obesità in età pediatrica ............................................................................................ 240
B. Castaldi, G. Di Salvo, G. Santoro, M.G. Russo
15 Aterosclerosi in età pediatrica ......................................................................... 254
F. Martino, C. Colantoni, E. Martino, C. Zanoni
16 Ipertensione arteriosa in età pediatrica ............................................... 275
I. Pollini, S. Favilli, E. Chiappa
17 Scompenso cardiaco in età pediatrica ..................................................... 287
D. Masarone, G. Limongelli, A. Rea, G. Pacileo, R. Calabrò
18 Gestione del soffio cardiaco .............................................................................. 305
M. Lombardi, S. Manieri, U. Vairo
19 Cuore e malattie reumatiche
del bambino e dell’adolescente ......................................................................... 317
F. Zulian
20 Farmaci cardiovascolari in età pediatrica ......................................... 327
R. Formigari, L. Ragni
21 Idoneità sportiva agonistica e non agonistica
in pediatria ................................................................................................................................. 343
P. Colonna, A. Capestro, E. Franchi
XVI
MARINO cardiologia pediatrica.indd 16
17/09/15 07:54
Cardiologia fetale
G. Tuo, M. Marasini
Introduzione
Le cardiopatie congenite (CC) rappresentano il
tipo più comune di malformazione congenita alla
nascita, con forme di gravità moderata e severa che
interessano circa lo 0,6% dei nati vivi 1. Benché rimangano una causa importante di morte nell’infanzia, non tutte le forme di CC maggiore si manifestano nel primo periodo neonatale tanto che, secondo
alcuni autori, approssimativamente il 25% dei bambini con forme severe di CC viene ancora oggi dimesso dalla neonatologia senza alcuna diagnosi 2, 3.
Nel Baltimore-Washington Infant Study, tra il 1981
e il 1989, è stato diagnosticato il 60% dei casi di
CC entro le quattro settimane di vita, l’80% entro
le dodici settimane e il 90% entro i due anni di
età 4. Nei nati morti, i difetti cardiaci sono osservati con una frequenza da quattro a cinque volte
più elevata 5. La vera incidenza delle CC in epoca
prenatale è, invece, ancora oggi difficile da valutare. Infatti, nonostante i progressi nella tecnologia
ultrasonografica e la diffusione della valutazione
con ultrasuoni del feto, le anomalie cardiache restano ancora le malformazioni più frequentemente
misconosciute in epoca prenatale e ciò può avere profonde conseguenze mediche, psicologiche,
socio-economiche e medico-legali. La probabilità di diagnosticare un difetto cardiaco in utero è
strettamente correlata all’esperienza dell’operatore,
all’epoca gestazionale in cui si esegue l’esame e
all’apparecchiatura che si utilizza, che necessariamente deve essere ad alta risoluzione 6, 7. Inoltre,
poiché alcune anomalie cardiache possono evolvere nelle loro caratteristiche ecografiche con la
progressione della gravidanza, un certo gruppo di
difetti non può essere escluso con certezza tramite un singolo esame, in particolare se questo è effettuato in un’epoca gestazionale molto precoce.
1
Negli ultimi anni si è focalizzata l’attenzione sullo
sviluppo di programmi di screening ecografici di
massa per la diagnosi prenatale delle CC indipendentemente dalla presenza nel singolo soggetto di
fattori di rischio più o meno specifici. È stato valutato, poi, l’impatto di questi programmi di screening sulla mortalità e sulla morbilità dei bambini
con diagnosi prenatale di CC rispetto alla popolazione generale, con risultati contrastanti 8, 9. Inoltre, la possibilità di intervenire in utero, che è ormai
codificata per quanto concerne il trattamento antiaritmico, ma ancora in fase sperimentale per altri
tipi di interventi, potrebbe ulteriormente migliorare
l’outcome 10, 11. Infine, non si può non prendere in
considerazione l’impatto che una diagnosi prenatale di cardiopatia ha sulla coppia e l’onere che ne
deriva per il cardiologo che deve fornire tutte le informazioni relative al tipo di anomalia, alle opzioni terapeutiche e alle possibilità di sopravvivenza.
Nelle pagine successive cercheremo di affrontare
nel dettaglio tutti questi aspetti della cardiologia
fetale rimandando quasi totalmente a testi specifici tutto ciò che riguarda le differenze rispetto al
postnatale, per quanto concerne gli aspetti tecnico,
teorico e pratico dell’esecuzione dell’esame sia in
un cuore normale che, e soprattutto, in un cuore
malformato.
Indicazioni all’Ecocardiografia
Fetale nella Popolazione
ad Alto Rischio di CC
Le indicazioni all’ecocardiografia fetale risultano strettamente collegate al riconoscimento e
alla quantificazione dei possibili fattori di rischio
e, conseguentemente, all’esatta conoscenza delle
cause della cardiopatia congenita. A oggi sono stati
individuati tre principali gruppi causali, sulla base
1
MARINO cardiologia pediatrica.indd 1
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
di dati empirici, genetici e di esperimenti su animali 4, 12-14. Questi sono: fattori genetici primari (aberranze cro­mosomiche, 5%; sindromi monogeniche,
3%), fattori esogeni primari (infezioni materne, 1%;
teratogeni/malattie metaboliche materne 1%) ed
eredità multifattoriale (90%). In quest’ultimo caso si
ritiene che l’anomalia cardiaca derivi dall’interazione tra una genetica aspecifica e fattori ambientali.
Questo presuppone una predisposizione genetica
e l’influenza di fattori scatenanti ambientali agenti
a un certo punto dell’embriogenesi; il periodo più
sensibile è intorno alla sesta settimana di gestazione 13. Con i progressi della genetica molecolare, il
concetto di eziologia multifattoriale è posto sempre più in discussione, in quanto numerosi difetti
cardiaci sono stati identificati come il risultato di
mutazioni monogeniche ereditate o ex-novo, e di
micro-delezioni ereditate o spontanee 14-16. In ogni
modo, a oggi l’eziologia della maggioranza delle
anomalie cardiache è ancora oggetto di ricerca,
e di conseguenza nella pratica clinica l’identificazione di popolazioni ad alto rischio è basata sulla
storia clinica del paziente e sulle anomalie o i marcatori riscontrati all’esame ecografico. A questa minoranza di feti affetti da cardiopatia con fattori di
rischio noti (circa il 10%) dovrebbe essere offerto
un esame ecocardiografico dettagliato che includa
anche una valutazione nel primo trimestre di gravidanza. In particolare si distinguono indicazioni
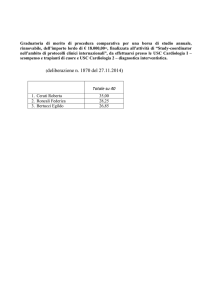
materno-familiari e indicazioni fetali all’ecocardiografia prenatale (Tab. 1-I) 17.
Indicazioni materno-familiari
Per malattie materne s’intendono in particolare
i difetti metabolici, le malattie autoimmuni, le infezioni o l’esposizione materna a sostanze teratogene.
Nell’ambito delle patologie metaboliche il diabete mel­lito insulino-dipendente, specie se non
compensato, comporta un aumento di circa cinque
volte del rischio di CC rispetto alla popolazione
generale 18. La fenilchetonuria è associata a un rischio 10-15 volte maggiore di CC, in particolare di
tetralogia di Fallot (TF) e di difetto interventricolare
(DIV), a seguito dell’esposizione del feto durante
l’organogenesi a valori elevati di fenilalanina materna (eccedenti 15 mg/dl) 19.
Le malattie autoimmuni quali il lupus eritematosus, la sindrome LLAC (Lupus Like Anticoagulant), la malattia di Sjogren o la semplice presenza
di anticorpi materni anti-RO e/o anti-LA, anche
in assenza di malattia, comportano un rischio di
blocco atrio-ventricolare completo fetale che va-
Tabella 1-I Indicazioni all’ecocardiografia fetale 17.
Indicazioni materne
•Malattie metaboliche materne, in particolare se scarsamente controllato in fase precoce di gravidanza:
–– diabete melito
–– fenilchetonuria
•Esposizione materna a teratogeni cardiaci:
–– anticonvulsivanti, acido retinoico, litio
–– infezioni virali e altre infezioni. Come rosolia, citomegalovirus, coxsackie, parvo e toxoplasma
•Malattie materne del collagene con anticorpi Ro/
SSA e/o La/SSB
•Malattie cardiache congenite materne e cardiomiopatia familiare
•Terapia materna con farmaci antinfiammatori non
steroidei dopo la venticinquesima fino alla trentesima settimana di gestazione
Indicazioni familiari
•Malattia cardiaca congenita paterna
•Precedente bimbo o feto con malattia congenita cardiaca o blocco cardiaco congenito
•Anomalie cromosomiche, disordini genetici o sindromi con malattie cardiache congenite o cardiomiopatie
Indicazioni fetali
•Sospetto di malformazione cardiaca durante l’esame
ostetrico
•Idrope fetale
•Idrotorace
•Polidramnios
•Malformazioni extracardiache, in particolare:
–– omfalocele
–– ernia diaframmatica
–– atresia duodenale
–– fistola tracheoesofagea
–– igroma cistico
•Anomalie cromosomiche
•Translucenza nucale superiore al 99°percentile per
lunghezza vertice-sacro
•Aritmie:
–– bradicardia sostenuta a meno di 100 battiti per
minuto
–– tachicardia, sia intermittente che sostenuta, se con
frequenza maggiore di 180-200 battiti per minuto
–– frequenti battiti ectopici
•Altre condizioni con rischio noto di scompenso cardiaco fetale:
–– tumori fortemente vascolarizzati
–– fistole artero-venose
–– assenza del dotto venoso
–– acardiac twin
–– sindrome da trasfusione feto-fetale
–– anemia
•Gravidanza gemellare monocoriale (per l’aumentato
rischio di malformazioni cardiache strutturali fetali)
ria dall’1 al 6%. Le pazienti affette producono
autoanticorpi antinucleari di tipo IgG (anti-RO e
anti-LA) che, oltrepassando la placenta a 16-18
settimane, possono interferire con il sistema di
conduzione cardiaca determinando una reazione
2
MARINO cardiologia pediatrica.indd 2
17/09/15 07:54
1 – Cardiologia fetale
infiammatoria che può portare a fibrosi del nodo
atrio-ventricolare 20.
Le infezioni materne, per lo più di natura virale, comportano un rischio di anomalie cardiache
che varia in funzione dell’epoca gestazionale di
esposizione: la rosolia contratta in gravidanza,
specie se prima della dodicesima settimana, comporta un rischio malformativo sino al 50%, ma
anche le infezioni da Coxsackie, da Parvovirus e
da Citomegalovirus sono associate a una maggiore incidenza di CC e di miocarditi fetali 22, 23. Infine, sebbene non si possa escludere la possibilità
che molte sostanze o farmaci possano interferire con la cardiogenesi, solo in pochi casi si sono
evidenziate associazioni significative. L’uso nel
primo trimestre di anticonvulsivanti, alcol, litio
e di derivati della vitamina A sembra comunque
aumentare il rischio di CC 24-28.
Per malattie familiari o ereditarie s’intende la
presenza su base familiare di sindromi a trasmissione mendeliana monogenica o secondarie a microdelezione, caratterizzate dalla presenza di difetti
cardiaci congeniti associati. L’ecocardiografia fetale
assume particolare importanza nei casi in cui tali
condizioni non siano identificabili o escludibili in
epoca prenatale con metodologie più precoci e
sensibili (tecniche di biologia molecolare; FISH) o
siano piuttosto caratterizzate da notevole variabilità
di espressione (es. fenotipo CATCH) 29-37.
In caso di familiarità per CC, il rischio di ricorrenza varia in funzione del tipo di lesione cardiaca
e del grado di parentela: in caso di un solo figlio
precedente affetto il rischio è dell’1-4% e aumenta fino a 3-4 volte (10%) in caso di due figli con
malformazione cardiaca. Quando invece è affetto
uno dei due genitori il rischio di ricorrenza, una
volta escluse malattie ereditarie su base mendeliana, è pari al 5-10%. Sebbene vi siano opinioni discordanti, tale rischio sembra essere maggiore se a
essere affetta è la madre o per alcune classi specifiche (patologie ostruttive del cuore sinistro, difetti di
settazione e anomalie del situs) 13, 38-40. Il rischio di
ricorrenza riguarda, di solito, lo stesso tipo o la stessa classe di alterazioni cardiache, per cui è molto
importante, in sede di ecocardiografia, conoscere il
difetto precedentemente diagnosticato.
Indicazioni fetali
Le alterazioni cromosomiche rappresentano
un’in­dicazione all’esame ecocardiografico in modo
da permettere un counselling adeguato. Ciò è particolarmente vero a seguito della diagnosi prenatale
di alterazioni cromosomiche maggiori (monosomie,
trisomie) associate a quadri sindromici severi per i
quali la coppia abbia deciso di proseguire la gravidanza per scelta o per diagnosi tardiva; a seguito della diagnosi prenatale di alterazioni cromosomiche associate a quadri fenotipici variabili o non
definiti in maniera assoluta, come nel caso della
sindrome di Turner o di alterazioni cromosomiche
di raro riscontro; a seguito della diagnosi prenatale
di alterazioni cromosomiche non associabili a precise alterazioni del fenotipo come nel caso di mosaicismi di II e III livello, di cromosomi marcatori,
o di alterazioni strutturali de novo apparentemente
bilanciate 41, 42.
Il sospetto di anomalia strutturale cardiaca nel
corso di ecografia di routine in pazienti a basso rischio deve rappresentare la principale indicazione
all’effettuazione di un’ecocardiografia fetale. I risultati degli studi ottenuti utilizzando come metodica di screening la sezione 4 camere evidenziano una sensibilità molto variabile (dal 4,5 al 63%)
in funzione dei diversi protocolli di studio adottati
e dell’esperienza dei diversi operatori coinvolti.
Alcune cardiopatie sono facilmente individuabili
mediante immagine a 4 camere (atresia mitralica
o tricuspidalica, canale atrio-ventricolare ecc.),
altre lo sono solo in caso di ampi difetti interventricolari. Altre ancora possono diventare evidenti
solo tardivamente nel terzo trimestre (fibroelastosi endocardica, stenosi aortica e polmonare,
coartazione aortica ecc.). Tutte le malformazioni
tronco-conali invece, che rappresentano circa il
30-40% delle cardiopatie congenite a emergenza
neonatale, non sono teoricamente evidenziabili
impiegando solo questa scansione. Per i motivi
sopra esposti, la detection-rate teorica mediante
immagine a 4 camere dovrebbe essere intorno
al 50-60% delle cardiopatie maggiori e questo
conferma come l’enorme discrepanza riscontrata
nei lavori pubblicati possa dipendere interamente
dall’esperienza e dal training del centro e del singolo operatore. Inserendo nel programma di screening la scansione degli “assi lunghi” per la valutazione degli efflussi ventricolari, possono essere
identificate anche le anomalie tronco-conali e la
detection-rate teorica dovrebbe salire a circa l’8090% delle cardiopatie maggiori e in particolare
dovrebbe permettere il riconoscimento in utero
di quelle cardiopatie che maggiormente possono
beneficiare della diagnosi prenatale (traposizione
delle grandi arterie e cardiopatie dotto arterioso
dipendenti) 43-50.
3
MARINO cardiologia pediatrica.indd 3
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
Il riscontro di aritmia fetale all’esame di screening è un’ovvia indicazione all’esame ecocardiografico che permette di valutare il tipo e il rilievo
emodinamico dell’aritmia e di escludere eventuali
anomalie strutturali cardiache associate (presenti
nel 30-50% dei casi di blocco atrio-ventricolare e
nel 5% dei casi di tachiaritmia) 50.
Il test della “traslucenza nucale” (NT) positivo
rappresenta un’altra indicazione all’ecocardiografia fetale, in quanto la prevalenza di difetti cardiaci maggiori aumenta in maniera esponenziale con
l’aumentare dello spessore della NT, in feti cromosomicamente normali. Hyett et al. in un lavoro retrospettivo hanno rilevato che il 56% dei feti affetti
da CC maggiore si collocava nella popolazione con
spessore NT superiore al 95°percentile, evidenziando un aumento di circa 8-10 volte del rischio per
anomalie cardiache maggiori (1,5% versus 1,7‰) 51.
Aumentando il valore di cut-off dal 95° al 99°percentile si riduceva il numero di pazienti da esaminare ma la sensibilità diminuiva dal 56 al 40%.
La frequenza di associazione tra CC e anomalie extracardiache dopo la nascita varia dal 25 al
45%, per cui il riscontro di anomalia extracardiaca in epoca fetale deve prevedere una valutazione specialistica del cuore fetale. È importante
ricordare come i diversi tipi di cardiopatie possano essere associate in percentuale variabile a
malformazioni di altri organi: ad esempio il canale
atrio-ventricolare è associato ad altre anomalie in
circa il 50% dei casi, mentre il difetto interventricolare, il difetto interatriale, la tetralogia di Fallot
e la malposizione cardiaca lo sono in circa il 30%
dei casi 52, 53. L’idrope fetale non immunitario può
essere l’epi-fenomeno sia di anomalie del ritmo
cardiaco che di anomalie strutturali cardiache 50.
Altre malformazioni frequentemente associate a
CC sono l’onfalocele, l’atresia esofagea più o meno
associata alla fistola tracheoesofagea, l’atresia
duodenale, l’igroma cistico (spesso in presenza di
aneuploidia), l’ernia diaframmatica e l’agenesia del
corpo calloso 54-57.
Il rischio di cardiopatie congenite, di solito associate ad aneuploidia o a sindromi complesse, è
aumentato anche in caso di difetto di crescita fetale
precoce, oppure di tipo simmetrico, a prescindere
dall’epoca gestazionale e in caso di gravidanza gemellare monozigotica 38, 50, 53, 58.
Infine, in questi ultimi anni, alcuni autori hanno
proposto di eseguire l’ecocardiografia fetale anche
a quelle pazienti che, pur presentando fattori di
rischio per anomalia cromosomica (età materna,
screening biochimico positivo, anamnesi), rifiutano l’effettuazione di metodiche prenatali invasive
e quindi a rischio di perdita fetale 50. In questi casi,
l’ecocardiografia fetale potrebbe rappresentare un
completamento diagnostico importante, dal momento che una cardiopatia si rileva nel 50% degli
affetti da Trisomia 21, nel 35% dei feti affetti da
sindrome di Turner e nel 90-99% di quelli con Trisomia 13 e 18. Le anomalie cardiache, inoltre, a
differenza di altri marcatori ecografici, non hanno
carattere transitorio bensì evolutivo, né risultano
dipendenti da particolari livelli di cut-off correlati
all’epoca gestazionale. In questi casi, un’accurata
ecocardiografia può aumentare in misura sostanziale anche la possibilità di riscontro di una cromosomopatia 59.
Aspetti tecnici
Per l’esecuzione dell’ecocardiografia fetale devono essere utilizzate apparecchiature con un sistema bidimensionale ad alta risoluzione, con sonde
aventi un numero elevato di cristalli che permettano di ottenere immagini nitide anche a ingrandimenti elevati. Inoltre, le apparecchiature devono
essere provviste di funzione Cine Loop, di zoom
e di funzioni quali l’M-mode, il Doppler pulsato e
continuo e infine del color Doppler.
Per quel che riguarda l’impostazione dell’apparecchio, è necessaria una regolazione più “dura”
dell’immagine (rispetto all’immagine ostetrica), ov­
vero un basso livello di “range” dinamico e un aumento del contrasto, utile per una migliore definizione del contorno parietale e delle altre strutture
anatomiche del cuore.
È necessario, inoltre, utilizzare un frame-rate
ele­vato per una corretta valutazione di strutture
in rapido movimento, quali quelle cardiache fetali.
Le sonde utilizzate dovrebbero essere la settoriale e la convessa, preferibilmente a multifrequenza
(3-8 MHz), in modo da ottimizzare la frequenza di
emissione in funzione della distanza del cuore fetale. Nel secondo trimestre si utilizzano frequenze
più alte (5-8 MHz), mentre si utilizzano quelle più
basse (3-5 MHz) nel terzo trimestre o in presenza di
adiposità materna.
Sebbene la valutazione del cuore fetale sia già
possibile a tredici settimane di gestazione, l’epoca
ideale in cui eseguire l’esame ai fini di ottenere una
diagnosi della cardiopatia precoce, ma il più completa e accurata possibile, è quella compresa tra diciotto e ventidue settimane 60.
4
MARINO cardiologia pediatrica.indd 4
17/09/15 07:54
1 – Cardiologia fetale
Esame bidimensionale:
piani di scansione
Lo studio del cuore fetale inizia con l’individuazione della posizione fetale determinando il lato
destro e sinistro del feto, la posizione della testa
e della colonna vertebrale. Una volta compresa la
posizione del feto, si passa all’esame morfologico
del cuore fetale sfruttando scansioni trasverse, oblique e longitudinali (Fig. 1.1-1.4).
La prima scansione è quella trasversa dell’addome che permette di evidenziare la posizione del­
l’aorta addominale, della vena cava inferiore, del fegato, della colecisti e della vena ombelicale rispetto
alla colonna vertebrale in modo da definire il situs
viscero-atriale e i rapporti spaziali tra le varie strutture. Inclinando di poco il trasduttore verso la testa
del feto si ottiene una scansione trasversa del torace a livello delle 4 camere cardiache e con questa
scansione è possibile riconoscere il 50-60% delle
cardiopatie maggiori 45, 61. Queste prime due sezioni permettono di definire il situs, l’asse cardiaco, la
posizione e le dimensioni delle camere cardiache,
Figura 1.1 Sezione del torace dimostrante la proiezione
4 camere apicale (ottenuta quando il feto ha dorso posteriore). Attraverso questa proiezione si visualizzano l’aorta
discendente (di fronte alla colonna vertebrale), l’atrio destro (AD) e sinistro (AD), il ventricolo destro (VD) e sinistro (VS), il setto interatriale e interventricolare, il forame
ovale, la valvola mitrale e tricuspide.
Figura 1.3 Scansione in asse lungo del VS che permette
di verificare che un ventricolo morfologicamente sinistro sia in relazione con l’aorta e che vi sia continuità tra
il setto interventricolare e la parete anteriore dell’aorta
(escludendo in particolare la presenza di difetti interventricolari perimembranosi con o senza “aorta a cavaliere”
del DIV.
Figura 1.2 Sezione del torace dimostrante la proiezione 4 camere trasversa (ottenuta quando il feto ha dorso
laterale) dove si evidenzia più accuratamente lo spessore
delle pareti e del setto interventricolare (che viene esaminato a un angolo di circa 90 gradi rispetto al fascio di
ultrasuoni). Si osserva inoltre l’apertura in atrio sinistro
della membrana della fossa ovale.
Figura 1.4 Sezione del mediastino superiore dimostrante:
proiezione dei tre vasi con scansione trasversa dell’arco
duttale e dell’arco aortico fino all’istmo. Attraverso questa
proiezione si visualizzano da sinistra verso destra la vena
cava superiore (VCS), l’aorta (AO) e il tronco dell’arteria
polmonare che si continua del dotto arterioso (DA). La trachea si riconosce come una struttura circolare con parete
ecogena alla destra dei due grandi vasi e dietro la VCS.
5
MARINO cardiologia pediatrica.indd 5
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
in assoluto e rispetto a quelle del torace 62. Il situs
descrive la distribuzione degli organi e dei vasi nel
torace e nell’addome e può essere solitus, inversus
o indeterminato. L’asse cardiaco descrive l’orientamento rotazionale del cuore nel torace ed è quindi
strettamente legato all’orientamento dell’apice cardiaco verso destra o verso sinistra, nonché al suo
grado di rotazione rispetto alla parete toracica. Può
essere misurato come l’angolo formato dal setto
interventricolare con il piano anteroposteriore che
biseca il torace: in tal modo esso definisce la rotazione del cuore all’interno della gabbia toracica.
Normalmente l’apice cardiaco è orientato a sinistra
della linea mediana (levocardia) e quest’angolo misura 40-45°; una levorotazione maggiore (>59°) o
minore (<28°) frequentemente si correla con una
patologia cardiaca primitiva (sindromi isomeriche,
trasposizione corretta delle grandi arterie) o, indipendentemente dall’associazione con cardiopatie,
a sindromi polimalformative 63. La posizione cardiaca descrive la collocazione del cuore all’interno
della gabbia toracica e identifica l’emitorace occupato in prevalenza dal cuore. Normalmente, due
terzi del cuore occupano l’emitorace sinistro e il
rimanente un terzo l’emitorace destro. Il ventricolo destro è posto sotto la parete toracica anteriore, mentre l’atrio sinistro è situato al davanti della
colonna vertebrale. Un’anomalia della posizione
cardiaca è spesso secondaria a una malformazione
extracardiaca (ernia diaframmatica, malformazione
adenomatoide cistica polmonare ecc.). Riguardo
alle dimensioni del cuore, esso usualmente occupa
circa un terzo dello spazio toracico. La misurazione dell’indice cardio-toracico, dato dal rapporto tra
circonferenza cardiaca e toracica, varia da 0,40 a
0,55, un valore superiore può essere dovuto a un
vero aumento delle dimensioni cardiache (cardiomegalia) o a una ipoplasia del torace 64. Tale scansione permette, infine, di studiare le singole strutture del cuore, definendone le dimensioni e le caratteristiche di ognuna. Rimandando a testi specifici la
descrizione, si ricorda che per definire una normale
anatomia cardiaca, le dimensioni delle sezioni sinistre e destre devono essere simili (rapporto 1-1,2
anche se nel terzo trimestre di gestazione si può osservare una maggiore prevalenza del ventricolo destro sul sinistro), almeno due delle quattro vene polmonari devono essere visualizzate in atrio sinistro,
il ventricolo sinistro deve essere posteriore e presentare una forma più allungata rispetto al ventricolo destro. Una corretta valutazione dell’integrità del
setto interventricolare richiede sempre l’effettuazio-
ne di una scansione trasversa in cui il setto venga
esaminato a un angolo di circa 90 gradi rispetto al
piano di scansione degli ultrasuoni per evitare fenomeni di falso “drop out” a volte visualizzabili nella
scansione apicale. Va comunque sottolineato che
la valutazione del tratto perimembranoso, sottoaortico del setto non è possibile neanche in questa
scansione, ma solo in asse lungo di sinistra, e che
un difetto perimembranoso isolato, anche di dimensioni non piccole, può sfuggire all’esame ecocardiografico fetale. Talvolta durante l’esame delle
4 camere si visualizzano delle zone iperecogene
puntiformi (“golf balls”) a livello dei muscoli papillari del ventricolo sinistro, del ventricolo destro o, più
raramente, sul setto e sulle pareti libere ventricolari che sono l’esito di un’eccessiva deposizione di
sali di calcio. Tali aree iperecogene possono essere
multiple o singole e non rappresentano un segno di
patologia cardiaca, anche se alcuni autori riportano, ma solo per particolari localizzazioni, un lieve
aumento del rischio di CC. Secondo alcuni autori la
presenza di “golf ball” può rappresentare un’indicazione al cariotipo fetale se in associazione ad altri
fattori di rischio di cromosomopatia 65.
Per l’analisi degli efflussi ventricolari (assi lunghi
di destra e di sinistra) si devono utilizzare scansioni oblique, ottenute con minimi movimenti del
trasduttore verso la spalla destra del feto, e scansioni assiali che richiedono invece un’ulteriore piccola inclinazione verso l’alto della sonda rispetto al
piano precedente. Queste scansioni permettono di
verificare l’assenza di ostacoli all’efflusso biventricolare, il tipo e il modo di connessione ventricoloarteriosa, il calibro dei due grandi vasi e la continuità tra il setto interventricolare e l’arteria che origina
dal ventricolo sinistro.
Con un’ulteriore inclinazione verso la testa del
feto si ottengono delle sezioni dell’arco aortico e
dell’“arco polmonare”, della vena cava superiore e
dell’origine e del decorso dei tronchi sovra-aortici,
sia in scansione longitudinale che trasversa 66. Oltre
alle scansioni elencate, esistono altre sezioni intermedie, come la 5 camere e l’asse corto dei ventricoli che permettono di ottenere ulteriori informazioni anatomiche.
Monodimensionale,
Doppler pulsato e color-Doppler
Tali metodiche vengono comunemente impiegate in ecocardiografia fetale per lo studio del ritmo
cardiaco fetale o per ottenere informazioni funzionali (gradienti di pressione, distribuzione dei flus-
6
MARINO cardiologia pediatrica.indd 6
17/09/15 07:54
1 – Cardiologia fetale
A
B
Figura 1.5 M-mode in corrispondenza della scansione
asse lungo del VS per la valutazione del ritmo cardiaco.
Sovrapponendo il color-Doppler su M-mode è possibile registrare oltre ai movimenti parietali anche le linee
di flusso atrio-ventricolare telediastolico (in basso, corrispondente alla contrazione atriale) e sistolico ventricoloarteriosa (in alto o al centro, corrispondente alla contrazione ventricolare). A) Calcolo dell’intervallo PR; B) calcolo dell’intervallo RR.
si attraverso una comunicazione anomala), sia nel
cuore strutturalmente normale che in caso di cardiopatia 67-69 (Fig. 1.5).
Con il monodimensionale si ottiene un’immagine grafica delle strutture che vengono attraversate
dal fascio di ultrasuoni in funzione del tempo. In
proiezione 4 camere trasversa si deve fare in modo
che il singolo fascio di ultrasuoni attraversi le cavità
ventricolari perpendicolarmente, subito al di sotto
dei lembi delle valvole atrio-ventricolari. L’immagine ottenuta consente una misurazione accurata
delle dimensioni della cavità, dello spessore delle
pareti e dei movimenti di queste ultime permettendo di ottenere tutta una serie di informazioni sulla
funzione contrattile e sulla funzione diastolica biventricolare 70, 71.
Con il color-Doppler è possibile visualizzare, sovrapposta alle immagini bidimensionali, una mappa
in tempo reale della distribuzione dei flussi all’interno delle cavità cardiache 67. I filtri colore saranno
modificati in base al distretto da esaminare: per i
flussi transvalvolari bisogna utilizzare un filtro “di
parete” e una PRF alti (200-300 Hz), in modo da
evitare disturbi del segnale dovuti ai movimenti delle valvole e dei setti; al contrario per i flussi lenti,
venosi, conviene utilizzare un filtro e una PRF bassi.
Considerando che durante la vita fetale raramente
le velocità massime di flusso eccedono 1 m/sec,
diventa agevole, grazie a un adeguato settaggio
dell’apparecchio, visualizzare la normale circolazione di flussi laminari durante il ciclo cardiaco all’in-
terno delle vene, delle cavità atriali, ventricolari e
delle grandi arterie. Utilizzando le stesse scansioni
dell’esame bidimensionale e ponendo attenzione
affinché l’angolo d’insonazione del flusso da esaminare sia prossimo allo zero (cioè parallelo), sarà
possibile ottenere utili informazioni riguardanti la
normale pervietà delle connessioni cardiache (veno-atriale, atrio-ventricolare e ventricolo-arteriosa)
così come un’eventuale anomalia nella presenza,
velocità o direzione del flusso.
Analogamente al monodimensionale, con il
Doppler pulsato si ottiene un’immagine grafica, in
funzione del tempo, della distribuzione del flusso
all’interno di un singolo punto (volume campione)
potendone valutare la direzione, la velocità e il pattern del flusso ematico 69. Per l’esame dei flussi attraverso le valvole atrio-ventricolari, il volume campione è posizionato, in 4 camere apicale, subito al
di sotto dei lembi delle valvole atrio-ventricolari,
cercando di ottenere una posizione il più parallela
possibile alla direzione del flusso. Si ottiene, così,
un’onda con una morfologia simile alla lettera M
(positiva o negativa, secondo la posizione delle camere cardiache rispetto alla fonte di ultrasuoni) con
un picco più basso in protodiastole (onda E) e un
picco più alto in telediastole (onda A, che rappresenta il riempimento ventricolare durante la contrazione atriale) 69, 72. A livello della valvola mitrale, a
causa della continuità fibrosa che esiste fra il lembo
anteriore della mitrale e la valvola aortica, si registra spesso un flusso sistolico-aortico a direzione
contraria rispetto a quello diastolico-mitralico. Variazioni della distensibilità e del precarico cardiaco,
insieme a cambiamenti della frequenza cardiaca
modificheranno le caratteristiche dell’onda a M.
Nel feto sano, l’onda A è usualmente più alta, anche se con il progredire della gravidanza, il rapporto E/A aumenta, avvicinandosi ai valori riscontrati in
epoca postnatale. Mentre esiste un generale accordo sul fatto che l’aumento del rapporto E/A sia dovuto all’aumentare della velocità dell’onda E rispetto all’onda A che rimane piuttosto costante durante
la gestazione, c’è ancora disaccordo circa l’andamento con cui aumenta la velocità dell’onda E che
secondo alcuni avviene in maniera lineare durante
la gestazione, mentre secondo altri solo nell’ultimo
trimestre 72, 73. Nel feto, è stata riportata la riduzione
del rapporto E/A di entrambe le valvole, mitrale e
tricuspide, nella sindrome da trasfusione feto-fetale,
associata ad altri segni di disfunzione diastolica 74.
Tuttavia, altri studi hanno dimostrato un aumento
del rapporto E/A in situazioni di compromissione
7
MARINO cardiologia pediatrica.indd 7
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
cardiaca quali nello IUGR e nell’idrope da malformazione adenomatoide-cistica congenita 75, 76.
Dopo aver esaminato i flussi attraverso la mitrale,
inclinando leggermente la sonda verso la testa del
feto e spostando il volume campione verso il setto
interventricolare si registra meglio il flusso aortico
sistolico, rappresentato da una curva con un unico
picco centrale a una velocità inferiore a un metro al
secondo. Un flusso del tutto analogo a quello aortico si registra in arteria polmonare quando questa
viene visualizzata in asse lungo, con l’aorta in asse
corto al centro (cosiddetto arco duttale).
Spostando poi il volume campione verso la parte
distale dell’arco della polmonare si evidenzia il flusso di sangue attraverso il dotto arterioso. A questo
livello si registrano i flussi a velocità più elevata durante la vita fetale con un picco sistolico (superiore
a 1 m/sec nel terzo trimestre di gravidanza), una
progressiva discesa della velocità nella seconda
metà della sistole e nella prima parte della diastole
e una persistenza del flusso a bassa velocità per tutta la fase terminale della diastole.
La funzione sistolica, dipendente dalla capacità
contrattile del cuore e dalle resistenze periferiche,
si può valutare esaminando la velocità di picco sistolico e il tempo di accelerazione nell’aorta, nella
polmonare e nel dotto arterioso77.
Posizionando il volume campione a livello delle
vene cave si ottiene un’onda con una morfologia
trifasica: la prima fase, di maggior ampiezza anterograda, corrisponde alla sistole, la seconda di ampiezza inferiore, anterograda, corrisponde alla diastole precoce e una terza, retrograda, sincrona con
la contrazione atriale al termine della diastole. La
suddetta morfologia varia con il variare della pressione atriale.
La misurazione del flusso nel dotto venoso si ottiene ponendo il volume campione a livello della
sua origine dalla vena ombelicale. La sua onda è bifasica costituita da due fasi anterograde corrispondenti rispettivamente alla sistole e alla diastole.
La misurazione del flusso nelle vene polmonari
si ottiene in scansione 4 camere apicale posizionando il volume campione a livello del loro ingresso in atrio sinistro. L’onda è costituita da due picchi
anterogradi relativi alla sistole e alla diastole rispettivamente.
In conclusione, pur lasciando alla tecnica bidimensionale il ruolo fondamentale nell’esame del
cuore fetale e nello screening delle cardiopatie in
utero, l’M-mode, il Doppler e il color-Doppler devono essere considerati un utile complemento di
indagine anche nello studio dei cuore normale in
quanto facilitano il riconoscimento di importanti
anomalie funzionali, particolarmente nel terzo trimestre di gravidanza.
Doppler tissutale, strain, strain-rate
Per Doppler tissutale (TDI) si intende l’applicazione dei principi del Doppler alla misurazione
della velocità del miocardio piuttosto che al flusso di sangue intracardiaco. Considerato che l’apice
del cuore rimane relativamente stazionario durante
il ciclo cardiaco, l’analisi del movimento dell’anello valvolare mitralico rispetto all’apice fornisce una
buona approssimazione della contrattilità longitudinale del ventricolo 78. Lo spettro del Doppler
tissutale pulsato a livello dell’annulus mitralico è
caratterizzato da tre onde: l’onda S’ (onda positiva
in proiezione 4 camere apicale nell’esame postnatale transtoracico, corrispondente alla funzione sistolica) descrive la velocità del movimento sistolico
dell’anello verso l’apice; l’onda È (onda negativa,
corrispondente alla funzione diastolica) descrive la
velocità del movimento proto-diastolico in allontanamento dall’apice e l’onda A’ (onda negativa, corrispondente alla funzione meccanica atriale) descrive la velocità del movimento dell’annulus associato
alla contrazione atriale. Dal 1999, anno in cui veniva pubblicato il primo lavoro relativo al Doppler tissutale nella valutazione del miocardio fetale, a oggi
sono stati riportati pareri discordanti circa l’utilità di
questa tecnica di analisi della funzione cardiaca fetale. Jamjureeruk nel 2001 riportava un giudizio negativo nell’utilizzo del TDI per la valutazione delle
velocità miocardiche di ciascuna parete cardiaca.
Gardiner nel 2006 sottolineava invece l’utilità del
TDI per dimostrare i progressivi cambiamenti che
si verificano a carico sia della funzione sistolica che
diastolica fetale durante il corso della gravidanza,
affermando inoltre che l’andamento del processo
maturativo della velocità tissutale miocardica è simile per i due ventricoli, nonostante condizioni di
carico differenti 79-81.
A differenza del TDI che misura il picco di velocità del segmento miocardico interrogato, il color
TDI ne misura la velocità media. L’applicazione del
color Doppler al TDI permette di valutare lo strainrate (ovvero i cambiamenti della lunghezza delle fibre per unità di tempo) e, per derivazione matematica, lo strain miocardico stesso (ovvero i cambiamenti della lunghezza delle fibre). Queste modalità
di analisi hanno il vantaggio di misurare direttamente i segmenti miocardici, anziché i cambiamenti a
8
MARINO cardiologia pediatrica.indd 8
17/09/15 07:54
1 – Cardiologia fetale
carico delle dimensioni delle camere ventricolari,
e in questo modo riflettono in maniera più accurata la contrattilità miocardica. Tuttavia, così come il
Doppler TDI pulsato, sono limitate dall’angolo-dipendenza e dalla valutazione del singolo segmento
piuttosto che della funzione globale 81, 82.
Ruolo dell’ecocardiografia
fetale nel cambiamento
dell’epidemiologia
e della prognosi
delle Cardiopatie Congenite
Il ruolo dell’ecocardiografia fetale è andato progressivamente modificandosi nel corso degli anni.
Nel 1972 veniva pubblicato uno dei primi studi relativi alla registrazione ecocardiografica M-mode
dell’attività cardiaca nel feto umano 83. Gli autori ipotizzavano di analizzare la funzione cardiaca
fetale con la misurazione della frazione di eiezione del ventricolo sinistro, sfruttando la registrazione M-mode del movimento di parete cardiaca
nel tempo. Gli stessi autori, tuttavia, prevedevano
che le piccole dimensioni, l’anatomia complessa e
la frequenza cardiaca elevata del feto umano non
avrebbero permesso di diagnosticare in epoca fetale le malformazioni congenite cardiache complesse. Nell’introduzione alla sua epocale monografia
del 1974, “Patologie congenite del Cuore”, il dottor Abraham M. Rudolph enfatizzava l’importanza
insita nella caratterizzazione dell’adattamento del
sistema cardiovascolare del feto durante il periodo
prenatale e perinatale, nel migliorare la comprensione della condizione clinica del neonato, e nel
contribuire alla formulazione di strategie di gestione delle diverse forme di CC, basate sulla logica e
sulla fisiologia 84. Gli studi di Rudolph sulla fisiopatologia della circolazione nel feto di agnello e il lavoro citato in precedenza servirono da stimolo per
l’inizio di un progetto di ricerca presso la Facoltà di
Medicina dell’Università di Yale, mirato all’applicazione in ambito clinico dell’ecocardiografia fetale,
per validare nel feto umano gli assunti tratti dal modello fetale ovino. Tale progetto servì a dimostrare
come l’ecocardiografia M-mode potesse fornire la
conferma non invasiva che quanto riscontrato sperimentalmente sul cuore fetale dell’agnello valeva
più o meno allo stesso modo anche per il cuore
fetale umano. Helen Taussig nel 1978 sottolineava
l’importanza di queste scoperte, intravedendo la
possibilità di diagnosi di CC in epoca gestazionale
precoce, oltre alla possibilità di un’eventuale inter-
ruzione di gravidanza per ridurre il peso individuale
e sociale della CC da cui erano affetti questi bambini. L’attenzione rivolta a stabilire il ruolo clinico
del­l’ecocardiografia fetale andava crescendo e nel
1980 iniziavano ad essere pubblicati i primi lavori sull’argomento. Kleinman et al. dell’Università di
Yale suggerirono le indicazioni all’ecocardiografia
fetale in gravidanze considerate ad alto rischio per
CC e riportarono l’utilizzo dell’ecocardiografia Mmode e bidimensionale per documentare i primi
casi di diagnosi prenatale di cardiopatie congenite
e aritmie fetali 85. Nello stesso anno, pubblicazioni da San Diego e da Londra documentavano la
possibilità di esaminare il cuore fetale con un’analisi sequenziale, segmentale delle strutture cardiache 86, 87. Nel 1985 al Congresso Mondiale di Cardiologia Pediatrica, Fermont da Parigi riportava la
propria esperienza nell’esecuzione dello screening
del cuore fetale (mediante la scansione 4 camere),
per la rilevazione delle malformazioni congenite,
e in seguito anche Allan forniva la descrizione del
programma regionale di screening impostato a Londra 88, 89. Dalla metà degli anni Ottanta a oggi, lo
screening ostetrico per le malformazioni cardiache
fetali, inteso come strumento attraverso cui si identificano i feti che necessitano di una valutazione
più approfondita con ecocardiografia fetale, è stato
riconosciuto universalmente come esame standard
di routine e la richiesta di esecuzione è aumentata
progressivamente nel tempo 89, 90. Considerato, infatti, che circa l’80% dei feti con CC si presenta in
gravidanze prive di fattori di rischio, allo stato attuale si ritiene che non sia sufficiente lo screening
basato esclusivamente sulla presenza di fattori di
rischio noti, ma che sia necessario effettuarlo anche in gravidanze a basso rischio per aumentare la
percentuale di casi diagnosticati in epoca prenatale 90, 91. Nel corso degli anni, il diffondersi dello
screening ostetrico ha consentito di aumentare la
percentuale di CC riconosciute all’ecocardiografia
fetale, con la probabilità più alta di diagnosi prenatale per le forme più severe di CC 92-94. I progressi
della tecnologia hanno permesso di effettuare diagnosi sempre più accurate e in epoca gestazionale sempre più precoce, aspetti che insieme farebbero presupporre un miglioramento nella gestione
clinico-terapeutica delle CC con diagnosi prenatale,
anche se restano ancora numerose controversie in
merito. Non è, in effetti, semplice provare che la
diagnosi prenatale abbia un impatto positivo sulla
prognosi di tutti i casi di CC. Una delle difficoltà
nel fare un confronto tra l’evoluzione dei casi con e
9
MARINO cardiologia pediatrica.indd 9
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
senza diagnosi prenatale è che la diagnosi prenatale
consente una valutazione dell’evoluzione a partire
dall’epoca fetale in cui si fa diagnosi, avendo dunque la possibilità di considerare l’eventuale morte
spontanea intrauterina e la morte precoce neonatale prima della chirurgia. Al contrario, i bambini con
una diagnosi postnatale sono quelli sopravvissuti
alla vita fetale e spesso al periodo neonatale precoce fino a raggiungere un centro di terzo livello dove
viene confermata la diagnosi ed effettuato il trattamento. Inoltre, lo spettro fetale delle anomalie cardiache è ulteriormente aggravato dalla presenza di
lesioni complesse e da quelle associate con anomalie extracardiache e cromosomiche 94, 95. Il confronto, quindi, non è tra popolazioni omogenee ed è
per questo motivo che solo pochi studi in letteratura hanno saputo dimostrare un vantaggio in termini
di sopravvivenza complessiva nelle serie prenatali
rispetto a quelle postnatali. D’altro canto, invece,
diversi autori hanno riportato un impatto positivo
da parte della diagnosi prenatale sulle condizioni
preoperatorie dei neonati con malformazioni cardiache complesse. La diagnosi prenatale sembra,
infatti, essere associata con una ridotta morbilità
preoperatoria, una ridotta acidosi e con l’evidenza
di una migliore perfusione d’organo 96-101. Le malformazioni per cui abbiamo a disposizione un numero più o meno ampio di dati pubblicati in questo
senso sono la sindrome del cuore sinistro ipoplasico, la trasposizione delle grandi arterie, la coartazione dell’aorta e l’atresia polmonare.
Eapen et al. hanno descritto una ridotta morbilità postnatale, un minor ritardo nella correzione
chirurgica e un periodo più breve di ricovero in
terapia intensiva nei pazienti con patologia ostruttiva severa del cuore sinistro diagnosticata in epoca
prenatale rispetto ai pazienti con diagnosi postnatale. La diagnosi prenatale ha, inoltre, condizionato la scelta della sede del parto nel 65% dei casi,
preferendo un centro di terzo livello all’ospedale
di periferia. Non si è, tuttavia, registrata differenza in termini di mortalità chirurgica tra i pazienti con e senza diagnosi prenatale, benché gli autori suggeriscano che le condizioni preoperatorie
più favorevoli possano contribuire a una migliore
prognosi neurologica 98. Anche Mahle et al. hanno studiato l’impatto della diagnosi prenatale sulla
sopravvivenza e sulla morbilità neurologica in neonati con la sindrome del cuore sinistro ipoplasico
sottoposti a chirurgia palliativa. Gli eventi neurologici perioperatori sono risultati inferiori nei pazienti
con diagnosi prenatale rispetto a quelli senza, an-
che se non si è riscontrata una parallela riduzione
della mortalità ospedaliera 9. Questi autori, come
Eapen in precedenza, suggeriscono che la diagnosi
prenatale possa migliorare l’evoluzione neurologica a lungo termine. Nemmeno lo studio di Kumar
dimostra un miglioramento della mortalità preoperatoria e postoperatoria precoce per i pazienti con
diagnosi prenatale di cuore sinistro ipoplastico, pur
presentandosi questi in condizioni preoperatorie
più favorevoli di quelli con diagnosi postnatale 100.
Tworetzky nel 2001 descrive, invece, l’assenza di
mortalità nel gruppo di 14 feti a cui era stata fatta diagnosi di cuore sinistro ipoplasico contro una
mortalità di 13 su 38 bambini con la stessa diagnosi effettuata in epoca postnatale, presso un singolo
centro negli Stati Uniti 99. Oltre al beneficio sulla
mortalità, la diagnosi prenatale di cuore sinistro
ipoplasico si associa a una migliore funzione ventricolare, a un’insufficienza tricuspidalica più lieve
e a una ridotta richiesta di inotropi e bicarbonato.
L’autore conclude, quindi, affermando che la diagnosi prenatale può migliorare la prognosi chirurgica, presumibilmente grazie al miglioramento dello
stato preoperatorio del paziente. Anche Franklin et
al., nell’unico lavoro che abbia esaminato l’impatto della diagnosi prenatale di coartazione severa
dell’aorta sulla prognosi postnatale, riportano un
impatto positivo della diagnosi prenatale in termini
di morbilità preoperatoria e mortalità, così come in
termini di funzione contrattile ventricolare alla presentazione iniziale del paziente 101.
Una conseguenza importante della diagnosi
prenatale sembra essere, dunque, quella di evitare
l’in­stabilità emodinamica nei difetti cardiaci dottodipendenti, come visto nella patologia ostruttiva
critica del cuore sinistro, ma anche nella trasposizione delle grandi arterie e nell’atresia polmonare.
La serie più ampia che ha preso in esame l’impatto
della diagnosi prenatale di trasposizione delle grandi arterie (TGA) sull’evoluzione postnatale è quella
pubblicata nel 1999 da Bonnet e dal gruppo francese dell’Ospedale Necker-Enfants Malades di Parigi 97. Questo studio ha incluso 68 neonati con diagnosi prenatale e 250 con diagnosi postnatale. La
mortalità preoperatoria e postoperatoria è risultata
rispettivamente del 6 e dell’8,5% nel gruppo con
diagnosi postnatale, mentre non sono stati registrati
decessi nel gruppo con diagnosi prenatale. Inoltre,
i pazienti con diagnosi postnatale si sono presentati
con un’acidosi metabolica e uno scompenso multiorgano più severo rispetto ai pazienti con diagnosi
prenatale. Nello stesso anno anche Maeno con il
10
MARINO cardiologia pediatrica.indd 10
17/09/15 07:54
1 – Cardiologia fetale
gruppo di Toronto, Boston e dell’Ontario ha sottolineato l’importanza della diagnosi prenatale nei feti
affetti da TGA, in particolare nel gruppo di trasposti con forame ovale e dotto arterioso restrittivi 102.
Secondo questi autori, e secondo lo stesso gruppo
del Necker che ha pubblicato un nuovo lavoro relativo alla gestione dei pazienti con TGA nel 2004,
l’identificazione in epoca prenatale dei feti a rischio più elevato di ipossiemia postnatale, e quindi
di morte precoce neonatale, offre la possibilità di
organizzare il parto in un centro di terzo livello e di
predisporre in maniera accurata la gestione perinatale del paziente, permettendo in questo modo una
prognosi migliore 102, 103. Recentemente, tuttavia, il
lavoro pubblicato da Raboisson et al. di Lione, relativo ancora una volta all’impatto della diagnosi
prenatale di TGA sulla prognosi, non ha confermato
la tesi di Bonnet e Maeno. La popolazione studiata
era composta da 121 neonati con trasposizione delle grandi arterie, di cui 48 con diagnosi prenatale.
Il confronto tra i due gruppi, rispettivamente con
e senza diagnosi prenatale, ha condotto gli autori
ad affermare che la diagnosi prenatale di TGA ha
un impatto solo sulle modalità del parto, incrementando la percentuale di parti indotti e di tagli cesarei, senza alcun impatto positivo sulla prognosi del
neonato, se non per la possibilità di effettuare una
settostomia atriale più agevolmente e di ottenere un
accesso in terapia intensiva più rapido 104.
A oggi, un numero più limitato di studi in letteratura ha preso in considerazione l’impatto della
diagnosi prenatale sulla prognosi dei neonati con
flusso polmonare dipendente dal dotto arterioso.
Nel lavoro pubblicato nel 1998, Daubeney ha analizzato la popolazione del Regno Unito affetta da
atresia polmonare a setto intatto, ponendo a confronto i pazienti con diagnosi prenatale con quelli diagnosticati dopo la nascita. In base ai risultati
raccolti, l’autore ha concluso che la cardiopatia in
esame è rara e che l’ecocardiografia fetale ha un
impatto importante sull’incidenza alla nascita per il
numero elevato di interruzione di gravidanza (IVG)
a seguito della diagnosi precoce (61%) 105. Tuttavia,
la diagnosi prenatale risulta influente anche nei casi
in cui l’interruzione volontaria di gravidanza non
è presa in considerazione, in quanto indirizza la
coppia di genitori a scegliere un centro di terzo livello per il parto e permette di organizzare meglio
la gestione perinatale del paziente, somministrando più precocemente le prostaglandine. In questo
studio è fallito, tuttavia, il tentativo di dimostrare
un vantaggio in termini di sopravvivenza a breve
e lungo termine rispetto ai pazienti senza diagnosi
prenatale. Successivamente, anche il gruppo inglese dell’Evelina Hospital di Londra ha pubblicato i
dati relativi ai pazienti con atresia polmonare 106.
I neonati con diagnosi prenatale si presentavano
con una saturazione arteriosa più elevata, ma questa condizione preoperatoria non si traduceva in
una mortalità o morbilità a breve termine migliore
di quelli senza diagnosi prenatale, a cui per altro
veniva fatta diagnosi entro una media di ventiquattro ore di vita. I dati raccolti recentemente presso
il nostro centro, relativi alla gestione dei pazienti
affetti da atresia polmonare a setto intatto, hanno
confermato alcuni possibili vantaggi della diagnosi
prenatale 107. Quest’ultima infatti ci ha permesso di
offrire un counselling dettagliato ai genitori, pianificando in epoca prenatale la strategia terapeutica
più appropriata per ogni singolo caso, in base alla
severità del difetto cardiaco riscontrato. Anche nella nostra esperienza non si è riscontrata una differenza significativa in termini di mortalità tra i pazienti con e senza diagnosi prenatale. Tuttavia, è
degno di nota che i pazienti diagnosticati in epoca
fetale e sopravvissuti al periodo critico neonatale
abbiano un’incidenza di mortalità e una necessità
di ulteriori interventi nel follow-up sovrapponibile
ai pazienti senza diagnosi prenatale, pur presentando alla nascita uno spettro significativamente molto più severo della cardiopatia rispetto a quelli con
diagnosi postnatale.
Un solo lavoro pubblicato nel 2009 ha posto,
infine, a confronto le caratteristiche e l’evoluzione
prognostica dei pazienti con truncus arteriosus diagnosticato in epoca pre- e postnatale 108. In utero,
la diagnosi di questa cardiopatia si conferma particolarmente insidiosa, essendo spesso impegnativo
differenziarla dalla tetralogia di Fallot con atresia
polmonare. In questo studio, tale cardiopatia è risultata, inoltre, associata a un’incidenza di IVG
complessiva (40%) più elevata rispetto ai precedenti dati riportati in letteratura, incidenza aumentata
fino al 61% quando vengono presi in considerazione solo i feti con meno di ventiquattro settimane di
età gestazionale alla diagnosi 109, 110. Pur non trattandosi di una lesione dotto-dipendente (a parte i
casi di truncus associati a un’interruzione dell’arco),
cardiopatie per le quali, come visto in precedenza,
la diagnosi prenatale si è dimostrata vantaggiosa ai
fini della gestione perinatale del paziente, in questo studio l’ecocardiografia fetale dimostra di essere comunque importante nella gestione della gravidanza e nel comprendere la storia naturale della
11
MARINO cardiologia pediatrica.indd 11
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
cardiopatia in utero, oltre a garantire la possibilità
di counselling per la coppia di genitori coinvolti.
L’impatto della diagnosi prenatale si è dimostrato
particolarmente rilevante nell’identificazione e nella successiva guida al trattamento, spesso salva-vita,
delle aritmie fetali. Per le tachicardie fetali, che per
lo più consistono in tachicardia sopraventricolare
da rientro o flutter atriale, esiste un’ampia evidenza
dell’efficacia della terapia prenatale per controllare
l’aritmia conducendo alla risoluzione dell’idrope fetale, se presente 111-113. Il farmaco da utilizzarsi deve
essere scelto in base al tipo di aritmia diagnosticato e alla presenza o meno dell’idrope (che influisce sulle capacità di passaggio transplacentare del
farmaco). Riguardo alle bradicardie fetali, dovute al
blocco di conduzione atrio-ventricolare completo,
esistono controversie circa la terapia da utilizzare.
Se esiste un difetto strutturale associato all’anomalia di conduzione, si tratta solitamente di una trasposizione corretta delle grandi arterie, di un isomerismo atriale sinistro, di cardiomiopatie o tumori
cardiaci. La prognosi per questi feti è solitamente
infausta indipendentemente dalla terapia, con una
minoranza di sopravvissuti alla nascita 114, 115. Se il
blocco congenito è isolato, per lo più consegue alla
presenza di anticorpi materni anti-Ro o anti-La. In
questi casi esistono pareri discordanti circa l’utilità
della terapia prenatale: alcuni raccomandano farmaci quali il desametasone e il salbutamolo in tutti
i feti affetti, mentre altri solo in quelli in cui ci sia
l’evidenza di scompenso emodinamico 116, 117. Di
certo il riconoscimento prenatale di una bradicardia
severa permette di organizzare la gestione perinatale del paziente mettendo a disposizione i farmaci
adeguati e il necessario per un’eventuale stimolazione cardiaca in sala parto.
Concludendo, dunque, l’ecocardiografia fetale
permette di identificare in epoca prenatale precoce difetti strutturali cardiaci anche molto complessi,
associati o meno ad anomalie elettriche o funzionali. Alcuni dei feti affetti raggiungono il termine di
gravidanza e su questi la diagnosi pre-natale può
influire nella decisione del timing, della sede e della
modalità del parto. Altri, invece, non sopravvivono
fino al termine della gravidanza, per morte endouterina da scompenso cardiaco o per decisione da
parte dei genitori di interrompere la gravidanza, alla
luce delle informazioni ricevute circa la prognosi a
breve e lungo termine della CC diagnosticata. Ne
consegue quindi, come descritto in precedenza,
che la diagnosi prenatale può incidere sull’evoluzione delle CC anche in termini di incidenza di in-
terruzione volontaria di gravidanza, determinando
di conseguenza una riduzione significativa dell’incidenza alla nascita di certi tipi di CC maggiori.
Dall’analisi eseguita da Bull nel 1999 circa gli effetti
della diagnosi fetale sul pattern delle CC di grado
severo giunte a termine nel Regno Unito si deduce
che tra il 1993 e il 1995 la frequenza media del riscontro di CC in utero è stata del 23% e metà delle
gravidanze patologiche è andata incontro a interruzione. Anche Todros, in una revisione della letteratura relativa alla diagnosi prenatale pubblicata nel
2000, ribadisce che l’unico vero impatto dello screening e della diagnosi prenatale delle CC è proprio
la riduzione dell’incidenza di nati vivi affetti, senza
alcun beneficio in termini di mortalità e morbidità 118, 119. In una revisione di diciassette registri condotti su popolazioni europee (database EUROCAT),
Garner et al. hanno rilevato che la frequenza media
di interruzione volontaria di gravidanza per sindrome del cuore sinistro ipoplastico è stata dell’85%
quando la cardiopatia era diagnosticata prima delle ventiquattro settimane di gestazione, rispetto al
37% se la diagnosi avveniva dopo le ventiquattro
settimane 120. Nella maggior parte dei Paesi, eccetto
la Francia, la ventiquattresima settimana rappresentava allora l’età gestazionale oltre la quale non era
più possibile interrompere. Recentemente Marek
et al., riportando i risultati dello screening e della
diagnosi prenatale effettuati nella Repubblica Ceca,
hanno confermato l’incidenza molto elevata di interruzione volontaria di gravidanza a seguito della
diagnosi fetale di CC complesse, non mancando
però di sottolineare come questa possa allo stesso
modo fornire un impatto positivo in termini di morbilità e di gestione perinatale del feto affetto, nel
caso in cui la coppia decida di portare a termine la
gravidanza 94.
Trasporto intrautero
Esistono molti difetti cardiaci per i quali la diagnosi prenatale ha un impatto limitato nella gestione terapeutica perinatale, come nel caso del difetto interventricolare o del canale atrio-ventricolare
senza ostruzione agli efflussi ventricolari, dove non
ci si aspetta di dover effettuare alcun trattamento,
almeno fino alla riduzione spontanea delle resistenze vascolari polmonari. Per questi casi non esiste
indicazione a modificare la sede e le modalità del
parto, purché sia possibile una valutazione cardiologica neonatale, che confermi e perfezioni precocemente la diagnosi.
12
MARINO cardiologia pediatrica.indd 12
17/09/15 07:54
1 – Cardiologia fetale
Al contrario, nel caso di feti con cardiopatie dot­
to-dipendenti o nei quali ci sia la potenziale necessità di un intervento chirurgico o percutaneo in epoca neonatale precoce, la diagnosi prenatale permette di prevenire lo scompenso emodinamico acuto
del paziente mediante la programmazione del parto
vicino o direttamente all’interno di un centro di terzo livello dotato di unità cardiologica dove siano
immediatamente a disposizione il neonatologo, il
cardiologo pediatra e il cardiochirurgo 97, 101, 102, 105.
In questi casi, il trasporto intrautero permette un’immediata valutazione postnatale del paziente, evitando così i rischi del trasferimento neonatale in urgenza dalla sede del parto all’unità cardiologica, oltre
al vantaggio di avere subito presenti i genitori per
spiegar loro la procedura terapeutica programmata
e far firmare il consenso informato.
In pratica, comunque, molti genitori scelgono la
sede del parto vicino o direttamente in un centro
di terzo livello anche se il difetto cardiaco riscontrato all’ecocardiografia fetale non richiederebbe
un intervento nei primi giorni di vita. Questo atteggiamento riflette in gran parte la preoccupazione
dei genitori che devono separarsi dal loro piccolo
appena nato, nel caso in cui sia stata posta indicazione a una valutazione cardiologica anche se non
urgente e questa debba svolgersi in un centro geograficamente lontano rispetto alla sede del parto.
Riguardo alla modalità del parto, considerata la
fisiologia della circolazione in epoca prenatale, non
esiste alcuna controindicazione a un parto normale
per via vaginale per la maggior parte delle cardiopatie. Nella pratica comune si è soliti riservare il
parto con taglio cesareo a una minoranza di casi in
cui si preveda di dover intervenire immediatamente
dopo la nascita. Appartengono a questa minoranza
di casi i feti con TGA che presentino in utero un
setto interatriale e un dotto arterioso restrittivi, i feti
con cuore sinistro ipoplasico con setto interatriale restrittivo e i feti idropici nei quali possa essere
indicato un drenaggio immediato dei liquidi accumulati nello spazio toracico e addominale e infine
alcuni tipi di aritmie che rendono impossibile il monitoraggio fetale mediante cardiotocografia convenzionale durante il travaglio.
Storia naturale
intrautero delle CC
La crescente esperienza nella diagnosi prenatale, supportata dai progressi avvenuti nella tecnica
ecocardiografica, ha reso possibile il riconoscimen-
to in utero della grande maggioranza dei difetti
cardiaci congeniti, oltre che una più approfondita
conoscenza dello sviluppo del cuore umano e del
meccanismo di formazione di molte forme di cardiopatia. I controlli ecocardiografici seriati sui feti
affetti da CC hanno dimostrato, inoltre, che molti
difetti a carico delle strutture cardiovascolari iniziano a svilupparsi intorno alla sesta-settima settimana
di gravidanza (ovvero nel periodo dell’embriogenesi del cuore) e continuano poi a evolvere nel secondo e nel terzo trimestre gestazionale, mentre altri
difetti possono svilupparsi o diventare evidenti solo
molto dopo il termine dell’embriogenesi. Durante il
corso della gravidanza si può assistere per esempio
allo sviluppo e/o alla progressione di un’ipoplasia
di una struttura cardiaca o di un’ostruzione a livello dell’inlet o dell’outlet ventricolare, o al contrario,
alla riduzione fino alla chiusura spontanea prenatale di un difetto interventricolare 121-130. Per molte
CC lo spettro di severità osservato nel neonato è
strettamente correlato al periodo gestazionale in
cui avviene l’alterazione della circolazione fetale.
Benché il feto cresca rapidamente durante tutta la
gravidanza, i cambiamenti maggiori nella struttura
corporea globale e in particolare nella struttura cardiaca avvengono nelle prime venti settimane di sviluppo prenatale 131. Questo spiega perché la riduzione del flusso sanguigno diretto a un ventricolo o
a una grande arteria possa creare conseguenze così
severe allo sviluppo e alla crescita di queste strutture, in particolare quando si verifichi prima della
ventesima settimana di gestazione 132. La stenosi
della valvola aortica o della valvola polmonare è un
esempio di lesione che, se compare in un’epoca gestazionale precoce, comporta lo sviluppo secondario di altre condizioni patologiche associate. Quando si verifica un’ostruzione a carico di una valvola
semilunare, inizialmente il ventricolo omolaterale
si ipertrofizza per poi dilatarsi e talvolta andare
incontro a disfunzione contrattile, con o senza insufficienza significativa della valvola atrio-ventricolare. In assenza di un rigurgito severo della valvola
atrio-ventricolare si assiste a una redistribuzione di
flusso attraverso il forame ovale verso il ventricolo
“sano”, mentre quello affetto, essendo ipoperfuso,
va incontro a una progressiva ipoplasia 122, 131, 132.
Se l’ostruzione critica dell’outlet ha luogo a meno
di quattordici settimane di gestazione, il ventricolo
coinvolto può presentarsi estremamente ipoplasico
già alla ventesima settimana, delineando un quadro
di classica atresia del cuore sinistro. Se la stenosi
critica della valvola semilunare si sviluppa, invece,
13
MARINO cardiologia pediatrica.indd 13
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
a ridosso della ventesima settimana gestazionale, il
ventricolo al termine della gravidanza sarà visibile,
anche se ipoplasico. Quando infine l’ostruzione a
carico dell’efflusso ventricolare progredisce lentamente e diventa severa solo nel terzo trimestre di
gestazione si osserva una crescita più regolare del
ventricolo, perché il suo riempimento è conservato
per un periodo più lungo. Nell’ostruzione critica del
cuore sinistro può anche verificarsi una progressiva
restrizione del forame ovale secondario all’aumento
della pressione in atrio sinistro fino a determinare
una condizione di ipertensione atriale sinistra che
può anche alterare il flusso nelle vene polmonari e
lo sviluppo del circolo vascolare polmonare 133. Nei
casi in cui l’ostruzione precoce dell’efflusso ventricolare si associa a una significativa insufficienza
della valvola atrio-ventricolare, l’aumento del precarico contribuisce solitamente a un normale sviluppo della camera ventricolare corrispondente.
Tuttavia questa condizione è molto meno ben tollerata dalla circolazione fetale, probabilmente per un
alterato riempimento del ventricolo sano.
Hornberger et al. hanno descritto anche il potenziale per lo sviluppo e/o la progressione in utero dell’ostruzione a carico di uno dei due efflussi
ventricolari nei feti affetti da lesioni tronco-conali e
in particolare da tetralogia di Fallot, ventricolo destro a doppia uscita e interruzione dell’arco di tipo
B. Come la stenosi valvolare aortica o polmonare,
anche l’ostruzione dell’outlet nelle lesioni troncoconali comporta una redistribuzione del flusso
che, attraverso il difetto interventricolare, è diretto
verso l’outlet non ostruito. In questa condizione,
può verificarsi la progressiva ipoplasia del vaso a
valle dell’ostruzione, che è più comune nelle lesioni tronco-conali associate a un’ostruzione severa
all’efflusso già presente in epoca precoce. Nel caso
di ostruzione del tratto di efflusso polmonare possono andare incontro a una ridotta crescita entrambi i rami polmonari. Poiché la progressiva ipoplasia
dei rami polmonari è osservata più frequentemente
nelle forme severe di tetralogia di Fallot che nella
stenosi polmonare o nell’atresia polmonare a setto
intatto, si ritiene che sia la fisiopatologia intrinseca
del letto vascolare polmonare nella tetralogia di Fallot a poter contribuire a questo pattern di crescita
anomalo, piuttosto che l’età gestazionale più precoce alla comparsa dell’ostruzione 123.
Come visto in precedenza anche la transposizione delle grandi arterie con setto interventricolare
intatto può evolvere in utero con una progressiva
restrizione del forame ovale e costrizione del dotto
arterioso nel corso del terzo trimestre di gestazione,
verosimilmente dovute alla fisiopatologia caratteristica della cardiopatia stessa 145. In questa condizione, infatti, le arterie polmonari ricevono sangue con
un contenuto di ossigeno più alto che può essere
sufficiente a ridurre le resistenze vascolari polmonari aumentando il flusso a livello del circolo polmonare e di conseguenza il ritorno venoso all’atrio
sinistro. L’aumentata pressione in atrio sinistro può
condurre alla restrizione del forame ovale, mentre
la redistribuzione del flusso attraverso i polmoni
e l’aumentata percentuale di ossigeno nel sangue
possono determinare una costrizione prematura del
dotto arterioso o almeno una riduzione del calibro
rispetto ai feti normali.
Anche l’insufficienza di una valvola semilunare
o atrio-ventricolare può evolvere in utero, comportando la comparsa di altre condizioni patologiche
associate. Hornberger in un lavoro pubblicato nel
1991 ha descritto la storia naturale intrauterina della malattia della valvola tricuspide associata a insufficienza valvolare, includendo sia l’anomalia di
Ebstein che la cosiddetta displasia della valvola tricuspide 134. Al controllo ecocardiografico seriato, si
è dimostrata una progressiva cardiomegalia interessante in particolare le sezioni destre, e talvolta un
quadro di idrope fetale con episodi subentranti di
flutter atriale correlati alla dilatazione atriale destra.
Nei feti affetti da insufficienza tricuspidalica importante è frequente anche il riscontro di ipoplasia dei
polmoni, considerato che il cuore, estremamente
ingrandito, va a occupare lo spazio intratoracico
necessario al normale sviluppo polmonare. Questo
studio fornisce, inoltre, un’ulteriore evidenza della
possibile evoluzione tardiva nel corso della gravidanza di una lesione cardiaca associata. Infatti, gli
autori documentano lo sviluppo di ostruzione a
carico del tratto di efflusso destro in feti che inizialmente avevano avuto diagnosi esclusivamente
di patologia tricuspidalica con flusso transpolmonare normale al Doppler pulsato e al color Doppler.
Secondo l’ipotesi eziologica proposta dagli autori,
il peggioramento dell’insufficienza valvolare può
determinare un’incapacità da parte del ventricolo
destro di sviluppare una pressione sistemica adeguata conducendo alla mancanza di flusso attraverso l’uscita ventricolare destra. Questa condizione comporterebbe, come risultato, la progressiva
ostruzione anatomica ovvero una stenosi polmonare o anche un’atresia polmonare che si manifestano nel secondo o terzo trimestre di gestazione.
In alternativa, può svilupparsi la cosiddetta atresia
14
MARINO cardiologia pediatrica.indd 14
17/09/15 07:54
1 – Cardiologia fetale
polmonare funzionale, in cui la valvola è anatomicamente pervia, ma non si apre in sistole proprio
per l’incapacità del ventricolo di generare una sufficiente pressione sistolica.
Counselling
Per counselling prenatale si intende il colloquio
che il cardiologo pediatra, esperto in cardiologia
fetale, ha con la coppia nel momento in cui avviene il riscontro di un’anomalia cardiaca fetale. Un
corretto counselling è importante quanto ottenere
una diagnosi precisa, anche se spesso gli è riservata un’attenzione inferiore a quella che merita. Gli
obiettivi principali del counselling sono di provvedere una diagnosi accurata della malformazione
(sempre che questa non sia già stata effettuata da
un’altra figura di esperto in cardiologia fetale), di
fornire un quadro chiaro e veritiero della prognosi a breve e lungo termine, e di esporre le diverse
possibilità di gestione e trattamento disponibili per
la cardiopatia in questione 135, 136. Il processo del
counselling è facilitato se preceduto da una preparazione adeguata della coppia da parte del collega
ostetrico che effettua l’esame di screening e che indirizza la donna in gravidanza dal cardiologo fetale. È indispensabile, infatti, che la coppia arrivi a
chi deve precisare la diagnosi e a chi deve fare il
counselling (che siano la stessa persona o due distinte poco importa), consapevole che potrà ricevere delle brutte notizie o che al contrario il sospetto
di malformazione potrà non essere confermato. Prima che inizi la valutazione specialistica mediante
ecocardiografia fetale, è opportuno informare la
coppia che l’esame ultrasonografico richiederà un
certo tempo e una certa concentrazione per esplorare tutte le strutture del cuore fetale e che quindi
non devono farsi intimorire da eventuali lunghi silenzi del medico. Utile, inoltre, che sappiano che
una spiegazione completa di quanto riscontrato e
delle conseguenti implicazioni sarà fornita solo al
termine dell’esame, una volta raccolti tutti i dati
rilevanti per la diagnosi. I medici in formazione e
gli osservatori presenti nell’ambulatorio dovrebbero essere dissuasi dal commentare l’esame in corso,
posticipando un eventuale insegnamento interattivo al momento della revisione delle immagini acquisite. È, inoltre, preferibile togliere il volume del
Doppler in presenza di un’anomalia fetale in quanto sentire il battito cardiaco rappresenta sempre
un’emozione molto forte per i genitori. Idealmente,
il counselling non dovrebbe aver luogo nella stessa
stanza in cui si è effettuato l’esame, ma in un ambiente diverso, tranquillo, dove il medico è seduto
di fronte alla coppia e dove non possa essere interrotto. Il counselling non deve essere un monologo,
ma un dialogo e per questo è utile iniziarlo chiedendo ai genitori che cosa è stato loro detto e che
cosa hanno capito fino a quel momento del percorso diagnostico intrapreso. Il “counsellor” deve
essere consapevole che l’ansietà e la paura provate dalla coppia possono spesso tradursi in ostilità
e rabbia, e deve inoltre tenere presente le possibili
difficoltà che la coppia stessa può avere nell’assorbire tante informazioni in uno stato di tale coinvolgimento emotivo. Dovrà, dunque, dimostrare
estrema disponibilità nel fornire la spiegazione più
chiara possibile, ripetendo eventualmente più volte i concetti fondamentali e accertandosi che siano
stati assimilati. Far ricorso a diagrammi o disegni
per spiegare il difetto cardiaco riscontrato, oltre che
fornire informazioni scritte relative alla diagnosi e
ai passaggi chirurgici principali, può essere utile ai
genitori aiutandoli a ricordare quanto spiegato loro,
una volta che abbiano lasciato l’ambulatorio. Dovrebbero inoltre essere anticipate domande molto
frequenti, quali il perché della presenza di un difetto cardiaco e il rischio di ricorrenza in un’ipotetica
nuova gravidanza. È importante, inoltre, rassicurare
i genitori sulla possibilità di ulteriori incontri con il
medico che ha effettuato il counselling, nel caso
in cui avessero nuove domande da porre una volta
terminato il colloquio iniziale. Molti cardiologi fetali preferiscono un approccio multidisciplinare che
coinvolga nella stessa seduta l’ostetrico, il genetista
e il cardiochirurgo in modo da fornire informazioni
addizionali. Tuttavia, per alcune coppie di genitori
questo approccio può risultare intimidatorio e può
essere più gradito incontrare singolarmente i vari
specialisti coinvolti. Un problema addizionale che
complica attualmente il processo del counselling,
è la consultazione da parte dei genitori di internet
che rappresenta una fonte di informazioni che talvolta può essere di aiuto alla comprensione della
patologia di cui è affetto il proprio bambino, ma
più spesso si rivela ingannevole, inappropriata e soprattutto fonte di confusione. Il “counsellor” deve
mettere in guardia la coppia e deve essere pronto a
spiegare ogni possibile discrepanza tra le informazioni ottenute da internet e quelle estrapolate dal
counselling stesso.
I dettagli del counselling dipenderanno dalla
precisa diagnosi cardiaca, tenuto conto dell’ampio
range di severità che contraddistingue le malforma-
15
MARINO cardiologia pediatrica.indd 15
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
zioni cardiache (da difetti che non richiedono alcun
trattamento come un piccolo difetto interventricolare muscolare, fino a cardiopatie complesse che
possono ricevere solo chirurgia palliativa), dalla
storia naturale della malformazione diagnosticata
durante la vita fetale, dalle opzioni chirurgiche a disposizione sia nel proprio ospedale che altrove, e
dall’eventuale associazione con altre malformazioni extracardiache. Accanto alla possibilità di continuare la gravidanza, il “counsellor” ha il dovere di
considerare anche la possibilità che la coppia decida di interrompere la gravidanza, qualunque sia
la propria idea al riguardo. L’interruzione volontaria
di gravidanza rappresenta una scelta legale nella
maggior parte dei Paesi sviluppati, anche se variano l’età gestazionale entro cui essa è consentita e
alcune impostazioni concettuali di legge. Generalmente è permessa fino alla ventiquattresima settimana di gestazione, ma in diversi Paesi europei o
stati americani è possibile un’interruzione oltre la
ventiquattresima settimana, sempre che venga documentata una malformazione fetale grave tale da
compromettere in modo significativo la durata e la
qualità di vita del nascituro. In Italia, è l’articolo 6
della legge 194/78 a disporre in materia di interruzione di gravidanza oltre i novanta giorni (ovvero
oltre le dodici settimane di età gestazionale). Dopo
i novanta giorni – recita l’articolo – una gestazione può essere interrotta solo per gravi problemi
di salute della gestante che possano mettere a repentaglio la sua sopravvivenza, o per la presenza
di severe anomalie del feto che alterano lo stato di
salute fisica o psichica della gestante stessa. Come
si evince dall’articolo riportato, in Italia non è permesso l’aborto “eugenetico”, ovvero l’aborto esclusivamente legato alle anomalie del feto.
La decisione di interrompere la gestazione deve
essere presa solo ed esclusivamente dalla coppia
di genitori sulla base delle informazioni diagnostiche e prognostiche fornite loro dall’equipe che ha
eseguito la seduta di counselling prenatale. Spesso,
invece, capita che il “counsellor” influenzi la decisione finale della coppia in base al proprio modo
di comunicazione che non risulta completamente
imparziale (directive counselling). È, invece, dovere
di chi fa il counselling sostenere i genitori in un momento tanto delicato quale quello di decidere sulla
successiva gestione della gravidanza, ascoltando i
dubbi e le perplessità di entrambi al fine di aiutarli
nella scelta a loro più consona. Indipendentemente dall’opinione della coppia circa l’interruzione di
gravidanza, questa non è mai una decisione che
viene presa con leggerezza ed è sempre causa di
grande angoscia. Per questo la coppia si aspetta,
comunque, un supporto totale e senza riserve da
parte del “counsellor”, indipendentemente da quella che può essere la sua opinione circa la decisione
da loro intrapresa.
Prospettive di terapia
intervenzionale intrautero
L’obiettivo comune della terapia fetale, sia essa
mirata alla cura di una patologia cardiaca o di altro
organo, è quello di fornire un trattamento precoce
del feto affetto per correggere il problema anatomico, o almeno per ridurre l’entità dei danni secondari che solitamente si verificano quando vi è una
condizione di anatomia o fisiologia anomala 137. Attualmente, il più comune intervento per squilibrio
circolatorio è quello praticato per evitare la morte fetale nella sindrome da trasfusione feto-fetale,
mentre resta limitato il numero di feti cardiopatici
sottoponibile a intervento in epoca prenatale, pur
potendo anch’essi andare incontro a idrope fino a
morte intrauterina. A oggi, gli interventi proponibili
in utero per cardiopatia sono la valvuloplastica nei
feti con stenosi critica o atresia della valvola aortica
o della valvola polmonare, e la settostomia atriale
con palloncino in quelli con chiusura precoce del
setto interatriale associata a cardiopatie nelle quali la sopravvivenza neonatale dipende dalla pervietà della stessa, quali la trasposizione semplice
delle grandi arterie o la sindrome del cuore sinistro ipoplasico con atresia della valvola aortica o
mitrale 10, 11, 138-142. Infine è stato riportato qualche
caso di efficace ma solo temporanea stimolazione
cardiaca in feto con scompenso cardiaco e idrope
secondario a blocco atrio-ventricolare totale da anticorpi materni anti-Ro/La 143.
Il razionale per l’intervento fetale di valvuloplastica è supportato da alcune specifiche convinzioni.
La prima deriva dai dati chirurgici che dimostrano
come il precoce ristabilimento di un normale flusso
anterogrado attraverso l’uscita ventricolare promuova la crescita delle strutture cardiache interessate.
In effetti, a conferma di questi risultati, lo Z score
calcolato per le singole strutture cardiache dei feti
con ostruzione all’efflusso ventricolare, è suggestivo per una ridotta crescita in utero del cuore rispetto agli altri organi 10, 144-146. È stato, inoltre, accertato
che un meccanismo di danno secondario a carico
del ventricolo interessato da ostruzione deriva dalla
ridotta perfusione coronarica al miocardio conse-
16
MARINO cardiologia pediatrica.indd 16
17/09/15 07:54
1 – Cardiologia fetale
guente proprio ai valori elevati di pressione endocavitaria. Infatti, i feti con stenosi aortica critica e
sindrome del cuore sinistro ipoplasico hanno un
flusso anterogrado attraverso la radice aortica ridotto o addirittura assente e la perfusione coronarica
dipende in modo esclusivo o prevalente dal flusso retrogrado proveniente dal dotto arterioso che,
oltretutto, è ostacolato da un’associata ipoplasia
estrema dell’arco aortico. Considerato che nei feti
almeno il 75% della resistenza al flusso coronarico
dipende dalla pressione intracardiaca, ne deriva che
l’aumento della pressione telediastolica ventricolare associata alla stenosi aortica è un determinante
importante di una ridotta perfusione coronarica,
oltre che dello sviluppo di fibrosi endocardica 137.
Un ulteriore meccanismo di danno ventricolare è
costituito, poi, dalla risposta del miocardio fetale
all’aumento del postcarico secondario alla chiusura
valvolare aortica. L’aumento della pressione atriale sinistra correlata alla progressione della stenosi
aortica è anche responsabile dell’alterato sviluppo
della vascolatura polmonare fetale 133, 147. Un’altra
convinzione su cui si basa il razionale per la valvuloplastica è che un feto con una circolazione biventricolare possa raggiungere una qualità di vita
migliore di uno con una circolazione univentricolare. Per qualità di vita migliore non s’intende solo
l’abilità funzionale superiore, ma anche la prognosi
a livello neurologico. Come detto in precedenza,
infatti, in caso di un’ostruzione a carico delle strutture sinistre del cuore, come nella stenosi aortica
critica o nella sindrome del cuore sinistro ipoplasico, si verifica un’inversione del flusso a livello
dell’arco aortico, per cui il cervello fetale è perfuso
in maniera retrograda con sangue meno ossigenato.
Nella stenosi aortica critica, in particolare, questa
ipoperfusione coinvolgerebbe soprattutto il lato sinistro, tributario di succlavia e carotide sinistre, il
che può spiegare la maggior frequenza in questi
pazienti di danni cerebrali che coinvolgono la porzione sinistra dell’encefalo 9, 98, 99, 148. L’intervento
di valvuloplastica in epoca fetale, migliorando la
perfusione cerebrale e ristabilendo il flusso anterogrado attraverso l’arco aortico, potrebbe dunque
ridurre teoricamente il rischio di danno neurologico
associato alla cardiopatia.
La settostomia atriale con palloncino rappresenta invece una procedura invasiva di palliazione nei
feti affetti da TGA semplice o da sindrome del cuore sinistro ipoplasico. In questi ultimi infatti l’ispessimento fino anche la chiusura precoce del setto
interatriale comporta talvolta lo sviluppo d’idrope
con conseguente morte intrauterina 97, 102, 139. Nei
feti che anche sopravvivano alla chiusura precoce
del setto interatriale fino al termine della gravidanza, si riscontrano comunque anomalie del circolo
venoso polmonare fino a un’ipertensione polmonare irreversibile, che rendono complesse le manovre
rianimatorie alla nascita, così come lo svezzamento
dal ventilatore 133, 147. Inoltre, nei feti con sindrome
del cuore sinistro ipoplasico destinati a chirurgia
palliativa, un setto chiuso o molto restrittivo, può
danneggiare a tal punto la vascolarizzazione polmonare da rendere controindicato l’intervento di
Norwood classico. Nei pazienti con TGA semplice, un setto interatriale chiuso può determinare un’ipossia neonatale severissima da mancato
mixing atriale ed edema polmonare emorragico
con conseguente morte immediatamente dopo la
nascita, prima che possa essere effettuata una settostomia atriale anche in caso di diagnosi prenatale.
Le tecniche di valvuloplastica fetale variano lievemente da un centro all’altro e vengono descritte
da ciascuno nei dettagli nelle rispettive pubblicazioni 9, 10, 140-142. La maggior parte delle procedure
è stata effettuata in feti di età gestazionale compresa tra ventuno e trenta settimane, mediante un approccio percutaneo e sotto guida ecografica, sfruttando la tecnica Seldinger. Alcuni hanno preferito
procedere sotto anestesia generale materna, altri in
anestesia locale con sedazione della madre. Tutti
hanno somministrato anestesia al feto, solitamente
Fentanyl, sia per via intramuscolo che direttamente
nel cuore del bambino al momento della puntura
fetale. Quando l’acquisizione delle immagini ecografiche risultava limitata dall’habitus materno o
dalla posizione fetale, il gruppo di Boston ha preferito eseguire la procedura in mini-laparotomia ottenendo contemporaneamente immagini dirette del
cuore fetale attraverso la parete uterina e una più
facile manipolazione del feto 10. In commercio non
è disponibile un’attrezzatura dedicata a questo tipo
di procedura e l’approccio alla circolazione fetale
nei casi riportati in letteratura è avvenuto passando
dalla parete dell’utero al torace del feto e direttamente nel ventricolo o nell’infundibolo (nel caso di
atresia polmonare). L’ago deve essere lungo almeno 15 cm con un introduttore flessibile che possa
essere lasciato nel cuore del feto mentre il pallone
viene fatto scorrere su guida al suo interno. Poiché
la valvola polmonare è più grande di quella aortica,
è necessaria un’attrezzatura di dimensioni maggiori
per interventi di valvuloplastica a destra. Il rapporto
pallone/valvola oggi utilizzato è maggiore di quel-
17
MARINO cardiologia pediatrica.indd 17
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
lo in epoca postnatale, sia per la natura più grossolana della stenosi valvolare in epoca fetale, che
rende più difficile la dilatazione e più facile la restenosi, sia per la documentata tolleranza e reversibilità dell’insufficienza valvolare postprocedurale
nel feto. Come accennato in precedenza, il pattern
di crescita del feto non è lineare ma molto più rapido nella fase precoce di gestazione, anche se in
realtà la mancata crescita e il danno secondario si
sviluppano in maniera più progressiva. Per quanto
riguarda, dunque, la tempistica di esecuzione della procedura, la tendenza comune ai diversi centri è quella di proporre l’intervento poco dopo la
diagnosi, tenuto conto del tempo necessario per
eseguire il counselling e per prendere una decisione da parte della coppia. Lo svantaggio di effettuare
la procedura precocemente è rappresentato dalla
probabilità che questa si debba ripetere in una fase
più tardiva della gravidanza per avvenuta restenosi.
Tuttavia più a lungo permane una condizione di sovraccarico pressorio intraventricolare e di anomala
emodinamica, più probabile è la comparsa di fibrosi, calcificazioni e ipoplasia irreversibile nel ventricolo affetto del feto.
Diversi centri hanno eseguito la valvuloplastica
aortica con variabili percentuali di successo e complicanze. La serie più ampia pubblicata è quella del
Children Hospital di Boston in cui 100 feti sono
stati sottoposti a valvuloplastica aortica per stenosi
aortica severa a un’età gestazionale media di 23.8
settimane. La procedura ha avuto successo in 77
casi e di questi 38 (43% dei nati vivi) ha raggiunto
una correzione biventricolare 141. Lo stesso gruppo
di Boston qualche anno prima aveva riportato i dati
preliminari circa la dilatazione percutanea del setto
interatriale nei pazienti con TGA od ostruzione del
cuore sinistro 139. Successivamente sempre lo stesso
gruppo ha riportato il posizionamento con successo
di uno stent a livello del setto interatriale in un feto
con ipoplasia del cuore sinistro che è poi sopravvissuto fino al tempo dell’intervento di Norwood 149.
Huhta et al. hanno invece proposto i criteri per
candidare a valvuloplastica i feti affetti da atresia
polmonare a setto intatto, proponendo di valutare
in particolare la vena ombelicale per verificare la
presenza di pulsazione venosa, il dotto venoso per
la presenza o meno di flusso retrogrado telediastolico, le dimensioni e la funzione sisto-diastolica del
cuore (sulla base della frazione di accorciamento,
del jet di rigurgito, di un dP/dT <400, o ancora di
una curva di riempimento diastolico monofasico o
la presenza di idrope) 150. La procedura di valvu-
loplastica ha dimostrato di far regredire l’idrope e
di prolungare la gravidanza, ma sono rari i feti che
presentino condizioni ideali per essere sottoposti
alla procedura secondo i criteri elencati 11. A oggi
sono stati riportati i risultati di sei procedure di questo tipo in Europa con quattro successi tecnici, permettendo una correzione biventricolare in due casi
e una palliazione univentricolare negli altri casi che
presentavano una grossa fistola coronarica 142.
Riassumendo, la progressione della patologia
cardiaca fetale è rapida nei casi di stenosi e atresia
aortica o polmonare. Un certo numero di bambini
va incontro a decesso nel periodo neonatale nonostante un intervento chirurgico tecnicamente efficace, a causa dei danni a carico del circolo polmonare e del miocardio secondari a queste forme severe
di cardiopatia. In casi accuratamente selezionati,
l’intervento in epoca fetale è sembrato promettente,
in particolare se programmato in un’epoca di gravidanza precoce piuttosto che tardiva.
Terapia intrautero delle aritmie
Tutte le aritmie fetali, a eccezione delle extrasistoli, sono associate a un rischio moderatamente
alto di distress fetale 151. Il riscontro di un’aritmia
cardiaca comporta dunque la necessità di stabilire
se sia opportuna una terapia e, in caso affermativo, quale tipo di terapia effettuare, avendo come
primo obiettivo la prevenzione o la risoluzione
di un’eventuale compromissione emodinamica e
quindi la prevenzione dell’idrope fetale. Le aritmie fetali possono essere considerate benigne o
maligne a seconda della loro potenzialità di causare morte fetale o morbilità. L’extrasistolia fetale
viene definita un’aritmia benigna che, come detto,
non richiede alcun tipo di terapia, anche in caso
di extrasistoli numerose. Queste ultime richiedono, tuttavia, un monitoraggio attento da parte del
ginecologo, tale da riconoscere tempestivamente
la comparsa di una tachicardia sopraventricolare
sostenuta (1-2% dei casi di extrasistoli). In caso di
tachicardia fetale (Fig. 1.6), prima di iniziare una terapia farmacologica, è innanzitutto necessario valutare lo stato emodinamico del feto; questo tipo
di aritmie può infatti essere ben tollerato durante
la vita fetale e frequenze cardiache elevate, tachiaritmie di lunga durata e cardiopatie congenite
associate rappresentano solo dei fattori di rischio
relativi. Qualsiasi terapia antiaritmica è inoltre gravata da rischi che in funzione del tipo di farmaco,
delle dosi e della via di somministrazione riguar-
18
MARINO cardiologia pediatrica.indd 18
17/09/15 07:54
1 – Cardiologia fetale
Figura 1.6 Flutter atriale. Color M-Mode. Il fascio di
ultrasuoni, allineato sul VS (in alto) e sull’AD, rivela la
presenza di un Flutter atriale con una conduzione atrioventricolare 2:1 risultante in una frequenza atriale di circa
460 bpm e di una frequenza ventricolare di circa 230
bpm.
dano sia il feto (effetto proaritmico) che la madre.
In ogni singolo caso si dovrà dunque valutare il
rapporto rischio/beneficio di utilizzare un farmaco “maggiore” per ottenere il ripristino del ritmo
sinusale rispetto a quello di utilizzare un farmaco
“minore” che consenta un semplice controllo della frequenza cardiaca pur persistendo l’aritmia. La
terapia con antiaritmici dovrebbe essere iniziata in
ambiente ospedaliero con basse dosi che possono
essere aumentate gradualmente, monitorando attentamente la risposta al farmaco del feto e della
madre. Prima di iniziare la terapia antiaritmica, dovrebbero essere escluse, tramite la storia clinica e
un elettrocardiogramma, persistenti aritmie materne e la presenza di QT lungo. Quando la conversione a ritmo sinusale non è raggiunta attraverso
la somministrazione materna di uno o più farmaci
antiaritmici, è possibile optare per la terapia fetale
diretta mediante la somministrazione intraombelicale, intra-amniotica, intraperitoneale o intramuscolare 152, 153. La via ombelicale permette un accesso diretto alla circolazione fetale e perciò una
rapida risposta alla terapia, ma sottopone il feto a
un rischio di morbilità e mortalità significativo. Le
altre modalità di iniezione sono meno rischiose per
il feto e permettono un rilascio più graduale del
farmaco. La terapia fetale diretta dovrebbe, in ogni
modo, essere utilizzata solamente in caso di tachicardia complicata da idrope resistente a una multiterapia transplacentare. In linea generale i farmaci
più spesso utilizzati nella pratica corrente sono:
–– digitale: permette un ripristino del ritmo sinusale
nel 60-80% dei casi senza scompenso, ma solo
nel 20-40% dei casi con idrope. In questi casi
tuttavia, da solo o associato ad altri farmaci, consente di controllare la FC e di migliorare lo stato
di scompenso. Non sono riportati effetti proaritmici, né importanti effetti collaterali materni
(dosi: per os, attacco 1-2 g/die in 2-3 somministrazioni, mantenimento 0,5 g/die in 1-2 somministrazioni; ev, attacco 0,5-1 g/die in 2-3 dosi,
mantenimento 0,125-0,25 g/die. Digossinemia
materna ideale 1,8 ng/l);
–– Flecainide da sola o associata a digitale, a dosi
dimezzate. Ha dimostrato un tasso di conversione in una settimana dell’80% dei casi di tachicardia sopraventricolare (TSV) associati a idrope;
se impiegata in caso di flutter atriale è tuttavia a
rischio di morte intrauterina per riduzione della
frequenza atriale con conduzione 1:1 e fibrillazione ventricolare. Necessita di attento monitoraggio materno (dosi per os attacco 150-300 mg/
die; mantenimento: 75-150 mg/die);
–– Amiodarone: ha un’efficacia complessiva in circa il 60% dei casi, indipendentemente dal tipo
di aritmia e dalla presenza o meno di scompenso cardiaco. In particolare, risulta utile in caso di
feti idropici con TSV da rientro refrattarie al trattamento transplacentare e/o diretto intramuscolare con digitale o con altri antiaritmici 154. Gli
svantaggi correlati all’utilizzo di questo farmaco
sono la necessità di alte dosi (attacco 800-1600
mg/die; mantenimento 200-800 mg/die), la massima azione del farmaco tardiva e la frequenza
elevata di effetti collaterali materni e fetali (ipotiroidismo neonatale nel 50% dei casi trattati, per
lo più transitorio);
–– Sotalolo: rappresenta il farmaco di prima scelta nel trattamento del flutter atriale associato o
meno a idrope fetale, con tasso di successo del
65% come terapia singola e dell’80% se associato
a digitale. Nelle TSV è molto meno efficace. Sono
stati tuttavia descritti casi di morte improvvisa
endouterina, per lo più in feti idropici con TSV,
forse per fibrillazione ventricolare. Per ridurre il
rischio proaritmico si raccomanda bassa dose iniziale, incrementandola gradatamente, sotto stretto monitoraggio (dosi per os 160-300 mg/die).
Sulla base della nostra esperienza e dei dati riportati in letteratura, siamo soliti applicare il seguente schema terapeutico:
–– in caso di tachiaritmia intermittente (periodi ripetuti ma di durata inferiore a 30 minuti) senza
scompenso cardiaco, ci limitiamo a controlli bi-
19
MARINO cardiologia pediatrica.indd 19
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
settimanali del feto per valutare l’eventuale comparsa di segni precoci di insufficienza cardiaca
(dilatazione degli atri, insufficienza valvole AV,
inversione del flusso a livello della vena cava inferiore);
–– in caso di tachiaritmia persistente o incessante
senza segni di scompenso, somministriamo digitale per os e in caso di successo della terapia
proseguiamo con dosi di mantenimento; in caso
invece di insuccesso, se la gravidanza è oltre la
trentaseiesima settimana e la FC è >220 bpm,
anticipiamo il parto (che sarà effettuato mediante taglio cesareo), mentre se la gravidanza è prima della trentaseiesima, la frequenza cardiaca
è controllata e non ci sono segni di scompenso
neppure iniziali, proseguiamo con la sola digitale, eseguendo controlli bisettimanali del feto;
in caso contrario, aggiungiamo il farmaco maggiore più adeguato (Flecainide se si tratta di una
tachicardia da rientro atrio-ventricolare; Sotalolo
se Flutter atriale; Cordarone se diagnosi dubbia),
dimezzando la dose della digitale;
–– in caso di tachiaritmia persistente o incessante
con scompenso cardiaco, se la gravidanza è oltre la trentacinquesima settimana, a seconda del
grado di idrope fetale può essere indicato sia il
parto anticipato con taglio cesareo che un tentativo di cardioversione della aritmia e di riduzione dello scompenso con digitale a metà dose e
il farmaco maggiore più adeguato; se prima della
trentacinquesima settimana tentiamo l’utilizzo
del farmaco maggiore più efficace per il tipo di
aritmia eventualmente associato in un secondo
tempo alla digitale.
Per quanto riguarda il blocco AV completo (Fig.
1.7-1.8) nel 40% dei casi fetali esso è associato a
malformazioni cardiache (quali canale atrio-ventricolare sbilanciato, cuore univentricolare, trasposizione corretta delle grandi arterie), mentre nei
restanti casi non si osservano malformazioni ma è
spesso presente una malattia autoimmune materna,
quale lupus o sindrome di Sjogren latente o manifesta 155. Dovrà quindi essere ricercata, in tutti i casi
di BAV congenito isolato, la presenza di anticorpi
anti-SSA e anti-SSB nel sangue materno. Il trattamento di un feto in uno stadio irreversibile di danno nodale AV è primariamente quello di mitigare o
prevenire la concomitante infiammazione miocardica e quello di aumentare la frequenza cardiaca
fetale. A differenza del prednisone, il desametasone e il betametasone sono solo minimamente me-
Figura 1.7 Falso BAV. Color M-mode in corrispondenza
della scansione 4 camere. Il fascio di ultrasuoni allineato
in modo da attraversare contemporaneamente il VS (in
alto) e l’AD (in basso), permette di evidenziare le contrazioni atriali e quelle ventricolari molto più lente delle
precedenti. Ogni contrazione atriale è seguita a breve
distanza da un’altra contrazione atriale (extrasistole atriale) tanto precoce da essere bloccata a livello del nodo
atrio-ventricolare e quindi non condotta ai ventricoli. Ne
risulta una frequenza cardiaca bassa (solitamente 70-100
bpm) apparentemente regolare ma con un pattern di contrazione atriale irregolare (breve-lungo).
Figura 1.8 BAV totale. M-mode in corrispondenza della
scansione 4 camere. Il fascio di ultrasuoni, allineato in
modo da attraversare contemporaneamente il VS e l’AD,
permette di visualizzare le contrazioni atriali (in basso) a
normale frequenza e quelle ventricolari (in alto) molto più
lente e dissociate dalle precedenti.
tabolizzati dalla placenta, e diventano utili quando
è desiderato un trattamento di tipo antinfiammatorio. La somministrazione materna di desametasone
si è rivelata in grado di migliorare il blocco AV fetale, l’idrope e la disfunzione miocardica 156-159. Vari
beta-stimolanti sono stati utilizzati per incrementare la frequenza cardiaca fetale e la contrattilità miocardica con vari gradi di successo. Recentemente
è stato introdotto un protocollo di trattamento del
20
MARINO cardiologia pediatrica.indd 20
17/09/15 07:54
1 – Cardiologia fetale
BAV completo che si è rivelato in grado di migliorarne la prognosi 157. Secondo tale protocollo non
appena viene posta la diagnosi di BAV completo
immuno-mediato deve essere instaurata una terapia
materna con desametasone (4 mg/die), mantenuta, quando possibile, per tutta la durata della gravidanza. Un agente beta-stimolante (salbutamolo
orale 10 mg x 3/die) viene aggiunto se la frequenza cardiaca media del feto è inferiore a 55 battiti
al minuto. La gravidanza deve essere strettamente
sorvegliata sia con ecografia ostetrica che con ecocardiografia fetale. È importante, in particolare, ricordare che la terapia steroidea prenatale comporta
il rischio di oligoidramnios e, conseguentemente, di
possibile parto pretermine nel 20% dei casi di trattamento cronico, per cui è necessario monitorare
con attenzione la quantità di liquido amniotico durante tutto il corso della gravidanza. Tale gestione
clinico terapeutica materno-fetale consente solitamente di arrivare al terzo trimestre di gravidanza,
programmando il parto attorno alla trentasettesima
settimana di gestazione. È, comunque, indispensabile che questo avvenga sempre in un centro di terzo livello che possa garantire la possibilità, anche in
sala parto, di una stimolazione cardiaca temporanea (esterna o endocavitaria), considerato che casi
di BAV ben tollerati durante la gravidanza possono precipitare subito dopo la nascita per marcata
riduzione della frequenza cardiaca o per severa
compromissione delle condizioni di compenso cardiocircolatorio 158. Poiché il BAV completo si manifesta solamente nell’1-2% delle madri portatrici di
anticorpi anti-Ro/La, viene ancora dibattuta la reale
necessità di una profilassi cortisonica in caso di prima gravidanza. La terapia cortisonica con eventuale plasmaferesi deve, al contrario, essere instaurata
immediatamente nella fase precoce di “miocardite”
e in ogni modo nei BAV di recente insorgenza nella
speranza di una regressione della lesione infiammatoria 159, 160. Anche il BAV di secondo grado si
può associare a malattie del collagene materno e
a difetti strutturali cardiaci. Il trattamento è simile a
quello raccomandato per il BAV completo in presenza di malattie del collagene; in questi casi, è necessario un attento monitoraggio per la possibilità
di progressione del grado di blocco.
BIBLIOGRAFIA
1. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002;39:
1890-900.
2. Abu-Harb M, Hey E, Wren C. Death in infancy
from unrecognized congenital heart disease. Arch
Dis Child 1994;71:3-7.
3. Wren C, Reinhardt Z, Khawaja K. Twenty-year
trends in diagnosis of life-threatening neonatal
cardiovascular malformation. Arch Dis Child Fetal
Neonatal Ed. 2008;93: F33-5.
4. Perry LW, Neill CA, Ferencz C, Rubin JD, Loffredo CA. Infants with congenital heart disease:
the cases. In: Ferencz C, Rubin JD, Loffredo CA,
Magee CA, eds. Epidemiology of Congenital Heart
Disease. The Baltimore-Washington Infant Study
1981-1989. Perspectives in Pediatric Cardiology.
Armonk, NY: Futura. 1993;4: 33-62.
5. Hoffman JIE, Christianson R. Congenital heart disease in a cohort of 19502 births with long-term
follow-up. Am J Cardiol 1978;42:641-7.
6. Allan LD. Cardiac anatomy screening: what is the
best time for screening in pregnancy? Curr Opin
Obstet Gynecol 2003;15:143-6.
7. Tegnander E, Eik-Nes SH. The examiner’s ultrasound experience has a significant impact on the
detection rate of congenital heart defects at the
second-trimester fetal examination. Ultrasound
Obstet Gynecol 2006;28:8-14.
8. Kumar RK, Newburger JW, Gauvreau K, Kamenir
SA, Hornberger LK. Comparison of outcome when
hypoplastic left heart syndrome and transposition of the great arteries are diagnosed prenatally
versus when diagnosis of these two conditions
is made only postnatally. Am J Cardiol 1999;86:
1649-53.
9. Mahle WT, Clancy RR, McGaurn SP, Goin JE,
Clark BJ. Impact of prenatal diagnosis on survival and early neurologic morbidity in neonates
with hypoplastic left heart syndrome. Pediatrics
2001;107:1277-82.
10. Tworetzky W, Wilkins-Haug L, Jennings RW, van
der Velde ME, Marshall AC, Marx GR et al. Balloon dilation of severe aortic stenosis in the fetus: potential for prevention of hypoplastic left
heart syndrome candidate selecion, technique,
and results of successful intervention. Circulation
2004;110:2125-2131.
11. Tulzer G, Arzt W, Franklin RC, Loughna PV, Mair
R, Gardiner HM. Fetal pulmonary valvuloplasty for
critical pulmonary stenosis or atresia with intact
septum. Lancet 2002;360:1567-1568.
12. Koren G, Edwards MB, Miskin M. Antenatal
sonography of fetal malformations associated with
drugs and chemicals: a guide. Am J Obstet Gynecol 2007;134:256-7.
13. Nora JJ. Causes of congenital heart diseases: old
and new modes, mechanism, and models. Am
Heart J 1993;125:1409-19.
14. Pierpoint ME, Basson CT, Benson W, Gelb BD,
Giglia TM, Goldmuntz E, et al. Genetic basis for
congenital heart defects: current knowledge: a
scientific statement from the American Heart congenital Cardiac Defects Committee, Council on
Cardiovascular Disease in the Young. Circulation
2007;115:3015-38.
21
MARINO cardiologia pediatrica.indd 21
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
15. Payne R, Johnson M, Grant J, Strauss A. Towards
a molecular understanding of congenital heart disease. Circulation 1995;91:494-504.
16. Calcagni G, Digilio MC, Sarkozy A, Dallapiccola
B, Marino B. Familial recurrence of congenital
heart disease: an overview and review of the literature. Eur J Pediatr 2007;166:111-16.
17. Allan L, Dangel J, Fesslova V, Marek J, Mellander
M, Oberhansli I, et al. Recommandations for the
practice of fetal cardiology in Europe. Cardiol
Young 2004;14:109-114.
18. Rowland TW, Hubbel JP Jr, Nadas AS. Congenital
heart disease in infant of diabetic mothers. J Pediatr 1973;83:815-820.
19. Levy HL Waisbren SE. Effects of untreated and
treated pregnancies. N Engl J Med 1980;303:12021208.
20. Levy HL Waisbren SE. Effects of untreated maternal phenilketonuria and hyperphenylalaninemia
on the fetus N Engl J Med 1983;309:1269 1274.
21. Lee LA, Coulter S, Erner S, Chu H. Cardiac immunoglobulin deposition in congenital heart block associated with maternal anti-RO antibodies. Am J
Med 1987;83:793-796.
22. Dudgeon JA. Maternal rubella and its effect on the
fetus. Arch Dis Child. 1967;45:63.
23. Sever JL. Viruses and the fetus. Int J Gynecol Obstet 1970;8: 763-769.
24. Tikkanen J, Heinonen OP. Maternal exposure to
chemical and physical factors during pregnancy
and cardiovascular malformation in the offspring.
Teratology 1991;43:591-600.
25. Jacobson SJ, Jones K, Johnson K, Ceolin L, Kaur
P, Sahn D, et al. Prospective multicenter study of
pregnancy outcome after lithium exposure during
first trimester. Lancet 1992;339:530-533.
26. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation, 4th ed. 1994.
27. Jones KL, Smith DW, Ulleland CN, Streissguth AP.
Pattern of malformation in offspring of alchoholic
mothers. Lancet 1973;1:7815.
28. Rosa FW. Retinoic acid embryopathy. N Engl J
Med 1986;315:262.
29. Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, et al. Congenital heart disease
caused by mutations in the transcription factor
NKX2-5. Science 1998;281:108-111.
30. Van der Burght I and Brunner H. Genetic heterogeneity in Noonan syndrome: evidence for
an autosomal recessive form. Am J Med Genet
2000;94:46-51.
31. De Luca A, Sarkozy A, Consoli F et al. Familial
transposition of the great arteries caused by multiple mutations in laterality genes. Heart 2010;
96:673-7.
32. Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins
JA, Soults J, et al. Mutations in human cause limb
and cardiac malformation in Holt-Oram sindrome.
Nat Genet 1997;15:30-34.
33. Fokstuen S, Arbenz U, Artan S, Dutly F, Bauersfeld
U, Brecevic L, et al. 22q11 deletions in a series of
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
patients with non selective congenital heart defects: incidence, type of defects and parental origin. Clin Genet 1998;53:63-69.
Yamagishi H, Garg V, Matsuoka R, Thomas T,
Srivastava D. A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science 1999;283:1158-61.
Berend SA, Spikes AS, Kashork CD, Wu JM, Daw
SC, Scambler PJ, et al. Dual-probe fluorescence in
situ hybridization assay fro detecting deletions associated with VCFS/Di George syndrome I and Di
George syndrome II loci. Am J Med Genet 2000;
91:313-317.
Wang SM, Schinzel A, Kotzot D, Balmer D, Balmer D, Casey R, et al. Molecular and clinical correlation study of Williams-Beuren syndrome. Am J
Med Genet 1999;86:34-43.
Goldmuntz E, Clark BJ, Mitchel LE, Jawad AF, Cuneo BF, Reed L, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am
Coll Cardiol 1998;32:499-501.
Emery and Rimoin’s principle of Medical Genetics.
Rimoin D, Connor JM, Pyeritz RE, eds. Churchill
Livingstone, 1997.
Nora JJ. From generational studies to a multilevel
genetic-enviromental interaction. J Am Coll Cardiol 1994;23:1468-71.
Burn J, Coffey R, Allan LD et al. Isomerism: a genetic analysis. Pp 1126-28. In Doyle E.F., Engle
MA, Gersony WM el al. (eds): Paediatric cardiology. Springer Verlag, New York, 1986.
Gardner RJM, Sutherland GR. Chromosome Abnormalities and Genetic Counselling. eds. Oxford
Monographs on Medical Genetics n. 29,1996.
Van Karnebeek CDM, Hennekam RCM. Associations between chromosomal anomalies and congenital heart defects. Am J Med Genet 1999;84:
158-166.
Buskens E, Grobbee DE, Frohn-Mulder IME, Stewart PA, Juttmann REM Wladimiroff JW, Hess J.
Efficay of routine fetal ultrasound screening for
congenital heart disease in normal pregnancy. Circulation 1996;94:67-72.
Todros T, Faggiano F, Chiappa E, Gaglioti P, Mitola
B, Sciarrone A, et al. Accuracy of routine ultrasonography in screening heart disease prenatally.
Prenat Diagn 1997;17:901-906.
The International Society of Ultrasound in Obstetrics and Gynecology Guidelines. Cardiac screening examination of the fetus: guidelines for performing the ‘basic’ and ‘extended basic’ cardiac
scan. Ultrasound Obstet Gynecol 2006;27:107-13.
Vergani P, Mariani S, Ghidini A, Schiavina R,
Cavallore M, Locatelli A, et al. Screening for congenital heart disease with the four-chamber view
of fetal heart. Am J Obstet Gynecol 1992;167:
1000-1003.
Allan L. Congenital Heart Disease: Antenatal diagnosis of heart disease. Heart 2000;83:367-370.
Paladini D. Prenatal screening of congenital heart
disease between ethics and cost-effectiveness.
Time for a change in current prenatal ultrasound
22
MARINO cardiologia pediatrica.indd 22
17/09/15 07:54
1 – Cardiologia fetale
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
screening policies? Ultrrasound Obst Gynecol
1999;4:225-28.
Oggè G, Gaglioti P, Maccanti S, Faggiano F, Todros T. Prenatal screening for congenital heart disease with four-chamber and outflow tract views:
a multicenter study. Ultrasound Obstet gynecol
2006;28:779-84.
Gembruch U. Prenatal diagnosis of congenital
heart disease. Prenat Diagn 1997;17:1283-1298.
Hyett JA, Perdu M, Sharland GK, Snijders RJM,
Nicolaides KH. Using fetal nuchal translucency to
screen for major congenital heart defects at 10-14
weeks of gestation: population based cohort study.
BMJ 1999;318:81-85.
Greenwood RD, Rosenthal A, Parisi L, Fyler DC,
Nadas AS. Extracardiac abnormalities in infants
with congenital heart disease. Pediatrics 1975;
55:485-492.
Copel JA, Pilu G, Kleinman CS. Congenital heart
disease and extracardiac anomalies: Associations
and indications for fetal echocardiography. Am J
Obstet Gynecol 1986;154:1121-1132.
Cannon C, Dildy GA, Ward R, Varner MW, Dudley DJ. A population based study of congenital
diaphragmatic hernia in Utah: 1988-1994. Obstet
Gynecol 1996;87:959.
Hughes MD, Nyberg DA, Mack LA, Pretorius DH.
Fetal omphalocele: prenatal detection of concurrent anomalies and other predictors of outcome.
Radiology 1989;173:371-75.
Mellins RB, Blumenthal S. Cardiovascular anomalies and esophageal atresia. Am J Dis Child 1964;
107:160-167.
Parrish ML, Roessmann U, Levinsohn MW. Agenesis of the corpus callosum: A study of the frequency of associated malformations. Ann Neurol
1979;6:349.
Anderson RC. Congenital cardiac malformation in
109 sets of twin and triplets. Am J Cardiol 1977;
39:1045.
DeVore GR, Alfi O. The use of color Doppler ultrasound to identify fetuses at increased risk for trisomy 21: an alternative for high risk patients who
decline genetic amniocentesis. Obstet Gynecol
1995;85:378-386.
Carvalho JS, Moscoso G, Ville Y. First trimester
transabdominal fetal echocardiography. Lancet
1998;351:1023-26.
Copel JA, Pilu G, Green J, Hobbins JC, Kleinman
CS. Fetal echocardiographic screning for congenital heart disease: the importance of the fourchamber view. Am J Obstet Gynecol 1987:157:
648-55.
Caruso G, Becker AE. How to determine atrial situs? Considerations initiated by 3 cases of absent
spleen with a discordant anatomy between bronchi and atria. Brtt Heart J 1979;41:559-567.
Crane JM, Ash K, Fink N, Desjardins C. Abnormal
fetal cardiac axis in the detection of intrathoracic
anomalies and congenital heart disease. Ultrasound Obstet Gynecol 1997;10:90-93.
64. Paladini D, Chita SK, Allan LD. Prenatal measurement of cardiothoracic ratio in evaluation of heart
disease. Arch Dis Child 1990;65:20-3.
65. Chaoui R, Bierlich A. Intracardiac echogenic focus
and fetal heart defects. Ultrasound Obstet Gynecol 2000;16 suppl 1:13-14.
66. Yoo SJ, Lee YH, Kim ES, Ryu HM, Kim MY, Choi
H-K, et al. Three vessels of the fetal upper mediastinum: an easy means of detecting abnormalities
of the ventricular outflow tracts and great arteries during obstetric screening. Ultrasound Obstet
Gynecol 1997;9: 173-82.
67. Chaoui R, Bollmann R. Fetal color-Doppler
echocardiography. 1. General principles and normal findings. Ultraschall Med 1994;15:100-4.
68. Maulik D, Nanda N, Saini VD. Fetal Doppler
echocardiography: Methods and characterization
of normal and abnormal hemodynamics. Am J
Cardiol 1984;53:572-578.
69. Shenker L, Reed KL, Marx GR et al. Fetal cardiac
Doppler flow studies in prenatal diagnosis of heart
disease. Am J Obstet Gynecol 1988;158:1267-1273.
70. DeVore GR. Assessing fetal cardiac ventricular function. Semin Fetal Neonatal Med 2005;10:515-541.
71. Carvalho JS, O’Sullivan C, Shinebourne EA and
Henein MY. Right and left ventricular long-axis
function in the fetus using angular M-mode. Ultrasound Obstet Gynecol 2001;18:619-622.
72. Reed KL, Sahn DJ, Scagnelli S, Anderson CF,
Shenker L. Doppler echocardiographic studies of
diastolic function in the human fetal heart: changes during gestation. J Am Coll Cardiol 1986;8:
391-395.
73. Harada K, Rice MJ, Shiota T, Ishii M, McDonald
RW, Reller MD et al. Gestational age- and growthrelated alterations in fetal right and left ventricular diastolic filling patterns. Am J Cardiol 1997;79:
173-177.
74. Stirnemann JJ, Mougeot M, Proulx F, Nasr B, Essaoui M, Fouron JC, et al. Profiling fetal cardiac
function in twin-twin transfusion syndrome. Ultrasound Obstet Gynecol 2010;35:19-27.
75. Mahle WT, Rychik J, Tian ZY, Cohen MS, Howell LJ, Cromblehome TM, et al. Echocardiographic
evaluation of the fetus with congenital cystic adenomatoid malformation. Ultrasound Obstet Gynecol 2000;16:620-624.
76. Crispi F, Hernandez-Andrade E, Pelsers MM, Plasencia W, Benavides-Serralde JA, Gratacos E. Cardiac dysfunction and cell damage across clinical
stages of severity in growth-restricted fetuses. Am J
Obstet Gynecol 2008;199:254 e251-258.
77. Rizzo G, Arduini D, Romanini C. Doppler echocardiographic assessment of fetal cardiac function.
Ultrasound Obstet Gynecol 1992;2: 434-445.
78. Ho CY and Solomon SD. A clinician’s guide to
tissue Doppler imaging. Circulation 2006;113:
e396-398.
79. Gardiner HM, Pasquini L, Wolfenden J, Barlow
A, Li W, Kulinskaya E and Henein M. Myocardial
tissue Doppler and long axis function in the fetal
heart. Int J Cardiol 2006;113:39-47.
23
MARINO cardiologia pediatrica.indd 23
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
80. Harada K, Tsuda A, Orino T, Tanaka T and Takada
G. Tissue Doppler imaging in the normal fetus. Int
J Cardiol 1999;71:227-234.
81. Jamjureek V. Evaluation of ventricular myocardial
velocities and heart motion of the fetal heart by
tissue Doppler image. J Med Assoc Thai. 2001;
84:1158-63.
82. Pellerin D, Sharma R, Elliott P and Veyrat C. Tissue
Doppler, strain, and strain rate echocardiography
for the assessment of left and right systolic ventricular function. Heart 2003;89 Suppl 3:iii9-17.
83. Winsberg F. Echocardiography of the fetal and
newborn heart. Invest Radiol 1972;7:152-158.
84. Rudolph AM. Congenital Disease of the heart.
Chicago: Yearbook, 1974.
85. Kleinman CS, Copel JA. Fetal cardiovascular physiology and therapy. Fetal Diagn Ther 1992;7:147-157.
86. Lange LW, Sahn DJ, Allen HD, Goldberg SJ, Anderson C and Giles H. Qualitative real-time crosssectional echocardiographic imaging of the human
fetus during the second half of pregnancy. Circulation 1980;62:799-806.
87. Allan LD, Tynan MJ, Cambell S, Wilkinson JL, Anderson RH. Echocardiographic and anatomical
correlates in the fetus. Br Heart J 1980;44:444-451.
88. Kaschaner J, Fermont L. Prenatal cardiology. Press
Med 1985;14:517-519.
89. Allan LD, Crawford DC, Chita SK, Tynan MJ. Prenatal screening for congenital heart disease. Br
Med J (Clin Res ED) 1986:292:1717-1719.
90. American College of Obstetricians and Gynecologist. Ultrasound in pregnancy. ACOG Tech. Bull.
1993;187:1-8.
91. Allan LD, Sharland GK, Milburn A, Lockhart SM,
Groves AM, Anderson RH, et al. Prospective diagnosis of 1006 consecutive cases of congenital
heart disease in the fetus. J Am Coll Cardiol 1994;
23:1452-1458.
92. Marasini M, Pongiglione G, Lituania M, Cordone
M, Porro E, Garello-Cantoni L. Aortic Arch Interruption: Two-dimensional echocardiographic recognition in utero. Pediatr Cardiol 1985;6:147-149.
93. Todros T. Prenatal diagnosis and management of
fetal cardiovascular malformations. Curr Opin Obstet Gynecol 2000;12:105-109.
94. Marek J, Tomek V, Skovrànek J, Povysilova V, Samanek M. Prenatal ultrasound screeenig of congenital heart disease in an unselected national population: a 21-year experience. Heart 2011;97:124-130.
95. Sharland G. Fetal cardiac screening: why bother?
Arch Dis Child Fetal Neonatal Ed 2010;95: F64-68.
96. Chang AC, Huhta JC, Yoon GY, Wood DC, Tulzer
G, Cohen A, et al. Diagnosis, transport, and outcome in fetuses with left ventricular outflow tract
obstruction. J Thorac Cardiovasc Surg 1991;102:
841-848.
97. Bonnet D, Coltri A, Butera G, Fermont L, Le Bidois
J, Kachaner J, Sidi D. Detection of transposition of
the great arteries in fetuses reduces neonatal morbidity and mortality. Circulation 1999;99:916-918.
98. Eapen RS, Rowland DG, Franklin WH. Effect of
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
prenatal diagnosis of critical left heart obstruction
on perinatal morbidity and mortality. Am J Perinatol 1998;15:237-242.
Tworetzky W, McElhinney DB, Reddy VM, Brook
MM, Hanley FL, Silverman. Improved surgical outcome after diagnosis of hypoplastic left heart syndrome. Circulation 2001;103:1269-1273.
Kumar RK, Newburger JW, Gauvreau K, Kamennir
SA, Hornberger LK. Comparison of outcome when
hypoplastic left heart syndrome and transposition of the great arteries are diagnosed prenatally
versus when diagnosis of these two conditions
is made only postnatally. Am J Cardiol 1999;83:
1649-1653.
Franklin O, Burch M, Manning N, Sleeman K,
Gould S, Archer N, et al. Prenatal diagnosis of coarctation of the aorta improves survival and reduces morbidity. Heart 2002;87:67-69.
Maeno YV, Kamenir SA, Sinclair B, van der Velde
ME, Smallhorn JF, Hornberger LK. Prenatal features
of ductus arteriosus constriction and restrictive foramen ovale in d-transposition of the great arteries.
Circulation 1999;99:1209-1214.
Jouannic JM, Gavard L, Fermont L, Le Bidois J,
Parat S, Vouhe PR, et al. Sensistivity and Specificity of Prenatal Features of Physiological shunts to
predict neonatal clinical status in transposition of
the great arteries. Circulation 2004;110:1743-1746.
Raboisson MJ, Samson C, Ducreux C, Rudigoz
RC, Gaucherand P, Bouvagnet P, Bozio A. Impact of prenatal diagnosis of transposition of the
great arteries on obstetric and early postnatal
management. European Journal of Obstetrics &
Gynecology and Reproductive Biology 2009;
142:18-22.
Daubeney PEF, Sharland GK, Cook AC, Keeton BR,
Anderson RH, Webber SA. Pulmonary atresia with
intact ventricular septum. Impact of fetal echocardiography on incidence at birth and postnatal outcome. Circulation 1998;98:562-566.
Tzifa A, Barker C, Tibby SM, Simpson JM. Prenatal
diagnosis of pulmonary atresia: impact on clinical
presentation and early outcome. Arch Dis Child
Fetal neonatal ED 2007;92:F199-203.
Tuo G, Volpe P, Bondanza S, Volpe N, Serafino
M, De Robertis V, et al. Impact of prenatal diagnosis on outcome of pulmonary atresia and intact
ventricular septum. J Matern Fetal Neonatal Med
2011;Jun 24.
Swanson T, Tierney ESS, Tworetzky W, Pigula F,
McElhinney DB. Truncus Arteriosus: diagnosis accuracy, outcomes, and impact of prenatal diagnosis. Pediatr Cardiol 2009;30:256-261.
Duke C, Sharland GK, Jones AM, Simpson JM.
Echocardiographic features and outcome of truncus arteriosus diagnosed during fetal life. Am J Cardiol 2001;88:1379-1384.
Volpe P, Paladini D, Marasini M, Buonandonna
AL, Russo MG, Caruso G, et al. Common arterial
trunk in the fetus: characteristics, associations, and
outcome in a multicentre series of 23 cases. Heart
2003;1437-1441.
24
MARINO cardiologia pediatrica.indd 24
17/09/15 07:54
1 – Cardiologia fetale
111. Simpson JM, Sharland GK. Fetal tachycardias:
management and outcome of 127 consecutive cases. Heart 1998;79:576-81.
112. van Engelen AD, Weijtens O, Brenner JI, Kleinman
CS, Copel JA, Stoutenbeek P, et al. Management
outcome and follow-up of fetal tachycardia. J Am
Coll Cardiol 1994;24:1371-5.
113. Simpson JM. Fetal arrhythmias. Ultrasound Obstet
Gynecol 2006;27:599-606.
114. Schmidt KG, Ulmer HE, Silverman NH, Kleinman
CS, Copel JA. Perinatal outcome of fetal complete
atrioventricular block: a miulticener experience. J
Am Coll Cardiol 1991;17:1360-6.
115. Jaeggi ET, Hornberger LK, Smallhorn JF, Fouron
JC. Prenatal diagnosis of complete atrioventricular block associated with structural heart disease:
combined experience of two tertiary centers and
review of the literature. Ultrasound Obstet Gynecol 2005;26:16-21.
116. Groves AM, Allan LD, Rosenthal E. Outcome of
isolated congenital complete heart block diagnosed in utero. Heart 1996;75:190-4.
117. Jaeggi ET, Fouron JC, Silverman ED, Ryan G, Smallhorn J, Hornberger LK. Transplacental fetal treatment improves the outcome of prenatally diagnosed
complete atrioventricular block without structural
heart disease. Circulation 2004;110:1542-8.
118. Bull C, for the British Paediatric Cardiac Association. Current and potential impact of fetal diagnosis on prevalence and spectrum of seriuous congenital heart disease at term in the UK. Lancet
1999;354:1242-1247.
119. Todros T. Prenatal diagnosis and management of
fetal cardiovascular malformations. Curr OPin Obstet Gynecol 2000;12:105-109.
120. Garner E, Loane M, Dolk H, De Vigan C, Scarano
G, Tucker D, et al. Prenatal diagnosis of severe
structural congenital malformations in Europe. Ultrasound Obstet Gynecol 2005;25:6-11.
121. Hornberger LK, Need L, Benacerraf BR. Development of significant left and right ventricular hypoplasia in the second and third trimester fetus. J Ultrasound Med 1996;15:655-659.
122. Hornberger LK, Sanders SP, Rein AJ, Spevak J, Parness IA, Colan S. Left heart obstructive lesions and
left ventricular growth in the midtrimester fetus. A
longitudinal study. Circulation 1995;92:1531-1538.
123. Hornberger LK, Sanders SP, Sahn DJ, Rice MJ, Spevak PJ, Benacerraf BR, McDonald RW, Colan S. In
utero pulmonary artery and aortic growth and potential for progression of pulmonary outflow tract
obstruction in Tetralogy of Fallot. J Am Coll Cardiol 2002. 23:739-745.
124. Marasini M, De Caro E, Pongiglione G, Ribaldone D, Caponnetto S. Left heart obstructive disease with ventricular hypoplasia: changes in the
echocardiographic appearance during pregnancy.
J Clin Ultrasound 1993;21:65-68.
125. Rici MJ, McDonald RW, Peller MD. Progressive
pulmonary stenosis in the fetus: two cases reports.
Am J Perinatal 1999;10:424-427.
126. Simpson JM, Sharland GK. Natural history and
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
outcome of aortic stenosis diagnosed prenatally.
Heart 1997;77:205-210.
Paladini D, Palmieri S, Lamberti A, Teodoro A,
Martinelli P, Nappi C. Characteristics and natural
history of ventricular septal defects in the fetus. Ultrasound Obstet Gynecol 2000;16:118-122.
Zosmer N, Bajoria R, Weiner E. Clinical and
echocardiographic features of in utero cardiac dysfunction in the recipient twin in twin-twin transfusion syndrome. Br Heart J 1994;72:74-79.
Kleinman CS, Donnerstein RL, DeVore GR, Jaffe
CC, Lynch DC, Berkowitz RL, et al. Fetal echocardiography for evaluation of in utero congestive
heart failure- A technique for study of nonimmune
fetal hydrops. N Engl J Med 1982;306:568-575.
Naheed ZJ, Starsburger JF, Deal BJ, Benson JrDW,
Gidding SS. Fetal tachycardia: mechanisms and
predictors of hydrops fetalis. J Am Coll Cardiol
1996;27:1736-1740.
Hornberger LK, Barrea C. Diagnosis, natural history and outcome of fetal heart disease. Ann Sem
Thoracic Surg 2001;4:229-243.
Rudolph AM, Heyman MA, Spitznas U. Hemodynamic considerations in the devolopment of narrowing of the aorta. Am J Cardiol 1972;30:514-25.
Better DJ, Apfel HD, Zidere V, Allan LD. Pattern of
pulmonary venous flow in the hypoplastic left heart
syndrome in the fetus. Heart 1999;81:646-649.
Hornberger LK, Sahn DJ, Kleinman CS, Copel JA,
Reed KL. Tricuspid valve disease with significant
tricuspid insufficiency in the fetus: diagnosis and
outcome. J Am Coll Cardiol 1991;17:167-173.
Sharland G. Fetal Cardiology. Semin Neonatol 2001;
6:3-15.
Mellander M. Perinatal management, counselling and outcome of fetuses with congenital heart
disease. Seminars in Fetal & Neonatal Medicine
2005;10:586-593.
Gardiner HM. In-utero intervention for severe
congenital heart disease. Best Practice and Research Clinical Obstetrics and Gynaecology 2008;
22:49-61.
Senat MV, Deprest J, Boulivan M, Paupe A, Winer
N, Ville Y. Endoscopic laser surgery versus serial
amnioreduction for severe twin-to-twin transfusion
syndrome. N Engl J Med 2004;351:136-144.
Marshall A, Levine J, Morash D, Silva V, Lock JE,
Benson CB et al. Results of in utero atrial septoplasty in fetuses with hypoplastic left heart syndrome. Prenat Diagn 2008;28:1023-8.
Makikallio K, McElhinney DB, Levine JC, Marx GR,
Colan SD, Marshall AC, Lock JE. Fetal aortic valve
stenosis and the evolution of hypoplastic left heart
syndrome, patient selection for fetal intervention.
Circulation 2006;113:1401-1405.
Freud LR, McElhinney DB, Marshall AC, Marx GR,
Friedman KG et al. Fetal aortic valvuloplasty for
evolving hypoplastic left heart syndrome. Postnatal outcome of the first 100 patients. Circulation
2014;130:638-45.
Tulzer G, Gardiner H. Cardiac interventions in the
25
MARINO cardiologia pediatrica.indd 25
17/09/15 07:54
CARDIOLOGIA PEDIATRICA
143.
144.
145.
146.
147.
148.
149.
150.
151.
fetus: potential for right-sided lesions. Fetal interventions in right heart disease. Progress in Pediatric Cardiology 2006;22:79-83.
Carpenter RJ, Strasburger JF, Garson A, Smith RT,
Deter RL, Engelhardt HT. Fetal ventricular pacing
for hydrops secondary to complete atrioventricular
block. J Am Coll Cardiol 1986;8:1434-1436.
Daubeney PEF, Delany DJ, Anderson RH, Sandor
GG, Slavik Z, Keeton BR, et al. Pulmonary atresia
with intact ventricular septum: range of morphology in a population-based study. J Am Coll Cardiol
2002;39:1670-9.
Hanseus K, Bjorkhem G, Laurin S. Cross-sectional
echocardiographic measuraments of right ventricular size and growth in patients with pulmonary atresia and intact ventricular septum. Pediatr
Cardiol 1991;12:135-42.
Gardiner H. Response of the fetal heart to changes
in load: from hyperpalsia to heart failure. Heart
2005;91:871-3.
Haworth S, Reid L. Quantitative structural study
of pulmonary circulation in the newborn with
aortic atresia, stenosis or coarctation. Thorax
1977;32:121-8.
Wernovsky G, Shillingford AJ, Gaynor JW. Central
nervous system outcomes in children with complex congenital heart disease. Curr Opin Cardiol
2005;2:94-9.
Kohl T, Muller A, Tchatcheva K, Achenbach S,
Gembruch U. Fetal transesophageal echocardiography: clinical introduction as a monitoring tool
during cardiac intervention in a human fetus. Ultrasound Obstet Gynecol 2005;26:780-785.
Huhta J, Quintero RA, Suh E, Bader R. Advances
in fetal cardiac intervention. Curr Opin Paediatric
2004;16:487-93.
Strasburger JF, Cuneo FB, Michon MM, et al. Ami-
152.
153.
154.
155.
156.
157.
158.
159.
160.
odarone therapy for drug-refractory fetal tachycardia. Circulation 2004;109:375-379.
Kleinman CS, Nehme RA. Cardiac arrhythmias in
the human fetus. Pediatric Cardiol 2004;25:234-251.
Hansmann M, Gembruch U, Bald R, et al. Fetal
tachyarrhythmias: transplacental and direct treatment of the fetus. A report of 60 cases. Ultrasound
Obstet gynecol 1991;1:162-170.
Krapp M, Kohl T, Simpson JM, et al. Review of diagnosis, treatment and outcome of fetal atrial flutter compared with supraventricular tachycardia.
Heart 2003;89:913-917.
Machado MVL, Tynan MJ, Curry PV Land Allan
LD. Fetal complete heart block. Br Heart J 1988;
60,512-15.
Martin TC, Arias F, Olander DS, et al. Successful
management of congenital atrioventricular block
associated with hydrops fetalis. J Pediatr 1988;
112:984-6.
Jaeggy ET, Fouron JE, Silverman, et al. Transplacental treatment improves the outcome of prenatally
diagnosed complete atrioventricular block without structural heart disease. Circulation 2004;110:
1542-8.
Groves AM, Allan LD, Rosenthal E. Outcome of
isolated congenital complete heart block diagnosed in utero. Heart 1996;75:190-194.
Trucco SM, Jaeggi E, Cuneo B, et al. Use of intravenous gamma globulin and corticosteroids in the
treatment of maternal auto-antibody mediated cardiomyopathy. J Am Coll Cardiol 2011;57:715-23.
Lamberti A, Vautjier-Brouzes D, Wechesler B.
Prednisone and prevention of fetal heart block in
mothers with connective tissue disease: report on
78 consecutive pregnancies. Proc 2nd World Congr
Pediatr Cardiol Cardiac Surg. Armonk NY Futura
Publishing CO.1998:559-61.
26
MARINO cardiologia pediatrica.indd 26
17/09/15 07:54