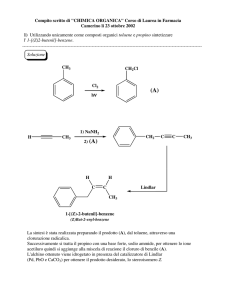
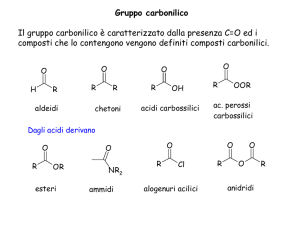
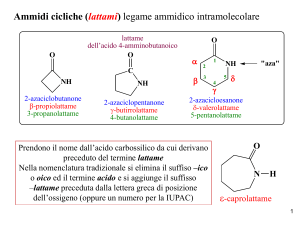
Dispense del Corso di
Chimica delle Sostanze Organiche Naturali
a.a. 2006-2007
1
Il materiale presente in queste dispense è tratto da:
P. M. Dewick
Chimica biosintesi e bioattività delle sostanze naturali
Edizione italiana
Piccin 2001
Clayden, Greeves, Warren, Wothers
Organic Chemistry
Oxford University Press 2001
K. B. G. Torssell
Natural Product Chemistry
John Wiley & Sons 1983
K. C. Nicolau, E. J. Sorensen
Classics in Total Synthesis
VCH 1996
J. R. Hanson
Natural products
Tutorial Chemistry Text
The Royal Society of Chemistry 2003
2
Tutti gli organismi hanno bisogno di sintetizzare e trasformare un gran numero di composti organici
per poter vivere, crescere e riprodursi. Essi devono procurarsi energia sotto forma di ATP e rifornirsi
delle materie prime per costruire i propri tessuti. A questo scopo utilizzano una serie di reazioni
chimiche mediate da enzimi ed accuratamente regolate, che nel loro insieme sono chiamate
metabolismo intermedio. Alcune delle molecole fondamentali per la vita sono i carboidrati, le
proteine, i grassi e gli acidi nucleici e, a parte i grassi, tutti questi composti sono polimerici. La
capacità degli organismi di sintetizzare e trasformare sostanze chimiche è molto varia. Per esempio le
piante sono molto efficienti nel sintetizzare, mediante fotosintesi, composti organici da materiali
inorganici presenti nell’ambiente, mentre altri organismi come gli animali od i microrganismi
ottengono le loro materie prime dalla dieta. Per questo molte vie metaboliche riguardano la
degradazione del materiale assunto come cibo producendo dei “mattoni molecolari” più semplici,
mentre altri processi sono richiesti successivamente per sintetizzare molecole specializzate dai
“mattoni” metabolici così ottenuti.
Nonostante le caratteristiche estremamente varie degli organismi viventi, le vie per modificare e
sintetizzare carboidrati, acidi nucleici e proteine sono essenzialmente le stesse in tutti gli organismi, a
parte piccole variazioni. Questi processi che dimostrano la sostanziale unità di tutta la materia
vivente sono descritti nel loro insieme come metabolismo primario ed i composti che sono compresi
in queste vie sono chiamati metaboliti primari.
In contrasto con le vie metaboliche primarie, che sintetizzano, degradano e più in generale
trasformano composti presenti in tutti gli organismi, esiste anche una parte del metabolismo che
riguarda composti con una distribuzione in natura molto più limitata. Questi composti, chiamati
metaboliti secondari, si trovano solo in specifici organismi o gruppi di organismi e sono espressione
dell’individualità della specie. I metaboliti secondari non sono necessariamente prodotti in tutte le
condizioni e nella stragrande maggioranza dei casi non sono ancora noti né la funzione di questi
composti né i vantaggi che essi apportano all’organismo che li produce. Alcuni sono prodotti per
ragioni facilmente comprensibili, ad esempio composti tossici che forniscono protezione contro la
predazione, composti volatili che servono come attrattori verso la stessa od altra specie, composti che
fungono da coloranti. E’ comunque logico pensare che tutti i metaboliti secondari abbiano un ruolo
importante per il benessere dell’organismo che li produce. È proprio l’area del metabolismo
secondario che fornisce la maggior parte delle sostanze naturali farmacologicamente attive.
Una buona definizione di metabolita secondario è la seguente: “non-nutritional chemicals controlling
the biology in other species in the environment or in other words secondary metabolites play a
prominent role in the co-existence and co-evolution of other species”. Ogni organismo ha adattato la
produzione di metaboliti alle sue condizioni di vita e questa produzione non può essere casuale.
3
I metaboliti secondari prodotti nelle piante e negli animali hanno un forte controllo sul
comportamento di altri individui od altre specie. È stato possibile individuare diversi tipi di controllo
chimico applicato alla lotta per la sopravvivenza:
1. attrazione sessuale (maschio-femmina)
È l’esempio più noto. Nel 1954 è stato isolato il primo feromone, il bombicolo, dalla
femmina del baco da seta, bombyx mori.
OH
bombicolo
Il potere di segnalazione di questi attrattori sessuali è elevatissimo. Il materiale prodotto da
una singola femmina può essere percepito, in condizioni di vento favorevole, fino a diversi
chilometri di distanza. Il maschio reagisce a livelli di concentrazione di circa 100 molecole
per cm3. E’ ovvio che un sistema di richiamo così efficace debba essere anche
estremamente selettivo.
Questa efficacia e selettività consente di utilizzare questi composti anche per il controllo
della popolazione di un certo insetto. Lo scarafaggio Ips typographus causa notevoli danni
nelle pinete della Scandinavia, ma è attratto da un semplice monoterpene, il verbenolo, che
può essere usato per attirare l’insetto in trappole predisposte.
La “sostanza della regina” è anch’essa un feromone sessuale che controlla le api operaie e
la costruzione della cella della regina
O
CO2H
sostanza della regina
2. feedants, antifeedants, repellenti e tossine (animali-piante, animali)
La farfalla cavolaia (Pieris brassicae) ha sviluppato il gusto per uno specifico composto, la
sinigrina, presente nel cavolo, che funziona da attrattore e ne stimola la nutrizione (feedant).
Questo stesso composto funziona da repellente ed è tossico per molti altri insetti. Il
vantaggio per la farfalla è che non deve competere per il cibo con altre specie ma non si
vede il vantaggio per la pianta. Alcuni insetti si sono adattati ad alcune tossine e le usano a
proprio vantaggio. Ad esempio la farfalla monarca si nutre, fin dal suo stato larvale, di
alcuni milkweeds ricchi di cardenolidi (vedi capitolo steroidi). Queste tossine si
accumulano nel bruco e successivamente nella farfalla. In questo modo un solo individuo
4
adulto può indurre un violento vomito in un uccello predatore che eviterà di ripetere la
cattura.
3. difesa ed allarme (animali)
Tipico è l’esempio della puzzola che produce mercaptani come sistema di difesa.
SH
SH
sostanze prodotte dalla puzzola
È noto che formiche sotto stress producono per difesa acido formico. Questo è anche un
segnale di allarme per il resto della colonia.
4. sviluppo, metamorfosi, soppressione della crescita (animali-piante, animali, piante)
Un tipo di salvia californiana produce monoterpeni, 1,8-cineolo e canfora, come inibitori
della crescita di altre piante e per questo è sempre circondata da una zona di terreno nudo.
O
1,8 cineolo
Durante la stagione delle piogge queste sostanze sono maggiormente dilavate dalla pianta e
questo porta ad una riduzione dell’area libera attorno ad essa.
5. comportamento sociale
Sempre messaggi chimici sono alla base del comportamento di termiti per quanto riguarda
la costruzione del nido, di api per la delimitazione del proprio territorio, di formiche per il
riconoscimento del cammino eseguito. L’ipsidienolo è il feromone di aggregazione di un
tipo di coleottero.
OH
ipsidienol
La distinzione tra metaboliti primari e secondari lascia una zona grigia di confine per cui alcune
sostanze potrebbero appartenere ad entrambe le categorie. Un esempio sono gli acidi grassi e gli
zuccheri: la maggior parte di essi sono descritti come metaboliti primari, ma alcuni di loro sono
molto rari e presenti solo in alcune specie. Analogamente, la biosintesi di steroidi produce una vasta
gamma di strutture molto diffuse, ma alcuni steroidi, spesso dotati di attività farmacologica, sono
presenti solo in alcuni tipi di organismi. La divisione è quindi per certi versi convenzionale e lascia
spazio a sovrapposizioni.
5
I metaboliti secondari, con alcune eccezioni, sono presenti in quantità inferiori allo 0.01% del peso
secco dell’organismo. L’estratto di 1 Kg di sostanza secca può contenere fino a 100 mg di un
prodotto naturale che spesso può essere instabile o far parte di miscele particolarmente complesse.
L’organismo deve essere identificato in modo preciso, perché all’interno della specie vi possono
esser chemotipi con composizioni diverse. Per quanto riguarda le piante, alcuni composti possono
essere presenti solo nella corteccia o nelle radici, foglie, fiori o frutti. Alcuni prodotti poi hanno un
ciclo stagionale. Per tutti questi motivi dovrebbe essere registrata la parte della pianta utilizzata, il
tipo di pianta, il luogo di origine e la data di raccolta.
Prodotti naturali possono essere ottenuti da materiale sminuzzato per estrazione con solventi quali
etere di petrolio, cloroformio, acetato di etile, metanolo. L’estrazione può essere fatta più volte
utilizzando solventi sempre più polari. In questo modo materiale lipidico (cere, acidi grassi, steroli,
carotenoidi e terpeni) può essere estratto con etere di petrolio, mentre sostanze più polari come i
glicosidi e gli alcaloidi devono essere estratti con metanolo, o addirittura con H2O. L’estrazione
iniziale viene in genere eseguita da una separazione in frazione acida, basica e neutra come
esemplificato nello schema seguente.
Estratto sciolto in etile acetato
sol. satura NaHCO3
estrazione in fase
acquosa di acidi forti
carbossilici e solfonici
sol. HCl
basi vengono
protonate ed estratte
in fase acquosa
sol. NH3
sol. HCl
riformazione degli acidi
indissociati estratti poi
in fase organica
Le basi vengono liberate
ed estratte in fase organica
sol. NaOH
estrazione acidi
deboli (fenoli)
composti neutri
sol. HCl
riformazione degli acidi
indissociati estratti poi
in fase organica
Nonostante alcuni prodotti più abbondanti possano essere ottenuti semplicemente per estrazione o
cristallizzazione, la maggior parte deve essere isolata dopo un attento processo di purificazione, per
lo più cromatografico. Il passo successivo è la determinazione strutturale che consiste in diversi
passaggi.
6
Caratterizzazione:
determinazione delle caratteristiche chimico-fisiche, la
formula molecolare ed i diversi gruppi funzionali
Determinazione scheletro carbonioso:
Utilizzo tecniche spettroscopiche e trasformazioni
chimiche.
Determinazione della posizione dei
gruppi funzionali e della stereochimica
relativa:
Utilizzo tecniche spettroscopiche
Determinazione della stereochimica
assoluta:
Alcune tecniche spettroscopiche, raggi X, sintesi totale.
L’ultimo passaggio, la determinazione della stereochimica assoluta, è probabilmente il più
complesso. Per quanto le tecniche di indagine spettroscopica siano strumenti indispensabili ed
efficaci per l’analisi strutturale, a volte non è possibile risolvere il problema in modo definitivo se
non ricorrendo alla sintesi totale del prodotto naturale. Ancora oggi si possono trovare in letteratura
lavori in cui la sintesi totale di un metabolita secondario ha consentito di correggere una struttura
assegnata erroneamente.
Individuazione della sequenza metabolica
Per milioni di anni la natura ha raffinato la sua capacità sintetica e vi è un forte desiderio, e necessità,
da parte dei chimici di individuare le modalità con cui la natura costruisce e degrada le sue molecole.
Inizialmente risultati accidentali contribuirono alla delucidazione di passaggi intermedi. Knoop
postulò fin dal 1904 che la degradazione degli acidi grassi avvenisse attraverso una -ossidazione
che avrebbe portato alla produzione di acido acetico, cioè la catena sarebbe stata tagliata di due atomi
per volta. Acidi grassi con un gruppo aromatico terminale ed a numeri pari di atomi carbonio
vengono metabolizzati e recuperati dalle urine come acido fenilacetico mentre acidi con numero di
carboni dispari danno l’acido benzoico. All’incirca negli stessi anni (1907) Collie postulò che la
reazione inversa, condensazione dell’acetato tipo Claisen, fosse all’origine di molti derivati fenolici
presenti in natura. Normalmente gli intermedi di una via biosintetica sono presenti ad una bassa
concentrazione dello stato stazionario, ma durante una malattia si può avere un accumulo di un
intermedio. Ad esempio un sintomo del diabete è la grande quantità di acetoacetato che appare nel
sangue e nelle urine che lo indica come prodotto di degradazione dall’ossidazione di acidi grassi.
Studi nutrizionali su organismi sani possono indicare l’inizio e la fine di un processo metabolico ma
non possono dare informazioni sugli stadi intermedi. Un vero progresso si è avuto con lo studio di
organismi geneticamente alterati, i cosiddetti mutanti, e con l’introduzione di composti
isotopicamente marcati.
7
Microrganismi mutanti possono sorgere spontaneamente o possono essere prodotti per azione di
agenti chimici o di radiazioni (raggi x, UV). Se la lesione così causata non è letale spesso accade che
un solo gene viene danneggiato, cioè la cellula è incapace di produrre uno specifico enzima. Un
danno nel metabolismo primario posta alla inibizione della crescita. Più difficile è metter in risalto un
danno nel metabolismo secondario, poiché questi prodotti non sono essenziali per lo sviluppo
dell’organismo, ma il principio alla base è lo stesso.
Supponiamo di studiare la sequenza essenziale A→E. L’organismo sano può completare l’intera
sequenza ed A è necessario per la crescita. Nel mutante 1 il passaggio D→E è bloccato, D si
accumulerà e la crescita può essere sostenuta solo aggiungendo E al nutriente. Nel mutante 2 il
composto C si accumulerà e l’aggiunta di D od E riporterà la normale attività dell’organismo. Nel
mutante 3 B→C è bloccato, ma non necessariamente B si accumulerà poiché B può, magari, essere il
substrato di un altro enzima che porterà ad altri metaboliti F o G. Inoltre il filtrato del terreno di
coltura di 1 può sostenere la crescita di 2 ma non viceversa. La via dell’acido shikimico è stata
analizzata per gran parte da Davis usando mutanti dell’escherichia coli.
Organismo sano
A
→
B
→
C
→
D
→
E
Mutante 1
A
→
B
→
C
→
D
no
E
Mutante 2
A
→
B
→
C
no
D
→
E
Mutante 3
A
→
B
no
C
→
D
→
E
→
G
↓
F
Un metodo estremamente potente è l’uso di isotopi. Le ricerche fondamentali sono state fatte
utilizzando isotopi radioattivi 3H, 14C, 32P, ma in anni più recenti è stata la spettroscopia 13C-NMR ad
essere la più impiegata.
I mattoni biosintetici
I mattoni biosintetici per i metaboliti secondari derivano da metaboliti primari. I metaboliti
provenienti da processi fondamentali quali la fotosintesi, la glicolisi ed il ciclo di Krebs possono
essere allontanati dai processi di generazione dell’energia ed originare intermedi biosintetici.
8
GLICOLISI
OH
glucosio
OH
O
OH
OH OH
OH
H2N
OH
Glicina
OH
OP OH
glucosio 6-P
O
O
HS
OH
HO
NH2
L-Cisteina
OH
O
O
CICLO DEL PENTOSO
FOSFATO
HO
PO
O
OH
HO2C
FOTOSINTESI
OH
O
OH
NH2
L-serina
OH
PO
acido
3-fosfoglicerico
NH2
L-fenilalanina
CO2H
HO2C
O
L-valina
NH2
O
L-leucina
HO2C
OH
NH2
acido shikimico
HO
acido
fosfoenolpiruvico
OH
L-alanina
OP
OH
OH
O
HO
O
O
OH
acido piruvico
NH2
L-tirosina
O
OH
NH2
OH
CoSA
O
N
H
NH2
L-triptofano
Acetil-CoA
Un altro grafico che può dare l’idea della relazione tra metabolismo primario e secondario può essere
il seguente tratto da Clayden.
9
Il numero di mattoni necessari è sorprendentemente piccolo e, come con i giochi delle costruzioni,
una grande varietà di oggetti può essere costruita con un limitato numero di mattoni base. Metaboliti
secondari possono essere sintetizzati combinando molti mattoni biosintetici dello stesso tipo, oppure
usando una miscela di mattoni biosintetici diversi. Questo accresce la varietà strutturale. I più
importanti mattoni biosintetici sono quelli riportati nello schema seguente.
10
MATTONI BIOSINTETICI
C
C 1 deriva da L-metionina
O
S
OH
NH2
C
5
2
deriva da Acetil-CoA o da malonil-CoA
CO2H
SCoA
SCoA
O
O
unità isoprenica, deriva da acido mevalonico che a sua volta deriva da tre unità di
acetl-CoA
HO
SCoA
3X
CC
O
CO2H
OH
deriva da fenilalanina e tirosina per perdita del gruppo amminico. I gruppi C C e C C
6 2
6 3
6 1
derivano da C C per frammentazione
6 3
O
OH
NH2
O
OH
NH2
HO
C C N deriva da fenilalanina e tirosina per decarbossilazione.
6 2
O
OH
NH2
NH2
O
OH
HO
NH2
11
Indolo. C N deriva da triptofano per decarbossilazione.
2
O
OH
NH2
N
H
N
H
NH2
C4N
Si trova di solito come sistema pirrolidinico e si forma
dall'amminoacido non proteinogenico L-ornitina. Ornitina non fornisce il
suo atomo di N
-amminico ma quello -amminico
O
H2N
OH
H2N
CH3
N
H
NH2
C N Analogo al precedente ma usando l'amminoacido L-lisina
5
O
H2N
OH
NH2
H2N
CH3
N
H
Ovviamente l’elaborazione ed il collegamento dei diversi mattoni biosintetici avviene in reazioni per
lo più catalizzate da enzimi ma che possono sempre essere descritte in termini di reattività chimica.
Di volta in volta cercheremo di approfondire la descrizione della reattività dei diversi mattoni
biosintetici e delle reazioni interessate.
12
VIA DELL’ACETATO: I POLICHETIDI
I polichetidi costituiscono una cospicua classe di prodotti naturali raggruppati sulla base della loro
comune biosintesi. Tutti derivano da -polichetoni formatisi per condensazione di molecole di acido
acetico (C2).
n CH3CO2H → CH3(COCH2)n-1CO2H
I metaboliti formatisi mediante questa via biogenetica sono gli acidi grassi, composti poliacetilenici
e le prostaglandine, ma anche gli antibiotici macrolidici ed i molti composti aromatici come gli
antrachinoni e le tetracicline.
La formazione di una catena -polichetonica può essere immaginata come una serie di reazioni di
Claisen. Si può immaginare che due molecole di acetil-coA diano luogo ad una condensazione di
Claisen formando l'acetoacetil-coA, e la ripetizione per n volte di questa reazione genera un polichetotioestere.
O
O
O
O
S-CoA
O
O
S-CoA
O
O
S-CoA
S-CoA
S-CoA
Un modello di questo tipo è però impreciso. E’ infatti noto che l’agente nucleofilo nella reazione di
Claisen è il malonil-SCoA secondo il meccanismo descritto nello schema seguente.
O
S-CoA
O
O
-H+
S-CoA
O
-CoAS-
O
S-CoA
S-CoA
O
O
S-CoA
O
H
Appare evidente il vantaggio dell’uso di un derivato del’acido malonico in questa condensazione di
Claisen. Questo reagente è infatti molto più reattivo e la concomitante decarbossilazione fa sì che il
protone che viene perso sia quello dell’acido carbossilico e non uno legato all’atomo di C, processo
decisamente più facile.
13
Il poli--chetotioestere così formato può andare incontro ad una diversa sequenza di reazioni a
seconda che si debba formare un polichetide aromatico oppure un acido grasso. Nel primo caso il
processo di allungamento della catena continua, generando una catena poli--chetidica altamente
reattiva, stabilizzata dalle interazioni con la superficie dell'enzima. Per quanto riguarda gli acidi
grassi, il gruppo carbonilico viene ridotto prima della successiva condensazione con un'altra
molecola di malonil-coA. Nella sintesi di macrolidi
sono invece possibili parziali riduzioni.
Acidi grassi
Nella biosintesi degli acidi grassi un ruolo importante è
svolto dall'enzima acido grasso sintetasi che negli
animali è un unica proteina multifunzionale contenente
tutti i siti catalitici, mentre nei batteri e nelle piante
diversi enzimi operano le stesse reazioni.
La biosintesi avviene attraverso il meccanismo riportato
a fianco (tratto da Lehninger). L’enzima acido grasso
sintetasi è schematizzato in grigio. Il gruppo acetilico è
legato ad un residuo di cisteina mentre il residuo di
acido malonico è legato ad una proteina (ACP acyl
carrier protein) che forma un complesso con l’enzima.
La successiva reazione di Claisen trasforma il malonilACP
in
acetoacetil-ACP
che
viene
ridotto
al
corrispondente -idrossi estere consumando NADPH.
La reazione è stereospecifica così che si forma solo
l'enantiomero R. La successiva eliminazione di H2O
porta alla formazione dell'acil-ACP - insaturo con
stereochimica E. Nuovamente NADPH riduce il doppio
legame fornendo l'acil-ACP saturo che costituisce il
materiale di partenza per un nuovo ciclo di reazioni
dopo essere stato trasferito sul residuo di cisteina. Ad
ogni ciclo si ha l'allungamento della catena dell'acilACP di 2 atomi di C. A questo punto l'acil-ACP può
essere rilasciato dall'enzima sotto forma di acil-coA o
14
come acido libero. Nello schema successivo viene mostrata la biosintesi dell’acido palmitico (ripresa
da Lehninger).
Una volta raggiunta la lunghezza di 16 atomi di C (acido palmitico) l’accrescimento della catena si
ferma e la catena viene espulsa dall’enzima.
Questa combinazione di unità acetato porta alla formazione di acidi lineari con un numero pari di
atomi di carbonio.
Acidi grassi a catena con n° dispari di atomi di C si possono formare da acido propionico che
sostituisce l'acetato.
BUTIRRICO
C4
STEARICO
C18
CAPRONICO
C6
ARACHIDICO
C20
CAPRILICO
C8
BEHENICO
C22
CAPRICO
C10
LIGNOCERICO
C24
LAURILICO
C12
CEROTICO
C26
MIRISTICO
C14
MONTANICO
C28
PALMITICO
C16
MELISSICO
C30
15
I principali grassi naturali sono i trigliceridi, ossia esteri del glicerolo con acidi grassi. Questi
prendono il nome di grassi se solidi o di oli, se liquidi.
Si parla di trigliceridi semplici se i tre acidi sono uguali, misti se diversi. La maggior parte dei grassi
e degli oli naturali sono composti principalmente da trigliceridi misti. I trigliceridi si formano a
partire da glicerolo 3-fosfato mediante esterificazione con Acil-CoA
O
O
OH
R
HO
O
R1
HO
OP
glicerolo-3-fosfato
P=
S-CoA
O
O
R
O
S-CoA R1
R
O
O
OP
1-acilglicerolo-3-fosfato
O
P
OH
OH
OP
1, 2-diacilglicerolo-3-fosfato
idrolisi
e terza esterificazione
O
O
R1
R
O
O
O
R2
O
1,2-diacilglicerolo-3-fosfato prende anche il nome di acido fosfatidico e costituisce lo scheletro dei
fosfolipidi. In questi composti il fosfato è esterificato con colina, etanolammina, serina.
HO
HO
HO
N
colina
etanolammina
NH2
CO2H
serina
NH2
A questo punto possiamo fare alcune valutazioni di carattere chimico legate alla natura dei composti
coinvolti nella biosintesi di acidi grassi. Una prima domanda può essere: perché sono coinvolti
tioesteri piuttosto che esteri?
La principale differenza con i normali esteri è che i doppietti di non legame dell’atomo di zolfo sono
in orbitali 3p. Questi orbitali sono troppo estesi per sovrapporsi in modo efficace con l’orbitale 2p
16
posto sull’atomo di carbonio del gruppo carbonilico. Per questo motivo vi è una minore
coniugazione.
O
O
O
O
R
O
R
O
S
buona sovrapposizione e coniugazione
R
S
R
scarsa sovrapposizione e coniugazione
Questa differenza influenza tutti e tre gli stadi della condensazione di Claisen nello stesso modo. I
tioesteri sono più facilmente convertiti nei loro enolati, sono più facilmente attaccati da nucleofili e
l’anione RS- è un miglior gruppo uscente di ROQueste proprietà possono essere sfruttate anche in processi chimici utilizzando tioesteri in
condensazioni di Claisen
COSEt
EtSH
COSEt
COSEt
O
91%
NaH
CH3OCH2CH2OCH3
Il tioestere derivante dall’acido adipico subisce una condensazione di Claisen a temperatura
ambiente, in condizioni più blande ed una resa superiore rispetto al corrispondente diestere.
E’ possibile spingere ancora più in là questo parallelo tra biosintesi e sintesi eseguendo la
condensazione di Claisen in condizioni neutre utilizzando reagenti specifici: il sale di Mg del
malonato mono-tioestere come nucleofilo ed una imidazolide come elettrofilo.
N
O
O
O
S
-CO2
O
R
O N
R
N
R
O
O
S
R
R
S
R
N
-
N
O
N
Semplicemente mescolando a temperatura ambiente i due reagenti si ottiene in modo efficace il
prodotto finale. L’uso della imidazolide è legato al valore di pKa dell’imidazolo (7). Questo fa sì che
l’anione dell’imidazolo sia un ottimo gruppo uscente e che l’imidazolide sia un forte elettrofilo.
L’imidazolide può essere preparata da un acido carbossilico ed il carbonil diimidazolo
17
O
N
N
O
N
N
R
O
O
O
N
O
N
-CO2
R
N
N
N
R
OH
HN
Acidi grassi insaturi
Gli acidi grassi insaturi possono essere sintetizzati attraverso più di una via biogenetica ma nella
maggior parte degli organismi essi sono formati per deidrogenazione dei corrispondenti acidi grassi
La maggior parte degli eucarioti possiede una 9-desaturasi che catalizza la formazione di un doppio
legame cis in un acido grasso saturo richiedendo NADPH ed ossigeno come cofattori. Il meccanismo
non è del tutto chiarito.
Il tioestere dell'acido stearico (C18) è l'acido grasso di partenza che, dopo deidrogenazione, viene
trasformato in acido oleico (individuato anche come 18:1 (9c), sotto forma di CoA è utilizzato da
animali e funghi ed in forma di ACP dalle piante.
COS-CoA
stearico
9 desaturasi
COS-CoA
oleico
La posizione dei doppi legami successivi dipende dall'organismo interessato. Nei non mammiferi (in
primo luogo batteri e piante), gli enzimi catalizzano l'introduzione di un ulteriore doppio legame tra
quello già esistente ed il terminale metilico e quindi si ha la trasformazione in linoleico ed linolenico.
COS-CoA
oleico
18:1 (9c)
COS-CoA
linoleico
18:2 (9c, 12c)
COS-CoA
-linolenico
18:3 (9c, 12c, 15c)
18
Si può notare come i nuovi doppi legami introdotti nei diversi passaggi non siano coniugati ma vi sia
sempre un gruppo metilenico ad interrompere la coniugazione. Questa caratteristica è comune a tutti
i principali acidi grassi insaturi.
Invece nei mammiferi il nuovo doppio legame viene formato verso la funzione carbossilica e quindi
l'oleato viene trasformato in 6,9 octadienoato.
COS-CoA
oleico
18:1 (9c)
COS-CoA
6,9 octadienoato
18:2 (6c, 9c)
Gli animali però necessitano di linolenato per sintetizzare il di-omo--linolenato (20:3, 8,11,14,
eicosatrienoato) e l'arachidonato (20:4, 5,8,11,14) precursori delle prostaglandine della serie uno e due.
L'acido linoleico deve quindi essere introdotto con la dieta e successivamente deidrogenato verso il
terminale carbossilico per ottenere il gamma-linolenico che viene utilizzato per estendere la catena.
Anche l'-linolenico deve essere introdotto con la dieta ed è il precursore dell'EPA che è precursore
delle prostaglandine della serie tre. Linoleico, DHA e EPA sono gli acidi grassi omega-3.
Nella figura seguente è riportata una tabella riassuntiva dei diversi passaggi biosintetici:
CO-SR
Stearico 18:0
CO-SR
linoleico 18:2 (9c, 12c)
CH3
CH3
tutti gli eucarioti
piante
funghi
animali
piante
funghi
CO-SR
oleico 18:1 (9c)
CO-SR
-inolenico 18:3 (6c, 9c, 12c)
CH3
CH3
+ C2 (malonato)
animali
18:2 (6c, 9c)
CO-SR
diomo--linolenico 20:3 (8c, 11c, 14c)
CH3
CO-SR
CH3
Prostaglandine
serie 2
CH3
-linolenico 18:3 (9c, 12c, 15c)
animali
CO-SR
stearidonico 18:4 (6c,9c, 12c, 15c)
CH3
+ C2 (malonato)
CO-SR
20:4 (8c, 11c, 14c, 17c)
CH3
animali
Prostaglandine serie 1
CO-SR
CO-SR
CO-SR
arachidonico 20:4 (5c, 8c, 11c, 14c)
eicosapentaenoico EPA 20:5 5c,8c,11c,14c,17c
CH3
CH3
Prostaglandine
+
C2
(malonato)
serie 3
CO-SR
CO-SR
docoesaenoico DHA 4c,7c,10c,13c,16c,19c
CH3
docosapentaenoico DPA 7c, 10c, 13c,16c,19c
CH3
19
Tra i derivati degli acidi grassi insaturi troviamo l'acido ricinoleico, ottenuto per ossidrilazione
diretta, in posizione 12 dell'acido oleico (di solito esterificato sotto forma di fosfolipide da una
ossidasi O2 e NADPH dipendente. E’ il componente principale dell’olio di ricino.
COOR
acido oleico
CH3
O2
NADPH
COOR
acido ricinoleico
CH3
OH
Per successive deidrogenazioni di prodotti insaturi si ha la formazione di composti contenenti uno o
più tripli legami.
CO-SR
linoleico 18:2 (9c, 12c)
CH3
CO-SR
CH3
acido crepeninico 18:2 (9c,12a)
CO-SR
acido deidrocrepeninico 18:3
(9c,12a, 14c)
CH3
OH
OH
CH3
cicutossina
Questi composti sono largamente diffusi in natura. Alcuni sono molto instabili ed addirittura
esplosivi. Mentre gli acidi grassi con molti doppi legami tendono a formare sistemi non coniugati,
al contrario molecole con tripli legami tendono a formare sistemi coniugati
Acidi grassi ramificati
Vari esempi di acidi grassi ramificati sono stati individuati nei mammiferi.
Una catena laterale metilica può essere introdotta utilizzando metilmalonil-CoA che deriva dalla
carbossilazione, biotina dipendente, dell'acido propionico.
Ad esempio l'acido 2,4,6,8
tetrametildecanoico viene biosintetizzato a partire da una unità di acetato e quattro di metilmalonilSCoA.
CO2, ATP
SCoA biotina
HO
O
O
SCoA
O
HO
SCoA
O
Per il meccanismo vedi più avanti
O
SCoA
+ 4X
CO2H
O
In altri casi una unità metilica può essere introdotta mediante un meccanismo di C-alchilazione
usando S-adenosilmetionina (SAM). L'acido turbeculostearico (isolato dal bacillo della tubercolosi)
deriva dall'acido oleico per alchilazione al C10. E’ possibile ipotizzare che si formi un intermedio
carbocationico, il quale può accettare un idruro dal NADPH
dando luogo all'acido
tuberculostearico. In alternativa , la perdita di un protone con la formazione di un anello
21
ciclopropanico può dar luogo alla formazione dell'acido diidrosterculico. Questo acido viene
deidrogenato ad acido sterculico. Quest'ultimo si trova nell'olio di semi di alcune piante. E' un
inibitore della 9-desaturasi, l'enzima che converte l'acido stearico in oleico ed è potenzialmente
pericoloso per l'uomo.
Ad
S+
R
COS-CoA
H
CH+
COS-CoA
NADPH
-H+
CO2H
acido tuberculostearico
COS-CoA
COS-CoA
acido sterculico
Prostaglandine
Gruppo di acidi grassi a venti atomi di carbonio isolati per la prima volta dallo sperma umano.
Inizialmente si riteneva che fossero secrete dalla prostata, adesso si sa che sono diffuse nei tessuti
animali ma solo in piccole quantità ed è dimostrato che esse hanno molteplici attività. Sono attive a
concentrazioni molto basse, simili a quelle ormonali e regolano la pressione sanguigna, la
contrazione della muscolatura liscia, la secrezione gastrica e l'aggregazione piastrinica. A differenza
degli ormoni non circolano con il flusso sanguigno, né vengono accumulate in alcuni tessuti, ma
vengono prodotte localmente in caso di necessità, svolgono un ruolo dipendente dal tessuto in cui
vengono prodotte e successivamente vengono disattivate attraverso un metabolismo molto rapido
Lo scheletro base è quello di un acido grasso a venti atomi di carbonio contenente un anello
ciclopentanico, una catena laterale C7 con funzione carbossilica ed una C8 con il terminale
metilico. Sono biosintetizzate da tre acidi grassi essenziali:
22
CO-SR
CO-SR
CH3
CH3
diomo- -linolenico 20:3 (8c, 11c, 14c) arachidonico 20:4 (5c, 8c, 11c, 14c)
O
O
CO2H
CO2H
CH3
HO
CH3
OH PGE
HO
PGE 1 OH
2
Dai tre precursori si formano
prostaglandine diverse solo per il
grado di insaturazione nelle catene
laterali
CO-SR
CH3
eicosapentaenoico EPA 20:5 5c,8c,11c,14c,17c
O
CO2H
CH3
HO
OH PGE
3
Lo scheletro base delle prostaglandine è l'acido prostanoico.
O
9
1 OH
12
20
La nomenclatura semisistematica è basata sui sostituenti presenti sull'anello a 5 termini, a cui ci si
riferisce con una lettera, mentre il numero dei doppi legami nelle catene laterali è indicato da un
numero riportato in pedice. Una lettera o (sempre in pedice) indicano la configurazione al C9:
sotto il piano (naturale) sopra il piano.
O
O
PGA
O
HO
O
R2
R2
R2
R1
R1
PGB
R1
PGC
R2
R2
HO
R2
R1
R1
R1
O PGD HO PGE
HO PGF
PGA, PCB, PGC non naturali, artefatti
Serie 1
CO2H
OH
Serie 2
CO2H
OH
Serie 3
CO2H
OH
23
Lo scheletro prostaglandinico è particolarmente poco stabile a pH al di fuori dell’intevallo 5-8, per
questo motivo sono stati spesso ritrovati anche composti insaturi come PGA, PCB e PGC che non
sono naturali ma artefatti dovuti al processo di isolamento
La biosintesi delle prostaglandine avviene tramite il meccanismo sotto descritto.
CO-SR
CO-SR
H
O O
Il primo passaggio è una estrazione di un atomo di idrogeno da una posizione allilica da parte
dell’ossigeno. L’atomo rimosso è tra due doppi legami così che il radicale risultante è doppiamente
allilico. Questo radicale allilico cattura una molecola di ossigeno al C11 per formare un nuovo
ossiradicale. La reazione avviene ad un estremo del sistema allilico così che il prodotto è un diene
coniugato ed il nuovo doppio legame è E.
CO-SR
CO-SR
O• O
O O
A questo punto l’intermedio subisce una elaborata modifica dovuta all’addizione dell’ossiradicale al
C9 (è su un doppio legame)e quindi una seconda addizione sul diene precedentemente formato.
CO-SR
O
CO2H
O
O• O
Tre nuovi centri stereogenici vengono formati in questa ciclizzazione e tutti sono sotto il controllo
sia del centro già presente sia dal modo in cui la molecola si ripiega sotto l’influenza dell’enzima.
A questo punto il radicale allilico reagisce con ossigeno per dare l’idroperossido PGG2,
estremamente instabile (tempo di dimezzamento a pH=7 di circa 5 minuti)
O
O
CO2H
PGG 2
O
O
O OH
HO
CO2H
O
Successivamente il gruppo perossidico viene ridotto per dare la prostaglandina PGH2che
successivamentesubisce la rottura omolitica del legame perossidico. Questo intermedio è
fondamentale per la biosintesi di diversi derivati.
24
O
O
O
CO2H
PGH 2
CO2H
CH3
O
OH
HO
+ 2H
HO
PGF2
HO
+H
-H
O
CO2H
PGE2
CH3
CH3
HO
HO
CO2H
HO
La migliore evidenza per questo meccanismo è fornita da un esperimento con O2 marcato. Se viene
fornita, al microrganismo che produce PGF2una miscela di
16
O2 ed
18
O2, la prostaglandina
prodotta avrà, come evidenziato dallo spettro di massa, entrambi gli atomi di ossigeno dei gruppi
ossidrilici sull’anello ciclopentanico come
16
O o come
18
O, ma mai misti, a dimostrazione che
entrambi derivano dalla stessa molecola.
L’enzima che catalizza questa trasformazione, la cicloossigenasi, è un importante bersaglio. Inibire
le prostaglandine vuol dire intervenire su processi infiammatori. Un inibitore della cicloossigenasi è
l’aspirina (acido acetilsalicilico). Il problema è che le prostaglandine regolano anche la secrezione
di succhi gastrici e quindi un eccesso di aspirina può portare all’ulcera. L’inibizione avviene in
seguito alla acetilazione selettiva di un residuo di serina presente nell’enzima. I corticosteroidi
(cortisone) invece funzionano da antinfiammatori perché bloccano il rilascio di acido arachidonico
dai fosfolipidi di riserva.
Appare quindi evidente il ruolo fondamentale svolto dagli acidi grassi essenziali, acido linolenico
ed -linolenico assunti con la dieta. Senza una fonte di acido arachidonico o di composti che
possono essere trasformati in esso, la sintesi delle prostaglandine è bloccata e ciò può provocare
notevoli danni metabolici. E’ necessario un costante rifornimento di precursori delle prostaglandine
poiché esse vengono di continuo sintetizzate e poi disattivate. La degradazione delle prostaglandine
avviene o per ossidazione dell’ossidrile in posizione 15 a gruppo carbonilico od in seguito a
riduzione del doppio legame fra il carbonio in posizione 13.
Come già riportato precedentemente, l’intermedio diradicalico è importante per la sintesi di più
composti naturali facenti parte, insieme alla prostaglandine, del gruppo degli eicosanoidi, in quanto
tutti caratterizzati da una catena a 20 atomi di carbonio.
25
O•
H
O
CO2H
O•
CO2H
CH3
H•
HO
HO
HO2C
CH3
HO
O
PGI 2 Prostaciclina
CH3
HO
HO
Mentre l’ossiradicale in posizione 11 viene spento a formare un gruppo ossidrilico, quello in
posizione 9 ciclizza sul doppio legame tra i carboni C5 e C6 a formare un nuovo anello
tetraidrofuranico fuso con l’iniziale anello ciclopentanico. Il radicale così ottenuto viene spento per
perdita di un atomo di idrogeno dando origine alla prostaciclina PGI2. Questo composto riduce la
concentrazione di ioni calcio ed è efficace nell’abbassare la pressione sanguigna ed inibire
l’aggregazione piastrinica. E’ utilizzato per inibire la coagulazione sanguigna durante la dialisi
renale, ma la sua breve emivita (circa 3 minuti) richiede una continua somministrazione per via
endovenosa. L’elevata instabilità è legata alla presenza di un gruppo enoletere che viene facilmente
idrolizzato a formare la 6 cheto prostaglandina F1
HO2C
Prostaciclina
O
HO
CO2H
PGI 2
H2O
H+
HO
HO
O
HO
HO
6 cheto PGF1
HO2C
HO
iloprost HO
L’Iloprost, un analogo carbociclico, per la sua maggiore stabilità può essere adoperato nel
trattamento delle malattie trombotiche.
Lo stesso intermedio diradicalico visto precedentemente, derivante dalla PGH2, può subire ancora
una diversa trasformazione.
26
O
O
CO2H
CO2H
CH3
O
CH3
O
HO
HO
OH
CO2H
HO
O
CH3
TXB2
HO
CO2H
O
O
H2O
CH3
TXA2 HO
Attraverso il riarrangiamento radicalico sopra descritto si ha la formazione del trombossano A2
(TXA2) che presenta un anello ossetanico su una funzione acetalica, molto reattivo. Infatti viene
velocemente idrolizzato a formare il TBX2. Questi due prodotti sono stati isolati dalle piastrine ed il
TXA2 è molto più attivo biologicamente. Il loro ruolo è di aumentare la concentrazione di calcio nel
citoplasma provocando una deformazione delle piastrine e la loro aggregazione. Il ruolo del TXA2 è
quindi opposto a quello della PGI2. Entrambi comunque derivano dalla PGH2 che nelle piastrine è
trasformata in TAX2 mentre nelle pareti dei vasi sanguigni in PGI2.
Sempre del gruppo degli eicosanoidi vi sono anche altri composti importanti come i leucotrieni,
isolati per la prima volta dai leucociti. Anche in questo caso il precursore è l’acido arachidonico che
viene trasformato nel radicale corrispondente dall’enzima lipoossigenasi. In questo caso la
formazione del radicale avviene in una posizione diversa, al C7, rispetto a quanto succede per la
cicloossigenasi per le prostaglandine (C13).
O OH
O OH
CO-SR
CO-SR
Il radicale formatosi reagisce per dare l’idroperossido. Successivamente questo subisce
trasposizione del doppio legame con formazione del leucotriene LTA4.
Enz
B
H Enz
O OH
CO-SR
O
H
H
CO-SR
LTA4
Il leucotriene LTA4 è il precursore di tutte gli altri derivati leucotrienici. Sono composti coinvolti
nella risposta allergica e nei processi infiammatori. Alcuni di questi leucotrieni sono potenti
broncocostrittori e vasocostrittori. Alcuni di questi favoriscono la migrazione di leucociti nei
processi infiammatori ed intervengono in patologie come la psoriasi e l’artrite.
27
La scoperta delle prostaglandine come potenti agenti farmacologica ha motivato lo sviluppo di
numerose strategie sintetiche per il loro ottenimento. Senz’altro la strategia sintetica di maggior
successo è stata quella sviluppata da E. J. Corey a partire dal 1969.
Analizziamo inizialmente la retrosintesi della prostaglandina PGF2così come è stata proposta da
Corey
Wittig
HO
7
9
10
4
8
6
14
11
5
2
18
12
13
HO
15
HO
17
CO2H
5
HO
1
CO2H
3
16
Ph3P
O
9
6
15
20
RO
19
RO
PGF2
O
O
OH
Horner-Wadsworth
Emmons reaction
O
6
9
15
RO
RO
O
O
O
(MeO)2OP
O
15
O
O
9
O
RO
RO
13
OMe
AcO
Corey lactone
Il doppio legame 5,6 della PGF2 rappresenta un ottimo punto di partenza per semplificare lo
scheletro molecolare. La rottura retrosintetica del doppio legame fornisce un’aldeide ed una ilide di
fosfonio, immaginando che il doppio legame possa formarsi attraverso una reazione di Wittig. C’è
da attendersi che l’aldeide sia in equilibrio con il lattolo ciclico ma questo non dovrebbe impedire di
eseguire la reazione di Wittig. Un punto importante è la stereochimica del doppio legame
interessato che è Z. Questo non è un problema perché l’ilide di fosfonio utilizzata è una ilide non
stabilizzata ed è noto che ilidi non stabilizzate forniscono in modo selettivo doppi legami con
stereochimica Z. Il passaggio retrosintetico successivo prevede soltanto di variare il grado di
ossidazione e non presenta particolari difficoltà. In questo modo però è possibile individuare sulla
molecola una funzione carbonilica - insatura che può essere convenientemente ottenuta
attraverso una reazione di Horner-Wadswords-Emmons (reazione tra un aldeide ed un fosfonato). In
questo modo il precursore sarà l’aldeide. In una forma leggermente diversa e protetta prende il
nome di Lattone di Corey, intermedio essenziale in questa sintesi ma che ha trovato applicazione in
28
molte altre sintesi, anche di prodotti naturali a causa della elevata quantità di informazione
stereochimica e di gruppi funzionali che contiene.
L’analisi retrosintetica svolta fino a questo momento ha semplicemente mirato alla semplificazione
delle catene laterali ma non ha affrontato il problema della funzionalizzazione dell’anello a cinque
termini e della stereochimica relativa dei diversi sostituenti. L’anello ciclopentanico porta 4 centri
stereogenici contigui. Come talvolta accade, una variazione strutturale che consente una retrosintesi
semplice è in realtà, almeno inizialmente, una complicazione.
O
O
iodolattonizzazione
O
O
I
OMe
AcO
10
HO
OMe
11
Corey lactone
O
MeO
9
8
11
10
6
O
6
HO
12
HO
O
OMe
11
Baeyer-Villiger
MeO
MeO
Diels-Alder
8
Cl
CN
11
O
OMe
Cl
CN
In effetti l’introduzione di un atomo di iodio sembra complicare la struttura. In realtà in questo
modo è possibile individuare una reazione di iodolattonizzazione che consente di disconnettere il
lattone al derivato ciclopentenico. Risulta semplice a questo punto riconoscere il lattone a 7 termini
come il precursore dell’idrossiacido. Il lattone a sua volta può essere ottenuto per reazione di
Baeyer-Villiger a partire dal chetone ciclico.
Polichetidi aromatici
Nella biosintesi di acidi grassi il processo di riduzione dopo ogni condensazione di Claisen fornisce
una catena carboniosa satura. In assenza del processo di riduzione, la catena carboniosa poli 29
carbonilica che si ottiene risulta particolarmente reattiva e deve essere stabilizzata sulla superficie
enzimatica fino a che non sia completato il processo di allungamento. Il poli--chetotioestere è
molto reattivo e ci sono varie possibilità di reazioni di Claisen od aldoliche intramolecolari che
avvengono a seconda del tipo di enzima e di come il substrato si ripiega ma comunque sempre a
formare cicli a sei termini.
Fenoli semplici
La catena polichetoesterea formatasi a partire da 4 unità acetato (in realtà da 1 acetato e 3 da
malonato) può piegarsi in almeno due modi, A o B.
CH3 OHO
CH3 O
S-CoA H
+
A O
H+
O
O
H
O
S-CoA
O
S-coA
-H2O O
O
enolizzazione
CH3 O
O
S-coA
HO
OH
acido orsellinico
S-coA
B
H O SCoA
O
O
O
O
O
O
O
H
O
+
H
-CoASH
O
O
O
enolizzazione
OH O
HO
OH
floracetofenone
Come nel caso della acido grasso sintetasi, l'intera sequenza è controllata da un complesso
enzimatico che trasforma acetilSCoA e malonilSCoA nel prodotto intermedio. Questa strada è stata
dimostrata somministrando al microrganismo malonilSCoA arricchito in
che si ottiene ha incorporato 3 atomi di
13
C. L’acido orsellinico
13
C come evidenzia il suo spettro di massa che mostra un
picco a M+3. Attraverso la spettroscopia NMR è stato possibile individuare la posizione dei tre
atomi di 13C.
O
O
3X HO
CO2H
S-CoA
13
C
HO
OH
Una caratteristica distintiva di un sistema aromatico derivante dall'acetato è che alcuni degli
ossigeni carbonilici del poli--chetotioestere si ritrovano nel prodotto finale legati in maniera
alternata ad atomi di carbonio del ciclo. Uno o più gruppi carbonilici possono prendere parte alla
30
reazione di ciclizzazione, come avviene per l'acido orsellinico. Tuttavia la presenza di funzioni
ossigenate su carboni alterni, cioè in meta, è facilmente riconoscibile e mette in evidenza l'origine
biosintetica della molecola. L'acido 6-metilsalicilico si differenzia dall'orsellinico per l'assenza di un
gruppo OH. Deriva anch'esso dalla via dell'acetato ma una delle funzioni carboniliche è rimossa
durante la biosintesi.
HO
S-CoA
O
O
O
in assenza di NADPH
O
O
lattone dell'acido triacetico
NADPH
S-CoA
O
CH3
OH O
-H2O
H
S-CoA
O
O
H+
O
S-CoA
O
OH
O
S-CoA
malonil-coA
O
SCoA
OH enolizzazione
O
SCoA
Condensazione
aldolica
O
O
S-CoA
O
acido 6-metilsalicilico
Dopo il processo di disidratazione si ottiene un alchene E che non potrà subire la ciclizzazione
finale. Occorre quindi procedere ad uno shift del doppio legame con controllo della stereochimica
per ottenere l’olefina Z.
Che la riduzione del gruppo carbonilico in posizione 3 sia un processo essenziale è stato dimostrato
coltivando il microrganismo (penicillum patulum) in assenza di NADPH ed ottenendo al posto
dell’acido acetilsalicilico il lattone dell’acido triacetico derivante dalla lattonizzazione della forma
enolica della catena lineare. Questo tipo di riduzione può avvenire su uno qualsiasi dei gruppi
carbonilici anche in catene più lunghe ed è uno dei modi per ottenere diversità strutturale nella
classe dei polichetidi.
Altri studi di marcatura isotopica sono stati effettuati anche su altri substrati come nel caso
dell’alternariolo.
31
O
O
*
*
O
7X
* OH
O
O
* *
*O *
*
OO
2 reazioni aldoliche
O
S-coA
O
O
CO2H
O
enolizzazione
OH
OH
OH
CO2H
OH
OH
HO
O
HO
O
formazione del lattone
alternariolo
È sufficiente fornire al microrganismo dell’acido acetico (sotto forma di acetato di sodio) marcato
per ottenere l’alternariolo che presenta nelle posizioni attese la marcatura isotopica al
13
C. Questo
anche perché è l’acetato (sottoforma di acetil-SCoA) il precursore del malonil-SCoA secondo il
meccanismo illustrato.
O
H N
H
O
O
N H
H
CO2
N
O
Enz
S
O
O
Mg2+
S-CoA
O
NH Biotina-Enzima
H
H
Enz
S
Biotina (Vitamina H)
O
O
1
H O
S-CoA
O
N
NH
H
H
N-Carbossibiotina-Enzima
Enz
S
O
O
+ Biotina-Enzima
S-CoA
HO
O
Malonil CoA
E’ possibile eseguire in laboratorio una reazione analoga per semplice reazione di una urea ciclica
con due equivalenti del reagente di Grignard MeMgBr.
BrMg
OMgBr
O
HN
N
H MeMgBr HN
N MeMgBr
OMgBr
BrMg
N
N
32
Questo derivato reagisce con due molecole di CO2 per dare un sale di Mg, stabile, come polvere
bianca.
MgBr
O
BrMg
N
N
O
O
C
O
BrMg
CO2
N
N
O
O
O
OMgBr
BrMgO
CO2
O
N
N
OMgBr
Il semplice riscaldamento in presenza di un chetone porta ad una efficace carbossilazione. Il
chetoacido così ottenuto è instabile ma può essere subito trasformato in estere per reazione con
diazometano.
O
O
Mg2+
O
O
O
N
Mg2+
O
N
O
O
Mg2+
O
O
CH2N2
O
CO2Me
Modificazioni strutturali
Gli antrachinoni sono un eccellente esempio di metaboliti derivati biosinteticamente dall'acetato.
L'endocrocina si forma da un polichetide contenente otto unità C2 che formano lo scheletro
carbonioso. Altre reazioni successive di ossidazione e decarbossilazione danno altri composti.
Le modificazioni strutturali possono essere raggruppate in due tipi fondamentali, a seconda del
momento in cui avvengono nel corso della sequenza biosintetica. Ad esempio le funzioni
ossidriliche sono ridotte prima che il polichetide ripiegato e ciclizzato venga rilasciato dall'enzima e
queste reazioni sono mediate da una reduttasi, parte di un complesso enzimatico, prima della
reazione di ciclizzazione. Invece reazioni come la decarbossilazione, la O-metilazione e la sequenza
di ossidazioni che porta dal metile al carbossile sono esempi di trasformazioni che avvengono dopo
la reazione di ciclizzazione. E' chiaro che l'assemblaggio dello scheletro antrachinonico (e delle
strutture policicliche correlate) avviene con una sequenza a più stadi. Dopo che la catena
polichetidica si è ripiegata, avviene prima la ciclizzazione in corrispondenza del centro della catena,
seguita dalla formazione degli altri 2 anelli. Molti di questi composti sono prodotti da specie di
penicillium. Vari derivati dell’emodina, del fiscione, del crisofanolo, dell’aloe emodina e della reina
sono i principi attivi presenti nei lassativi derivati da senna, cascara, frangola, rabarbaro ed aloe. Gli
stessi antrachinoni hanno una debole attività terapeutica, ma necessitano di essere trasformati in
glucosidi, derivati solubili in acqua, per esercitare la loro azione.
33
O
1) NADPH HO
2) Aldoliche
CH3
O
O
O
S-coA
S-CoA
O
O
O
O
O
-3 H2O reazioni aldoliche
O
S-CoA
O
O
O
O
ossidazione ed enolizzazione
O
HO
O
O
-H2O ossidazione
enolizzazione
O
CH3
crisofenolo
CO2H
OH O
OH
CH3
O
CH3
O
CH3
endocrocina
ossidazione
OH
CO2H
CO2H
OH O
OH
-CO2
OH O
OH
aloe emodina
O
HO
CH3
emodina
O
CO2H
OH O
CH3
O
O
OH
SAM
CO2H
CH3
OH O
reina
Fiscione
OH O
OH
OH
E' necessario notare che molti altri antrachinoni naturali non sono formati a partire dall'acetato ma
mediante vie biogenetiche più complesse che coinvolgono lo shikimato ed il mevalonato. Tali
antrachinoni non presentano funzioni ossidriliche in meta ed in molti casi presentano funzioni
ossigenate solo su uno dei due anelli aromatici.
La diversa lunghezza della catena poli--chetotioesterea e le numerose modificazioni strutturali
consentono una elevata diversità strutturale dei diversi composti. Questa diversità strutturale è
aumentata anche dalla possibilità di utilizzare unità iniziali alternative all'acetato. Flavonoidi e
stilbeni (di cui parleremo più avanti) sono esempi di prodotti derivanti da un gruppo iniziale
differente dall'acetato ed in particolare dal cinnamoil CoA (il quale a sua volta deriva dall'acido
shikimico).
OH
OH
O
HO
O
3 x malonil-coA
CoA-S
O
O
OH
S-CoA
O
OH
resveratrolo
(stilbene)
OH
HO
O
OH O
naringenina
(flavonoide)
34
Anche acidi grassi sotto forma di esteri del CoA, possono funzionare da unità iniziali.
S-coA
palmitoleil-coA
O
3 x malonil-coA
O
O
acido anacardico
aldolica + enolizzazione
O
Aoc-S
riduzione
O
-CO2
ossidrilazione
HO
urusciolo
O
OH
HO
OH
Particolarmente complessa e
non ancora chiara la sintesi
delle aflatossine a partire da
esanoil-coA
O
O
O
H
S-coA
O
O H
O
O
L’urusciolo è un prodotto ad attività allergenica isolato da alcune piante rampicanti delle
anacardiacee, mentre le aflatossine sono delle micotossine prodotte da alcuni funghi del genere
aspergillus. Ne esistono diverse ma la più comune e tossica è la aflatossina B1, quella riportata nella
figura superiore, che possiede una elevata attività carcinogenica. Queste tossine sono spesso
presenti nelle noccioline americane, nei pistacchi, nel mais, nel riso e sebbene presenti in tutto il
mondo, sono particolarmente diffuse nelle piantagioni tropicali e subtropicali.
O
O
O
O
O
O
HO
H
O
O
O
O H
aflatossina M1
O
O
O H
aflatossina B1-epossido
L’aflatossina M1 è la forma ossidrilata della B1, ritrovata nel latte di mucche alimentate con
mangimi infetti da B1. Anche questa possiede attività carcinogenica ma può facilmente essere
monitorata per la sua fluorescenza.
L’attività carcinogenica di questi composti si esplica a livello del fegato a causa della loro
ossidazione ai corrispondenti epossidi che possono intercalarsi nel DNA ed alchilare un residuo di
guanina. In questo modo si ha inibizione della replicazione del DNA.
35
Tetracicline
Sono un gruppo di antibiotici a largo spettro, somministrabili per via orale, prodotti da specie di
streptomyces, largamente utilizzati in terapia. Sono costituite da uno scheletro tetraciclico di origine
polichetidica, la cui unità iniziale è rappresentata da malonamil-CoA
O
NH2 8 x malonil-coA
Aoc-S
O
S-coA
O
O
O
O
NH2
O
O
O
O
O
O
N
HO
OH
D
C
B
A
OH
O
OH O
Tetraciclina
NH2
O
Cl HO
N
OH
NH2
OH
O
OH O
O
Clortetraciclina
Macrolidi e polieteri
Gli antibiotici macrolidici sono un esempio di composti naturali derivanti dalla via biogenetica
dell'acetato, ma composti principalmente da unità propionato o miscele di unità derivanti da acetato
e propionato. I macrolidi costituiscono una vasta famiglia di prodotti naturali, molti dei quali aventi
attività antibiotica, costituiti da un anello lattonico macrociclico, tipicamente a 12, 14, o 16 termini,
a seconda del numero di unità utilizzate. Lo zearalenone, prodotto da alcuni funghi ha una struttura
semplice interamente formata da unità acetato e malonato. Le necessarie riduzioni e disidratazioni
che coinvolgono la catena avvengono durante il suo allungamento, come nella biosintesi degli acidi
grassi e prima che ulteriori unità malonato si aggiungano alla catena.
36
O
S-CoA
O
CH3
HO
S-CoA S-CoA S-CoA
O
O
O
S-CoA S-CoA S-CoA S-CoA
S-CoA
O
O
O
O
O
O
CH3
HO
O
CH3
HO
O
O
O
O
O
OH O
O
CH3
HO
O
CH3
O
CH3
HO
HO
O
CH3
HO
O
O
CH3
HO
zearalenone
CH3
HO
CH3
In questa classe un altro composto importante è l’Eritromicina, prezioso antibatterico, che presenta
due unità saccaridiche.
O
O
OH
OH
O
S-coA
+ 6X
OH
CH3
O
eritromicina
HO
O
O
O
NMe2
O
OH
S-coA
CH3
O
O
OMe
CH3
OH
La stereochimica della catena è controllata dalle reazioni di condensazione e riduzione, e comunque
tutti i macrolidi noti presentano le stesse caratteristiche stereochimiche.
Macrolidi aventi cicli più grandi di quelli visti fino ad ora, costituiscono il gruppo dei macrolidi
polienici, molti dei quali sono antifungini ma non antibatterici. L'anello macrociclico può avere da
26 a 38 termini con una funzione polienica che può contenere fino a 7 doppi legami in
configurazione E. I gruppi metilici sono relativamente pochi, da cui si deduce che principalmente
unità di malonil-coA sono utilizzate per allungare la catena.
37
OH
OH
O
O
CH3
OH
OH OH O
OH OH
OH
O
HO
H2N
amfotericina B
HO
O
3x
S-coA
15 x
S-coA
CH3
O
O
HO
S-coA
O
OH
O O
O
I sistemi macrolidici sopra descritti sono prodotti dalla formazione di un legame estereo
intramolecolare. Non sempre si ha la formazione di un ciclo e si possono avere composti lineari.
Composti di questo tipo sono il lasalocide A e la monensina A isolati da Streptomyces,
rappresentativi di una grande classe di composti chiamati antibiotici polieterei.
HO
HO
CO2H
HO
HO
OH O
lasalocide A
O
O
O
MeO
O
O
O
O
monensina A
HO
HO
HO2C
OH
HO
O
S-coA
4x
S-coA
O
O
HO
7x
HO
O
O
S-coA
O
S-coA
O
Questi farmaci sono utilizzati in medicina veterinaria, poiché sono attivi nel prevenire e curare la
coccidiosi ed aumentano l'efficienza nella conversione del cibo nei ruminanti. Questi antibiotici
macrolidici sono caratterizzati dalla presenza nella catena di numerosi cicli tetraidrofuranici e
tetraidropiranici. Il meccanismo d'azione dei farmaci polieterei deriva dalla possibilità di agire nei
parassiti come ionofori, aumentando il flusso di ioni sodio e causando così un fatale aumento della
pressione osmotica.
La brevetossina A è associata alla marea rossa dovuta alla fioritura dei dinoflagellati che ha
provocato gravi danni economici alla pesca ed al turismo, specialmente in Florida e nel Golfo del
Messico. Queste tossine sono prodotte dal Gymnodium breve e sono gli agenti responsabili
dell'avvelenamento neurotossico da molluschi che provoca problemi neurologici e gastrointestinali.
38
Si pensa che questi composti siano sintetizzati a partire da un acido grasso poliinsaturo in seguito ad
epossidazione dei doppi legami, seguita da una sequenza concertata di aperture degli anelli
epossidici.
HO
CoA-S
O
O
H+
HO
O
O
O
-O2C
O
O
O
O
O
O
O
HO
O
O
O
O
O
O
O
O
O
O
O
O
39
LA VIA BIOGENETICA DEL MEVALONATO: TERPENI E STEROIDI
I terpenoidi costituiscono una vasta famiglia di sostanze naturali strutturalmente molto diverse tra
loro, derivanti da unità isopreniche C5 unite in modo testa coda.
scheletro
isoprenico
isoprene
Tipiche strutture contengono scheletri carboniosi costituiti da unità (C5)n e sono catalogati come
emiterpeni (C5), monoterpeni (C10), Sesquiterpeni (C15), Diterpeni (C20), triterpeni (C30) e
tetraterpeni (C40).
CH3 OH
acido mevalonico
HO
O
OH
Emiterpeni (C5)
OPP
OPP
DMAPP
IPP
C10
Monoterpeni (C10)
IPP
C15
Sesquiterpeni (C15)
C20
Diterpeni
(C20)
C25
Sesterpeni
(C25)
C30
Triterpeni
(C30)
C40
Tetraterpeni (C40)
IPP
2X
IPP
2X
Steroidi
Carotenoidi
A questi composti che, ad eccezione degli steroidi, si riscontrano prevalentemente nelle piante
superiori fu dato il nome di terpeni che deriva dal terebinto (Pistacia terebinthus, da cui la
turpentine = trementina). La loro classificazione risale alla cosiddetta regola biogenetica
dell’isoprene di Ruzicka (1953) che, sebbene fosse basata su conoscenze biogenetiche ancora
frammentarie, stabiliva formalmente che tali sostanze erano multipli di unità C5 uniti testa coda.
40
CH3
CH3
OH
CH3
CH3
Geraniolo
OH Farnesolo
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
squalene
CH3
H
H
OH
H
mentolo
L’isoprene viene prodotto naturalmente ma non è coinvolto nella biosintesi di questi prodotti. Sono
state, invece, identificate come unità isopreniche biologicamente attive gli esteri difosfato
dimetilallil difosfato (DMAPP) ed isopentenil difosfato (IPP). Queste unità derivano dal
metabolismo dell’acetato attraverso la formazione di acido mevalonico (MVA).
OH
O
CoAS
CoAS
O
Claisen
-CoASH
O
O
OH O
aldolica
CoAS
CoAS
SCoA
OH
idrolisi
enantioselettiva
CoAS
OH
O
O
O
O
OH
NADPH
HO
OH
acido mevalonico
mevalonolattone
O
H
O
OH O
HO
SCoA
HMGCoA
HMGCoA
reductase 3S-3-hydroxy-3-methylglutarylCoA
O
HO P O ADP
OH
OH
OPP
OPP
-CO2
isopentenildifosfato
-ADP
IPP
-H3PO4
isomerasi
OPP
DMAPP
dimetilallildifosfato
Nella biosintesi dell’acido mevalonico vengono utilizzate tre molecole di acetil-CoA. Due si
combinano inizialmente in una reazione di Claisen per dare acetoacetil-CoA, ed una terza molecola
è incorporata attraverso una addizione aldolica stereospecifica che porta alla formazione dell’estere
a catena ramificata beta-idrossi-beta-metilglutaril-CoA. Il composto che viene a formarsi è
otticamente inattivo ma l’idrolisi enantioselettiva regolata dall’enzima porta all’ottenimento di uno
41
solo dei due isomeri dell’acido corrispondente. La via biogenetica del mevalonato non utilizza
malonil derivati a differenza della via dell’acetato. La trasformazione di HMG-CoA in MVA
implica una reazione a due stadi che trasforma il tioestere in un gruppo alcolico primario, un
processo lento ed irreversibile, favorito anche dalla maggiore elettrofilicità del tioestere rispetto al
gruppo CO2H L’inibizione farmacologica dell’enzima coinvolto è un mezzo per regolare la
biosintesi di mevalonato ed, in ultimo, del colesterolo (meccanismo azione delle Statine).
Il composto MVA a sei atomi di carbonio viene trasformato nelle unità isopreniche fosforilate C5
mediante una serie di reazioni che iniziano con la fosforilazione dell’alcool primario. La
decarbossilazione e la disidratazione successiva è aiutata da una terza molecola di ATP. Il composto
IIP viene isomerizzato a DMAPP da un enzima isomerasi che rimuove stereospecificamente il
protone pro-R. Sebbene l’isomerizzazione sia reversibile, l’equilibrio è molto spostato a destra. Il
processo di isomerizzazione avviene in modo concertato ed è il protone al di sotto del piano della
molecola che viene eliminato, mentre l’attacco di un secondo protone avviene sopra al piano come è
stato dimostrato attraverso una marcatura isotopica. Sempre attraverso la marcatura isotopica (13C)
si può mettere in evidenza la diversa origine dei due gruppi metilici del DMAPP.
O
H
OH
O
OPP
OPP
OPP
Perchè sono utili sia IIP che DMAPP? Possiamo provare a razionalizzare in questo modo: DMAPP
funziona bene come elettrofilo e quindi come agente alchilante poiché possiede un ottimo gruppo
uscente, il difosfato, e può generare attraverso un meccanismo SN1, un carbocatione allilico che è
stabilizzato per delocalizzazione della carica.
SN1
OPP
DMAPP
Invece IPP, con il suo doppio legame terminale è più adatto a reagire come nucleofilo a formare un
carbocatione terziario (vedi oltre). Queste differenti reattività costituiscono le basi nella biosintesi
dei terpenoidi ed i carbocationi giocano un ruolo molto importante nella razionalizzazione dei
meccanismi di reazione che sono alla base delle varie vie biogenetiche. Anche se quanto detto è una
accettabile razionalizzazione del processo e verrà utilizzato anche negli esempi successivi, in realtà
appare che la reazione tra IPP ed DMAPP avvenga in un processo concertato.
Emiterpeni
In natura sono relativamente rari e l’esempio più importante è proprio l’isoprene, un composto
volatile presente in diversi tipi di alberi e che si forma per perdita di un protone dal catione allilico.
42
Monoterpeni
La giunzione tra DMAPP ed IPP ad opera dell’enzima preniltransferasi porta alla formazione del
geranil difosfato (GPP).
SN1
OPP
DMAPP
OPP
OPP
H HS
R
H H
R S
GPP
OPP
Si ritiene che questa biosintesi coinvolga la ionizzazione di DMAPP a catione allilico che si
addiziona ad IIP con perdita finale di un protone in modo stereoselettivo. Questo porta ad un
monoterpene difosfato in cui il nuovo doppio legame è E. Il linalilPP ed il nerilPP sono isomeri del
geranilPP ed è probabile che essi si formino per ionizzazione a catione allilico del geranilPP.
Questo composti in seguito a modificazioni relativamente semplici, possono dare luogo ad una serie
di monoterpeni lineari che si ritrovano come componenti degli oli volatili e che vengono utilizzati
nella produzione di aromi e profumi. I composti che ne derivano possono essere idrocarburi, alcoli,
aldeidi od anche esteri, specialmente acetati.
OPP
GPP
-OPP
OPP
OPP
-OPP
OPP
OPP
Linalil PP
Geranil PP
Neril PP
OH
OH
geraniolo
(geranio)
O
geraniale
(limone)
linalolo
(olio di coriandolo)
OH
citronellolo
(olio di rosa)
O
OH
beta mircene
(luppolo)
nerolo
(olio di rosa)
nerale
(limone)
O
citronellale
(olio di citronella)
La varietà di monoterpeni che si riscontrano in natura viene considerevolmente aumentata dalla
possibilità che avvengano reazioni di ciclizzazione che possono portare alla formazione di sistemi
monociclici e biciclici. Non è presumibile che tali ciclizzazioni avvengano con il precursore geranil
43
PP poiché la stereochimica E del doppio legame terminale è sfavorevole alla formazione
dell’anello. Neril PP o linalil PP invece hanno una stereochimica favorevole. E’ stato dimostrato
che gli enzimi monoterpene ciclasi accettano tutti e tre i difosfati e sembra che essi abbiano sia la
capacità di isomerizzarli che di ciclizzarli. E’ quindi conveniente considerare la specie coinvolta
nella ciclizzazione come un catione allilico delocalizzato fortemente legato all’anione difosfato e la
formazione del legame che ne consegue avviene grazie alla vicinanza degli elettroni del doppio
legame.
OPP
OPP
OPP
GPP
LPP
NPP
catione mentile\-terpinile
Il catione mentile può poi subire tutte le possibili trasformazioni già discusse precedentemente per i
cationi. Può reagire con nucleofili (essenzialmente H2O), può perdere un protone per formare un
nuovo doppio legame oppure subire ulteriori processi di ciclizzazione.
-terpineolo
H2O
HO
catione mentile\-terpinile
-H+
limonene
ciclizzazione
O ox
canfora
OH
Borneolo
ciclizzazione
H2O
-H+
catione bornile
catione pinile
-H+
-pinene
HO
luce, O2
-pinene
HO
sobrerolo
-Terpineolo è presente in tutti gli oli essenziali degli agrumi. -Pinene si ritrova negli oli di
trementina (dal 55 all’80%. L’essenza di trementina è un liquido incolore con proprietà solvente.
L’essenza greggia è detta acqua ragia. Il residuo di distillazione viene detto colofonia. Per la
44
raccolta della oleoresina (trementina) si provocano incisioni nel tronco dell’albero e si raccoglie il
liquido denso di colore biondo trasparente. Per irraggiamento in presenza di ossigeno a-pinene dà
vari composti, tra cui il sobrerolo (sobrepin) mucolitico.
La canfora viene estratta dall’albero omonimo, spontaneo in Cina e giappone. E’ una pianta molto
longeva (1000 anni e 50 metri di altezza). La canfora veniva estratta dal tronco sminuzzato per
distillazione in corrente di vapore.
Anche se non è stato mai messo in evidenza negli schemi precedenti, la sterochimica relativa ed
assoluta dei diversi stereocentri è di primaria importanza. La maggior parte dei monoterpeni sono
otticamente attivi ed esistono molti esempi in cui i due diversi enantiomeri dello stesso composto
vengono isolati da fonti diverse.
(+)-canfora
salvia
(+)-carvone
cumino
(-)-canfora
tanaceto
(-)-carvone
Menta verde
(+)-limonene
odore arance
(-)-limonene
odore limone ma menta piperita contiene entrambi gli enantiomeri.
Anche il pino contiene entrambi gli enantiomeri dell’-pinene.
I monoterpeni così ottenuti possono quindi essere ulteriormente metabolizzati dando luogo and una
vasta serie di composti analoghi
Sesquiterpeni
L’addizione di una ulteriore unità C5 IPP al geranil PP porta alla formazione del precursore
fondamentale dei sesquiterpeni, il farnesildifosfato.
OPP
OPP
GPP
H
H
IPP
OPP
Farnesil PP
nerolidil pp
E,E
E,Z
OPP
OPP
Il Farnesil PP (FPP) può dare origine ad una serie di sesquiterpeni sia lineari che ciclici.
Il numero dei possibili modi di ciclizzazione aumenta all’aumentare della lunghezza della catena e
del numero di doppi legami. Il doppio legame più vicino al gruppo di fosfato può assumere
45
configurazione E o Z attraverso una ionizzazione (analogo a quanto visto per il geraniolo). Come
nel caso dei monoterpeni le reazioni standard dei carbocationi forniscono una spiegazione razionale
della maggior parte dei comuni scheletri carbonioso che si incontrano in questa classe. Le strutture
incontrate sono generalmente molto complesse e vale la pena solo di ricordare il catione bisabolile.
-bisabolene (nell'aroma di zenzero)
-H+
catione bisabolile
-bisabolo
camomilla tedesca (matricaria)
H2O
HO
Diterpeni
Derivano dal geranilgeranil difosfato, ottenuto per addizione di IPP al farnesil PP sempre attraverso
lo stesso meccanismo.
Il diterpene più semplice è il fitolo in cui tutti i doppi legami sono ridotti salvo l’ultimo.
OPP
Geranilgeranil PP
OH
fitolo
Il fitolo è un residuo lipofilico unito alla clorofilla. Il geranilgeraniolo viene ridotto a fitolo solo
dopo l’ancoraggio sulla clorofilla.
Un altro scheletro diterpenico importante è quello che fa capo al principio attivo “taxina” isolato dal
tasso comune (taxus baccata) e che prende il nome di scheletro taxadienico.
Tratto da Dewick (Medicinal Natural Products.)
46
O
O
O
NH
O OH
O
Ph
O
OH
O
H
HO
O
O
O
O
47
48
Altra grande classe di diterpenoidi è costituita dagli acidi delle resine, composti di formula bruta
C19H29CO2H. Quando si incidono gli alberi di pino, si ottiene una oleoresina la cui frazione volatile
è la trementina mentre il residuo vetroso è la colofonia: miscela complessa di acidi resinici labili
che isomerizzano ad acido abietico.
H
HO2C
acido abietico
H
49
Triterpeni
Due molecole di farnesil PP si uniscono coda-coda per dare l’idrocarburo squalene. Il nome
squalene deriva dall’isolamento nel fegato di squalo. Successivamente è stato isolato anche da topo
e poi ci si è resi conto che è estremamente diffuso in natura (è presente anche nell’olio di oliva). Il
meccanismo biosintetico è relativamente complesso e non del tutto chiaro. L’ipotesi meccanicistica
si basa sull’isolamento da topo del presqualene e dal dato sperimentale che il presqualene può
essere trasformato in squalene.
Durante il processo di accoppiamento, che sulla carta richiede soltanto la rimozione dei due gruppi
fosfato, viene perso un protone dalla posizione C-1 di una molecola di FPP e viene inserito un
idruro ad opera di NADPH. Se in una preparazione cellulare non viene fornito NADPH non si ha la
formazione di squalene, ma solo l’accumulo di presqualenedifosfato.
PPO
OPP
-OPP
OPP
H
H
-H+
OPP
presqualene
--OPP
NADPH
squalene
50
La formazione del ciclo a 3 termini del presqualene è una reazione particolare e non può essere
riprodotta in laboratorio, ma solo in ambito biologico perché è l’enzima che posiziona la molecola
nella giusta conformazione. Lo squalene verrà poi ripreso nella parte dedicata alla biosintesi degli
steroidi.
Tetraterpeni
Sono rappresentati da un solo gruppo di composti, i carotenoidi, comprendenti alcune centinaia di
varianti strutturali naturali. Giocano un ruolo nella fotosintesi ma si ritrovano anche nei tessuti di
piante non fotosintetiche oltre che in funghi e batteri. La formazione dello scheletro tetraterpenico
del fitene coinvolge un accoppiamento coda-coda di due molecole di geranilgeranil PP in una
sequenza analoga a quella vista per lo squalene.
Il composto ciclopropanico prefitene difosfato è un intermedio della biosintesi. Evolve in modo
diverso dal presqualene perché invece di accettare H- da NADPH perde un H+ per dare un doppio
legame in un sistema coniugato. Nelle piante e funghi questo nuovo doppio legame è Z, nei batteri è
E. Questo sistema trienico previene ogni ciclizzazione.
OPP
prefitene
-H+
Z-fitene
licopene
51
La coniugazione viene poi estesa da una sequenza di reazioni che danno alla fine il licopene che,
come la maggior parte dei carotenoidi, ha la configurazione tutto-trans. L’esteso sistema di elettroni
conferisce il colore ai carotenoidi. Il licopene è il pigmento caratteristico dei pomodori maturi.
Un tipo di ciclizzazione possibile è quella del doppio legame non coniugato presente alle due
estremità della molecola. La ciclizzazione procede per catalisi acida e porta a diversi tipi di anelli,
sempre a sei termini, in funzione del tipo di eliminazione che a cui va incontro l’intermedio
carbocationico.
Si ottiene così un discreto numero di derivati, molti dei quali sono pigmenti, la cui struttura
completa è riportata nello schema seguente.
52
53
Il più importante è senz’altro il -carotene che dà origine alle vitamine A secondo lo schema
seguente.
54
55
Steroidi
Triterpeni modificati contenenti il sistema tetraciclico del lanosterolo mancante di tre gruppi
metilici di cui 2 al C4 ed uno al C11.
Come visto nei terpenoidi il precursore del lanosterolo è lo squalene. Questo viene trasformato in
squalene ossido da un enzima, una flavoproteina che utilizza O2 e NADPH come cofattore.
CH3
CH3
CH3
H3C
CH3
Squalene
CH3
CH3
CH3
CH3
CH3
CH3
O
H3C
CH3
Squalene ossido
CH3
CH3
CH3
Se lo squalene ossido è opportunamente posizionato e ripiegato sulla superficie dell’enzima la
struttura triterpenica ciclica formata può essere razionalizzata come derivante da una iniziale
ciclizzazione concertata mediata da carbocationi seguita da una serie di migrazioni concertate W.M.
di gruppi metilici e di ioni idruro.
H3C
CH3
CH3
CH3
H3C
CH3
CH3
CH3
CH3
H3C
CH3
CH3
CH3
CH3
CH3
+
C
CH3
+
HO
CH3
C
H
CH3
HO
CH3
O
+
CH3
CH3
H
H3C CH3
CH3
H3C
H3C
H3C
+
CH3
CH3
CH3
CH3
CH3
CH3
C
CH3
CH3
CH3
+
CH
+
CH3
C
H
CH3
H
H
H
H3C CH3
CH3
CH3
CH3
CH3
HO
H
H3C CH3
HO
H
H3C CH3
Catione protosterile
Il ripiegamento avviene attraverso una conformazione sedia-barca-sedia-barca.
56
O
barca
sedia
sedia
barca
Il catione protosterile subisce poi gli shift di idruri ed alchili.
H3C
H
+
CH3
C
H
CH3
CH3
CH3
H
CH3
HO
H
H3C CH3
Catione protosterile
-H
+
CH3
H3C
CH3
CH3
CH3
H
CH3
HO
H
H3C CH3
Lanosterolo
La conformazione che porta al lanosterolo è solo una delle possibili, infatti esistono molte altre
strutture triterpenoidee diffuse in microrganismi e piante.
18
12
11
19
1
2
HO
4
28 29
D
14
H
16
15
8
5
27
17
C
B
A
3
25
13
9
10
23
20
26
24
22
21
30
7
6
H
Numerazione del lanosterolo e dei suoi anelli
57
Il colesterolo costituisce una struttura fondamentale ma ulteriori modificazioni specialmente in
catena laterale danno luogo ad un ampia varietà di prodotti naturali biologicamente importanti come
gli steroli, saponine steroidee, glucosidi cardioattivi, acidi biliari, corticosteroidi ed ormoni sessuali
dei mammiferi.
Per molti composti caratterizzati da una stessa struttura scheletrica è stata riscontrata una attività
biologica marcatamente diversa: ciò è dovuto alla presenza di gruppi funzionali legati al nucleo
steroideo ed, in parte al riarrangiamento molecolare complessivo derivante dalla stereochimica delle
fusioni degli anelli. I sistemi ciclici contenenti anelli a 6 termini possono presentare una fusione
trans come nella trans-decalina, oppure cis come nella cis declina.
H
H
H
H
cis-decalina
trans-decalina
Una fusione di tipo trans nei due anelli in conformazione a sedia produce un appiattimento della
molecola. Entrambi gli idrogeni sulle giunzioni sono assiali rispetto a entrambi gli anelli. La
decalina cis invece è una molecola curva con maggior libertà conformazionale che comunque viene
persa quando si introduce un terzo anello.
21
20
18
H
10
14
15
1
8
14
3
15
22
20
12
9
14
10
1
15
2
10
3
5
5
18
11
15
1
H
9
2
7
10
H
17
26
13
16
14
15
H
5
7
4
27
25
8
H
3
colestano
24
23
H
19
6
6
22
20
12
8
4
7
4
21
16
14
H
H
28
27
26
13
9
7
25
H
8
H
3
11
19
16
H
1
2
24
23
17
12
17
13
15
6
H
18
23
H
18
22
20
24
14
H
pregnano
21
21
16
8
5
4
Androstano
Estrano
19
9
H
3
6
13
H
10
H
7
5
4
11
1
2
8
H
7
6
11
19
9
17
12
16
H
10
2
H
5
4
13
11
19
H
3
16
18
17
12
13
H
9
1
2
18
17
12
11
ergostano
6
colano
28
29
28
21
22
20
24
23
21
27
25
21
26
18
18
11
1
2
13
H
19
9
10
5
4
16
H
14
11
15
H
2
9
10
7
5
4
23
12
11
26
13
16
14
15
2
9
10
5
4
7
6
stigmastane
27
25
29
26
16
H
14
15
8
30
H
3
17
13
H
19
1
H
24
23
18
25
H
17
22
20
27
8
H
3
campestane
24
H
19
1
6
H
12
8
H
3
20
17
12
22
7
6
H
28
lanostano
Negli steroidi ci sono esempi di giunzione tra anelli A e B sia di tipo trans che di tipo cis ed
insaturazioni sia 4 che 5. Addirittura negli estrogeni l’anello A è aromatico ed in questo caso
58
manca il metile sul C10. Tutti gli steroidi naturali hanno la giunzione tra B e C di tipo trans. La
giunzione Ce D è generalmente trans anche se ci sono eccezioni. La nomenclatura sistematica è
basata su una serie di idrocarburi capostipiti.
La stereochimica è riportata come (alfa, sotto il piano) o (beta, sopra il piano).
Colesterolo
Il lanosterolo negli animali viene convertito in colesterolo attraverso un processo che richiede non
solo la perdita di tre gruppi metilici, ma anche la riduzione di un doppio legame in catena laterale e
la generazione di doppio legame 5 al posto del 8. Generalmente si ha prima la perdita del metile
sul C14 che viene rimosso come acido formico attraverso successive reazioni di ossidazione ed
ossidazione finale tipo Bayer-Villiger.
R
14
15
R
R
O2
O2
NADPH
NADPH
O
OH
O2
NADPH
R
R
R
O
O
NADPH
H2O
O
O
H
Trasposizione
Bayer-Villiger
O
H
R
Enzima
H
La perdita dei due gruppi metilici in 4 avviene in sequenza attraverso un meccanismo
decarbossilativo facilitato dalla contemporanea ossidazione del gruppo ossidrilico C3 a chetone.
H
HO
+
O
O
H
H
O
H
H
O
H
H
1)Ox
HO
HO
2) decarbox
3) rid
59
Il doppio legame 8 migra al 5 formando prima un 7 e poi un diene 5,7. Si ottiene così il
colesterolo.
21
22
20
18
23
12
11
9
2
10
3
5
14
4
H
26
16
27
15
8
H
colesterolo
H
HO
25
13
H
19
1
24
17
7
6
Il colesterolo è il principale sterolo animale: è stato ritrovato in tutti i tessuti animali dove svolge il
ruolo di costituente delle membrane cellulari.
Nell’uomo i calcoli biliari sono costituiti interamente da un precipitato di colesterolo prodotto dalla
bile. Il colesterolo è attualmente ottenibile in grossa quantità dal midollo spinale e dal cervello dei
bovini, sottoprodotti della lavorazione della carne. Questi sono quindi una fonte di substrato utile
nella sintesi parziale di farmaci steroidei. Grosse quantità di colesterolo sono attenute dalla lanolina:
un materiale grasso che ricopre la lana delle pecore. E’ una miscela complessa di esteri di acidi
grassi con steroli ed alcoli a lunga catena. La saponificazione della lanolina grezza fornisce una
frazione alcolica che contiene circa il 34% di colesterolo ed il 38% di lanosterolo/diidrolanosterolo.
Nei mammiferi troviamo anche la forma ridotta del colesterolo, cioè senza il doppio legame in C5.
Poiché per riduzione del doppio legame si forma un nuovo stereocentro si possono ottenere due
diversi derivati:
CH3
H3C
CH3
H3C
CH3
CH3
CH3
CH3
H
H
H
H
HO
CH3
CH3
H
H
H
H
HO
H
colestanolo
(3,5)-cholestan-3-ol
H
coprostanolo
(3,5)-cholestan-3-ol
Glicosidi Cardiaci
Questa classe di composti è costituita da una serie di steroidi C21-C24 che portano sul C3 un
residuo glucidico che assicura la solubilità in acqua e che può essere facilmente eliminato. Tali
composti di origine vegetale si ritrovano in piante che crescono in climi tropicali o temperati, sono
tossici ed hanno un potente effetto sul muscolo cardiaco. Hanno trovato impiego come veleni
(punte di freccia), ma in bassa concentrazione anche come stimolanti cardiaci. L’esempio più
60
classico è rappresentato dagli estratti di foglie di una specie di digitale il cui principio attivo è il
glucoside digitonina contenente l’aglicone digitossigenina.
O
O
O
O
CH3
CH3
H
H
H
H
OAc
OH
H
cis-trans-cis
H3C
H
H
digitossigenina
H3C
H
OH
cis-trans-cis
bufotalina
Gli agliconi con anello eterociclico a 5 terminisul C17 sono detti cardenolidi, mentre i meno
abbondanti composti con un ciclo a sei termini che si ritrovano principalmente nelle rane e nei rospi
sono dette bufadienolidi o bufotossine. Un esempio è la bufotalina.
E’ noto che le farfalle regina e le loro larve accumulano nel loro corpo una certa varietà di
cardenolidi che assumono attraverso la dieta a base di piante come la genziana. Questa caratteristica
le rende inappetibili alla maggior parte dei predatori.
Saponine
Si ritrovano in molte famiglie di piante, spesso in associazione con i glucosidi cardiotonici. Con il
termine sapogenine si intendono gli agli coni delle saponine e sono caratterizzati dal possedere una
catena laterale spirochetalica . Un’importante sapogenina è la diosgenina usata come materiale di
partenza per la sintesi parziale di altri steroidi. Il nome saponine deriva dalla capacità di piante che
contengono questi prodotti di formare schiume. In alcuni casi sono state utilizzate come saponi
naturali. Non sono tossiche per uso esterno o nell’alimentazione umana, ma se entrano nel circolo
sanguigno alterano la permeabilità cellulare e lo divengono.
H
H3C
CH3
H
CH3
H
O
H
CH3
O
H
H
HO
diosgenina
Si può fare un parallelo con le saponine triterpenoidiche anche loro costituite da un aglicone
triterpenoidico e da una catena, più o meno ramificata di residui zuccherini. Saponine di questo tipo
sono contenute nella liquirizia e sono responsabili del sapore gradevole. L’acido glicirrizico (residui
61
zuccherini+acido glicirretico) sotto forma di sali di potassio e di calcio (vedi pag 203 Dewick) è
responsabile del gusto. Il colore giallo brillante della radice di liquirizia è dovuto alla presenza di
flavonoidi liquiritina ed isoliquiritina.
CO2H
O
HO2C
O
HO
HO
HO2C
O O
HO
HO
OH
H
O
CHO
acido glicirretico
acido glicirrizico
OH
OH
GlcO
O
GlcO
OH
isoliquiritina
liquiritina
O
O
Fitosteroli
I principali fitosteroli sono l’ergosterolo (provitamina D) e lo stigmasterolo.
CH3
H3C
CH3
CH3
CH3
H3C
CH3
CH3
H
CH3
CH3
CH3
H
CH3
H
H
H
H
H
HO
HO
stigmasterolo
ergosterolo
L’ergosterolo si ritrova nell’amido, mentre il secondo principalmente nell’olio di semi di soia. Da
notare la presenza di atomi aggiuntivi sulla catena laterale in posizione 24 (prendono il nome di 24 1
e 242), inoltre, l’ergosterolo possiede un ulteriore doppio legame in posizione 7, mentre entrambi
hanno un doppio legame in posizione 22. Questa insaturazione in posizione 22 è caratteristica di
molti fitosteroli, ma mai di steroli animali. Questi fitosteroli si ritrovano sia come alcoli liberi che
come esteri di acidi grassi e come glucosidi. Sono i costituenti delle membrane cellulari nelle piante
nelle alghe e nei funghi.
62
La fonte carboniosa per il metile o l’etile extra in catena laterale è, in entrambi i casi, la Sadenosilmetionina (SAM) che in un meccanismo visto già altre volte alchila il doppio legame ed il
carbocatione risultante può eliminare H+ e dare una nuova olefina che può venire ridotta od essere
nuovamente alchilata.
Acidi Biliari
Sono acidi steroidei a 24 atomi di carbonio che si trovano in forma di sali nella bile e nell’intestino
per emulsionare i grassi e facilitare la digestione. Questi agiscono come detergenti in virtù del loro
nucleo steroideo, relativamente poco polare, e della catena laterale polare che contiene una funzione
carbossilica generalmente legata ad una glicina od a una taurina. Così l’acido colico viene trovato
come glicolato di sodio e taurocolato di sodio. La via metabolica che porta agli acidi biliari è quella
attraverso la quale principalmente i mammiferi degradano il colesterolo introdotto con la dieta.
Questi composti si formano nel fegato a partire dal colesterolo attraverso una catena ossidativa che
elimina dalla catena laterale tre atomi di carbonio. I sali biliari possono poi essere riassorbiti ed
immagazzinati nella cistifellea, oppure essere escreti dal corpo. Questa è la principale via di
eliminazione del colesterolo in eccesso. L’incapacità di rimuovere il colesterolo attraverso gli acidi
biliari e la loro escrezione è uno dei fattori che contribuiscono all’insorgenza di patologie come
l’aterosclerosi e la calcolosi biliare.
OH
H3C
O
CH3
NH
H
CH3
H
HO
CO 2Na
H
OH
H
H3C
O
CH3
OH
NH
H
Glicocolato sodico
H
CH3
SO3Na
H
taurocolato sodico
H
HO
H
OH
H
Caratteristiche strutturali tipiche degli acidi biliari:
Scheletro colanico (C24);
Fusione cis degli anelli A/B;
Catena laterale C5 con funzione carbossilica terminale;
Gruppo ossidrilico in 3-alfa e 7-alfa.
63
Vitamine del Gruppo D
E’ la cosiddetta vitamina antirachitica; è essenziale per la formazione dell’osso, dato che la sua
funzione è quella di controllo dei metabolismi del calcio e del fosforo.
Nel 1924 fu notato come l’irraggiamento UV di alcuni cibi per animali da esperimento facesse
acquistare loro proprietà antirachitiche; si osservò inoltre un miglioramento nei soggetti affetti da
rachitismo quando fossero esposti alla luce solare o venissero somministrato loro olio di fegato di
merluzzo od altri oli di fegato di pesci. Sono tre i composti che vanno sotto il nome di vitamina D:
1. La vitamina D2, detta calciferolo o ergocalciferolo che proviene dall’ergosterolo (estratto dal
lievito) attraverso una sequenza di reazioni fotochimiche e termiche; si assume con il cibo.
2. La vitamina D1: in realtà è una miscela 1:1 dell’ergocalciferolo con il lumisterolo
3. La vitamina D3, detta colecalciferolo, considerata la vitamina D per eccellenza.
CH3
CH3
CH3
H3C
CH3
CH3
CH3
luce ultravioletta
reazione elettrociclica
conrotatoria
H
H
H
CH3
H3C
CH3
CH3
H
CH3
H
pre-ergocalciferolo
HO
HO
ergosterolo
trasposizione
sigmatropica 1,7
luce ultravioletta
CH3
CH3
H3C
CH3
CH3
28
H3C
21
23
20
25
27
CH3
CH3
12
1
18
13
9
14
2
10 CH2
19
8
3
5
7
H
CH3
17
11
CH3
CH3
24
22
26
H
15
H
16
HO
H
lumisterolo
H
HO
4
6
ergocalciferolo
Ricerche successive avevano messo in luce che la vitamina D contenuta negli oli dei pesci o
formata nella pelle per esposizione ai raggi ultravioletti non fosse identica all’ergocalciferolo. Essa
infatti deriva dal 7 deidrocolesterolo (provitamina D3) presente nella cute che subisce per azione
della luce, l’apertura elettrociclica di un anello con formazione di un triene che a sua volta
attraverso la reazione sigmatropica-1,7 dà luogo al colecalciferolo.
64
H3C
H3C
CH3
CH3
CH3
CH3
H
CH3
H3C
H
CH2
H
H
H3C
H
HO
HO
colecalciferolo
7-deidrocolesterolo
Sia la vitamina D2 che la D3 si trovano, generalmente, in un’altra rappresentazione grafica.
CH3
H3C
H3C
CH3
CH3
CH3
CH3
CH3
CH3
H
H
H
H
H
H
H
H
CH2
CH2
HO
HO
colecalciferolo
ergocalciferolo
Esempio generico di reazione elettrociclica
Il cis-1,3,5 esatriene subisce una facile trasformazione per riscaldamento per dare 1,3-cicloesadiene.
Questa tipo di isomerizzazione è noto come reazione elettrociclica. La reazione è periciclica, cioè
procede attraverso uno stato di transizione ciclico e non prevede intermedi.
CH2
CH2
La presenza di sostituenti sui doppi legami terminali pone dei problemi di stereochimica. In un
sistema a [4n+2] elettroni la reazione per via termica avviene con moto disrotatorio, la reazione
per via fotochimica con moto conrotatorio.
CH3
H
H
CH3
disrotatoria
CH3
CH3
CH3
CH3
H
H
CH3
conrotatoria
CH3
65
Ormoni Sessuali
Gli ormoni sessuali dei mammiferi derivati dal colesterolo sono stati e sono ancora oggetto di
numerosi studi. Essi sono suddivisi in tre classi: gli estrogeni e gli androgeni che regolano,
rispettivamente, le caratteristiche sessuali primarie femminili e maschili ed i progestinici (gestageni)
che regolano invece varie funzioni del ciclo riproduttivo femminile (ormoni del corpo luteo). Le
loro strutture chimiche sono molto simili e vengono prodotti in quantità estremamente ridotte e
bilanciate sotto il controllo degli ormoni gonadotropinici (ormoni peptidici secreti dal lobo anteriore
della ghiandola pituitaria) nelle gonadi. Il primo estrogeno, l’estrone, fu isolato nel 1929 dall’urina
di una donna incinta; successivamente (1930) fu isolato l’estriolo ed infine nel 1935 l’estradiolo da
ovaie di scrofe. Quest’ultimo è l’ormone primario più attivo e gli altri due ne sono i metaboliti.
CH3
O
CH3
H
H
H
H
OH
OH
H
HO
H
HO
estrone
CH3
OH
estriolo
H
H
H
HO
estradiol
Caratteristiche: scheletro Estrano C18, aromatizzazione anello, assenza catena laterale
Il primo androgeno isolato (1931) è stato l’androsterone, che in realtà è un metabolita dell’ormone
maschile primario cioè il testosterone, isolato nel 1935.
CH3
CH3
O
CH3
H
H
HO
H
androsterone
CH3
H
OH
H
H
H
O
testosterone
Il principale ormone progestinico è il progesterone (1934, da scrofa); questo ormone viene secreto
dalle ovaie e prepara l’utero per la gravidanza e può sopprimere l’ovulazione. Questa possibilità lo
rendeva interessante come contraccettivo, ma la sua scarsa assimilabilità per via orale ne ha limitato
l’uso.
66
H3C
O
CH3
CH3
H
H
H
O
progesterone
Caratteristiche strutturali: scheletro Pregnanico C21. Sistema 3-oxo-4
Ormoni della Corteccia Surrenale
Una grande quantità di ormoni steroidei è stata isolata e caratterizzata dalle ghiandole surrenali.
Poiché vengono prodotti dalla corteccia surrenale vengono detti anche ormoni adrenocorticali o
corticosteroidi. Sono caratterizzati da uno scheletro pregnanico (C21).
CH3
20
H3C
12
11
1
19
10
3
5
9
16
14
15
8
H
H
4
17
13
H
CH3
2
18
21
7
6
Pregnano
Si suddividono in base alla loro attività in due gruppi: i glucocorticoidi, coinvolti nella sintesi
proteica dei carboidrati e nel deposito di glicogeno nel fegato, e mineralcorticoidi, che presiedono il
controllo del bilancio elettrolitico (rilascio di Na+ e Cl- ed escrezione di K+). I glucocorticoidi
giocano un ruolo importante nei processi infiammatori. Tra i più importanti:
OH
OH
OH
OH
CH3
O
OH
CH3
H
H
H
H
idrocortisone
HO
CH3
H
H
H
H
H
O
O
O
CH3
CH3
CH3
HO
O
O
O
Cortisone
corticosterone
Caratteristiche tipiche dei glucocorticoidi: Scheletro pregnanico (C21), catena laterale
17-COCH2OH, funzione 4-3-cheto (comune), ossidrile 17 (comune).
Biogeneticamente derivano dal colesterolo per successive ossidazioni.
67
VIA BIOGENETICA DELLO SHIKIMATO
La via dello shikimato fornisce una via alternativa verso i composti aromatici, in particolare gli
amminoacidi aromatici L-fenilalanina, L-tirosina ed l-triptofano. Questa via è impiegata da piante e
microrganismi ma non da animali e di conseguenza gli amminoacidi aromatici figurano per l'uomo
tra quelli essenziali, cioè da assumere con la dieta. Un intermedio centrale di questa via biosintetica
è l'acido shikimico.
OH
O
HO
OH
OH
L’acido shikimico è stato isolato la prima volta da piante del genere Ilicium, in giapponese dette
shikimi. La maggior parte degli intermedi di questa via sono stati identificati grazie allo studio di
mutanti di Escherichia Coli ottenuti per irradiazione UV. Un ceppo mutante capace di crescere in
genere differisce dal suo progenitore solo in un singolo gene e l’effetto è di solito la sintesi di un
enzima danneggiato. Di solito un mutante in cui è bloccata la trasformazione del composto A nel
composto B, richiede il composto B per crescere, mentre il composto A si accumula nel suo mezzo
di coltura. In questo modo è stata tracciata la via che porta agli amminoacidi aromatici.
La via dello shikimato comincia con l'accoppiamento del fosfoenolpiruvato (PEP) con il D-eritrosio
4-fosfato per dare l'intermedio a sette atomi di carbonio acido 3-deossi-D-arabino-eptulosonico 7fosfato DAHP.
Successivamente si ha una nuova reazione aldolica dopo l’eliminazione di acido fosforico.
68
Il meccanismo sopra descritto però prevede l’uscita di un protone (quello verde) che in realtà non è
affatto acido. Più coerente è il meccanismo che prevede una iniziale ossidazione del gruppo
ossidrilico adiacente, l’eliminazione con meccanismo E1cb e la successiva riduzione che porta poi
alla condensazione attesa.
Infine, sempre attraverso un meccanismo E1cb, favorito in ambito fisiologico, si ha la formazione
dell’acido deidroshikimico.
L’acido deidrochinico può essere trasformato in acido chinico che non appartiene alla via
metabolica dello shikimico, ma è un composto naturale molto comune in natura e si può trovare in
combinazione con alcaloidi come la chinina.
Un derivato diretto dell’acido shikimico, l'acido gallico, figura come componente di molti materiali
tanninici, presenti nelle piante, materiali che sono stati usati per migliaia di anni nella concia delle
pelli animali per la loro capacità di legare tra di loro molecole proteiche. Inoltre i tannini
contribuiscono al gusto amaro di cibi e bevande come il tè, il caffè ed il vino.
69
CO2H
CO2H
CO2H
-H2O
Ox
HO
OH
O
HO
OH
OH
acido gallico
OH
acido protocatechico
OH
Un composto molto importante sulla via dello shikimato è l'acido corismico che incorpora una
ulteriore molecola di PEP come catena laterale di tipo etere enolico. PEP si combina con acido
shikimico 3-fosfato per dare l'acido 5-enolpiruvilshikimico 3-fosfato (EPSP).
H
+
CO 2H
CO 2H
CO 2H
CH2
H
ATP
PO
HO
PO
OH
CO 2H
OH
PO
PO
OH
OH
O
CO 2H
OH
EPSP
O
Sintetasi
P
NH
HO 2C
CO 2H
OH
OH
Glifosate è inibitore della EPSP e viene usato come
diserbante. Non potendo sintetizzare gli amminoacidi
aromatici la pianta muore
CH2
PO
O
CO 2H
OH
-HOP
CO 2H
CH2
O
CO 2H
OH
Acido corismico
L'acido 4-idrossi benzoico si può formare da acido corismico per eliminazione di acido enolpiruvico
(nei batteri). I tre acidi fenolici incontrati, il 4-idrossibenzoico, il protocatechico ed il gallico
CO 2H
CO 2H
HO
OH
CO 2H
OH
HO
OH
OH
70
dimostrano alcuni dei pattern di ossidrilazione caratteristici dei metaboliti derivati dall'acido
shikimico, cioè un singolo gruppo ossidrilico para rispetto alla catena laterale, 2 gruppi OH orto tra
di loro, e di solito in posizione 3,4 rispetto alla catena laterale, e tre gruppi ossidrilici ancora orto tra
di loro e di solito in posizione 3,4,5 rispetto alla catena laterale. La singola ossidrilazione para e le
poliossidrilazioni orto contrastano con la disposizione meta degli ossidrili caratteristica dei fenoli
derivati dalla via dell'acetato ed in molti casi permettono di dedurre l'origine biosintetica. L'acido
2,3 diidrossi benzoico ed il 2-idrossibenzoico (salicilico) derivano, nei microrganismi, dall'acido
corismico attraverso il suo isomero acido isocorismico.
H
CO 2H
O
H
CO 2H
OH
CH2
O
CH2
CO 2H
O
CO 2H
OH
acido
isocorismico
acido corismico
idrolisi e disidrat.
CO 2H
CO 2H
OH
OH
OH
acido salicilico
acido 2,3-diidrossibenzoico
Semplici derivati amminici degli acidi fenolici si ottengono dall'acido corismico utilizzando
l'ammoniaca (derivante dalla glutammina) come nucleofilo. L'acido corismico può essere amminato
in posizione 4 dando l'acido 4-ammino-4-deossiscorismico, od in C2 dando l'acido 2-ammino-2deossiisocorismico che può essere trasformato nell'acido antranilico
CO2H
NH3
O
CO2H
NH2
CO2H
O
OH
CO2H
NH2
CO2H
acido antranilico
intermedio sulla via
del triptofano
NH3
O
CO2H
NH2
CO2H
p-amminobenzoico
PABA
CO2H
NH2
71
Proseguendo il corso principale della via dello shikimato troviamo la trasformazione dell'acido
corismico in acido prefenico attraverso una trasposizione di Claisen che trasferisce la catena laterale
derivata dal PEP in modo che esso diventi direttamente legato al carbociclo. In questo modo si
genera lo scheletro carbonioso di base della fenilalanina e tirosina.
O
O
Trasposizione di Claisen
La Trasposizione di Claisen è una reazione periciclica: avviene attraverso uno stato di transizione
ciclico senza la formazione di intermedi.
CO2H
CO2H
HO 2C
O
CO2H
O
OH
OH
Acido Corismico
Acido Prefenico
HO
O
O
CO2H
Stato di transizione a sedia
La trasposizione è catalizzata dalla Corismato mutasi, che rende 106 volte più veloce la reazione, la
quale potrebbe avvenire anche per riscaldamento, però al di sopra dei 100 °C.
Non vi è catalisi acida o basica e l’aumento della velocità di reazione è legato alla favorevole
conformazione che l’enzima impartisce al substrato. Interessante è la struttura dell’inibitore della
corismato mutasi che mima lo stato di transizione, pur non potendo evolvere ad un prodotto finale.
Il percorso che porta alla formazione degli amminoacidi naturali fenilalanina e tirosina da acido
prefenico può variare a seconda dell'organismo e può essere presente più di un processo all'interno
di uno stesso organismo.
72
Sono fondamentali 3 passaggi: aromatizzazione decarbossilativa, transaminazione e, nel caso della
tirosina, l'ossidazione. Ciò che differenzia le varie strade è l'ordine con cui avvengono le diverse
reazioni.
NH2
O
CO2H
CO2H
PLP
fenilalanina
acido
fenilpiruvico
decarb.
O
H
O
CO2H
CO2H
CO2H
HO 2C
O
PLP
NH2
acido
CO2H
O
OH
arogenico
H
+
OH
OH
Ox
decarb.
O
NH2
CO2H
CO2H
acido
PLP
4-idrossi fenilpiruvico
tirosina
OH
OH
E' noto che in alcuni microrganismi enzimi a largo spettro accettano sia l'acido prefenico che
l'arogenico.
Quanto visto fino ad ora vale per microrganismi e piante. Negli animali la via dello shikimato
manca, e si può avere ossidrilazione diretta della L-fenilalanina a L-tirosina od anche ad L-DOPA.
NH2
NH2
NH2
CO2H
CO2H
CO2H
Precursore
noradrenalina ed
adrenalina
fenilalanina
tirosina
OH
HO
L-DOPA
OH
73
Acidi Cinnamici
La fenilalanina e la tirosina come mattoni sintetici C6C3, sono i precursori di una vasta gamma di
sostanze naturali.
Nelle piante il primo stadio osservato è l'eliminazione di ammoniaca (enzima phenylalanine
ammonia liasi, PAL) per generare l'acido cinnamico E (trans). Dalla fenilalanina si ottiene l’acido
cinnamico, dalla tirosina l’acido 4-cumarico.
Tutte le piante sembrano avere la capacità di deaminare la fenilalanina attraverso PAL, ma la
corrispondente trasformazione della tirosina è limitata alle graminacee (enzima TAL). Le specie che
non trasformano la tirosina ottengono il 4-cumarico per ossidazione del cinnamico. Altri acidi
cinnamici sono ottenuti per ulteriore ossidrilazione e metilazione, che in sequenza ricostruiscono la
disposizione di sostituenti tipica dello shikimato.
CO2H
CO2H
NH2
CO2H
CO2H
CO2H
O2
O2
O2
NADPH
NADPH
SAM
NADPH
O
HO
acido
OH
acido
cinnamico
OH
acido
4-cumarico
caffeico
acido
OH
ferulico
CO2H
1) O2, NADPH
O
O
2) SAM
OH
acido sinapico
Questi acidi si possono ritrovare sia liberi che in forma esterificata. Ad esmpio l'acido caffeico è
presente nel caffé come acido clorogenico, esterificato con l'acido chinico.
HO
CO 2H
O
HO
O
OH
Acido clorogenico
OH
OH
74
Lignani e Lignina
Gli acidi cinnamici figurano anche nelle vie verso altri metaboliti basati sui mattoni biosintetici
C6C3 . Tra queste estrema importanza ha il polimero vegetale Lignina, un materiale che rinforza le
pareti delle cellule vegetali, funzionando da matrice per la microfibra di cellulosa. La lignina
rappresenta un vasto serbatoio di molecole aromatiche quasi mai utilizzato per le difficoltà che si
incontrano nel liberare questi metaboliti. La lignina si forma per accoppiamento ossidativo fenolico
di alcoli idrossicinnamilici derivati dagli acidi cinnamici.
acido
acido
acido
4-cumarico
ferulico
sinapico
CO2H
CO2H
O
O
O
OH
OH
OH
OH
OH
OH
O
O
O
OH
CO2H
OH
OH
alcool
alcool
coniferilico
sinapilico
I dimeri che si formano possono reagire ancora a formare il polimero lignina.
75
Si trovano in natura anche i semplici dimeri che vanno sotto il nome di lignani o neolignani in
funzione della struttura.
Attraverso una complessa serie di trasformazioni l’alcool coniferilico viene trasformato, in piante
del genere Podophyllum diffuse sia in Asia che in America, in podofillotossina.
76
La podofillotossina, insieme ad altri composti, viene estratta dalle radici di queste piante. Per
estrazione con alcool delle radici sminuzzate si ottiene una soluzione omogenea dalla quale, per
aggiunta di acqua, precipita la cosiddetta resina di podofillo. A lungo questa resina è stata utilizzata
come purgante, ma più recentemente la scoperta delle proprietà citotossiche della podofillotossina
ha reso questa pianta interessante dal punto di vista commerciale. La podofillotossina è un potente
antimicotico ed agisce legandosi alla tubulina ed impedendo la formazione dei microtubuli durante
la scissione cellulare. Questo composto non ha trovato applicazione come antitumorale per i suoi
gravi effetti collaterali, ma suoi analoghi sintetici hanno trovato applicazione in tal senso.
Fenilpropani
La sequenza riduttiva da un acido cinnamico ad alcool cinnamilico non è limitata alla biosintesi di
lignani e lignina, ma è anche utilizzata per la produzione di vari derivati del fenilpropano.
La cinnamaldeide è la componente principale dell'olio estratto dalla corteccia della cannella
(cynnamomum zeilanicum). La corteccia fresca contiene acetato di cinnamile e la cinnamaldeide
viene ottenuta per idrolisi enzimatica ed ossidazione durante la fermentazione.
O
O
OH
O
cinnamaldeide
Le foglie di cannella invece contengono eugenolo, che è anche il componente essenziale dei chiodi
di garofano.
O
HO
Eugenolo
77
La catena alchilica presenta un doppio legame terminale. Questa posizione può essere giustificata
per riduzione con NADPH del carbocatione allilico che si ottiene per uscita di H20 dall'alcool
cinnamilico.
H
R
+
+
R
+
R
OH
HO
R
R
O
O
NADPH
NADPH
R
R
R
R
O
O
eugenolo
O
anetolo
Biosintesi analoga all'eugenolo ha la miristicina. Presente nella noce moscata, ha un debole effetto
allucinogeno.
Miristicina
O
O
O
L'anetolo invece si trova nei semi di anice (pimpinella anisum), anice stellato e finocchio. L'utilizzo
di questi composti come aromi è limitato per la loro natura di deboli cancerogeni.
78
Acidi Benzoici da Fenilpropani
Oltre che da intermedi iniziali della catena dello shikimico, gli acidi benzoici possono essere
ottenuti da derivati cinnamici.
CO2H
COSCoA
COSCoA
COSCoA
O
HO
HSCoA
H2O
NAD
+
ATP
R
R
R
R
OH
OH
OH
OH
H2O
+ HSCoA
COOH
HO
CHO
COSCoA
COOH
NAD
Retro
aldolica
R
R
retro-Claisen
-CH3COSCoA
+
R
R
OH
OH
OH
OH
R = OMe Vanillina
Questo è anche il processo per la sintesi dell'acido salicilico nelle piante.
COOH
COOH
COOH
OH
salicilico
OH
Ox
CHO
CHO
2-cumarico
GlcO
HO
glucosilazione
riduzione
OH
OH
O
O
HOHO
OH
salicina
corteccia del salice
79
Cumarine
L'ossidrilazione degli acidi cinnamici in orto alla catena laterale è essenziale nella biosintesi delle
cumarine. L'ossidrilazione diretta dell'anello aromatico dell'acido cinnamico è frequente, ma di
solito avviene in posizione 4, in para alla catena laterale, e le successive ossidrilazioni avvengono
in orto a questo sostituente. Nel caso delle cumarine invece l'ossidrilazione dell'acido cinnamico o
del 4-cumarico avviene in orto alla catena laterale. L'acido 2,4-diidrossicinnamico sembra
possedere la disposizione meta degli ossidrili caratteristica dei fenoli derivanti dalla via dell’acetato
e può causare confusione. Il riconoscimento dello scheletro C6C3 aiuta ad effettuare la corretta
assegnazione.
COOH
COOH
COOH
COOH
OH
OH
2,4-diidrossicinnamico
4-cumarico
2-cumarico
OH
OH
Successivamente i due acidi subiscono la variazione di configurazione al doppio legame che passa
da trans a cis consentendo poi la lattonizzazione. In questo modo si ha biosintesi della cumarina e
dell'umbelliferone.
COOH
COOH
OH
OH
O
O
cumarina
COOH
HO
OH
COOH
HO
OH
HO
O
O
umbelliferone
Successivamente altre reazioni di ossidrilazione ed alchilazione danno origine a molti altri derivati,
tutti sotto il nome generico di cumarine. Le cumarine sono molto diffuse nelle piante, sia in forma
libera che come glicosidi. Nel meliloto (pianta diffusa nei prati) si ritrova come glicoside dell'acido
2-cumarico.
80
Quando la pianta viene tagliata si trasforma in cumarina che ha il caratteristico odore di fieno. Se il
meliloto viene fatto fermentare si produce la 4-idrossi cumarina che per reazione con formaldeide
produce il dicumarolo, forte anticoagulante, letale per gli animali, usato come topicida.
OH
4-idrossicumarina
O
O
O
O
CH2O
OH
OH
OH
O O
O
O
CH3
O
dicumarolo
O
warfarina
O
Questa sostanza interferisce con gli effetti della vitamina K nella coagulazione del sangue e così
anche piccoli traumi portano a gravi emorragie interne. Il dicumarolo è stato utilizzato come
anticoagulante orale nel trattamento delle trombosi. Adesso è stato sostituito dalla warfarina che ne
è un analogo sintetico. La warfarina è stata inizialmente sviluppata come rodenticida ed è stata
utilizzata largamente. Sempre più spesso i roditori stanno diventando resistenti alla warfarina,
capacità attribuita ad una maggiore produzione di vitamina K da parte della loro flora intestinale.
Alcune cumarine naturali hanno uno scheletro più complesso ed incorporano altre catene. L'anello
aromatico dell'umbelliferone è attivato ad alchilazioni con DMAPP (dimetilallildifosfato).
OPP
HO
O
HO
O
O
O
O2
NADPH
HO
ulteriori
biotrasformazioni
O
O
O
Psoralene
(Furanocumarina)
O
O
Marmesina
O
La sintesi della marmesina passa presumibilmente, ma non è dimostrato, attraverso un intermedio
epossidico. Questo potrebbe giustificare la formazione del ciclo a cinque termini e di quello a sei
che viene riscontrato in alcuni derivati.
81
O
a
a
b
HO
O
b
HO
O
O
O
O
HO
O
O
O
Gli psoraleni sono furanocumarine lineari, molto diffusi in tutte le piante, ma in particolare nelle
umbelliferae e nelle rutaceae. Gli esempi più comuni sono lo psoralene, il bergaptene, la
xantotossina e l’isopimpinellina.
H3C
O
O
O
O
O
O
O
bergaptene
psoralene
H3C
O
O
O
O
O
O
O
O
xantotossina
H3C
O
isopimpinellina
H3C
Le piante contenenti psoraleni sono state impiegate per uso esterno ed interno per promuovere la
pigmentazione della pelle e l’abbronzatura. L’olio di bergamotto, il bergamotto è una rutacea, può
contenere fino al 5% di bergaptene ed è usato nella preparazione di abbronzanti per uso esterno. Lo
psoralene grazie al suo esteso cromoforo assorbe nel vicino UV e fa sì che questa radiazione stimoli
la produzione di pigmenti melaninici. La xantotossina (o metossalene) viene utilizzato per via orale
insieme a trattamenti UV nel caso di vitiligine (trattamento PUVA cioè psoralene +UV-A).
A causa della loro struttura planare gli psoraleni si intercalano facilmente nel DNA e questo
permette una reazione di cicloaddizione promossa dalla radiazione UV tra le basi pirimidiniche dal
DNA e l’anello furanico degli psoraleni. La reazione con gli psoraleni inibisce la duplicazione del
DNA e riduce la velocità di divisione cellulare.
82
Questi effetti possono creare dei problemi nella manipolazione di piante che contengono livelli
significativi di furanocumarine. Il sedano normalmente non contiene psoraleni ma l’infezione da
parte di un fungo induce come reazione la sintesi di furanocumarine. Gli agricoltori che toccano
queste piante infette diventano molto sensibili alla luce UV e soffrono di una forma di scottatura
solare chiamata fotofitodermatite. Anche il prezzemolo può avere effetti simili.
Flavonoidi
Gli acidi cinnamici od i loro esteri possono essere utilizzati come unità iniziali per l'allungamento
della catena con unità di malonil CoA, unendo così la via dello shikimato e dell'acetato. Nella
maggior parte dei casi sono aggiunte tre unità C2 di malonato per dare polichetidi, che a seconda
della natura dell'enzima utilizzato possono essere ripiegate in due maniere diverse. Possono così
avvenire condensazione aldoliche o di Claisen che generano anelli aromatici.
OH
SCoA
O
OH
-3 CO2 3 X malonil-CoA
SCoA
O
O
O
O
O
OH
SCoA
OH
O
O
O
O
O
SCoA
O
calcone
sintasi
OH
sintasi
stilbene
-CO2
O
HO
OH
OH
Naringenin-calcone
HO
(un calcone)
OH
resveratrolo
O
OH
(uno stilbene)
OH
HO
O
Naringenina
OH
O
(un flavanone)
I calconi fungono da precursori di un ampia gamma di flavonoidi ritrovati in tutto il regno vegetale.
La maggior parte di questi composti contiene un anello eterociclico, formato per attacco nucleofilo
di tipo Michael del gruppo fenolico sul chetone insaturo. Questa reazione dà origine ai flavanoni.
Questa isomerizzazione può avvenire chimicamente, e condizioni acide favoriscono il flavanone,
mentre condizioni basiche favoriscono il calcone. La reazione è comunque regolata da un enzima.
I flavanoni possono poi dare numerose varianti di questo scheletro di base, come i flavoni, i
flavonoli, le antocianidine e catechine.
83
Consumiamo notevoli quantità di flavonoidi nella nostra dieta vegetale. C'è la crescente
convinzione che alcuni flavonoidi siano particolarmente benefici, poiché agiscono come
antiossidanti e proteggono contro malattie cardiovascolari e certe forme di cancro. I flavonoidi del
vino rosso (quercetina, kaempferolo ed antocianidine) si sono dimostrati efficaci antiossidanti
contro i radicali liberi. I flavonoidi contribuiscono al colore delle piante, giallo per calconi e
flavonoli, rosso, blu e violetto per le antocianidine. Anche le sostanze incolori come i flavoni
assorbono fortemente le radiazioni UV e sono percepibili dagli insetti. La neoesperidina, presente
nell’arancia amara, e la naringina, presente nella buccia di pompelmo sono glucosidi flavanonici
estremamente amari, ma la loro trasformazione in diidrocalconi per idrogenazione in soluzione
alcalina porta a diidrocalconi con un potere dolcificante fino a mille volte superiore a quello dello
zucchero.
84
Isoflavonoidi
Formano una sottoclasse ben separata dai flavonoidi, essendo una variante strutturale in cui l'anello
aromatico derivato dallo shikimato si è spostato sul carbonio adiacente. Questa trasposizione è
effettuata da un enzima che dipende dal citocromo P-450. E' stato proposto un meccanismo
radicalico
OH
HO
ox
O
OH
HO
HO
O
O
migrazione
1,2 di arile
R
O
R = H, liquiritigenina
R = OH, narigenina
R
R
O
O
OH
H2O
HO
O
R
O
R = H, daidzenina
R = OH, genisteina isoflavoni
-H2O
OH
HO
O
R
O
OH
OH
85
Questa trasposizione è piuttosto rara in natura e gli isoflavonoidi sono limitati quasi esclusivamente
alle leguminose anche se questo non ha impedito l’esistenza di molte centinaia di composti
isoflavonoidi e la complessità strutturale è ottenuta attraverso reazioni di ossidrilazione ed
alchilazione, variando lo stato di ossidazione dell’anello eterociclico o formando nuovi anelli.
Il cumestrolo che si ottiene da erba medica e trifoglio ha attività estrogenica sufficiente ad
influenzare la riproduzione di animali da pascolo ed è un fitoestrogeno. Questa molecola planare
imita la forma dell’estradiolo. Per questo motivo è necessario limitare il consumo di foraggio da
leguminose. Si ritiene che gli isoflavonoidi nella dieta umana, per esempio da prodotti derivanti
dalla soia, diano una certa protezione verso i tumori dipendenti da estrogeni come il cancro al seno.
Il rotenone, ed altri composti analoghi, hanno capacità insetticida e pesticida e storicamente sono
stati usati per catturare facilmente i pesci. Il veloce metabolismo a cui vanno incontro una volta
ingeriti da mammiferi li rende innocui per l’uomo.
86
ALCALOIDI
Gli alcaloidi sono basi organiche azotate ritrovate principalmente nelle piante superiori, ma in
misura minore anche in microrganismi ed animali. Queste molecole sono caratterizzate dalla
presenza di uno o più atomi di azoto, normalmente come ammine primarie, secondarie o terziarie e
ciò conferisce loro una certa basicità che è spesso utilizzata per l’isolamento e la purificazione,
poiché consente di formare sali idrosolubili in presenza di acidi. Il grado di basicità è molto
variabile e dipende dalla struttura dello specifico alcaloide ed in particolare dalla presenza e dalla
localizzazione di gruppi funzionali. Sono stai infatti ritrovati in natura alcuni alcaloidi
essenzialmente neutri ed altri contenenti sali di ammonio quaternario. L’attività biologica di molti
alcaloidi ha spesso una netta correlazione con la trasformazione della funzione amminica in sale di
ammonio a pH fisiologico. L’atomo di azoto degli alcaloidi deriva sempre da un amminoacido e,
normalmente lo scheletro carbonioso della molecola di partenza è conservato intatto nella struttura
finale con l’eccezione dell’atomo di carbonio della funzione carbossilica che viene quasi sempre
perso per decarbossilazione. Per questa ragione l’approccio più razionale per la classificazione degli
alcaloidi è quello basato sul precursore amminoacidico. I più importanti amminoacidi precursori di
di alcaloidi sono: ornitina, lisina, acido nicotinico, tirosina, triptofano, acido antranilico ed istidina.
Nelle strutture alcaloidiche sono inoltre incluse subunità derivanti dalla via dell’acetato, dello
shikimato o del mevalonato. Inoltre è stato dimostrato che alcuni alcaloidi ricavano i propri atomi di
azoto da reazioni di transaminazione e quindi solo l’atomo di azoto deriva da un amminoacido.
Questi composti sono detti pseudoalcaloidi.
Alcaloidi da Ornitina
CO 2H
H2N
L-ornitina
NH2
E’ un amminoacido non proteinogenico che negli animali entra a far parte del ciclo dell’urea dove è
prodotto a partire da L-arginina grazie ad una reazione catalizzata dall’enzima arginasi. Nelle piante
deriva invece da L-glutammato. L’ornitina contiene due gruppi amminici, nelle posizioni e , ed è
proprio l’azoto in che entra nello scheletro degli alcaloidi di questa classe. L’ornitina fornisce
quindi una unità C4N sotto forma di ciclo pirrolidinico o come parte del tropano. Le reazioni
dell’ornitina trovano il loro corrispettivo con quelle della L-lisina che invece dà l’unità C5N.
87
Alcaloidi pirrolidinici o tropanici
Il nucleo pirrolidinico si forma inizialmente come catione 1-pirrolidinio che deriva dall’ornitina:
una decarbossilazione mediata da PLP dà la Putrescina che viene alchilata (SAM) a Nmetilputrescina. Questa viene deamminata da una deamminoossidasi che fornisce l’aldeide
corrispondente. Una condensazione intramolecolare fornisce poi il catione pirrolinio. Esiste anche
una via alternativa per ottenere la putrescina a partire dall’arginina e che prevede anche una
reazione di idrolisi per allontanare il gruppo guanidinio.
NH
NH
PLP
CO 2H
H2N
NH
H2N
-CO 2
L-Arginina
animali
NH
agmantina
NH2
NH2
O
Arginasi
H2N
- urea
NH
N-carbamoilputrescina
NH2
CO 2H
HO 2C
CO 2H
PLP
H2N
acido glutammico
NH2
H2N
Putrescina
L-ornitina
piante
-CO 2
NH2
NH2
SAM
HN
HN
+
CH3
N
O
diammina
ossidasi
CH3
NH2
N-Metilputrescina
CH3
catione N-metil-1-pirrolidinio
Ottenuto il catione pirrolinio, gli altri atomi di carbonio necessari derivano dall’acetato via acetilCoA in una sequenza che vede l’aggiunta successiva di due unità di acetil-CoA. Nel primo stadio
l’anione enolato agisce da nucleofilo verso lo ione pirrolinio portando a prodotti con la
stereochimica R o S. La seconda addizione è una condensazione di Claisen che allunga la catena
laterale il cui prodotto è una pirrolidina 2-sostituita che contiene ancora il gruppo tioestereo della
seconda unità di acetil-CoA. L’Igrina e molti degli alcaloidi tropanici non hanno questo carbonio
tioestereo che viene perso grazie ad una sequenza di idrolisi-decarbossilazione.
O
Catione
N-metil-pirrolidinio
S-CoA
N
CH3
R
N
CH3
O
AcS-CoA
S-CoA
O
O
S-CoA
N
CH3
idrolisi decarbossilativa
O
S
N
CH3
S-CoA idr. decarb.
O
O
AcS-CoA
N
CH3
(-)-Igrina
N
CH3
(+)-Igrina
88
La struttura biciclica dello scheletro tropanico di iosciamina e cocaina si ottiene per una seconda
reazione di tipo Mannich che richiede prima un passaggio ossidativo per generare il nuovo catione
pirrolinio e la rimozione del protone in al gruppo carbonilico. La reazione di Mannich
intramolecolare sull’enantiomero R seguita da decarbossilazione genera il tropinone da cui la
riduzione stereospecifica del carbonile genera la tropina che presenta un ossidrile La iosciamina
è l’estere della tropina con l’acido (s)-tropico. La iosciamina racemica va sotto il nome di atropina,
mentre la scopolamina racemica va sotto il nome di atroscina.
O
N
CH3
O
S-CoA
Ox
idrolisi
O
N
CH3
O
OH
enolato
- CO2 H C N
3
O
tropinone
NADPH
H3C
N
H3C
OH
N
OH
O
tropina
O
(-)-Iosciamina
Ox
Scopolamina
(-)-Ioscina
H3C
OH
N
O
O
O
Atropa Belladonna
89
La Belladonna (atropa Belladonna, Solanacee) è una pianta nota da lungo tempo per la sua estrema velenosità. Il nome
deriva da Atropos, che nella mitologia greca è la parca che taglia il filo della vita. In effetti le bacche di questa pianta
sono particolarmente pericolose ma anche tutte le altre parti della pianta contengono alcaloidi tossici e quindi anche
semplicemente toccando la pianta si possono avere le manifestazioni dovute a questa tossicità. A ciò va aggiunto il
fatto che gli alcaloidi contenuti sono facilmente assorbiti attraverso la pelle. L’uomo è estremamente sensibile a queste
tossine mentre animali come pecore, maiali capre e conigli lo sono meno. E’ una pianta erbacea perenne molto alta che
produce fiori a campana color porpora scuro seguiti da abbondanti frutti nero brillante e delle dimensioni di una
piccola ciliegia. E’ una pianta tipica del centro e sud europa anche se non troppo diffusa. Viene coltivata a scopo
medicinale. Contiene lo 0.3-0.6% di alcaloidi, principalmente iosciamina. Dalle radici vengono estratti alcaloidi
minori come la (-)ioscina (scopolamina) e cuscoigrina. La miscela di alcaloidi estratti dalla pianta è ancora usata per
sedare dolori gastrointestinali. Il nome belladonna deriva dalla caratteristica della iosciamina e scopolamina di avere
un effetto midiatrico, cioè un effetto dilatatore della pupilla. Le antiche cortigiane applicavano negli occhi il frutto di
questa pianta ottenendo pupille dilatate ed un aspetto che sicuramente colpiva anche se la vista era annebbiata per
l’impossibilità di mettere a fuoco.
Stramonio:
Lo stramonio (Datura stramonium, solanacee, in inglese thornapple “melaspinosa” per la forma dei frutti) veniva
utilizzato come droga nel medioevo per drogare le vittime prima di derubarle: la vittima appariva normale, non reagiva
anzi aiutava il ladro e dopo non ricordava niente di quanto accaduto. Attualmente la pianta è coltivata a scopo
farmaceutico in europa e sudamerica. Le foglie di stramonio contengono lo 0.2-0.45% di alcaloidi, principalmente (-)iosciamina e (-)-scopolamina. Il nome Datura deriva da dhat, il veleno indiano usato dai Thugs.
Iosciamina, Scopolamina:
Questi alcaloidi competono con l’acetilcolina per l’occupazione del recettore muscarinico del sistema parasimpatico,
impedendo il passaggio degli impulsi nervosi e sono classificati come anticolinergici. L’acetilcolina può legarsi a due
tipi di recettori, quello muscarinico e quello nicotinico, nomi che derivano dagli alcaloidi muscarina e nicotina (vedi
più avanti). La somiglianza strutturale fra acetilcolina e muscarina è facilmente apprezzabile mentre la iosciamina è
capace di occupare lo stesso sito recettoriale grazie al fatto che la distanza tra l’atomo di azoto ed il legame estereo è
simile a quella dell’acetilcolina.
OH
H3C
CH3
H3C
O
+
+
CH3
N
N
H3C
H3C
CH3
O
muscarina
CH3
O
acetilcolina
O
H
+
HN
H3C
O
OH
Tutte le piante che producono alcaloidi tropanici sono molto tossiche, gli alcaloidi diffondono velocemente anche
attraverso la pelle raggiungendo il flusso sanguigno.
La Scopolamina butilbromuro (sale ammonio quaternario) (Buscopan) è uno spasmolitico gastrointestinale
90
Più raramente si ha una variazione strutturale se il carbonio carbossilico della catena laterale
acetoacetilica non viene perso come nella formazione della tropina: in questo caso lo scheletro
tropanico includerà la funzione carbossilica. E’ un processo raro, ma importante, perché l’unico
esempio è la formazione dei derivati dell’ecgonina, come la cocaina. La sua biosintesi è analoga a
quella
vista
per
la
iosciamina,
ma
deve
partire
dall’enantiomero
S
del
l’N-
metilpirrolidinacetoacetilCoA. La funzione tioesterea diventa un semplice metilestere e la
metilecgonina si forma successivamente dal metossicarboniltropinone grazie ad una riduzione
stereospecifica del gruppo carbonilico. Va notato che in questo caso la riduzione del carbonile
avviene dalla parte opposta rispetto a quanto visto per la tropina, infatti nell’ecgonina si ottiene una
configurazione La cocaina è il diestere dell’ecgonina.
O
O
O
Ox
N
S
N
Base
CO 2Me
CO 2Me
N
O
H3C
S-CoA
CH3
CH3
N
-
CH
+
S-CoA
O
COS-CoA
OH
H3C
SAM
N
H3C
NADPH
cocaina
O
O
metilecgonina
benzoil-S-CoA
La coca contiene anche quantità di cinnamoilcocaina, in cui
l’acido cinnamico sostituisce il benzoico, ed anche
tropacocaina in cui è assente il gruppo metilestere, ma si
conserva il gruppo estereo di tipo che conferma l’origine
biosintetica analoga a quella della cocaina.
O
O
CH3
O
N
H3C
H
cinnamoilcocaina
O
N
H3C
O
H
O
tropacocaina
Pianta di coca
91
Le foglie di coca sono prodotte da piante del genere Erythroxylum, piccoli arbusti originari delle
regioni andine del sud America (Colombia, Ecuador, Perù e Bolivia) e viene coltivata anche in
Indonesia. Le piante coltivate vengono mantenute piccole grazie a continue potature che
consentono di raccogliere una grande quantità di foglie almeno tre volte l’anno. Le foglie di coca
vengono masticate dagli indiani del Sud America da tantissimi anni. La foglia viene mescolata con
calce allo scopo di liberare il principale alcaloide cocaina come base libera e la pasta ottenuta da
questa miscelazione viene masticata.. La cocaina ha una potente azione nell’allontanare la
sensazione di fatica è ciò consente ai lavoratori di ignorare fame, fatica e freddo. E’ stato stimato
che circa il 25% del raccolto viene consumato dai lavoratori locali, ognuno dei quali consuma circa
50 g di foglie al giorno, che corrispondono a 350 mg di cocaina. Solo una piccolissima percentuale
della coca prodotta (1-2%) viene utilizzata a scopo farmaceutico, il resto serve ad alimentare il
traffico illegale. La foglia di coca contiene 0.7-2.5% di alcaloidi, di cui il principale componente è
la (-)-cocaina, diestere della (-)ecgonina. Solo i derivati dell’ecgonina sono di importanza
commerciale e per la produzione legale di cocaina può essere seguito un approccio semisintetico.
Questo prevede l’idrolisi totale degli alcaloidi in modo da ottenere ecgonina che viene poi
riesterificata.
Farmacologicamente la cocaina è un importante anestetico locale per applicazione topica. Viene
rapidamente assorbita dalle mucose e paralizza le terminazioni periferiche dei nervi sensoriali. La
parte farmacoforica della cocaina è costituita dall’estere di un acido carbossilico aromatico e da un
gruppo amminico basico separati da una catena carboniosa lipofila. Questo ha consentito di
sintetizzare una serie di molecole più sicure come anestetici.
NH2
H2N
H3C
O
N
O
CH3
O
O
H3C
benzocaine
procaine
CH3
H3C
O
N
CH3
NH
CH3
lignocaine
92
Le calistegine sono un gruppo di derivati poliidrossinortropanici (manca il metile) idrosolubili
recentemente scoperti nelle foglie e nelle radici di molte piante delle solanacee tra cui Atropa e
Mandragora.
OH
HN
HO
OH
OH OH
HN
OH
HO
Calistegina B 2
Calistegina A 3
Sono composti interessanti perché inibitori delle glicosidasi ed hanno un elevato potenziale
farmacologico per la cura di HIV.
Alcaloidi pirrolizidinici
N
nucleo pirrolizidinico
Due molecole di ornitina possono dare luogo alla formazione dello scheletro biciclico degli
alcaloidi pirrolizidinici, attraverso una serie di reazioni che passa sempre attraverso la putrescina.
Due molecole di putrescina vengono condensate insieme in una reazione NAD+ dipendente
fornendo una immina che è poi convertita in omospermidina per riduzione con NADH. Lo scheletro
pirrolizidinico si forma dall’omospermidina per deamminazione ossidativa, formazione dello ione
imminio, nuova deamminazione ossidativa e reazione di Mannich intramolecolare. Un esempio
classico di alcaloide a struttura pirrolizidinica è la retronecina che può essere derivata dall’aldeide
pirrolizidinica grazie a semplici passaggi ossidoriduttivi.
Le pirrolizidine raramente si trovano come basi libere mentre più spesso sono presenti come esteri
di rari acidi mono o dicarbossilici, gli acidi necici: ad esempio la senecionina è un diestere della
retronecina con l’acido senicico che deriva dall’isoleucina.
Molti alcaloidi pirrolizidinici mostrano una forte tossicità epatica e sono responsabili di numerosi
casi di avvelenamento di bestiame. Le strutture delle molecole tossiche possiedono una
insaturazione nelle posizioni 1,2 del nucleo pirrolizidinico ed una funzione esterea in catena
laterale. Le ossidasi presenti nel fegato dei mammiferi trasformano l’alcaloide nativo in strutture
pirroliche estremamente reattive, potenti reagenti alchilanti che reagiscono con una serie di
nucleofili presenti nella cellula come gli acidi nucleici o proteine. La presenza di alcaloidi
93
pirrolizidinici in alcune preparazioni medicinali erboristiche ne ha evidenziato i potenziali pericoli
nell’uso come rimedio per problemi infiammatori reumatici e gastrointestinali. Il loro uso
prolungato può dare seri danni epatici.
NH2
NH2
NH2
NH2
NH2
NAD
O
NH2
NH2
+
NADH
Deamminazione
NH2 H2N
putrescina
NH
N
putrescina
ossidativa
NH
omospermidina
O
H
O
H
7
6
7a
Deamminazione
1
+
+
N
5
NH2
O
4
+
N
ossidativa
N
2
N
ione imminio
3
hexahydro-1H-pyrrolizine-1-carbaldehyde
H
CH3
H
HO
H3C
O
2X
CH3
CH3
O
H3C
OH
H
HO
OH
O
O
NH2
H
O
H
N
retronecine
N
senecionine
Alcaloidi da Lisina
La L-lisina è l’omologo superiore della L-ornitina ed anch’essa può funzionare da precursore degli
alcaloidi, usando reazioni del tutto analoghe a quelle viste per l’ornitina. L’ulteriore gruppo
metilenico presente nella struttura della lisina implica per questo amminoacido la formazione di
anelli a sei termini di tipo piperidinico. Anche in questo caso il gruppo carbossilico viene perso,
l’atomo di azoto che rimane è quello terminale () e si ottiene così una unità C5N.
O
H2N
OH
NH2
N
H
94
Alcaloidi piperidinici
La N-metilpelletierina è un alcaloide isolato dalla corteccia del melograno dove sono presenti
anche la pelletierina e la pseudo pelletierina e la miscela di questi alcaloidi ha dimostrato attività
tenifuga. La N-metil pelletierina e la pseudopelletierina sono omologhi superiori dell’igrina e del
tropinone e per la loro biosintesi può essere proposta una via analoga che prevede l’uso di
cadaverina.
O
PLP
H2N
OH
deamminazione
ossidativa
-CO 2
NH2
NH2
NH2
NH2
O
cadaverina
O
O
N
H
H3C
CH3
pelletierina
O
-
CH
Mannich
idrolisi
decarbox
+
N
H
O
N
S-CoA
+
H
+
N
H
N
Delta- 1-piperina
Mannich
intermolecolare
CH3
O
CH3
Mannich
intramolecolare
H3C
N
H
anaferina
N
H
N
O
Pseudopelletierina
Se, nella reazione di Mannich iniziale, la controparte è il
benzoilacetil-CoA, si ha l’accesso agli alcaloidi della Lobelia
(lobelia inflata).
95
O
O
-
CH
S-CoA
O
Mannich
idrolisi
decarbox
+
N
H
N
H
Ox.
riduzione
O
O
N
H
+
N
H
sedamina
Mannich
idrolisi
decarbox.
O
O
SAM
O
O
N
N
H
CH3
lobelanina
O
OH
N
CH3
Lobelina
La lobelina stimola il recettore nicotinico in modo simile alla nicotina ed è stata impiegata come
deterrente per fumatori. Ha anche effetto contro asma e bronchiti ma in dosi eccessive è molto
tossica. Il sapore piccante dei frutti del pepe nero è legato all’alcaloide piperina che contiene il
nucleo, appunto, piperidinico.
O
O
N
piperina
O
Alcaloidi Chinolizidinici
Gli alcaloidi del lupino responsabili delle proprietà tossiche associate a queste piante, sono
caratterizzati da uno scheletro chinolizidinico.
N
Scheletro chinolizidinico: octahydro-2H-chinolizine
96
H
+
NH
Ginestra dei carbonai
H
cadaverina
N
+
N
H
OH
H
H
idrolisi
CHO
CHO
CHO
H
H
N
NH
H
-H 2O
(-)-Lupinina
deamminazione
+
NH
N
NH
ossidativa
O
NH2
+
N
CHO
H
N
H
+
H
N
?
+
N
N
N
H
N
Aldrich 46 euro 10 ml
N
(-)sparteina
H
Questo sistema biciclico è strettamente correlato al sistema pirrolizidinico, ma deriva da due
molecole di lisina. La lupinina presenta uno scheletro particolarmente semplice, ma altri alcaloidi
del lupino come la sparteina, alcaloide principale anche della ginestra, presentano una struttura più
complicata: uno scheletro tetraciclico che incorpora una terza molecola di lisina. Gli alcaloidi
chinolizidinici sono stati ritrovati principalmente nelle piante della famiglia delle leguminose.
Scoraggiano o respingono gli animali erbivori per i quali risultano tossici. Alcune piante, come il
laburno (vedi foto) contengono quantità significative di alcaloidi e possono essere pericolose anche
per l’uomo.
Il laburno è particolarmente rischioso perché tutte le sue parti,
compresi i semi, contengono quantità elevate di alcaloide. I
cosidetti lupini dolci sono varietà selezionate con un
contenuto di alcaloidi basso (circa un quarto delle varietà
amare) e sono coltivati per il loro alto contenuto proteico.
Laburno (Maggiociondolo comune)
97
Alcaloidi indolizidinici
Gli alcaloidi indolizidinici sono caratterizzati da anelli fusi a cinque e sei membri con un atomo di
azoto a testa di ponte. Sebbene derivino dalla lisina, si discostano molto dal punto di vista
biosintetico dagli altri alcaloidi. Infatti l’acido pipecolico, intermedio biosintetico, mantiene come
atomo di azoto quello in gruppo carbossilico.
O
O
deamm Ox
H2N
O
OH
OH
NH2
NH2
-H 2O
O
+
HSCoA
S-CoA
O
H
N
NH
acetil-CoA
NH
CO 2H
CO 2H
acido L-pipecolico
OH
O
OH
H
HO
N
N
HO
castanospermina
Molecole a struttura poliidrossiindolizidinica hanno dimostrato una attività contro HIV grazie alla
loro capacità di inibire gli enzimi glicosidasici impiegati nella biosintesi delle glicoproteine. Poiché
l’espressione delle glicoproteine è essenziale per la proliferazione del virus dell’AIDS, ne risulta
un’attività antivirale che ha stimolato diverse ricerche mirate all’elaborazione di strutture correlate
ed all’indagine del loro meccanismo di azione. L’estere 6-O-butanoilcastanospermina è in fase
clinica come agente anti-AIDS. Va notato che c’è una forte somiglianza strutturale tra la
castanospermina e lo ione ossonio formato dalla rottura idrolitica di un glucoside. Questi alcaloidi
sono tossici per gli animali provocando vari disturbi intestinali e malnutrizione dovuta alla loro
interferenza con le idrolasi intestinali. Si trovano nelle leguminose.
R
O
H
+
OH
O
HO
OH
HO
OH
HO
OH
OH
OH
O
+
OH
OH
+
HN
OH
castanospermina
98
Alcaloidi da Acido Nicotinico
Alcaloidi piridinici
I più importanti alcaloidi del tabacco sono la nicotina e l’anabasina. Queste molecole contengono
un anello piridinico legato ad uno pirrolidinico o piperidinico derivanti rispettivamente da ornitina e
lisina. L’unità piridinica ha invece origine da acido nicotinico (vitamina B3 detta anche niacina).
N
N
N
CH3
H
N
N
O
nicotinic acid
anabasine
nicotine
OH
La vitamina B3 è una vitamina idrosolubile stabile, ampiamente distribuita negli alimenti,
specialmente carne, fegato, farina e lievito. In alcuni cibi, come ad esempio nel mais, essa può essere
presente in una forma non facilmente disponibile: diete basate principalmente sul mais possono
dunque portare a deficienza di acido nicotinico. L’amminoacido triptofano può essere convertito
nell’organismo in acido nicotinico e può provvedere ad una quantità richiesta della vitamina. Sotto
il nome di vitamina B3 va anche la combinazione nicotinammide-acido nicotinico. Sotto forma di
coenzima NAD+, la nicotinammide gioca un ruolo vitale nei processi ossidoriduttivi. La carenza di
nicotinammide causa la pellagra che si manifesta con dermatiti, diarrea e demenza. Il sintomo più
evidente della pellagra è la lingua rossa e le lesioni orali. L’acido nicotinico è sintetizzato negli
animali a partire da triptofano passando attraverso l’acido 3-idrossiantranilico.
CO 2H
NH2
N
H
dell'anello aromatico
NH2
O
HO
CO 2H
scissione ossidativa
O
HO 2C
NH2
OH
CO 2H
CO 2H
-CO 2
N
N
CO 2H
acido chinolinico
acido nicotinico
Invece piante come Nicotiana usano una via biosintetica differente che impiega gliceraldeide 3fosfato ed acido L-aspartico come precursori. L’acido chinolinico compare in entrambe le vie.
OP
HO
-
CO 2H
HO
CO 2H
CO 2H
HC
O
H2N
CO 2H
N
CO 2H
N
CO 2H
acido chinolinico
99
Nella biosintesi della Nicotina, un anello pirrolidinico derivante dall’ornitina, probabilmente come
catione N-metil-1-pirrolinio è legato all’anello piridinico dell’acido nicotinico sostituendo il
gruppo carbossilico.
H
+
H
CO 2H
O
H
H
Putrescina
-CO 2
NADPH
HNADPH
N
+
H
H
N
N
H
O
N
H
H
+
H
acido diidronicotinico
CH3
1, 2 -diidropiridina
H
N
N
H
CH3
H
CH3
N
H
NADP
N
+
Attraverso un processo analogo avviene la biosintesi della anabasina, ovviamente a partire dalla
lisina.
Approfondimento: Il tabacco è costituito da foglie conciate ed essiccate di nicotiana tabacum (solanacee), una pianta
annuale indigena dell’america tropicale ma ormai diffusa in tutto il mondo. Nella foglia gli alcaloidi sono presenti come
Sali di acido malico e citrico ma la nicotina pura è una sostanza oleosa e volatile. In piccole dosi può agire come
stimolante respiratorio, ma in grosse dosi causa depressione respiratoria. Le foglie di tabacco in polvere sono state per
lungo tempo usate come insetticidi ed utilizzate sia per scopi agricoli che in orticoltura. La base libera è molto più tossica
dei suoi sali e nelle formulazioni vengono inclusi anche dei saponi proprio per assicurare il pH basico. Comunque oggi si
preferiscono usare sostanze meno tossiche. La nicotina è tossica nell’uomo a causa del suo effetto sul sistema nervoso,
cioè l’interazione con i recettori nicotinici dell’acetilcolina. Studi recenti suggeriscono che la nicotina può migliorare la
trasmissione degli impulsi nervosi e questo potrebbe spiegare la bassa incidenza del morbo di Alzheimer tra i fumatori.
Comunque qualsiasi beneficio è abbondantemente sopravanzato da un aumentato rischio di patologie cardiache,
polmonari e respiratorie.
Alcaloidi Derivanti dalla Tirosina
La
decarbossilazione
PLP-dipendente
della
L-Tirosina
fornisce
il
semplice
derivato
feniletilamminico Tiramina che per doppia N-metilazione dà l’ordenina (alcaloide isolato dall’orzo
che possiede attività inibitrice della germinazione). Più comunemente le molecole a struttura
feniletilamminica sono derivati 3,4-diossidrilati o 3,4,5-triossidrilati e si formano a partire dalla
dopamina, prodotto di decarbossilazione della L-DOPA.
100
Le più importanti tra queste molecole sono sicuramente le catecolammine: noradrenalina,
neurotrasmettitore nei mammiferi, e adrenalina, ormone
rilasciato nell’uomo dalle ghiandole surrenali in risposta
a situazioni di stress. Questi composti vengono
sintetizzati per successive reazioni di -ossidrilazioni ed
N-metilazioni sulla dopamina. Reazioni di ossidrilazione
aromatica ed O-metilazione in un cactus (peyote)
portano alla conversione della dopamina in mescalina,
alcaloide con proprietà psicotrope ed allucinogene.
CH3
NH2
CO 2H
N
CH3
PLP
SAM
NH2
HO
-CO 2 HO
HO
ordenina
tiramina
tirosina
O
NH2
NH2
OH
HO
HO
PLP
HO
HO
O2
-CO 2
HO
NH
CH3
SAM
HO
HO
OH
NH2
HO
noradrenalina
dopamina
L-DOPA
HO
adrenalina
Ox
CH3
SAM
O
H2N
O
CH3
mescalina
O
CH3
Approfondimento: Le catecolammine dopamina, noradrenalina ed adrenalina sono prodotte dalle ghiandole surrenali e
dal tessuto nervoso e sono importanti neurotrasmettitori nei mammiferi. Sono stati identificati diversi recettori grazie
ai quali queste molecole esplicano la loro azione fisiologica. I recettori sono normalmente eccitatori e producono la
contrazione del muscolo liscio vasale, uterino ed intestinale. I recettori sono normalmente inibitori sul muscolo liscio
ma stimolatori su quello cardiaco. La dopamina può agire sui recettori vascolari che sui cardiaci , ma ha anche
recettori propri in diversi organi. Nel morbo di Parkinson si verifica una deficienza di dopamina dovuta ad una
disfunzione nervosa. Il trattamento con L-DOPA aiuta ad aumentare i livelli di dopamina nel cervello. La
noradrenalina è un potente vasocostrittore periferico che agisce soprattutto sui recettori ed è utile nell’aumentare la
pressione sanguigna in casi di ipotensione acuta. L’adrenalina agisce sia con i recettori che e tra le risposte dei
primi c’è la costrizione del muscolo liscio della pelle. Le risposte dei recettori comprendono un aumento delle
contrazioni del muscolo cardiaco. Un’ampia serie di agenti bloccanti i recettori dell’adrenalina (-bloccanti) sono
stati sviluppati per controllare la velocità e la forza delle contrazioni cardiache nell’ipertensione od in altre patologie.
101
Altri alcaloidi isolati dal cactus peyote e correlate con la mescalina sono la analamina, analodina ed
analodinina che possono essere classificati come alcaloidi tetraidroisochinolinici semplici.
O
H3C
H3C
SAM
NH2
H3C
H3C
O
H3C
acido piruvico
NH2
O
HO
O
OH
O
O
H3C
N
O
+
H3C
-H 2O
OH
CO 2H
OH
OH
H
acido gliossilico
CH3
O
O
CH3
O
H3C
H3C
NH
NH
OH H3C
O
OH
analamina
CH3
NH
O
O
H3C
+
H3C
H
OH H3C
CO 2H
decarbox.
ossidativa
CO 2H
CH3
CH3
O
O
O
Riduzione
H3C
N
O
SAM
NH
H3C
H3C
O
NH
O
OH
CH3
OH
CH3
analonidina
H3C
O
CH3
analonina
La ciclizzazione avviene con un meccanismo tipo Mannich: l’effetto mesomerico di un sostituente
ossigenato esalta la nucleofilicità dell’anello. Questa reazione è l’equivalente sintetico della
reazione di Pictet-Spengler che utilizza semplici aldeidi e che quindi non necessita del passaggio di
decarbossilazione. Nella reazione biosintetici possono essere usati sia chetoacidi che aldeidi
(specialmente se più complessi).
La reazione tra una unità feniletilica ed una feniletilamminica dà luogo ad uno scheletro
benziltetraidroisochinolinico. Questo scheletro può subire ulteriori
NH
modificazioni dando luogo ad un’ampia serie di alcaloidi vegetali,
alcuni di grande interesse farmacologico. Molti degli esempi di
alcaloidi
benziltetraidroisochinolinici
contengono
una
orto
diossigenazione in ciascuno degli anelli aromatici la cui origine è
dunque potenzialmente ascrivibile all’utilizzo di due unità di DOPA.
scheletro benziltetraidroisochinolinico
Sebbene nella biosintesi vengano usate due molecole di tirosina, è
solo il frammento feniletilamminico a formarsi da DOPA mentre gli altri atomi di carbonio
derivano da tirosina via acido 4-idrossifenilpiruvico e 4-idrossifenilacetaldeide. Il risultato della
reazione di Mannich è perciò il triidrossialcaloide norcoclaurina, formato come enantiomero S.
102
NH2
-CO 2
HO
NH2
Ox
HO
HO
tiramina
tirosina
dopamina
O
CO 2H
HO
O
transaminazione
NH
HO
(S)
-CO 2
HO
HO
HO
norcoclaurina
O
H3C
N
O
papaverine
O
CH3
H3C
H3C
N
HO
CH3
O
HO
O
H3C
H3C
O
S-reticulina
Il concetto di accoppiamento fenolico ossidativo è fondamentale per comprendere le modificazioni
cui vanno incontro gli scheletri base benziltetraidroisochinolinici per formare una serie di alcaloidi
ascrivibili a differenti classi. I principali alcaloidi dell’oppio, morfina, codeina e tebaina, derivano
da questo tipo di accoppiamento, sebbene la successiva riduzione di uno degli anelli aromatici
mascheri la loro origine. La R-reticulina è stata identificata come il precursore di questi alcaloidi
morfinanici.
O
H3C
O
O
H3C
H3C
N
HO
+
CH3
NADP
+
HO
N
N
HO
CH3
CH3
NADPH
HO
HO
HO
H3C
O
H3C
O
S-reticulina
H3C
O
R-reticulina
103
O
O
H3C
O
H3C
H3C
HO
HO
HO
O
O2
CH3
CH
CH3
N
C
CH3
N
N
H
H3C
H
H3C
O
H3C
O
R-reticuline
O
OH
O
accoppiamento
radicalico
O
O
O
H3C
H3C
H3C
HO
HO
HO
CH3 Acetil-CoA
H
H
H
H3C
H3C
O
O
O
H3C
N
N
H3C
O
O
CH3
NADPH
CH3
N
H3C
CH3
O
salutaridina
OH
salutaridinolo
H3C
O
O
O
Ox.
O
O
O
N
N
CH3
CH3
N
H
CH3
H
H
O
O
H
tebaina
+
O
H
CH3
neopinone
O
H3C
H3C
H3C
O
O
O
O
O
NADPH
O
demetilazione
N
N
CH3
N
H
CH3
CH3
H
H
O
HO
HO
morfina
codeinone
codeina
Approfondimento: L’oppio è il lattice essiccato ottenuto per incisione delle capsule non ancora mature del
papavero da oppio. La pianta è un arbusto autunnale con grossi fiori solitari di colore bianco, rosa o rosso porpora.
Per la produzione di oppio, le capsule in maturazione, che stanno appena cambiando colore dal blu-verde al giallo,
vengono accuratamente incise con un coltello per recidere i condotti lattiferi senza penetrare all’interno della
capsula. Poiché tali condotti sono tutti interconnessi non è necessario tagliarli tutti. Il latte che trasuda
velocemente ha inizialmente una consistenza lattiginosa ed un colore bianco, ma rapidamente diventa marrone e
coagula. Questo materiale, detto oppio grezzo, viene rimosso la mattina successiva, e dopo essere stato raschiato
viene deposto in recipienti che gli conferiscono la forma sferica o a blocchi. L’oppio grezzo è usato sin
dall’antichità come analgesico, per indurre il sonno (narcotico) e per il trattamento della tosse.
Sebbene le capsule di papavero possano contenere poco più dello 0.5 % di alcaloidi, l’oppio ne è una forma molto
più concentrata, arrivando a contenere fino al 25 % in massa di alcaloidi. Molti sono gli alcaloidi identificati
nell’oppio (più di quaranta) ma solo sei sono i principali: morfina, codeina, tebaina, papaverina, noscapina (o
narcotina) e narceina.
104
H3C
CH3
O
O
O
O
H
N
HO
CH3
N
O
codeina
HO
CH3
tebaina
H3C
O
CH3
O
H3C
O
CH3
N
H
CH3
O
HO
N
morfina
H3C
O
O
papaverine
O
N
CH3
O
O
H
H
CH3
H3C
O
N
O
CH3
H3C
O
O
O
H3C
O
O
Noscapine
narceina
CH3
HO
CH3
O
OH
O
O
O
O
OH
O
CH3
OH
O
acido meconico
Gli alcaloidi sono spesso salificati con acido meconico e l’oppio stesso contiene fino al 3-5% di questa sostanza.
L’acido meconico si ritrova sempre nell’oppio ma, a parte altre specie di papavero, non viene riscontrato in
nessuna altra fonte naturale. L’acido meconico dà un complesso rosso fuoco con FeCl3 e questa reazione è stata
usata come saggio rapido ed abbastanza specifico per identificare l’oppio.
La morfina è un potente analgesico-narcotico e rimane uno dei migliori analgesici per alleviare il dolore forte. La
morfina induce anche uno stato di euforia e di indifferenza mentale insieme a nausea, vomito, stipsi, tolleranza e
dipendenza. La codeina è uno dei più usati alcaloidi dell’oppio e data la sua scarsa quantità, viene preparata per
semisintesi da morfina. L’azione della codeina dipende dalla parziale demetilazione che subisce nel fegato
(producendo morfina) e che le conferisce effetti analgesici di tipo morfinico. Come analgesico la codeina possiede
circa un decimo delle proprietà della morfina. Possiede inoltre un notevole capacità antitussiva, sia per sedarla che
per prevenirla, agendo direttamente sul centro della tosse. La tebaina è praticamente priva di attività analgesica ma
può essere utilizzata come antagonista della morfina. E’ il punto di partenza per la semisintesi di altri farmaci. La
papaverina ha proprietà ipnotiche ed analgesiche quasi nulle. Ha capacità vasodilatatorie e spasmolitiche. Usata
come espettorante e nel trattamento degli spasmi intestinali. La noscapina è stata usata a lungo come antitussivo
ma la scoperta della sua teratogenicità né ha fermato l’uso. Più del 90% della morfina estratta viene sottoposta a
trasformazione in altri derivati. La maggior parte della codeina usata è ottenuta per monoalchilazioine della
morfina che avviene soprattutto sull’ossidrile fenolico acido. Analogamente la folcodina, un efficace e sicuro
antitussivo, può essere ottenuta per alchilazione. La diidrocodeina è una forma ridotta della codeina con simili
proprietà analgesiche. L’eroina, detta anche diamorfina, è un potente analgesico ed ipnotico. L’aumentato
105
carattere lipofilo conferisce un miglior trasporto ed assorbimento anche se la molecola effettivamente attiva è la 6acetilmorfina, poiché il gruppo acetifico in posizione 3 viene idrolizzato dalle esterasi presenti nel cervello.
L’eroina fu inizialmente sintetizzata allo scopo di ottimizzare le proprietà antitussive della morfina. Viene usata
nei malati terminali per le proprietà analgesiche ed antitussive. La nalorfina è un analgesico ma anche
un’antagonista dei diversi oppiacei. La apomorfina, ottenuta per trattamento acido a caldo della morfina, non ha
nessuna caratteristica analgesica od ipnotica ma è un forte emetico e viene utilizzato in casi di emergenza per
avvelenamenti.
HO
HO
O
O
H
H
N
NH
CH2
HO
HO
nalorfina
normorfina
O
CH3
CH3
HO
O
O
O
O
O
H
H
N
N
N
H
CH3
O
morfina
HO
CH3
CH3
HO
eroina
O
codeina
CH3
O
N
CH3
O
O
HO
O
O
H
N
HO
N
CH3
H
CH3
CH3
H
N
HO
HO
diidrocodeina
pholcodine
apomorfina
Esistono svariati farmaci modellati sullo scheletro della morfina, ma di origine artificiale. L’anello piperidinico
tipico della morfina non è presente nel metadone,
che comunque può mimarla.
CH3
CH3
N
O
CH3
H
CH3
alla morfina ma dà meno euforia ed ha una durata
CH3
N
CH3
OH
CH3
HO
metadone
Il metadone è attivo per via orale, ha attività simile
O
etorfina
O
CH3
di azione maggiore. Viene usato per il trattamento e
la riabilitazione di eroinomani. Può dare dipendenza
in modo simile alla morfina ma i sintomi da
astinenza sono molto meno gravi. Esistono anche altri derivati semisintetici della morfina, in cui il potere
analgesico viene esaltato. Ad esempio l’Etorfina risulta fino a 10000 volte più potente della morfina, troppo per
uso umano, e trova applicazione in veterinaria con animali di grossa taglia.
106
Alcaloidi Derivanti dal Triptofano
L-triptofano è un amminoacido aromatico contenente un sistema indolico che deriva dalla via
biosintetici dell’acido shikimico attraverso l’acido antranilico. Ci limiteremo a vederne alcuni della
vasta serie di questi prodotti.
Alcaloidi indolici semplici
Sono la triptamina ed il suo idrossiderivato serotonina formati da una serie di reazioni di
decarbossilazione, mutilazione ed idrossilazione.
NH2
O
NH2
-CO 2
OP
NMe 2
SAM
HO
ox
N
H
triptofano
triptamina
N
H
N
H
psilocibina
ox
-CO 2
HO
NH2
N
H
serotonina
Questi composti sono ampiamente distribuiti nel regno vegetale. La serotonina si riscontra anche
nei tessuti animali dove funziona da neurotrasmettitore. La serotonina è un potente vasocostrittore.
La psilocibina è responsabile delle proprietà allucinogene di alcuni funghi ampiamente distribuiti in
tutto il mondo. Particolarmente noti sono quelli presenti in Messico. Sono stati infatti utilizzati dalle
popolazioni locali in antichissime cerimonie.
Alcaloidi dell’ergot
La segale cornuta (ergot) è una affezione che si verifica principalmente su piante delle graminacee,
sia coltivate che selvatiche, ed è causata da un fungo appartenente alla specie Claviceps. Le sue
manifestazioni sono caratterizzate dalla comparsa al posto dei normali semi della pianta, di ergot,
cioè strutture molto resistenti detti anche sclerozi, che costituiscono uno stato di riposo del fungo.
Le proprietà tossiche degli ergot dei cereali della specie segale, per uomo ed animali, sono note da
tempo e sono dovute ad alcaloidi.
Gli alcaloidi farmacologicamente più interessanti sono derivati dall’acido lisergico che si lega con
un amminoalcol a formare l’ergometrina o con un peptide per formare ergotamina. Le unità base
per la formazione dell’acido lisergico sono il triptofano ed una unità isoprenica.
107
OH
O
O
CH3
OH
H3C
N
H
NH
N
H
H
O
N
CH3
H
HO
H
O
N
H3C
O
H
NH
O
CH3
N
H
N
H
acido lisergico
O
N
H
H
ergometrina
OH
N
H
CH3
N
H
H
ergotamina
N
H
Approfondimento: L’ergot è lo sclerozio essiccato del fungo Claviceps purpurea che si sviluppa sull’ovario della
segale. E’ una patologia fungina di diverse specie di graminacee selvatiche e coltivate e inizialmente colpisce i
fiori. Successivamente uno sclerozio scuro si forma al posto del normale seme. Questa nuova formazione
fuoriesce dalla testa del seme ed infatti il nome ergot deriva dal
francese argot (sperone). Gli sclerozi cadono poi nel terreno,
germinando a primavera ed infettando tutto il raccolto di cereali. Gli
ergot possono poi essere raccolti insieme al cereale ed infettare la
farina ed il cibo di uomini ed animali. Il consumo di segale infetta
risulta nella malattia nota come ergotismo. Tre sono le manifestazioni
cliniche importanti: problemi gastrointestinali come diarrea, vomito e
dolori addominali; problemi circolatori con raffreddamento di mani e
piedi a causa di un effetto vasocostrittore, una diminuzione del
diametro dei vasi sanguigni, specialmente periferici; sintomi
neurologici come mal di testa, vertigini, convulsioni, disturbi psicotici
ed allucinazioni.
Questi effetti normalmente scompaiono eliminando la fonte
dell’avvelenamento, ma l’ingestione continua o l’assunzione di dosi
massicce di cibi contaminati dall’ergot possono provocare problemi
seri. L’effetto vasocostrittore porta ad una diminuzione del flusso sanguigno nelle arterie terminali con necrosi del
tessuto, sviluppo di gangrena che può provocare la perdita dell’arto. L’ergotismo gangrenoso era noto come fuoco
di Sant’Antonio, poiché tradizionalmente nel medioevo era l’ordine di Sant’Antonio ad occuparsi delle persone
che ne soffrivano. Gli effetti neurologici si manifestavano spesso con gravi e dolorose convulsioni. Scoppi di
epidemie erano frequenti nell’Europa medioevale, ma una volta stabilita la causa diventava relativamente
semplice evitare la contaminazione. La separazione dell’ergot dal frumento sano e l’uso di pesticidi hanno
108
eliminato molti dei rischi anche se le infestazioni dei raccolti sono ancora frequenti. Gli sclerozi contengono dallo
0.15% allo 0.5% di alcaloidi (circa 50 tipi diversi), quelli utili farmacologicamente sono solo i derivati dell’acido
lisergico. Sono stati anche messi a punto metodi fermentativi che portano alla produzione di quest alcaloidi in alte
rese. Nonostante gli effetti sgradevoli dell’ergot , preparazioni che lo contengono sono state usate fin dal XVI
secolo per indurre contrazioni uterine durante il parto e per ridurre le emorragie dopo il parto. L’effetto ossitocico
(ossitocina è un peptide ciclico che stimola il muscolo uterino) è ancora utile. L’ergometrina (nota anche come
ergonovina od ergobasina) è infatti usata come ossitocico e viene iniettata nella fase finale del parto specialmente
in presenza di emorragie. Spesso si somministra insieme alla ossitocina. L’ergotamina non è invece adatta perché
induce anche una forte vasocostrizione periferica. Questa sua capacità viene usata nel trattamento acuto
dell’emicrania in cui riesce a combattere la vasodilatazione dei vasi del cranio. Il trattamento prolungato con tutti
gli alcaloidi dell’ergot è comunque da evitare per i problemi di necrosi che può portare alle estremità.
Il piu famoso derivato dell’acido lisergico è la lisergide (cioè la dietilammide o LSD): è un allucinogeno molto
abusato ed è il più attivo tra gli psicotomimetici noti. E’ stato sintetizzato a partire dall’acido lisergico ed anche le
piccole quantità assorbite accidentalmente durante la preparazione dal ricercatore (Hoffman nel 1943) causarono
fortissime allucinazioni.
109
Alcaloidi Purinici
I derivati purinici caffeina, teobromina e teofillina vengono normalmente considerati alcaloidi
purinici. Come tutti gli alcaloidi essi hanno una distribuzione piuttosto limitata ma la loro origine
biogenetica è strettamente correlata a quella delle basi puriniche adenina e guanina componenti
fondamentali di nucleosidi, nucleotidi ed acidi nucleici. La caffeina assunta sotto forma di bevande
come te, caffe e cola è uno degli stimolanti naturali più ampiamente consumati e socialmente
accettati.
O
O
H3C
N
N
O
N
N
N
HN
H
N
H3C
N
O
N
N
O
CH3
CH3
N
N
CH3
teofillina
teobromina
caffeina
O
CH3
CH3
O
N
HN
O
N
N
xantina
Approfondimento: Questi alcaloidi sono derivati xantinici e normalmente sono presenti
contemporaneamente nelle piante. Le principali fonti di questi composti sono quelle
piante da cui si ricavano bevande come tè, caffè, cacao e cola e che devono le loro
proprietà stimolanti proprio a questi alcaloidi idrosolubili. Questi composti inibiscono
l’enzima fosfodiesterasi con conseguente aumento dell’AMP ciclico e successivo
rilascio di adrenalina. Questo produce come effetti principali una stimolazione del
sistema nervoso centrale, un rilassamento del muscolo liscio bronchiale ed una
induzione della diuresi. La caffeina, tra questi, è il miglior stimolante. Un caffè
espresso può fornire in media 40-60 mg di caffeina (2-4 mg per decaffeinato), una tazza
di tè circa 40 mg, tazza di cioccolata 5 mg, bevande a base di cola 25-100 mg. La
quantità giornaliera non dovrebbe superare 1g per evitare effetti collaterali. Gli effetti
biologici prodotti dalla caffeina ingerita tramite le differenti bevande possono variare
molto, poiché la biodisponibilità del principio attivo varia a seconda degli altri
costituenti della bevanda da cui viene assunto.
110
ANTIBIOTICILATTAMICI
Penicilline
Sono gli antibiotici più vecchi tra quelli utilizzati clinicamente, ma il loro uso è ancora ampiamente
diffuso. La prima ad essere utilizzata in modo significativo è stata la penicillina G
(benzilpenicillina).
Ph
H
N
H
5
6
O
O
7
1
S
2
N4
3
CO2H
Anello -lattamico
Anello tiazolidinico
E’ ottenuta dal fungo Penicillum chrisogenum per fermentazione in un brodo di cultura contenente
liquido di macerazione di granturco. Questi composti hanno una struttura -lattamica-tiazolidinica
ed hanno origine da un tripeptide i cui componenti amminoacidici sono: l’acido L-amminoadipico
(ottenuto da 2-oxoglutarico ed acetil-CoA), la L –cisteina, e la L -valina.
O
O
OH
OH
SH
H2N
O
OH
NH2
O
O
NH2
O
HO
HO
L-cisteina
O
acido 2-oxoglutarico
OH
L-valina
acido
2-amminoadipico
Gli studi più recenti indicano che la L-cisteina condensa con la L-valina in modo tale che l’enzima
che catalizza questa reazione provochi anche l’inversione di configurazione del residuo di valina
durante la formazione di questo dipeptide. L’acido L-amminoadipico condenserà poi con questo
dipetide per dare il tripeptide noto come ACV.
SH
OH
O
NH2
L-cisteina
NH2
+
O
OH
L-valina
H2N
SH
H
N
O
HO2C
acido
L-amminoadipico
H
N
NH2
H
N
O
O
COSEnz
ACV
SH
COSEnz
L-cys-D-val
111
ACV ciclizza per dare la isopenicillina N ed un unico enzima catalizza la formazione dei due anelli
del sistema biciclico delle penicilline. La reazione è ossidativa e richiede ossigeno molecolare. E’
stato accertato che il primo a formarsi è l’anello beta-lattamico.
H
N
HO2C
NH2
O
O
N
O
H
N
HO2C
NH2
H
N
HO2C
NH2
O
O
COSEnz
Enz
S
H
N
HO2C
O2
H
N
O
ACV
NH2
SH
NH2
O
Enz
N
H COSEnz
Enz
S
H
N
Ossidazione HO2C
Formazione di
un radicale libero
COSEnz
S
N
O
COSEnz
S
O
N
O
Isopenicillina N
CO2H
Il passaggio da isopenicillina N a penicillina G avviene in quel particolare brodo di coltura, perché
la feniletilammina presente viene trasformata dal fungo in acido fenilacetico che poi reagisce
sottoforma di estere del Coenzima A per dare la penicillina G. In vie biosintetiche alternative si può
avere prima la idrolisi a formare l’acido 6-amminopenicillanico (6-APA) che poi viene trasformato
nel prodotto finale.
H
N
HO2C
NH2
- acido L-amminoadipico
O
N
O
CO2H
Isopenicillina N
SCoA
Ph
S
Idrolisi legame amminico
O
SCoA
Ph
H2N
S
O
Penicillina G
N
O
CO2H
acido 6-amminopenicillanico
6-APA
Altri tipi di penicillina possono essere ottenuti semplicemente fornendo al brodo di coltura acidi
diversi.
La benzilpenicillina è ancora un farmaco utile per il trattamento di alcune infezioni da grampositivi. E’ sensibile all’acidità gastrica e per questo non viene somministrata per via orale; si
112
somministra invece il suo sale sodico, idrosolubile, per iniezioni intramuscolari od endovenose. Il
problema della sua stabilità è comune a molte altre penicilline. A pH basici(>8) l’anello betalattamico si apre a dare l’acido penicilloico mentre a pH acidi si trasforma in acido penicillenico o
penillico.
Ph
H
N
O
Ph
H
S
N
O
S
O
CO2H
HOpH>8
H
N
H+
N
H
O
CO2H
Ph
H
N
O
O
S
HN
OH
N
O
pH 4
H
S
CO2H
O
H
N
O
N
HS
O
CO2H
Acido penicilloico
pH 2
H
N
CO2H
acido penicillenico
HO2C
S
N
N
CO2H
Ph
Acido penillico
Quale è il motivo di tale instabilita? In fin dei conti una semplice ammide non viene mai idrolizzata
in condizioni così blande. La risposta è nella particolare natura del ciclo -lattamico dovuta
all’elevato strain a cui è sottoposto. Motivi strettamente geometrici non consentono una efficace
coniugazione tra l’atomo di azoto ed il gruppo carbonilico, che è alla base della inerzia del gruppo
ammidico, e quindi l’anello -lattamico subisce facilmente attacco da parte di molti nucleofili.
Questa spiegazione della particolare instabilità degli antibiotici -lattamici fu proposta all’inizio
degli anni 40 da R. B. Woodward. In particolare Woodward metteva in evidenza come i 4
sostituenti marcati rosso non potessero trovarsi sullo stesso piano a causa della fusione dei due
anelli. Infatti le penicilline risultano ancora più instabili dei semplici -lattami monociclici. Il potere
acilante dell’anello -lattamico delle penicilline è paragonabile a quello di un cloruro acilico.
113
O
O
N
Ph
H
N
O
Ph
H
N
H
S
H
S
O
N
O
efficace
N
N
O
CO2H
non efficace
CO2H
Quindi non appare strano che anche un ambiente debolmente basico (pH>8) possa avere una
sufficiente concentrazione di ioni HO- da idrolizzare un anello -lattamico. Più complesso è il
meccanismo che porta alla disattivazione in ambiente acido. In questo caso è la protonazione
dell’azoto lattamico (essendo scarsamente coniugato con il C=O, manterrà una residua basicità) che
innesca l’attacco nucleofilo da parte del C=O ammidico presente in catena laterale. L’intermedio
che ne deriva subirà poi diversi destini in base al pH della soluzione. Tale reattività impedisce una
somministrazione orale della Penicillina G in quanto verrebbe immediatamente disattivata dai
succhi gastrici. La più importante nuova penicillina prodotta è stata la fenossimetilpenicillina
(penicillina V), risultato dell’addizione di acido fenossiacetico alla coltura batterica.
Questa penicillina ha il grande vantaggio di essere acido resistente a causa dell’introduzione di un
eteroatomo elettron attrattore nella catena laterale che impedisce la degradazione rendendo meno
nucleofilo il gruppo carbonilico presente in catena laterale. Per questo motivo la penicillina V può
essere somministrata per via orale ed è utile per il trattamento di infezioni delle vie respiratorie e
delle tonsille.
H
N
PhO
O
S
N
O
CO2H
Penicillina V
Molte altre penicilline sono state prodotte per semisintesi dall’acido 6-amminopenicillanico.
L’acido può essere ottenuto per idrolisi della benzilpenicillina che può essere chimica (più
conveniente industrialmente) o enzimatica.
114
115
Le penicilline contenenti un gruppo amminico basico come l’ampicillina e l’amoxicillina sono
acido resistenti dal momento che a pH acido quel residuo viene protonato e quindi diviene un
ottimo gruppo elettron attrattore. Inoltre questi farmaci hanno uno spettro d’azione ampio: sono
attivi anche verso alcuni gram-negativi non sensibili alla penicillina G e V.
Le penicilline, come gli altri antibiotici -lattamici, esplicano il loro effetto antibatterico legandosi
ad alcune proteine (le cosiddette proteine penicillino-liganti) che intervengono nelle ultime tappe
della biosintesi della parete cellulare batterica. Un costituente importante della membrana batterica
è la rete costituita dal polimero peptidoglicano. Questo conferisce rigidità e resistenza alla parete
cellulare. Il peptidoglicano è composto da lunghe catene polisaccaridiche (composte da Nacetilglucosammina ed acido N-acetil murammico uniti da legami 1→4) tenute insieme da catene
116
peptidiche. La formazione del polimero finale avviene per cross-linking di singole catene
polisaccaridiche del peptidoglicano e coinvolge un intermedio acil-D-ala-D-ala che mostra una
stretta analogia strutturale con la molecola della penicillina.
H
N
Polisaccaride-peptide
H
N
O
H
N
H+
-D-ala
O
O
O
O
CO2H
Peptide-polisaccaride
H2N
OH
O
H+
Ser
Ser
Resido di serina
presente nel sito attivo dell'enzima
H
N
Polisaccaride-peptide
Peptide-polisaccaride
O
O
Il residuo dipeptidico
D-ala-D-ala
N
H
O
viene attaccato dal gruppo ossidrilico di un residuo di serina
presente nel sito attivo dell’enzima coinvolto (una delle proteine penicillino liganti) e si forma un
nuovo estere con la serina. L’intervento del residuo di glicina derivante da una seconda catena di
peptidoglicano idrolizza l’estere e forma il ponte tra due catene di peptidoglicano.
La conformazione in cui è stato scritto il residuo
D-ala-D-ala
mette in evidenza la analogia
strutturale con i derivati -lattamici. A causa di tale analogia la penicillina è in grado di occupare il
sito attivo dell’enzima e di legarsi ad esso in modo irreversibile causando il blocco della biosintesi
della parete cellulare.
H
N
Ph
O
H
S
O
N
O
OH
H+
H
N
Ph
CO2H
Ser
H
S
HN
O O
Ser
CO2H
complesso irreversibile
con il sito attivo.
Il microrganismo quindi muore per fuoriuscita del materiale cellulare dalla membrana incompleta.
Le penicilline, a causa del loro meccanismo di azione, sono antibiotici sicuri per la maggior parte
delle persone, infatti non esiste nelle cellule dei mammiferi una struttura analoga a quella delle
cellule batteriche e quindi l’azione è specifica.
E’ noto che i batteri sviluppano abbastanza facilmente resistenza alle penicilline; questo è dovuto,
tra le altre cause, alla produzione di -lattamasi, cioè di enzimi atti a idrolizzare il ciclo -lattamico.
Il meccanismo di azione è del tutto analogo a quanto visto nello schema precedente ( attacco di un
residuo di serina ed apertura dell’anello, ma in questo caso il complesso che si forma è reversibile e
l’acido penicilloico viene espulso dal sito che diventa di nuovo pronto all’idrolisi di una nuova
molecola di penicillina.
117
Importanti, quindi tutti i tentativi atti ad eludere l’attività delle -lattamasi che risultano molto
specifiche per il tipo di penicillina. Per questo motivo variazioni strutturali della catena laterale
della catena laterale delle penicilline possono ovviare a problemi di resistenza. Quest’azione ha dato
vita a numerosi tipi di penicilline ed i più importanti sono elencati nelle due tabelle allegate.
Una via alternativa è quella di utilizzare antibiotici -lattamici insieme ad acido clavulanico o suoi
analoghi sintetici. L’acido clavulanico è prodotto da colture di streptomyces clavuligerus ed è un
mediocre antibiotico, ma la sua caratteristica è di reagire molto velocemente con una larga serie di
lattamasi.
Enz
acido clavulanico
H
OH
O
N
O
OH
Ser
H+
H
OH
H+
O
HN
O O
CO2H
HN
O O
CO2H
Ser
-lattamasi
Formazione
di un legame irreversibile
Nu con il sito attivo della lattamasi
OH
O
CO2H
Ser
La presenza del doppio legame esociclico sull’anello ossazolidinico, rende l’atomo di ossigeno
parte di un enoletere e facilita l’apertura dell’anello a cinque termini con formazione di uno ione
imminio, forte elettrofilo, che può facilmente attaccare un residuo nucleofilo del sito attivo della
lattamasi. In questo modo si ha la formazione di un legame irreversibile ed inibizione completa
della lattamasi. La funzione dell’acido clavulanico quindi è quella di bloccare le lattamasi e di
aumentare il tempo di vita dell’antibiotico con cui viene somministrato. Una tipica associazione è
amoxicillina-acido clavulanico (co-amoxiclav, uno dei nomi commerciali è augmentin). La
particolarità dell’acido clavulanico, quindi è quella di avere una funzione enoletere che rende un
ottimo gruppo uscente l’atomo di ossigeno coinvolto, a causa delle trasformazione dell’enolo in
chetone. Una volta capito il principio questo può essere applicato alla sintesi di nuovi farmaci con la
stessa attività biologica. Un esempio è il sulbactam.
Enz
sulbactam
H O O
S
N
O
OH
Ser
-lattamasi
H+ CO2H
O
O
S
HN
O O
Ser
CO2H
Nu
HN
O O
O
S O
CO2H
Ser
Formazione
di un legame irreversibile
con il sito attivo della lattamasi
In questo caso è stato sufficiente cambiare il grado di ossidazione dell’atomo di zolfo presente
sull’anello tiazolidinico. Infatti il gruppo solfone è già un gruppo uscente sufficientemente efficace.
118
Sintesi totale della Penicillina V (J. C. Sheehan 1957) (tratto da Classics in Total Synthesis di
K.C. Nicolau e E.J. Sorensen)
Lo studio della sintesi della penicillina V fu iniziato dal prof. Sheehan nel 1948 al MIT è fu
coronato da successo solo dieci anni dopo, ed il tentativo di sintetizzare questo composto
utilizzando i metodi della chimica organica classica fu paragonato dallo stesso Sheehan come
tentare di aggiustare la molla di un orologio prezioso utilizzando un incudine, un martello e delle
tenaglie.
Analisi retrosintetica
H
N
PhO
O
O
N
O
Lattonizzazione
O
CO2H
t-Butil ftalimmidomaloaldeidato
S
HO2C N
H
HS
N
CHO
O
t-BuO2C
H
N
PhO
S
H3N
CO2H
Cl
D-penicillamina cloridrato
CO2H
O
N
Formazione dell'anello
S
O
t-BuO2C N
H
CO2H
La sensibilità dell’anello -lattamico impone che la sua costruzione avvenga in uno stadio avanzato
della sintesi, possibilmente proprio nell’ultimo stadio. Per questo motivo la rottura retrosintetica
dell’anello -lattamico è proprio il primo passo e porta all’acido penicilloico come primo
precursore. Il problema della lattamizzazione era cruciale. All’inizio del lavoro di ricerca furono
tentate altre vie sintetiche che non prevedevano la lattamizzazione e si basavano su tutti gli studi
precedenti eseguiti nel periodo bellico che ne dimostravano la difficoltà. Era quindi necessario
sviluppare metodi più blandi di lattamizzazione rispetto a quelli noti all’inizio del lavoro di
Sheehan. Gli altri passaggi erano più semplici ed il passaggio cruciale era la sintesi dell’anello
tiazolico che poteva essere ottenuto per condensazione della penicillamina su un aldeide
opportunamente sostituita.
Sintesi totale
Il primo sforzo sintetico è quindi diretto verso la sintesi della
D-penicillamina,
un composto
ottenibile anche per degradazione delle penicilline. La sua sintesi inizia dalla valina racemica e
procede attraverso due reazioni che riassumono una serie di passaggi sintetici che possono apparire
abbastanza complicati
119
O
CO2H Cl
COCl
CO2H
HN
NH2
HN
rac. valina
O
O
O
O
N
Cl
Cl
Cl
AcO-
O
isomerizzazione
O
N
O
H
O
N
O
N
Cl
N
NaHS, MeONa
MeOH
-
Cl
SH
O
O
O
O O
O
O
Ac2O
O
O
O
Addizione
Michael
HS
N
MeOO
O
HS
O
O
HS
N
O
+
N
H
N
H
CO2Me
1. HCl, H2O, reflux
2. acetone
HS
H3N
Cl-
1. risoluzione
con brucina
CO2H
D-penicillamina cloridrato
N
2. 2N HCl
H
HCO2H
Ac2O
S
CO2H
O
S
N
CO2H
H
isopropilidene-DL-penicillamina
La reazione della valina con cloroacetilcloruro dà la corrispondente ammide che in anidride acetica
si trasforma nell’ossazolone corrispondente. Questa serie di passaggi, per quanto complessa, viene
attivata dalla formazione di una anidride mista che risulta abbastanza elettrofila da portare alla
formazione dell’anello ossazolico. L’ossazolone così ottenuto è un buon accettore di Michael e
viene sostituto con un atomo di zolfo per trattamento con NaHS. L’ammide che viene così ottenuta
viene trasformata nel derivato ciclico solo perché questo, una volta trasformato in derivato
formammidico, risulta facilmente risolvibile nei nsuoi due enantiomeri per trattamento con brucina,
una base enantiomericamente pura che forma sali diastereomerici con il derivato racemo della
penicillamina. Infine la deprotezione finale fornisce la
D-penicillamina
cloridrato in forma
enantiopura. Il passaggio successivo riguarda la sintesi del derivato aldeidico che può essere
ottenuto facilmente per reazione del t-butilftalimidoacetato con t-butilformiato in presenza di tbutossido di sodio.
120
O
t-BuONa
t-BuOCHO
O
N
N
O
t-BuO2C
O
t-BuO2C
CHO
Il prodotto così ottenuto può essere fatto reagire con la penicillamina cloridrato in etanolo acquoso
tamponato con acetato di sodio e porta alla formazione di due derivati, epimeri al C-6, in un
rapporto equimolecolare.
O
N
O
+
S
6
O
CO2H
O
NH H
S
6
t-BuO2C HN
t-BuO2C HN
CO2H
D--6
H3N
Cl-
CHO
O
t-BuO2C
O
NH H
HS
+
D--6
CO2H
La reazione potrebbe portare alla formazione di 4 diastereisomeri, formandosi 2 centri stereogenici
interessati alla reazione, ma la selettività è tale che solo due ne vengono formati. Il distereoisomero
con la corretta configurazione è il D--6. Particolarmente interessante appare la possibilità di
riequilibrare il D--6 per semplice riscaldamento di una sua soluzione in presenza di piridina. Il D-6 formatosi in soluzione precipita per raffreddamento mentre il D--6 può essere ulteriormente
riciclato.
A questo punto è necessario rimuovere il gruppo ftalimmido che può essere allontanato utilizzando
idrazina (quale è la reazione e perché è così favorita?). Adesso è possibile introdurre il gruppo
fenossiacetico per semplice reazione con il corrispondente cloruro acilico e rimuovere il gruppo
estereo t-butilico per ottenere il precursore chiave per la reazione di lattamizzazione.
121
O
NH H
O
OPh
1. N2H4
2. HCl
HCl . H2N H H
PhOCH2COCl
S
t-BuO2C HN
S
6
t-BuO2C HN
NEt3
CO2H
H
NH
S
O
t-BuO2C HN
CO2H
CO2H
HCl, CH2Cl2
D--6
OPh
NH H
S
O
HO2C HN
CO2H
Siamo quindi arrivati al punto probabilmente più difficile per la sintesi, almeno per quanto riguarda
le conoscenze acquisite alla fine degli anni 40. E’ solo nel 1955 che proprio il gruppo di Sheehan
pubblicherà una metodologia per la formazione di legami ammidici che prevede l’uso della
dicicloesilcarbodiimmide (DCC). Questa procedura viene effettuata a temperatura ambiente a pH
neutro in ottime rese sfruttando l’attivazione del gruppo carbossilico da parte della DCC. Dopo due
anni viene quindi infine completatat la sintesi della penicillina V aggiungendo 4 equivalenti di DCC
ad una soluzione diossano acqua del sale monopotassico del precursore ottenendo con una resa del
12% l’attesa penicillina V.
OPh
OPh
NH
H
NH
1.KOH
2. DCC
S
O
HO2C HN
O
O
C6H11N
CO2H
+
H
H
S
N
O H
NHC6H11
Penicillina V
CO2H
Appare particolarmente istruttivo chiedersi il perché di una resa così bassa. La motivazione risiede
nel carattere nucleofilo del gruppo carbonilico dell’ammide in catena laterale. L’intermedio attivato
può infatti subire attacco nucleofilo non solo dall’atomo di azoto dell’anello tiazolidinico ma anche
dal gruppo carbonilico andando così incontro al processo detto di azlattonizzazione.
NH
OPh
OPh
OPh
H
O
HO2C HN
S
CO2H
NH
1.KOH
2. DCC
O
O
C6H11N
H+
H
N
O H
NHC6H11
S
azlattonizzazione O
O
CO2H
NH
H
S
HN
CO2H
122
Questo tipo di reattività può trovare un confronto nel processo di degradazione delle penicilline in
ambiente acido.
Nonostante la scarsa resa del passaggio finale, la sintesi della penicillina V era quindi riuscita.
Negli anni successivi lo stesso Sheehan ottimizzerà la sintesi di altri derivati delle penicilline
pubblicando
la
sintesi
dell’acido
6-amminopenicillanico
in
cui
utilizzava
la
diisopropilcarbodiimmide come reagente attivante ed il gruppo tritile come protettore della funzione
amminica in posizione 6.
H2N
N C N
H
S
HO2C HN
Ph3CCl
Et2NH
Ph3C NH
H
S
H
S
N
HO2C HN
CO2Bn
H
Ph3C N
O
CO2Bn
CO2Bn
1. H2, Pd
2. HCl
H2N
H
S
N
O
CO2Bn
L’acido 6-amminopenicillanico così ottenuto (67% di resa) è un utile precursore per la sintesi di
altre penicilline con altri gruppi in catena laterale.
L’interesse per questa sintesi è tra l’altro legato anche alla capacità del gruppo di Sheehan di
sviluppare nuove reazioni per superare problemi sintetici importanti. E’ infatti questo gruppo che
per primo introduce l’uso di carbodiiimidi alifatiche e quello che sviluppa l’uso di esteri t-butilici
come utili gruppi protettori rimuovibili in ambiente anidro attraverso l’uso di acidi di Lewis.
123
Altri antibiotici -lattamici
Per quanto le penicilline rivestano, anche dal punto di vista storico, una grande importanza, la classe
dei -lattami presenta anche altri composti naturali che presentano le stesse caratteristiche di attività
biologica o comunque possono risultare complementari all’azione delle penicilline. Questi composti
possono essere classificati sulla base del loro scheletro con i nomi riportati nello schema seguente.
S
S
NH
O
-lactam
N
O
penam
N
H R
N
R
N
O
N
O
SO3H
monobactam
O
carbapenem
N
O
penem
N
O
cephem
O
O
S
clavam
Abbiamo già visto l’acido clavulanico come esempio dello scheletro clavam, adesso vedremo le
cefalosporine come esempio dello scheletro cephem.
Molto diffuse come antibiotici sono caratterizzate dalla presenza di un anello tiazinico, un anello a
sei termini contenente un atomo di azoto ed uno di zolfo. Il loro nome è legato al fatto di essere
prodotte da specie del genere Cephalosporium. Il composto capostipite di questa classe è la
cefalosporina C la cui biosintesi parte proprio dalla isopenicillina N, che era un intermedio nella
biosintesi delle penicilline.
H
N
HO2C S
NH2
epimerasi
S
O
NH2
O
penicillina N O
N
O
Isopenicillina N
CO2H
H
N
NH2
O
S
N
NH2
O
HO2C
NH2
O
O
O
desacetilcefalosporina C CO2H
S
N
H
N
SCoA HO2C
S
N
CO2H
O
CO2H
H
N
N
H
N
HO2C
O
O2
Enz
S
O2
Enz
meccanismo proposto
espansione anello
HO2C
H
N
HO2C R
OH
NH2
CO2H
S
O
N
O
O
O
Cefalosporina C CO2H
Il meccanismo biosintetico non è ancora definitivo e questa è una possibile proposta. Le
cefalosporine risultano in genere più stabili agli acidi delle penicilline e quindi possono essere
124
somministrate per via orale, anche se, specialmente i primi esempi noti, risultano poco assimilabili a
livello intestinale. Questo ha dato vita ad un numero molto elevato di nuove cefalosporine che si
differenziano per entrambe le catene laterali presenti su i due anelli eterociclici.
125
Come è possibile vedere nelle tabelle precedenti le cefalosporine vengono divise per “generazioni”.
Questo è legato allo spettro antibatterico che ciascuna di loro possiede ed in parte al periodo di
produzione. E’ quindi possibile che una cefalosporina di seconda generazione possa essere stata
introdotta dopo alcune cefalosporine di terza. Attualmente vengono utilizzate cefalosporine di tutte
e tre le generazioni. Un problema abbastanza diffuso riguarda la difficoltà di assimilazione di questi
126
composti a livello intestinale. Una possibile soluzione è stata fornita dall’uso di pro-farmaci (prodrugs) come cefuroxime-axetil e cefpodoxime-proxetil (vedi tabella seguente) in cui i gruppi esterei
sul carbossile C4 vengono idrolizzati dopo l’assimilazione dalle esterasi.
Le cefalosporine rappresentano una risorsa molto importante come antibiotici in quanto meno
sensibili alle lattamasi ed utile alternativa in pazienti allergici alle penicilline. Infatti solo il 10% dei
pazienti allergici alle penicilline risulta anche allergico alle cefalosporine.
Sono stati sviluppati anche analoghi non naturali delle cefalosporine, i cosiddetti carbacefem, in cui
l’atomo di zolfo è stato sostituito con un atomo di carbonio. Il primo esempio è stato il loracarbef
che ha una attività antibatterica analoga al cefaclor ma presenta una maggiore stabilità, una emivita
più lunga ed un migliore assorbimento per via orale.
Una variazione analoga può avvenire anche sullo scheletro delle penicilline (penam) dando origine
ai carbapenem che sono però prodotti naturali in cui al posto di un atomo di zolfo è presente un
atomo di carbonio. L’esempio più notevole è la tienamicina che contiene un atomo di zolfo nella
catena laterale.
127
Questa caratteristica strutturale si ritrova anche negli acidi olivanici di cui è riportata sotto una
struttura a titolo di esempio
Gli acidi olivanici non sono buoni antibiotici ma svolgono lo stesso ruolo dell’acido clavulanico nei
confronti delle cefalosporinasi verso cui il clavulanico è molto poco efficace.
Infine un accenno può essere fatto per i monobattami. Il composto più semplice noto è quello che va
sotto il nome di SQ 26-180 la cui struttura è riporatata nello schema seguente. Molti di questi
composti naturali non hanno particolari proprietà antibatteriche ma la modifica delle loro catene
laterali ha portao alla sintesi di composti molto importanti come l’aztreonam attivo verso
Pseudomonas aeruginosa, Haemophilus influenzae, e Neisseria meningitidis.
La biosintesi dei monobattami è stata proposta iniziare dalla serina attraverso una trasformazione
del gruppo ossidrilico in un buon gruppo uscente. Il gruppo N-sulfonato deriva da solfato.
128