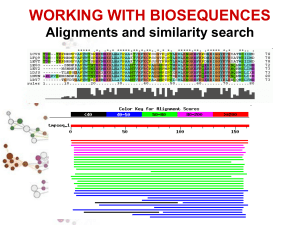
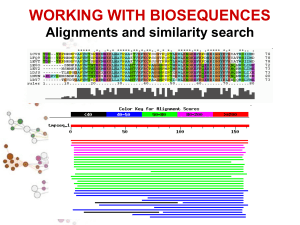
Z-score
lo Z-score è definito come:
Z-score = (opt query - M random)/ deviazione standard random
è una misura di quanto il valore di opt si discosta dalla deviazione standard media. indica di
quante dev. Standard si discosta (4 indica già che siamo fuori della distribuzione)
Deviazione standard
è l’indice di
dispersione della
distribuzione
(x
N
)2
Calcoli statistici
Statistica dei confronti locali tra due sequenze
• Per stimare se il punteggio di allineamento tra due sequenze è
dovuto al caso oppure è significativo si fanno un certo numero
di confronti (sequence space) di una delle due sequenze (la
Query) di stessa composizione AA ma con ordine casuale
contro la seconda sequenza (il Subject). L’equazione che mette
in relazione il punteggio grezzo per capire se è significativo o no
è l’E-value:
E( x
S)
E ( S ) kmne
S
S è il punteggio grezzo
K dipende dal numero di allineamenti prova effettuati (sequence space)
λ dipende dalla matrice di sostituzione
m ed n sono le lunghezze delle due sequenze
Punteggio grezzo S
E-value E(S)
Le prove di raccolta dei punteggi avranno una distribuzione simile a quella normale ma che in
realtà si chiama “distribuzione dei valori estremi” (Gumbel distribution o EVD)
0.4
A.
0.2
Yev
-2
-1
0
1
2
X
3
4
5
Se si vuole sapere quanto significativo è il punteggio ottenuto dal mio allineamento reale
in confronto alla distribuzione ottenuta secondo quanto detto prima, allora si ricorre all’Evalue che indica:
Il numero di differenti allineamenti con uno score (x) equivalente o migliore di
quello ottenuto dal mio allineamento (chiamato S) che possono capitare per caso
in una ricerca in database. Più basso è tale valore e più significativo è il mio
allineamento
E( x
S)
E ( S ) kmne
S
Bit-score
Il punteggio grezzo S ha in sé scarso significato perché è come un valore numerico
che indica la similarità tra le due sequenze ma senza una unità di misura che possa
essere utilizzata per il confronto con altre ricerche.
1. In pratica non riassume l’essenza statistica del sistema di punteggio utilizzato per calcolare
se quello che si osserva è veramente significativo o no.
2. Infatti uno stesso allineamento può avere punteggi S diversi se si utilizzano matrici di
sostituzione diverse che attribuiscono a match, mismatch e gap valori diversi
3. A tale scopo è stato introdotto il bit-score che consente di ottenere una normalizzazione
dei punteggi. S’ si normalizza come segue:
S'
Da cui deriva che l’E-value è:
E
S ln K
ln 2
mn2
S'
E, di conseguenza, dipende solo dai parametri di lunghezza delle sequenze.
Esiste infine un’altra misura che è il P-value molto simile all’E-value
Calcolo E-value nel caso di ricerca in banca
dati
• Nel caso di ricerca in banca dati le equazioni
precedenti sono:
E( x
E
S'
S)
mn2
E ( S ) kmne
S'
S ln K
ln 2
S
L’unica differenza è nel significato.
Nel caso di FASTA se m è la
lunghezza della sequenza query
n è è il numero delle sequenze
della banca dati
K e λ sono calcolati
dinamicamente per ogni singola
ricerca
La distribuzione dei veri e falsi positivi – la ricerca della giusta soglia
BLAST
Blast (Basic Local Aligment Search Tool) è un programma che cerca similarità locali utilizzando
l’algoritmo di Altschul et al. Anche Blast, come FASTA, funziona:
1. scomponendo la sequenza query in “parole” di pochi amminoacidi, di solito 2 o 3 (parametro W) e
generando una lista di parole affini (diverso da FASTA) con la matrice di sostituzione (BLOSUM). Le
parole affini conservate dovranno avere uno score superiore ad una soglia fissata T
2. Le parole affini sono ricercate nella banca dati per match esatti ed una volta trovate le sequenze che li
contengono questi vengono estesi a dx e sx dell’allineamento per una certa profondità stabilita dal
parametro X e le coppie di segmenti, presenti nella stessa coppia di sequenze, che totalizzano un
punteggio di similarità statisticamente significativo, superiore ad una soglia S, vengono definiti HSP
(High scoring Segment Pairs).
3. Nella stessa coppia possono esserci più HSP di cui é anche possibile calcolare la probabilità di
occorrenza (Karlin & Altschul, 1993).
W = word-size
T = threshold
X = elongation
S = HSP threshold
Si definisce MSP (Maximal scoring
Segment Pair) la coppia di
segmenti, di eguale lunghezza,
che realizza il massimo punteggio
di similarità nel confronto di due
sequenze; l’algorimo ne valuta in
modo rigoroso la significatività
statistica (Karlin & Altschul, 1990,
1993).
BLAST
two hit method
le versioni attuali di Blast adottano il Two-hit method che deriva dall'osservazione che il tempo
di esecuzione dell'algoritmo e' principalmente impiegato nell'allungamento degli Hits per
ottenere gli HSPs.
L'algoritmo allora considera solo i casi in cui esistono due hit sulla stessa diagonale ad una
distanza inferiore ad un parametro A prima di cercare gli HSPs.
Per non perdere in sensibilità e' stata abbassata la soglia di T.
L'algoritmo è più veloce e non ha perso in precisione
Nella sua attuale implementazione, inoltre, BLAST considera anche i gap nel tentativo di unire,
quindi, degli HSP "ungapped" che sono spazialmente vicini nella matrice di allineamento e la
cui unione in un unico frammento (contenente gap ed inserzioni) non comporta un
peggioramento dello score finale ma un miglioramento complessivo.
Il tutto secondo dei nuovi parametri che regolano i costi e le penalità della presenza di gap
nell'allineamento.
Il parametro A
BLAST
I vari algoritmi differiscono molto per il metodo con cui definiscono una sequenza casuale. BLAST calcola
a priori la probabilità che un certo punteggio sia significativo sulla base della dimensione e composizione
della banca dati applicando:
E (S ) kmne
S
m è la lunghezza della sequenza query e
n è la lunghezza in residui della banca dati
e K sono precalcolati secondo una distribuzione standard interna al contrario di FASTA.
Il punteggio è simile a quello di FASTA
La significatività di un risultato è espressa come valore E(S) (Expectation). Più basso il valore di E più
significativo è l’allineamento. Un valore di 1.0e-5 per esempio vuol dire che la probabilità di avere per
caso una sequenza con lo stesso score della mia “query” è uguale a 1.0e-5; ovvero l’attesa è che ogni
100000 sequenze se ne possa, in media, trovare una (1/100000) che totalizzi un punteggio uguale o
migliore di 1.0e-5.
CONFRONTO BLAST E FASTA
fasta3 proteina o DNA contro banca dati o proteici o DNA rispettivamente
fastx/y3 DNA contro banca dati proteico, traduzione nei 6 frame
tfastx/y3 proteina contro banca dtai di DNA tradotto
blastn query DNA banca dati DNA
blastp query proteina banca dati proteine
blastx query DNA (tradotta nei sei frame di lettura) banca dati proteine
tblastn query proteina banca dati DNA (sequenze tutte tradotte nelle sei fasi di lettura)
tblastx query DNA (tradotta nei sei frame di lettura) banca dati DNA (tradotta nei sei frame di lettura)
FASTA
SIMILARITA’
BLAST
Locale (e' in genere riportato solo il
miglior allineamento locale)
Locale (e' riportata la serie di allineamenti locali sopra il valore
soglia tra query e subject: al contrario di FASTA si riescono ad
individuare repeat e zone eventuali di overlap tra gli allineamenti
locali che sono eliminate da FASTA nella fase C)
Durante la estensione nella fase B.
In questo caso il calcolo si effettua
nella prima fase solo per identità
Fase di scansione per W e fase di estensione per gli HSP
l'algoritmo e' ottimizzato per ricercare parole W "simili" e non
esatte. Si traduce il tutto in una maggiore sensibilità di ricerca
rispetto a FASTA per le proteine.
K-TUPLE
1-2 aa / 4-6 nt
3 aa / 11-12nt. A livello nucleotidico, non essendo applicate matrici
di similarità che perdono di significato avendo solo 4 simboli
(A,C,G,T), BLAST perde in sensibilità avendo W=11
GAP
Consentiti nella fase C
Consentiti nella versione attuale
VELOCITA'
Da 1/2 ad 1/5 di BLAST
Da 2 a 5 volte maggiore di FASTA
SPECIFICITA'
Migliore per il confronto di sequenze
nucleotidiche
Migliore per il confronto
di sequenze proteiche
USO DELLA
SCORING
MATRIX
CONFRONTO BLAST E FASTA
Calcolo statistico
FASTA
Keλ
E-value
Calcolati dinamicamente
Precalcolati per quella determinata
matrice di sostituzione e valore di
gap penalties
m -> lunghezza sequenza query
n -> numero sequenze della banca
dati
m -> lunghezza sequenza query
n -> numero residui o nucleotidi
totali della banca dati
E (S ) kmne
S
E( x
S)
E ( S ) kmne
S'
S ln K
ln 2
E
mn2
S'
BLAST
S
Alcuni esempi di
interfacce web
• FASTA (http://www2.ebi.ac.uk/fasta3/)
• BLAST
(http://www.ncbi.nlm.nih.gov/BLAST/)