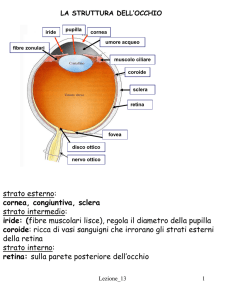
Contributi scientifici
IL PANORAMA
INTERNA2i5IONALE
Gene di trasmissione
e gene cl’cspressione dell’occhio
La ricerca si pone due obiettivi: capire i meccanismi della degenerazione retinica; elaborare una teoria in modo da sviluppare un potenziale trattamento terapico genetico per la Retinite Pigmentosa.
1 ricercatori, per studiare quali geni sono attivi o non attivi durante
la degenerazione retinica, hanno
osservato una cavia che soffriva di
un particolare tipo di R.P., causata da un gene difettoso per un enzima coinvolto nel processo visivo.
La cavia era anche affetta da una
forma recessiva di R.P. umana.
Usando una tecnica che evidenziasse lo schema dei geni attivi
nella degenerazione retinica, come un codice a barre, i ricercatori
hanno identificato geni che erano
stati attivati precedentemente nella vita della cavia, ma in tempi
successivi a secondo che la cavia
abbia avuto o meno la degenerazione.
1 ricercatori sono rimasti particolarmente sorpresi dal trovare i geni gammacristallini nella retina
dal momento che prima di allora si
pensava fossero attivi solo nel cristallino. Non è ancora chiara la loro presenza nella retina; si pensa
che abbiano qualche tipo di ruolo
nella risposta allo stress.
Forse questi geni anti-stress o simili potrebbero essere utili nell’aiutare i fotorecettori a sopravvivere.
Con una maggiore conoscenza di
queste alterazioni genetiche, potrebbero intentare approcci terapeutici più efficaci per il trattamento delle R.P.
La rice’rca in continuo progresso,
sta identificando quei geni la cui attivazione può risultare particolarmente benefica alla retina affetta.
1 ricercatori proveranno a breve la
preparazione migliorata del virus
adenoassociato (AAV) il quale trasporta la forma corretta del gene
degenerato nel topo. Intanto stanno incrementando gli sforzi per assembleare nuovi virus (AAV) che
trasporteranno altri geni potenzialmente terapeutici, i quali, come
si spera potrebbero definitivamente essere benefici a dispetto delle
cause nascoste della R.P.
Il Lavoro e stato portato avanti dal
Dr. Jomery con l’assistenza di Mr.
John Crist e Mr. Mike Thomas.
Si ringrazia il BK.P.S e «Iris Fund
for Prevention of Blindness» e la
«Guide Dogs» e la «Blind Association».
Occhio per occhio:
nnovi campioni di malattia
oculare genetica
Campioni animali di distrofie retiniche ereditate, geneticamente costruite, saranno alla base dei futuri progressi nella conoscenza delle
cause della cecità genetica. In
questo lavoro Petters et Al descrivono lo sviluppo di un maiale
transgenetico come modello di retinite pigmentosa. Questo rappresenta un notevole passo avanti rispetto ai modelli precedenti di
distrofia retinica.
Le mutazioni della rodopsina - il
gene dei fotopigmenti dei bastoncelli e centro del lavoro di Petters
et Al - si sono mostrate particolarmente prevalenti. Questa conoscenza è stata sfruttata usando
l’ingegneria genetica per produrre
modelli animali della malattia che
può essere studiata molto più dettagliatamente.
Molti di questi modelli già esistono. Si tratta per lo più di roditori.
L’utilita di questi modelli è comunque limitata dalle notevoli differenze che vi sono tra gli occhi dei
roditori e quelli umani.
Le due differenze principali e pertinenti sono: il numero e la distribuzione dei fotorecettori nella retina, specialmente i coni ed il fatto
che i l’occhio dei roditori è molto
più piccolo di quello umano. Nella
retina umana i coni si trovano in
abbondanza nella zona centrale
della retina chiamata macula. Nella retina dei roditori questi si presentano molto più sparsi.
Sebbene il difetto genetico nelle rodopsine mutanti i: confinato ai fotorecettori bastoncelli, l’handicap visivo nei casi umani di, R.P. è
ampiamente riconducibile ad una
secondaria perdita delle funzioni dei
coni. Pur non avendo una regione
maculare formata, il numero e la distribuzione dei coni tregli occhi del
maiale sono molto piìr simili a quelli
umani e quindi un campione costituito dal maiale dovrebbe rispondere in modo più o meno simile all’uomo alle stesse difese genetiche.
La validità di tutto ciò è stata confermata da Petters et Al.
Questi animali (maiali) mostrano
degenerazioni relativamente rapide dei bastoncelli ed una lenta degenerazione dei coni. Questi risultati sono molto simili all’evoluzione della malattia che è stata osservata negli studi istopatologici del
tessuto umano R.P.
L’altro grande vantaggio del campione suino su tutti gli altri disponibili, è costituito dalla somiglianza delle dimensioni oculari con
quelle umane.
Cecith genetica: concetti
correnti nella patologia della
distrofia retinica esterna
umana
La distrofia retinica esterna, è una
delle maggiori cause di cecita nel
mondo occidentale.
Le distrofie esterne della retina
possono essere classificate in due
tipi: quelle che colpiscono la retina periferica come la R.P.; quelle
che inizialmente colpiscono la retina centrale. La R.P. è la distrofia
retinale maggiormente riconosciuta e caratterizzata da una progressiva bilaterale e simmetrica degenerazione del tessuto retinico.
Gli studi di genetica molecolare ad
oggi hanno identificato un largo
insieme di geni coinvolti nel processo di sviluppo della retinopatia.
Tre grandi aree possono essere definite come importanti nella patofisiologia della distrofia retinica
esterna:
1) fotorecettori;
2) le protein,e strutturali;
3) le mutazioni che conducono a
difetti metabolici nei fotorecettori
R.P.E.
Nei bastoncelli la rodopsina assorbe la luce ed ì: la proteina più frequentemente associata alla distrofia retinica esterna.
Studi in vitro hanno dimostrato
che le rodopsine mutanti possono
legarsi con il reticolo endoplasmatico dei bastoncelli; questo conduce all’inibizione nel trasporto delle
proteine e quindi alle patologie
collegate alla distrofia retinica. Le
mutazioni, inoltre, possono condurre a variazioni nelle forme delle molecole proteiniche tali da ucciderle.
Un altro meccanismo patogeno è
la mutazione indotta nella proteina da una costante esposizione alla luce che inibisce l’attività dell’opsina e conduce parimenti alla
morte delle cellule.
Alcuni esperimenti condotti su
gatti e topi, benché riproducenti
situazioni simili al cor.p.0 umano,
hanno condotto a risultati che
vanno presi con beneficio d’inventario.
Nei pazienti con retinite periferica
1.d presenza dl mutazioni nella rodopsina ha comportato una crescita rilevante nei bastoncelli fino al
limite estremo della membrana:
Ciìj comporta serie difficoltà nel
definire terapie efficaci a causa di
un tasso di crescita delle cellule
troppo rapido.
Alcune ricerche genetiche dimostrano che nella distrofia maculare
di Stargardt la causa principale
del deficit risiede nella debolezza
dei coni. Questo interessa anche la
degenerazione della macula in relazione all’età (AMRD). La AMRD è
la causa più comune della cecità
retinica.
La sindrome di Usher è una condizione recessiva autosomica considerata la causa più comune di cecità c sordità. Il suo principale
sottotipo USHTB è associato con le
mutazioni di un particolare tipo di
miosina, la VII A, il cui deficit
comporta carenze di trasporto intracellulare.
La matrice extracellulare nella retina e regolata da una serie di proteine. 1 disturbi della retina collegati al deficit di queste proteine so-
dovuti ad un inibitore,
TIMPS. Questa inibizione conduce
ad una perdita della vista centrale
e ad un’atrofia maculare.
Nonostante si sia rilevata una grossa varieta di anomalie biochimiche
che conducono alla R.P., i danni
della retina sembrano essere gli
stessi. È inoltre poco chiaro perche
una anomalia che colpisce una
parte non essenziale per la sopravvivenza dei coni, possa portare le
cellule stesse alla morte. Inoltre
può una mutazione nei bastoncelli
e specificatamente nella rodopsina
portare alla morte dei coni?
Una risposta viene da recenti ricerche che individuano un meccanismo comune di morte dei fotorecettori nell’Apoptosis. Strumento
d’osservazione sono state delle cavie animali.
Benché si siano usate cavie che
presentavano diverse caratteristiche, in tutti gli esperimenti, partendo dalla mutazione base della
rodopsina, il processo di morte
delle cellule ha presentato caratteri assai similari.
Le anomalie genetiche che sottostanno a molte retiniti distrofiche
devono ancora essere spiegate.
La maggior parte degli studi istopatologici evidenziano che il gene
mutante scatenante le retinopatie
sta nei bastoncelli. Proprio questa
evidenza sembra indicare nell’ingegneria genetica, la corretta strategia su cui puntare per risolvere
in un futuro più 0 meno prossimo
i problemi connessi con le retinopatie.
1 n,o
Decisione dell’Assemblea generale dell ’ A s s o c i a z i o n e svizzcru di retinite pigmentosn del 28 marzo 1998
No nll’iniziutivn
protezione genetica
L’Associazione svizzera di retini& pigmentosa è un’associazione di aiuto reci~IYICO di persone con retinite pigmentosu e analoghe malattie degenerative della retina. Queste malattie sono ereditarie e R tutt’oggi non ancora trattabili, se
non in qualche rarissimo caso. 1 primi
sintomi che si mnnifestnno sono cecità
notturna, limitazioni del campo visivo e
un’elevata sensibilità all’abbagliamento. Le malattie degenerative della retina
possono portare R cecità completa. Una
forma p a r t i c o l a r e i: l a S i n d r o m e d i
Usher, una malattia in cui ai problemi
visivi sono associati dei difetti dell’udito. Oggi la Sindrome di Ushrr k la principale causa di soldo-cwità. In SvizzeTU, sono circa 5.000 le persone affette
da malattie degenerative di tipo ereditorio interessanti la retina.
In occasione della loro Assemblea Generale del 28 tnarzo 1998, i membri dell ’ A s s o c i a z i o n e svizzera di retinite pigmentosa hanno espresso la loro pwoccupnzione per le potenziali ripercussioni che
I’nccettnzione d e l l ’ i n i z i a t i v a
protezione genetica potrebbe comportare. Essi fanno appello alla popolazione
affinché non decida in modo dn privare
le persone con malattie genetiche, della
libertk di ricorrere u una futura terapia
genica. Gli attuali risoltati della riccrcn
lasciano presupporre che, una volta
mess8 a punto una simile terapia, una
parte delle persone malate p o t r a n n o
trarne giovamento. Fino a quel momento esse rivendicano lu liberti1 di ricercare le cnuse genetiche della loro malattia.
Dalle ricerche si aspettano per prima
cosa una migliorata consulenza genetica, come pure delle basi scientifiche per
sviluppare una terapia che possa rallentare il decorso della loro malattia o addirittura un suo arresto al fine di prevenire la cecit& La ricerca sulle cause genetiche delle malattie degenerative della retina e In verifica preclinica di una
potenziale terapia non sono fattibili
s e n z a l ’ i m p i e g o di cnvic transgeniche.
Se I’iniziativu protezione genetica venisse accettata in votazione popokwe, in
Svizzera tali riccwhe n o n v e r r e b b e r o
più permesse.
19