TRASMISSIONE GABAERGICA
Il GABA identificato per la prima volta nel 1950 è il neurotrasmettitore inibitorio più
diffuso.
Le seguenti numerose evidenze sperimentali indicano che il GABA a livello del sistema
nervoso centrale (SNC) svolge funzione di neurotrasmettitore:
a) presenza di alte concentrazioni di GABA e dell'enzima deputato alla sua sintesi, la
glutammato decarbossilasi (GAD) in preparati sinaptici del SNC (sinaptosomi).
b) liberazione selettiva di GABA sia spontanea che evocata da stimolazione nervosa
(rilascio Ca2+ dipendente);
c) esistenza di siti di legame specifici che mediano iperpolarizzazione postsinaptica;
d) presenza nei sinaptosomi di un sistema di ricaptazione (uptake) ad alta affinità.
Nei vertebrati la maggior parte di questo aminoacido è confinata nel SNC: cervello e
midollo spinale.
La distribuzione del GABA e della GAD nel SNC non è uniforme; le massime
concentrazioni si trovano nella substantia nigra, nel globo pallido, nell'ipotalamo, nei corpi
quadrigemini, nella corteccia cerebrale, nel cervelletto e nell'ippocampo.
Il GABA è presente anche nelle cellule gliali dove uno specifico sistema di uptake e di
catabolismo contribuisce all'allontanamento di questo aminoacido dallo spazio sinaptico.
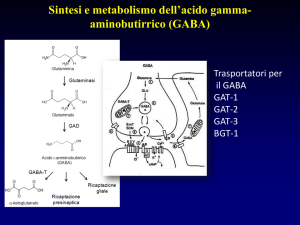
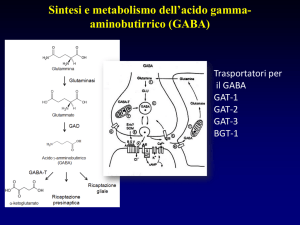
Fig.1. Schema di una sinapsi GABAergica dove sono rappresentati il terminale sinaptico che libera
GABA su recettori postsinaptici e una cellula gliale. Il processo di sintesi e di catabolismo dei
GABA è riportato sia a livello neuronale che nella cellula gliale.
SINTESI E METABOLISMO DEL GABA:
152
Il GABA si forma per decarbossilazione dell'acido glutammico
Questa reazione è catalizzata dalla GAD (Glutamico Acido Decarbossilasi), enzima
altamente specifico che ha come cofattore il piridossal-fosfato (PLP o Vit B6 (*)) ed è inibito
da diversi antagonisti del piridossal-fosfato, come isoniazide, tiosemicarbazide, ecc che
bloccano il gruppo aldeidico della Vit B6 sotto forma di derivati idrazonici:
→
R-CH=O + H2N-NH-R'
R-CH=N-NH-R'
. L'accumulo vescicolare del GABA è effettuato da un trasportatore specifico che utilizza,
come fonte di energia, sia il gradiente elettrico che di pH presente tra lume vescicolare e
citoplasma e generato dalla H+-ATPasi (pompa protonica) vescicolare.
GAD (PLP)
-
O
C
O
CH CH2
CH2 COOH
H2N CH2 CH2
+
CH2 COOH + CO2
NH3
GABA
Ac. Glutamico
GABA_T (PLP)
OHC
CH2
HO
CH2 COOH
C C CH2
O
Emialdeide succinica
al ciclo dell'acido citrico
O
CH2
COOH
Ac. a _ chetoglutarico
dal ciclo dell'ac. citrico
(*)
Interessante duplice ruolo coenzimatico della Vit B6: nella decarbossilazione, di qui il nome di cocarbossilasi
dato alla vit; nella transaminazione prendendo parte attiva nel trasferimento dell’ NH 3 dal gaba al chetoglutarato
(si consulti un libro di Biochimica). Il ruolo attivo del piridossal fosfato e illustrato dal seguente schema. Si
vede chiaramente come a partire da Vit B6 e AA si ottenga un chetoacido e l’immino-Vit B6, reazione verso
destra; mentre nel senso opposto un cheto acido viene trasformato in AA.
R C COOH
H
--
O PO3
HO
N
NH2
H3C
N
+
H
NH
H2O
CH
CH
CH
HO
O
R C COOH
R C COOH
R CH COOH
CHO
O
-PO3
HO
O
-PO3
--
O PO3
HO
N
+
H3C
Vit B 6 + Aminoacido
N
H
Base di Shiff
H3C
N
H
Struttura chinonica
H3C
N
Chetoacido + Immina di
Vit B 6
153
Il GABA viene degradato dall'enzima GABA-a-chetoglutaricotransaminasi (GABA-T) che
lo deammina a semialdeide succinica. [Quest'ultima viene ossidata ad acido succinico ad
opera di una semialdeide-succinico-deidrogenasi NAD-dipendente e infine entra a far parte
del ciclo di Krebs del’acido citrico]. Il gruppo amminico viene trasferito dalla GABA-T ad
una molecola di a-chetoglutarato per formare l'acido glutammico che viene riutilizzato per la
sintesi di nuovo GABA.
Il catabolismo del GABA può essere bloccato da sostanze che inibiscono l'attività
dell'enzima GABA-T quali il -vinil-GABA o vigabatrina , l'acido valproico, il valproato di
sodio e la valpramide. Queste sostanze prolungando l'emivita del GABA sono provviste di
attività antiepilettica.
COOH
H2N
COOH
H3C
CONH2
H3C
COOH
H2N
GABA
Ac. 2-propil.valerianico
Vigabatrina
-vinil.GABA
Valpramide
LIBERAZIONE E RICAPTAZIONE
Studi in vitro hanno dimostrato che il GABA viene liberato sia spontaneamente che in
seguito a stimolazione nervosa. La liberazione di GABA indotta dalla depolarizzazione, a
differenza di quella spontanea, è Ca2+-dipendente.
A livello delle sinapsi GABAergiche esistono specifici meccanismi di ricaptazione che
rimuovono rapidamente il GABA dallo spazio sinaptico ponendo così fine alla sua azione
inibitoria postsinaptica.
Numerosi composti sono capaci di bloccare l’uptake neuronale e gliale del GABA e di
potenziare quindi i meccanismi GABAergici a livello centrale. L'acido nipecotico e i suoi
derivati, gli analoghi del GABA (2-idrossi-GABA, 4-metil-GABA) e la guvacina sono i più
potenti inibitori dell'uptake neuronale. Specifici bloccanti dell'uptake gliale sono invece la
prolina e l'acido omo-nipecotico. Questi farmaci non hanno un impiego terapeutico.
CH3
H2N
COOH
COOH
COOH
HN
HN
COOH
H3C
Acido cis-3-aminocicloesan carbossilico
Ac. nipecotico
N
S
S
Tiagabina
RECETTORI PER IL GABA
Studi elettrofisiologici e biochimici hanno dimostrato l'esistenza di due differenti siti di
legame al GABA convenzionalmente denominati: GABAA e GABAB che differiscono fra loro
per profilo farmacologico, struttura molecolare e meccanismo di trasduzione del segnale. I
recettori GABAA sono recettori-canale permeabili allo ione Cl¯ mentre i recettori GABAB
sono accoppiati a proteine G inibitorie.
154
I recettori GABAA sono caratterizzati da una elevata sensibilità alla bicucullina e al
muscimolo, rispettivamente antagonista selettivo e agonista selettivo ad alta affinità per il sito
di legame del GABA, e contengono siti specifici di legame per le benzodiazepine ed i
barbiturici che ne modulano la funzione. I recettori GABAB sono attivati selettivamente dal
derivato del GABA, p-clorofenil-GABA (baclofen) e al contrario dei recettori GABAA
sono insensibili alla bicucullina e al muscimolo.
O
O
O
COOH
H2N
O
O
H2N
O
NH
N
Muscimolo
agonista diretto
O
CH3
O
Bicucullina
antagonista diretto
Cl
Baclofen
Agonista
Recettore GABAB
Recettore accoppiato a proteine G
a) sono insensibili all'azione della bicucullina e dei GABA-mimetici
b) sono attivati in modo stereospecifico dal GABA e dal p-clorofenil-GABA o baclofen
il cui (-)-isomero ha la stessa potenza del GABA, mentre il (+)-isomero è 100 volte
meno attivo
c) non sono modulati dalle benzodiazepine e dai barbiturici
d) non sono funzionalmente associati al canale allo ione eloro.
L'interazione del GABA o del baclofen con i recettori GABAB attiva principalmente una
proteina G specifica a carattere inibitorio (Gi), che produce una inibizione dell'enzima
adenilato ciclasi. La conseguente riduzione della concentrazione di cAMP si traduce in una
riduzione dei livelli di fosforilazione ed inibizione funzionale dei canali calcio/voltaggio
dipendenti implicati nel controllo presinaptico del rilascio di neurotrasmettitori.
Nella Tab..1 sono riportati numerosi effetti cellulari, ormonali e comportamentali attribuiti
all'attivazione dei recettori GABAB.
Recenti risultati sperimentali suggeriscono che gli antagonisti dei recettori GABAB
potrebbero migliorare i processi cognitivi e la memoria e avrebbero un effetto positivo sulle
crisi epilettiche. Un altro potenziale uso terapeutico degli antagonisti dei recettori GABAB
suggerito dai dati sperimentali potrebbe aversi nelle sindromi depressive. E’ stato dimostrato
infatti che il trattamento cronico con antidepressivi aumenta i siti di legarne dei recettori
GABAB in varie aree del SNC. In base a queste osservazioni è stato ipotizzato un possibile
effetto antidepressivo delle molecole capaci di legarsi ai recettori GABAB
Tab. 1. Conseguenze dell'attivazione dei recettori GABAB.
Effetti ormonali
Fattori di rilascio corticotropo
Produzione MSH
Effetti comportamentali
Epilettogenesi
Attacchi di panico
Catatonia
Secrezione gastrica
Fattore di rilascio della prolattina
Rilascio LH
Memoria
Ipotensione
Motilità gastrica
Effetti cellulari
Conduttanza K+
Conduttanza Ca++
Inibizioneattività adenilato
ciclasi
Inibizione idrolisi fosfoinositidi
155
Rilascio androgeni
Sintomatologia di astinenza da
diazepam e etanolo
Recettore GABAA
Il recettore GABAA è un recettore-canale permeabile agli ioni cloro. Poiché il cloro è
l'unico ione permeante attraverso il recettore GABAA, la sua attivazione «fissa» il
potenziale di membrana a quello d'equilibrio del che normalmente è di ca. -70 mV.
L'attivazione di questo recettore riduce quindi l'eccitabilità cellulare.
A livello del complesso recettoriale macromolecolare GABAA sono presenti i siti di legame
specifici per le seguenti molecole (vedi Fig. 3):
1) Sito di legame per il GABA, per i farmaci GABA mimetici (muscimolo) e GABAantagonisti (bicucullina). Questo sito di legame è situato sulla subunità del complesso
macromolecolare. L'interazione con il GABA o con un GABA mimetico si traduce
nell'apertura del canale ionico con conseguente iperpolarizzazione della membrana. La
bicucullina blocca con meccanismo competitivo l'interazione GABA-recettore.
2) Sito di legame per le benzodiazepine ed altre molecole benzodiazepino-mimetiche
(ciclopirroloni, imidazopiridine, triazolopiridine, -carboline, ecc.). Questo sito è posto
sulla subunità a ed è riconosciuto anche da ligandi ad azione agonista inversa (carboline) cioè molecole capaci di ridurre l'interazione del GABA col proprio sito di
riconoscimento ed indurre effetti (ansia-convulsioni) opposti alle benzodiazepine. Il sito
di legame delle benzodiazepine e riconosciuto anche da farmaci antagonisti competitivi
(flumazenil), privi di attività intrinseca ma capaci di antagonizzare sia l'azione degli
agonisti che quella degli agonisti inversi. Questo sito, oggi denominato recettore
centrale per le benzodiazepine, ha la capacità di mediare effetti opposti (ansioliticoansiogenico; anticonvulsivante-convulsivante; ipnotico-sonnolitico) quando viene
attivato rispettivamente dagli agonisti o dagli agonisti inversi. Questi effetti opposti
sono dovuti alla modulazione allosterica dell'interazione del GABA col proprio sito di
riconoscimento (facilitazione per gli agonisti, inibizione per gli agonisti inversi) e la
conseguente attivazione o riduzione di attività del canale ionico.
3) Sito di legame per i barbiturici e per il loro antagonista, la picrotossina e siti di
legame per alcuni derivati organofosforici quali, ad es., il t-butilbiciclofosfotionato
(TBPS). I siti di legame per i barbiturici e per il TBPS si trovano all'interno del canale
per lo ione cloro: i barbiturici, al contrario delle benzodiazepine, sono perciò capaci di
indurre influsso di cloro indipendentemente dal legame del GABA con il recettore
mentre la picrotossina ed il TBPS (farmaci ad azione antagonistica rispettivamente
diretta e allosterica sui barbiturici), sono in grado di bloccare la funzione del canale e
produrre effetti farmacologici opposti (ansia, convulsioni). Il recettore GABA A è un
importante sito d'azione anche per molti anestetici generali sia solubili che volatili, per
l'etanolo e per numerosi derivati steroidei. In particolare, questi ultimi composti
sembrano possedere dei siti di legame specifici a livello del canale ionico. I siti di
legame per il GABA, le benzodiazepine, i barbiturici e gli steroidi pur essendo entità
distinte sono legati tra loro in modo funzionale: l'attivazione e l'inibizione di uno di
questi siti da parte di uno specifico agonista o antagonista determina una variazione
nella capacità degli altri siti ad interagire con i propri ligandi specifici con il risultato
finale di una modulazione positiva (facilitazione) o negativa (inibizione) dell'attività del
recettore-canale.
156
(A)
(B)
Fig. 21.3. Schema ipotetico della struttura molecolare dei recettore GABAA. Sono schematizzati due differenti
momenti funzionali dei canale: (A) attivato, (B) inibito. Sono indicate le tre differenti subunità a necessarie
per costituire un recettore funzionalmente sensibile sia all'azione dei barbiturici che delle benzodiazepine. Sulle
subunità e a livello del canale sono riportati i siti recettoriali di differenti modulatori positivi e negativi. In (A)
sono riportati i siti di legame dei modulatori positivi dell'attività del recettore e in (B) quelli negativi capaci di
inibire la funzione dei canale allo ione cloro.
Ad es., l'interazione di una benzodiazepina col proprio sito di legame determina una
modificazione allosterica nella conformazione della subunità che contiene il sito di legame
per il GABA tale da favorirne l'interazione con questo composto e la conseguente apertura del
canale agli ioni cloro. Questo evento determina un aumentato flusso di cariche negative (Cl¯)
all'intemo della membrana postsinaptica con conseguente iperpolarizzazione della stessa. Al
contrario, se un agonista inverso interagisce con il sito di legame per le benzodiazepine, si ha
il fenomeno opposto cioè la subunità che contiene il sito di riconoscimento per il GABA
assume una conformazione «negativa» tale da sfavorirne l'interazione col GABA e quindi
ridurre la capacità di apertura dello ionoforo. E’ importante sottolineare che l'attivazione del
sito di legame per le benzodiazepine favorisce sia l'interazione del GABA col proprio sito che
quello dei barbiturici con il rispettivo sito di legame. Allo stesso modo la presenza di una
molecola di barbiturico sul sito di legame localizzato a livello del canale favorisce il legame
sia del GABA che delle benzodiazepine con i rispettivi siti di riconoscimento. Al contrario sia
le benzodiazepine che i barbiturici sfavoriscono l'interazione dei modulatori negativi della
funzionalità del canale ionico quali la picrotossina ed il TBPS. Questi effetti a livello
molecolare spiegano il sinergismo farmacologico tra benzodiazepine, barbiturici e steroidi.
Il recettore GABAA è formato da più subunità
157
Il recettore GABAA è probabilmente un pentamero costituito da almeno due differenti
subunità polipeptidiche (Fig.3); finora sono state identificate sei isoforme di subunità a (a1
– a6), tre (1 – 3), tre (1 – 3) e una . La molteplicità delle subunità e delle loro
possibili combinazioni e la varietà della loro distribuzione in aree diverse del SNC
suggeriscono che differenti sottopopolazioni di recettori possono controllare funzioni diverse
e presentare differente sensibilità alle molteplici azione dei farmaci ansiolitici e
anticonvulsivanti. In altre parole si sono posti le basi per poter intervenire in modo selettivo
sulle differenti funzioni del sistema gabaergico. Quindi in un prossimo futuro si potrebbe
disporre di farmaci ad alta specificità in grado di agire selettivamente come ansiolitici o
miorilassanti o anticonvulsivanti o ipnotici o antidepressivi.
H3C
R
COOR
N
N
N
O
N
N
N
Benzodiazepina
F
Flumazenil
(antagonista delle BDZ)
F
O
R
O
O
R
O
NN2
N
H
C
Triptofano
N
O
H
CH3
acetaldeide
N
H
CH3
-Carbolina
Le -carboline sono composti endogeni che si formano per reazione fra un estere del
triptofano ed una aldeide, comunemente acetaldeide, che è molto diffusa in circolo soprattutto
nei consumatori di bevande alcoliche.
IMIDAZOPIRIDINE, farmaci ad elevata specificità: ipnoinducenti, facilitando un sonno
molto vicino a quello fisiologico, senza effetti miorilassanti e anticonvulsivanti e con debole
attività ansiolitica.
Cl
CH3
N
N
H3C
N
O
Cl
N
CH2CH2CH3
CH3
Zolpiden
N
CH3
O
N
Alpidem
CH2CH2CH3
158