: quali sfide per il tumore più")
Prospettive in Pediatria
Aprile-Giugno 2016 • Vol. 46 • N. 182 • Pp. 127-134
Oncologia pediatrica
La Leucemia Linfoblastica
Acuta (LLA):
quali sfide per il tumore
più frequente?
Valentino Conter1
Antonella Colombini1
Francesco Ceppi2
Clinica Pediatrica, Università
Milano-Bicocca, Fondazione
MBBM-Ospedale San Gerardo,
Monza; 2 Pediatric HematoOncology Unit, Department
of Paediatrics, University
Hospital of Lausanne, Lausanne,
Switzerland
1 La Leucemia Linfoblastica Acuta (LLA) è il tumore più frequente in età pediatrica. La prognosi è progressivamente migliorata con l’intensificazione della chemioterapia, raggiungendo una sopravvivenza libera da malattia a 5 anni dalla diagnosi di circa l’80% e una
sopravvivenza di circa il 90%. Un’ulteriore intensificazione della terapia non è ritenuta ragionevole per il rischio di tossicità eccessiva. In questo scenario le nuove sfide per la LLA
sono concentrate sull’identificazione di trattamenti chemioterapici parimenti efficaci ma a
minor rischio di tossicità acuta e/o sequele come le complicanze infettive, l’osteonecrosi
e i danni da radioterapia encefalica, e sull’introduzione di terapie innovative e “mirate”. In
questo contesto sono di grande interesse i farmaci che agiscono direttamente su proteine
o meccanismi specifici delle cellule leucemiche e l’immunoterapia con anticorpi e terapie
cellulari. Va infine ricordato che circa l’80% dei bambini con LLA vive in paesi con risorse
limitate, in cui l’accesso alle cure è scarso e talora inesistente. La sfida maggiore per la
LLA (e non solo) rimane quindi la globalizzazione delle cure convenzionali.
Riassunto
Acute lymphoblastic leukaemia (ALL) is the most common childhood malignancy. Progressive improvement of results has been obtained thanks to intensification of chemotherapy, reaching a 5-year event-free survival and survival of approximately 80% and 90%
respectively. Further intensification of chemotherapy, however, is not considered feasible
due to the risk of excess of toxicity. The new challenges for ALL have thus changed markedly. The need is for chemotherapeutic treatments that are equally effective and that are
associated with a lower risk for acute or late toxicity such as infectious complications or
osteonecrosis, as well as for innovative “targeted” therapies. In this frame drugs targeted
for proteins or mechanisms specific for leukaemic cells and immunotherapy with antibodies or cellular therapies are of great interest. Last but not least, about 80% of children
live in low income countries where access to healthcare for ALL may be scarce or not
available at all. The major challenge for ALL (and not only) remains the globalisation of
adequate treatment.
Summary
Metodologia della ricerca
bibliografica effettuata
La ricerca degli articoli rilevanti sulla Leucemia Linfoblastica Acuta è stata effettuata sulla banca bibliografica Medline, utilizzando come motore di ricerca
PubMed e come parole chiave “ALL, Treatment, Innovative therapies and Global Medicine”. È stato utilizzato il filtro “children”.
Introduzione
I progressi nel trattamento della Leucemia Linfoblastica Acuta (LLA) sono un grande successo della
medicina moderna. Il tasso di sopravvivenza libera
da malattia, a 5 anni dalla diagnosi per i pazienti di
età inferiore a 18 anni, è passato gradualmente da
meno del 10% nei primi anni ’60 all’attuale 80%, con
una sopravvivenza a lungo termine del 90%. (Pinkel,
1971; Henze et al., 1981; Conter et al., 2010; Vora et
al., 2014; Möricke et al., 2016) Questo successo è
127
V. Conter et al.
iniziato negli anni ’40 con l’identificazione di singoli
agenti chemioterapici ed è proseguito, a partire dagli anni ’60, con lo sviluppo di strategie basate sulla
combinazione di farmaci per il trattamento sistemico e
di terapie specifiche per la prevenzione della ricaduta
nel Sistema Nervoso Centrale (SNC); nel corso degli ultimi quattro decenni i risultati sono progressivamente migliorati con la graduale intensificazione del
trattamento. Purtroppo i risultati rimangono del tutto
insoddisfacenti nei paesi con risorse limitate, dove
vive l’80% dei bambini. Negli ultimi due decenni tuttavia molte iniziative hanno permesso di migliorare le
prospettive anche in questo ambito (Navarrete et al.,
2014).
Incidenza
L’incidenza della LLA in età pediatrica, che rappresenta i 3/4 di tutte le leucemie, è di circa 30 casi/anno/
milione di soggetti di età 0-17 anni. Il numero stimato
di nuovi casi/anno è di circa 400 in Italia, 5.000 in
Europa e 100.000 nel mondo. Il picco di incidenza è
compreso tra i 2 e i 5 anni. L’incidenza è lievemente
più alta nel sesso maschile che in quello femminile, e
questa differenza è più marcata durante l’adolescenza e per le LLA a cellule T.
Nei paesi a risorse limitate l’incidenza della LLA può
solo essere stimata. In alcuni paesi del Nord Africa,
del Medio Oriente, in India e in Cina, l’incidenza sembra essere inferiore rispetto ai paesi industrializzati
ed è particolarmente bassa in alcuni paesi dell’Africa
Sub-sahariana, in cui vi è un’incidenza molto alta di
casi di linfoma di Burkitt.
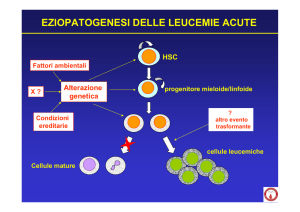
Aspetti eziopatogenetici
I fattori eziopatogenetici nella LLA sono tuttora largamente sconosciuti. Solo una frazione minoritaria può
essere associata a fattori genetici, ad agenti fisici e
chimici o a infezioni virali. Tra i fattori genetici la più
comune anormalità costituzionale associata a LLA
è la trisomia 21 (sindrome di Down). I bambini con
sindrome di Down hanno infatti un rischio 15 volte
superiore di sviluppare la malattia, rispetto alla popolazione generale. Altre alterazioni cromosomiche costituzionali associate alla leucemia sono la sindrome
di Klinefelter, la sindrome di Bloom e l’anemia di Fanconi. Anche le radiazioni ionizzanti e alcuni agenti chimici potrebbero giocare un ruolo nello sviluppo della
leucemia; ciò è stato dimostrato dall’elevata incidenza
in seguito allo scoppio della bomba atomica nella popolazione di Hiroshima e Nagasaki. Assai controverso
è il ruolo dell’esposizione a emissioni basali di radiazioni ionizzanti da centrali nucleari o come risultato di
dispersione dopo test nucleari atmosferici. Più recentemente è stato anche suggerito che l’esposizione a
campi elettromagnetici (CEM) potrebbe essere correlata a sviluppo di LLA in età pediatrica; in letteratu128
ra esistono studi contrastanti e la possibile rilevanza
eziologica non trova riscontro nella plausibilità biologica. (Magnani et al., 2014; Pisani et al., 2013) Altri
fattori studiati come possibili agenti causali di LLA
includono l’abitudine dei genitori al fumo di sigaretta,
l’esposizione paterna a erbicidi e pesticidi, l’assunzione materna di alcol, contraccettivi e dietilstilbestrolo,
l’esposizione familiare al radon e contaminazione chimica dell’acqua nel sottosuolo. In generale si ritiene
tuttavia che, con l’eccezione delle radiazioni ionizzanti, i fattori ambientali siano poco rilevanti.
Il possibile ruolo delle infezioni virali nella patogenesi
delle leucemie umane è stato estesamente studiato.
Alcuni autori hanno evidenziato un aumentato rischio
di LLA in bambini nati da madri recentemente infettate da virus dell’influenza, della varicella o da altri virus,
ma nessun legame tra l’esposizione virale prenatale e il
rischio leucemico è stato confermato eccetto per l’associazione tra il virus di Epstein-Barr (EBV) e casi di leucemia-linfoma di Burkitt endemici (LLA B-matura, sottotipo
morfologico L3, secondo la classificazione FAB).
Aspetti clinici e laboratoristici
all’esordio
Nella LLA qualunque organo o tessuto può essere infiltrato dalle cellule tumorali. Il midollo leucemico è generalmente infiltrato da una popolazione omogenea
di cellule che nella maggior parte dei casi (85%) è
costituita da piccoli linfoblasti rotondeggianti, L1 secondo la classificazione FAB (Fig. 1). Gli altri casi presentano una morfologia differente (cellule più grandi
con abbondante citoplasma e/o nucleoli intranucleari), definita come L2 o L3 (Bennet et al., 1981).
La durata dei sintomi può essere molto variabile, da
alcuni giorni a mesi. La febbre è presente approssimativamente nel 50-60% dei pazienti; l’anemia
(Hb < 7gr/100 ml), generalmente normocromica, normocitica con conta reticolocitaria normale o bassa,
Figura 1. Striscio midollare di LLA, L1 secondo la classificazione FAB (da Bennet et al., 1981, mod.).
La Leucemia Linfoblastica Acuta (LLA): quali sfide per il tumore più frequente?
e la piastrinopenia (PTL < 20.000/mmc) si riscontrano rispettivamente nel 40% e 30% dei casi circa. A
causa dell’infiltrazione del periostio, delle ossa, delle
articolazioni o dell’espansione dello spazio midollare
da parte delle cellule leucemiche, oltre 1/3 dei pazienti può presentare dolori ossei e/o artralgie. Segni e
sintomi meno comuni includono cefalea, vomito, dispnea, oliguria e anuria. In circa 1/3 dei casi si riscontra splenomegalia o epatomegalia, solitamente asintomatiche, con organi palpabili a più di 2 cm al di sotto
dell’arcata costale o linfoadenopatia, solitamente non
dolente, localizzata o sistemica. All’esordio di LLA
i livelli serici di acido urico e di lattato deidrogenasi
frequentemente risultano incrementati e correlati alla
massa tumorale leucemica. Talvolta può essere difficoltoso eseguire un aspirato midollare al momento
della diagnosi, a causa dell’elevata densità dei blasti
nel midollo. In queste situazioni si rendono necessari
aspirati midollari multipli o biopsie osteo-midollari.
gole cellule leucemiche esprimono simultaneamente
sia antigeni di superficie linfoidi B e T che mieloidi,
mostrando caratteristiche proprie di più linee emopoietiche. (Bene et al., 1995) Queste leucemie sono
state classificate come “bifenotipiche”, “a linea-mista”,
o “leucemie ibride”. In rapporto ai criteri applicati, l’incidenza delle LLA bifenotipiche in età pediatrica varia
dal 7% al 25%. La terminologia “leucemia bifenotipica” non deve essere confusa con “leucemia biclonale
o bilineare”, in cui coesistono due distinte popolazioni
cellulari.
La distinzione tra LLA con interessamento linfonodale
e linfoma Non-Hodgkin (NHL) con invasione del midollo osseo (Stadio IV) è convenzionalmente basata
sulla quota di infiltrazione midollare di blasti leucemici. La malattia è classificata come LLA al riscontro di
linfoblasti ≥ 25% nel midollo e come NHL allo Stadio IV in presenza di masse associate a una quota
midollare di linfoblasti ≥ 5 e < 25%.
Classificazione immunologica
Citogenetica convenzionale
e genetica molecolare
La trasformazione leucemica e l’espansione clonale
possono avvenire a differenti stadi del processo di
differenziazione e maturazione dei linfociti B e T normali. I blasti leucemici, pertanto, rappresentano la
controparte neoplastica dei normali linfociti B e T a
vari stadi di differenziazione (Fig. 2). In alcuni casi sin-
Le alterazioni citogenetiche riportate nella LLA coinvolgono sia il numero dei cromosomi (ploidia) che
riarrangiamenti strutturali. La LLA può essere sottoclassificata in 4 categorie, sulla base del numero
dei cromosomi: iperdiploide con più di 46 cromosomi
Figura 2. Classificazione immunofenotipica delle LLA. Linea B: pro-B CD19 pos; common: CD19 e CD10 pos; pre-B:
CD19 e Ig-citoplasmatiche M pos; B-matura: CD19 e Ig di membrana K o λ pos.
Linea T: pro-T Cd3 e CD7 pos; pre-T: CD3 o CD5 o CD8 pos; T corticale: CD3 e CD1a pos; T-matura: CD3 e TCR αβ o γδ pos.
129
V. Conter et al.
(35-45% dei casi, con DNA index (DI) > 1,0); diploide (46 cromosomi: 10-15% dei casi; DI = 1,0); pseudodiploide (46 cromosomi con anomalie strutturali o
numeriche: circa il 40% dei casi; DI = 1,0); ipodiploide
(meno di 46 cromosomi: circa l’8% dei casi; DI < 1,0)
(Pui et al., 2010).
La traslocazione t(9;22)(q34;q11) – denominata anche Cromosoma Philadelphia (Ph), per il suo derivativo der(22)-, giustappone i geni BCR e ABL creando
un gene di fusione BCR-ABL, è riscontrabile nel 3-5%
dei casi di LLA pediatrica, ed è stata associata a una
prognosi sfavorevole prima dell’introduzione di farmaci con attività inibitoria nei confronti della proteina di
fusione (inibitori di tirosin-chinasi, TKI).
Il gene MLL, mappato sul cromosoma 11 banda q23,
che codifica per la proteina nucleare mixed-lineage
leukemia (MLL), è frequentemente coinvolto in riarrangiamenti strutturali (traslocazioni, delezioni e duplicazioni parziali) soprattutto nella leucemia infantile
(età < 1 anno) o nei casi di leucemie secondarie dopo
terapia con epipodofillotossine.
La traslocazione t(12;21) è dimostrabile nel 25% circa
dei casi di LLA pediatrica di linea B (gene di fusione
ETV6-AML1) ed è associata a prognosi favorevole
(Pui et al., 2009).
mane, consente di ottenere un tasso di remissione
completa (RC) > 95%; la RC può non essere ottenuta per mortalità in induzione (1%) o per resistenza al
trattamento (~ 2%).
La terapia diretta al SNC si basa sulla somministrazione di farmaci per via intratecale e farmaci ad alte
dosi per via endovenosa. La radioterapia craniale può
essere somministrata nei pazienti con un rischio elevato di ricaduta al SNC.
La terapia di consolidamento/reinduzione è costituita
da una o più fasi di trattamento intenso dopo l’ottenimento della RC. La durata di queste due fasi può
essere estremamente variabile, da uno a diversi mesi.
Per i pazienti ad alto rischio (15-25%) possono essere
utilizzati schemi di chemioterapia intensiva a blocchi.
Il trapianto di cellule staminali emopoietiche (HSCT)
viene riservato ai casi con maggior rischio di ricaduta
(circa il 5% nei protocolli AIEOP-BFM).
Nella maggior parte degli schemi terapeutici di mantenimento vengono somministrati methotrexate settimanalmente e 6-mercaptopurina (6-MP) giornalmente. La durata ottimale della chemioterapia di mantenimento non è ancora stata definitivamente stabilita.
Tuttavia la maggior parte dei gruppi tratta i pazienti
per un totale di 24-30 mesi (a partire dalla diagnosi).
Fattori prognostici
Recidiva di LLA
La prognosi dei bambini con LLA è drasticamente migliorata nelle ultime quattro decadi grazie alla progressiva
intensificazione del trattamento. In questo contesto il valore prognostico di molti fattori convenzionali quali l’età, la
conta leucocitaria alla diagnosi, il sesso, l’immunofenotipo e la localizzazione SNC alla diagnosi è progressivamente diminuito o addirittura annullato. Attualmente i fattori prognostici indipendenti che vengono utilizzati anche
per la stratificazione per i gruppi di rischio sono la ploidia, le aberrazioni cromosomiche e genetico-molecolari
e soprattutto la risposta precoce al trattamento misurata
come malattia residua minima (MRM).
Globalmente ancora oggi circa il 15-20% dei bambini
può presentare una recidiva di malattia che può essere midollare isolata e/o extramidollare (SNC, testicoli, ovaie sono le sedi più comuni) oppure combinata
(midollare + extramidollare). La maggior parte delle
recidive (80%) si presenta durante la terapia o nei
primi due anni successivi alla sospensione della chemioterapia. La prognosi della ricaduta è influenzata
sfavorevolmente dalla brevità della durata della prima
remissione e dall’immunofenotipo a cellule T.
Trapianto di cellule staminali
L’HSCT da donatore familiare, o da donatore da banca, viene attualmente eseguito nella maggior parte
dei pazienti dopo una o più ricadute della malattia. I
risultati sono simili per donatori compatibili familiari o
da banca (Fagioli et al., 2013).
In passato nella gran maggioranza dei regimi di preparazione al trapianto si è utilizzata la radioterapia total body (TBI) (eccetto per i bambini più piccoli). Sono
attualmente in corso studi in cui si cerca di valutare
se si possano ottenere risultati altrettanto buoni con
regimi di condizionamento che non includono la TBI,
o con trapianto da donatore familiare aploidentico per
pazienti per i quali non sia disponibile un donatore
compatibile.
Terapia convenzionale
La caratterizzazione immunofenotipica, citogenetica
e molecolare e la precoce risposta al trattamento,
permettono di stratificare i pazienti in differenti gruppi di rischio che vengono trattati con strategie terapeutiche specifiche (Henze et al., 1981; Conter et al.,
2010; Vora et al., 2014; Moericke et al., 2016; Pui et al.,
2009; Hunger et al, 2012).
Il trattamento della LLA “non B-matura” in età pediatrica nei protocolli di chemioterapia adottati dai maggiori
gruppi cooperativi internazionali si articola in 4 fasi:
induzione, terapia diretta al SNC, consolidamento/
reinduzione e mantenimento. In genere, gli schemi di
terapia sono disegnati allo scopo di minimizzare lo
sviluppo di farmaco-resistenza.
La chemioterapia iniziale, somministrata in 4-6 setti130
Le nuove sfide
Nel contesto dei risultati riportati sopra anche gli
obiettivi della ricerca e la metodologia da utilizzare
La Leucemia Linfoblastica Acuta (LLA): quali sfide per il tumore più frequente?
per migliorare ulteriormente i risultati nella LLA pediatrica sono notevolmente modificati. A partire dagli
anni ’80, per circa 30 anni, si è attuata una progressiva intensificazione della terapia, resa possibile dai
progressi nella terapia di supporto e una graduale riduzione dell’uso della radioterapia craniale per ridurre
i rischi di danni cognitivi e di tumori cerebrali secondari. L’intensità terapeutica attuale è ormai arrivata al
limite della tollerabilità, per cui un ulteriore aumento
sarebbe associato a una tossicità inaccettabile. Le
nuove sfide sono quindi concentrate sulla riduzione
delle complicanze da chemioterapia, sulla miglior conoscenza della biologia della LLA in toto o in specifici
sottotipi della malattia, sull’identificazione di terapie
innovative mirate e/o alternative alla chemioterapia e
sulla globalizzazione della terapia. Anche per l’HSCT
si stanno cercando trattamenti che riducano la tossicità da terapia di condizionamento e le sequele.
Riduzione delle complicanze secondarie
al trattamento
Una delle sfide per i protocolli attuali è la riduzione
delle complicanze secondarie agli episodi infettivi e
all’osteonecrosi. Le infezioni possono essere lifethreatening o letali, come lo shock settico e l’ARDS,
e/o causare sequele importanti, come danni neurologici o perdita di sostanza nel caso della cellulite e
delle infezioni fungine. È quindi di fondamentale importanza associare alla terapia più intensa misure di
prevenzione e/o terapeutiche ottimali per il controllo
delle infezioni. L’osteonecrosi colpisce soprattutto gli
adolescenti e può causare limitazioni funzionali permanenti con necessità di applicazione di protesi per
le articolazioni più importanti (Mattano et al., 2012).
Nell’eziopatogenesi sono coinvolti il trattamento steroideo, polimorfismi genetici, l’età e l’intensità della
terapia. La ricerca clinica in questo campo è mirata a
identificare fattori di rischio e schemi terapeutici che
comportano un minor rischio di necrosi asettica vascolare e a valutare l’efficacia di procedure medicochirurgiche innovative, incluso l’uso di cellule staminali. Altre complicanze importanti che si cerca di prevenire o ridurre riguardano la cardiotossicità, la pancreatite, i danni cerebrali e/o i deficit neuro-cognitivi.
Terapie innovative
Per definizione, quando parliamo di terapie innovative, ci riferiamo a trattamenti per i quali ci sono ragionevoli probabilità di migliorare i risultati, ma che
possono essere associati a effetti collaterali non ancora sufficientemente documentati. L’introduzione delle terapie innovative avviene quindi per gradi. Esse
vengono pertanto sperimentate in clinica solo dopo
aver superato i test preclinici, inizialmente in pazienti
adulti e a seguire in età pediatrica. Nel corso degli
ultimi 15 anni abbiamo assistito alla realizzazione di
idee profondamente innovative per la terapia dei tu-
mori in generale e della LLA in particolare, che hanno
permesso di sperimentare terapie che ora sono già
parte della terapia convenzionale per alcuni sottotipi
di LLA (terapie mirate o targeted therapies), o che
sono in fase di sperimentazione molto avanzata per
poterlo diventare.
Inibitori di tirosin-chinasi
Il trattamento con inibitori di tirosin-chinasi (Imatinib,
dasatinib, nilotinib) è ormai consolidato per la LLA
BCR-ABL1 (o Philadephia) positiva. Il sequenziamento
del DNA ha però permesso negli ultimi anni di identificare nuove lesioni e meccanismi che possono attivare
le chinasi in maniera simile alle lesioni BCR-ABL1. Vi
sono quindi sottogruppi di LLA (Ph-like) che presentano riarrangiamenti del DNA nei geni ABL, JAK2, ed
EPOR, per i quali è già stata dimostrata una risposta a
inibitori di TKI in modelli sperimentali e anche in clinica
in casi aneddotici che presentavano resistenza alla terapia convenzionale (Waibel et al., 2013).
Inibitori di proteasoma
Gli inibitori del proteasoma inibiscono l’attività di enzimi che degradano le proteine cellulari, causando un
accumulo di proteine all’interno della cellula, e favorendo la morte della stessa cellula (apoptosi), particolarmente quando sono somministrati in associazione
alla chemioterapia. Il bortezomib è il primo inibitore
di proteasoma approvato per il trattamento di pazienti
affetti da mieloma e linfoma, e recentemente è stato
somministrato anche in pazienti con LLA refrattaria o
recidivata, in associazione con la terapia convenzionale (Messinger et al., 2012). Gli effetti indesiderati
più frequenti comprendono astenia, nausea, vomito e
diarrea, trombocitopenia, anemia, neutropenia e neuropatia periferica. Sono attualmente in fase di studio
altre molecole di seconda generazione, come il carfilzomib. Non è ancora possibile prevedere quale ruolo
avranno questi farmaci nell’armamentario terapeutico
per la LLA.
Anticorpi monoclonali
Gli approcci immunoterapici più recenti si sono concentrati soprattutto sulla LLA di linea B, sia perché
queste cellule esprimono regolarmente sulla superficie antigeni non espressi dalle cellule normali, per i
quali sono già disponibili anticorpi monoclonali sperimentati in clinica, sia perché è dimostrato che i linfociti T sono coinvolti nei meccanismi di controllo immunologico dei tumori. Tra gli anticorpi monoclonali più
utilizzati per la LLA in pazienti adulti c’è il rituximab,
che si lega all’antigene CD20 determinando una lisi
complemento mediata dei linfociti B, con un importante miglioramento della sopravvivenza libera da malattia (Thomas et al., 2010). Il suo uso in età pediatrica
è stato finora limitato ai linfomi di linea B ed è ancora
in fase di studio per l’efficacia, ma potrebbe essere
131
V. Conter et al.
esteso alla LLA B-matura. Il rituximab è generalmente
ben tollerato, anche se si possono verificare reazioni
infusionali, e raramente mucositi e dermatiti gravi, o
riattivazione dell’epatite B o leucoencefalopatia multifocale progressiva.
Un altro anticorpo interessante è l’epratuzumab, che
si lega all’antigene CD22, viene rapidamente internalizzato, causa una citotossicità diretta e inibisce la
proliferazione cellulare. La tossicità di questo anticorpo può essere aumentata mediante la coniugazione
con una tossina (Carnahan et al., 2007). Attualmente
l’epratuzumab viene usato anche in Italia nei bambini
trattati con il protocollo IntReALL SR 2010, per le ricadute di LLA a rischio standard (studio randomizzato).
Anticorpi bispecifici
Gli anticorpi bispecifici sono denominati BiTE (bispecific T cell engagers) e sono stati introdotti nella
clinica recentemente. Tra questi, il più utilizzato nella
clinica, è il blinatumomab, che è un anticorpo a catena singola che combina due siti di legame: uno per
CD3, parte del recettore della cellula T (TCR) e l’altro
per CD19, presente sulle cellule tumorali nella LLA di
linea B. Il farmaco, quindi, agisce facendo da “ponte”
e favorendo l’attivazione delle cellule T del paziente,
che esercitano la propria attività citotossica sulle cellule leucemiche di linea B (target), determinandone
la morte cellulare sia negli organi linfatici che nel midollo osseo. Il blinatumomab è stato studiato in pazienti con recidiva di LLA, somministrando il farmaco
in infusione continua per 28 giorni ogni 6 settimane
e dimostrando che questo trattamento consente di
ottenere remissioni complete di lunga durata anche
in casi di LLA refrattaria alla chemioterapia o di negativizzare la MRM anche in pazienti con MRM persistentemente elevata, durante trattamenti chemioterapici intensivi (Topp et al., 2011 e 2012). Gli eventi
avversi più frequenti associati alla terapia con blinatumomab sono dovuti al rilascio di citochine (cytokine
release syndrome – CRS), in particolare di TNFα, IL10, IFN-γ e IL-6, che causano febbre, brividi, cefalea e
complicanze neurologiche, e sono ridotti con l’utilizzo
di steroidi. In rari casi la CRS può essere una complicanza life-threatening. Nelle forme severe di CRS è
stato utilizzato con beneficio l’anticorpo monoclonale
anti-IL6 tocilizumab.
tale antigene (target). Per la preparazione di queste
cellule si devono anzitutto prelevare dal paziente i linfociti T, modificarli geneticamente ex vivo, inserendo
nel loro DNA il materiale genetico che codifica per
l’espressione del recettore (chimerico) mediante un
vettore virale o con una tecnica alternativa, espandere (moltiplicare) ex vivo queste cellule modificate e
poi infonderle per via endovenosa allo stesso paziente (Fig. 3). I trial iniziali che hanno utilizzato questo
approccio hanno prodotto risultati di estremo interesse, permettendo di ottenere la RC in una percentuale molto elevata di pazienti con LLA CD19 positiva
in ricaduta o resistenti alla chemioterapia (Maude et
al., 2014). In questo contesto il trattamento con CART è prevalentemente finalizzato a ottenere la RC e
consentire al paziente di essere avviato al trapianto
di cellule staminali con maggiori probabilità di successo. Teoricamente tuttavia l’immunoterapia con CAR-T
potrebbe sostituire trattamenti chemioterapici e/o il
trapianto con cellule staminali. Questo deve tuttavia
essere dimostrato. Anche il trattamento con cellule
CAR-T è associato alla CRS, che è secondaria a una
over-expression di citochine legata alla proliferazione
di cellule T, e che nella maggior parte dei casi (circa 2/3) è caratterizzata da sintomi lievi simili a quelli
dell’influenza, ma che in rari casi può essere estremamente grave e anche mortale.
CAR-T Cells
L’utilizzo dei linfociti T (T cells), che sono coinvolti nella sorveglianza immunitaria, per eradicare un tumore
nel proprio organismo, fino a poco tempo fa poteva
essere considerato solo un’ipotesi affascinante, ma
ora è un sogno che sta diventando realtà. Ciò è stato
reso possibile dalla manipolazione genetica, per cui le
cellule T “ingegnerizzate” esprimono recettori chimerici per antigeni (chimeric antigen receptors – CARs)
con una elevata specificità e quindi altamente specializzate per legarsi a cellule tumorali che presentano
132
Figura 3. La cellula T ingegnerizzata (cellula più piccola rosata sulla sinistra) esprime sulla propria superficie un
recettore (CAR, vedi testo), che crea una sinapsi immunologica (riquadro in alto a snx) con la cellula leucemica
CD19 + (cellula più voluminosa scura a destra). Il legame
specifico del CAR all’antigene bersaglio (CD19) stimola
l’attività citotossica, proliferativa e secretiva (citochine proinfiammatorie) da parte della cellula T geneticamente modificata, che determina la morte della cellula leucemica.
La Leucemia Linfoblastica Acuta (LLA): quali sfide per il tumore più frequente?
Globalizzaione
dell’emato-oncologia pediatrica
Come accennato nell’introduzione, ancora oggi,
purtroppo la grande maggioranza dei bambini vive
in paesi con risorse limitate (Low Income Countries – LIC), in cui non solo le terapie innovative, ma
anche le terapie convenzionali riportate sopra non
sono disponibili. Nei paesi più poveri, come Haiti o
alcuni paesi in Africa, la maggior parte dei bambini affetti da LLA non viene neppure diagnosticata.
Negli ultimi 30 anni sono stati attivati numerosi progetti di collaborazione tra Centri di paesi sviluppati
e Centri di LIC. Un’esperienza molto interessante
in questo contesto è stata quella del Centro America, in cui si è formata una rete di Centri di ematooncologia pediatrica con protocolli comuni per tutti
i paesi dell’area, grazie a una iniziativa del Centro
di emato-oncologia pediatrica di Monza, supportata
da altre istituzioni tra le quali l’Istituto dei Tumori di
Milano, AMCA (Bellinzona, Svizzera) e l’International Outreach Program (IOP) del St. Jude Children’s
Research Hospital di Memphis (USA) (Masera et
al., 1998; Barr et al., 2014). La sfida più grande da
vincere nella LLA in età pediatrica rimane quindi la
globalizzazione del trattamento di eccellenza.
Box di orientamento
• Cosa sapevamo prima
La Leucemia Linfoblastica Acuta (LLA) è il tumore più frequente in età pediatrica, con un picco di incidenza nella fascia di età 2-5 anni. La somministrazione di chemioterapia intensiva consente di ottenere una
sopravvivenza libera da malattia a 5 anni dalla diagnosi del 80% e una sopravvivenza del 90%.
• Cosa sappiamo adesso
Il fattore prognostico più importante è la risposta precoce alla terapia, misurata come malattia residua
minima (MRM), durante o dopo la terapia di induzione della remissione. La MRM viene ampiamente
usata per definire il rischio di ricaduta e di conseguenza l’intensità del trattamento. Oltre alla MRM, sono
importanti le caratteristiche biologiche e le alterazioni genetiche delle cellule della LLA.
• Quali ricadute sulla la pratica clinica
Le nuove sfide nella LLA si concentrano sull’identificazione di terapie mirate sulle caratteristiche biologiche della malattia e sull’utilizzo dell’immunoterapia, in associazione o in sostituzione della chemioterapia
intensiva. Un altro obiettivo molto importante è l’estensione della terapia efficace per la LLA anche per i
bambini che vivono in paesi con risorse limitate.
Bibliografia
Barr RD, Antillon Klussmann F, Baez F
et al. Asociacion de Hemato-Oncologia
Pediatrica de Centro America (AHOPCA):
a model for sustainable development in
pediatric oncology. Pediatr Blood Cancer
2014;61:345-54.
Bene MC, Castoldi G, Knapp W, et al.
Proposals for the immunological classification of acute leukemias. European
Group for the Immunological Characterization of Leukemias (EGIL). Leukemia
1995;9:1783-6.
Bennett JM, Catovsky D, Daniel MT, et al.
The morphological classification of acute
lymphoblastic leukaemia: concordance
among observers and clinical correlations.
Br J Haematol 1981;47:553-61.
Carnahan J, Stein R, Qu Z. Epratuzumab, a CD22 targeting-recombinant
humanized antibody with a different mode
of action from rituximab. Mol Immunol
2007;44:1331-41.
Conter V, Aricò M, Basso G, et al. Long-
term results of the Italian Association
of Pediatric Hematology and Oncology
(AIEOP) Studies 82, 87, 88, 91 and 95 for
childhood acute lymphoblastic leukemia.
Leukemia 2010;24:255-64.
* Viene descritta dettagliatamente
l’evoluzione della terapia per la LLA nei
protocolli AIEOP dal 1980 al 2000, con i
risultati a lungo termine ottenuti.
Fagioli F, Zecca M, Quarello P, et al. Hematopietic stem cell transplantation for
children with high-risk acute lymphoblastic leukemia in first complete remission: a
report from the AIEOP registry. Haematologica 2013;98:1273-81.
Henze G, Langermann HJ, Ritter J, et al.
Treatment strategy for different risk groups
in childhood acute lymphoblastic leukemia:
a report from the BFM Study Group. Haematol Blood Transfus 1981;26:87-93.
Hunger SP, Lu X, Devidas M, et al.
Improved survival for children and adolescents with Acute Lymphoblastic Leukemia between 1990 and 2005: a report
from the Children’s Oncology Group. JCO
2015;30:1663-72.
Magnani C, Mattioli S, Miligi L, et al.
SETIL: Italian multicentric epidemiological
case-control study on risk factors for childhood leukaemia, non hodgkin lymphoma
and neuroblastoma: study population and
prevalence of risk factors in Italy. Ital J
Pediatr 2014;40:103.
Masera G, Baez F, Biondi A, et al. NorthSouth twinning in paediatric haemato-oncology: the La Mascota programme, Nicaragua. Lancet 1998;352:1923-26.
* Viene descritta la problematica
dell’oncologia pediatrica nei paesi con risorse limitate e una metodologia efficace
per affrontare questo problema.
Mattano LA Jr, Devidas M, Nachman
JB, et al. Alternate-week versus continuos
dexamethasone scheduling on the risk
of osteonecrosis in acute lymphoblastic leukemia: results from the CCG-1961
randomized cohort trial. Lancet Oncol
2012;(9):906-15.
133
V. Conter et al.
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained
remissions in leukemia. New Engl J Med
2014:371:1507-17.
Messinger YH, Gaynon PS, Sposto R, et
al. Bortezomib with chemotherapy is higly
active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances
in Childhood Leukemia Lymphoma (TACL)
Study. Blood 2022;120:285-90.
Möricke A, Zimmermann M, Valsecchi
MG, et al. Dexamethasone vs prednisone
in induction treatment of pediatric ALL:
results of the randomized trial AIEOP-BFM
ALL 2000. Blood 2016;127:2101-12.
* Vengono riportati i risultati di un protocollo moderno e cooperativo basato su
una diagnostica avanzata e un trattamento
intensivo in pazienti trattati in Italia, Germania, Austria e Svizzera.
Navarrete M, Rossi E, Brivio E, et al.
Treatment of childhood acute lymphoblastic leukemia in central America: a lowermiddle income countries experience. Pediatr Blood Cancer 2014;61:803-9.
Pinkel D. Five-year follow-up of “total
therapy” of childhood lymphocytic leukemia. JAMA 1971;216:648-52.
Pisani P, Parodi S, Magnani C. Causes
and risk factors for childhood cancer. Epidemiol Prev 2013;37(Suppl 1):234-54.
Pui CH, Campana D, Pei D, et al. Treatment of childhood acute lymphoblastic
leukemia without prophylactic cranial irradiation. N Engl J Med 2009;360:2730-74.
Pui CH, Pei D, Sandlund JT, et al. Longterm results of St Jude Total Therapy studies 11, 12, 13A, 13B and 14 for childhood
acute lymphoblastic leukemia. Leukemia
2010;24:371-82.
Thomas DA, O’Brien S, Faderl S, et al.
Chemoimmunotherapy with a modified
hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia
chromosome-negative precursor B-lineage
acute lymphoblastic leukemia. J Clin Oncol
2010:28:3880-9.
Topp MS, Kufer P, Gokbuget N, et al.
Targeted therapy with the T-cell-engaging
antibody blinatumomab of chemotherapyrefractory minimal residual disease in
B-lineage acute lymphoblastic leukemia
patients results in high response rate and
prolonged leukemia-free survival. J Clin
Oncol 2011:29:2493-8.
Topp MS, Goekbuget N, Zugmaier G, et
al. Long-term follow-up of hematologic
relapse-free survival in a phase 2 study of
blinatumomab in patients with MRD in Blineage ALL. Blood 2012:120:5185-7.
Vora A, Goulden N, Mitchell C, et al. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with
clinical standard-risk and intermediate-risk
acute lymphoblastic leukaemia (UKALL
2003): a randomised controlled trial. Lancet Oncol 2014;15:809-18.
Waibel M, Solomon VS, Knight DA,
et al. Combined targeting of JAK2 and
Bcl-2/Bcl-xL to cure mutant JAK2-driven
malignancies and overcome acquired resistance to JAK2 inhibitors. Cell reports
2013:5:1047-59.
Corrispondenza
Valentino Conter
Clinica Pediatrica, Università Milano-Bicocca, Fondazione MBBM-Ospedale San Gerardo, via Pergolesi 33, 20900 Monza E-mail: [email protected]
134
: quali sfide per il tumore più")