Corso di Laurea in Fisica
Corso di Struttura della Materia - G. Rinaudo
Nota 4 – Transizioni radiative e spettroscopia
La spettroscopia (breve storia)
Gli inizi
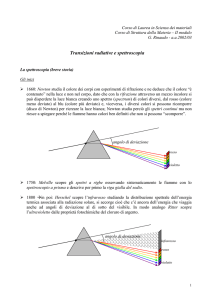
Ø 1660: Newton studia il colore dei corpi con esperimenti di rifrazione e ne deduce che il colore “è
contenuto” nella luce e non nel corpo, dato che con la rifrazione attraverso un mezzo incolore si
può disperdere la luce bianca creando uno spettro (spectrum) di colori diversi, dal rosso (colore
meno deviato) al blu (colore più deviato) e, viceversa, i diversi colori si possono ricomporre
(disco di Newton) per ricreare la luce bianca; Newton studia perciò gli spettri continui ma non
riesce a spiegare perché le fiamme hanno colori ben definiti che non si possono “scomporre”.
angolo di deviazione
rosso
violetto
Ø 1750: Melville scopre gli spettri a righe osservando sistematicamente le fiamme con lo
spettroscopio a prisma e descrive per primo la riga gialla del sodio.
Ø 1800 àin poi: Herschel scopre l’infrarosso studiando la distribuzione spettrale dell’energia
termica associata alla radiazione solare, si accorge cioè che c’è ancora dell’energia che viaggia
anche ad angoli di deviazione al di sotto del visibile. In modo analogo Ritter scopre
l’ultravioletto dalle proprietà fotochimiche del cloruro di argento.
angolo di deviazione
infrarosso
rosso
violetto
Dall’osservazione qualitativa alle misure e classificazioni su base quantitativa
1
Ø 1801: Young calcola per la prima volta una lunghezza caratteristica associabile a ogni colore,
utilizzando vecchi dati di Newton che descrivevano l’associazione fra il colore di pattern
colorati che si osservano su lamine sottili a cuneo e lo spessore locale d della lamina.
2d = differenza di cammino fra il raggio (2) e il raggio (1)
→ si assegna al colore la “lunghezza” λ = 2d
(1)
raggio incidente
(2)
d
Ø 1810 àin poi: Fraunhofer sviluppa nuovi tipi di spettroscopio, basati sul principio del reticolo
a diffrazione; studia così e descrive ben 700 righe identificate nello spettro solare, separandole
in righe chiare (di emissione) e righe scure (di assorbimento); inoltre classifica con lettere
maiuscole le 8 righe più intense (di qui la lettera D assegnata alla riga gialla del sodio, che
venne però riconosciuta come proveniente da transizioni del sodio solo molto più tardi). Altri
contributi a questa classificazione sistematica vengono dati in questo periodo da Brewster e da
Herschel e varie righe vengono via via associate alle diverse sostanze: si delinea così
l’associazione fra lunghezza d’onda della riga e sostanza che la emette e nasce quindi la
spettroscopia come tecnica di riconoscimento delle sostanze chimiche. Restano però dei casi
irrisolti, come quello della riga D che compare negli spettri di assorbimento di moltissime
sostanze a causa della difficoltà di eliminare completamente la presenza di contaminazioni di
sodio nelle sostanze analizzate.
Ø 1848: Foucault risolve il rompicapo della riga D studiando l’assorbimento della luce solare da
parte di vapori di sodio; associa quindi definitivamente la riga D al sodio e dà sicurezza alla
analisi spettroscopica come tecnica di analisi chimica.
Ø 1859: Kirchoff enuncia le leggi della spettroscopia:
- la lunghezza d’onda a cui una sostanza emette dipende unicamente dalla sostanza,
- le lunghezze d’onda a cui si verificano i picchi di emissione di una sostanza coincidono con
quelle a cui si verificano i picchi di assorbimento della stessa sostanza,
- una sostanza trasparente non emette nel visibile.
Kirchoff inoltre:
- spiega le righe scure osservate da Franhofer negli spettri della corona solare come righe di
assorbimento dovute alle stesse sostanze che producono le righe chiare di emissione,
- spiega perché la riga D del sodio si osserva in molti spettri anche di altre sostanze
giustificandola con la presenza diffusa di impurità di sodio,
- inizia a usare la spettroscopia non solo per riconoscere le sostanze in base ai loro spettri ma
anche per scoprire sostanze nuove o proprietà nuove dagli spettri: ad esempio predice
l’esistenza del rubidio estrapolando le caratteristiche ricorrenti in modo regolare degli spettri
del sodio e del potassio (nella tabella periodica di Mendeleiev, il rubidio, Z=37, è infatti il
quarto “metallo alcalino” dopo il litio, Z=3, il sodio, Z=11, e il potassio, Z=19).
Verso la fisica dei quanti
2
Ø 1885: Balmer scopre quasi casualmente la “serie” che porta il suo nome, cioè una relazione
“numerologica” tra le frequenze f delle righe spettrali dell’idrogeno che può essere espressa in
modo proporzionale alla differenza fra gli inversi al quadrato di due numeri interi,
1
1
f = R 2 − 2
n
1 n2
dove R≈ 3 ⋅ 10-15 s-1. È la prima volta che numeri interi vengono associati agli spettri di
radiazione.
Ø 1901: Planck spiega la distribuzione spettrale dell’emissione di corpo nero ipotizzando che
materia e radiazione scambino energia in modo quantizzato, cioè che l’energia scambiata E sia
proporzionale alla frequenza con costante di proporzionalità data dal quanto di azione h, E=hf.
Ø 1905: Einstein interpreta i dati sull’effetto fotoelettrico associando a una radiazione di frequenza
f una energia E data dalla relazione di Planck, E=hf. La radiazione consiste quindi di fotoni
(anche se il termine “fotone” venne coniato molto più tardi), cioè di quanti di energia. I fotoni
trasportano anche quantità di moto, p=E/c, con c = velocità della luce, come atteso dalla
relazione relativistica E2=p2c2+m2c4 per particelle di massa nulla (la verifica sperimentale venne
più tardi con gli esperimenti sull’effetto Compton).
Ø 1908: Ritz enuncia il principio di ricombinazione, secondo il quale a ogni atomo possono essere
associati dei termini spettrali, cioè delle frequenze caratteristiche discrete, tali che tutte le righe
spettrali emesse o assorbite dall’atomo si possono esprimere combinando in tutti i modi tali
frequenze, cioè calcolando le differenze fra i termini spettrali, come appunto avviene nella
relazione di Balmer.
Ø 1913: Bohr ipotizza che l’emissione e l’assorbimento da parte di un atomo sono dovuti a
transizioni fra stati stazionari dell’elettrone, in cui la frequenza della radiazione emessa o
assorbita, e quindi l’energia del fotone associato, è legata alla differenza di energia fra i due stati
dalla legge di conservazione dell’energia.
E2
assorbimento
E2
Eγ + E1 = E2
E1
emissione spontanea
E1
E2 = Eγ + E1
Ø 1917: Einstein ipotizza, sulla base dell’analisi statistica dello spettro di corpo nero, l’esistenza
di un secondo processo di emissione, l’emissione stimolata, cioè l’emissione indotta da un
fotone di energia risonante con la differenza fra le energie dei due livelli: i fotoni “uscenti” dalla
transizione sono perciò due e sono “identici” al fotone incidente
E2
Eγ + E2 = 2 Eγ + E1
emissione stimolata
E1
Ø 1954: Gordon, Zeiger e Townes realizzano per la prima volta un dispositivo che funziona sul
principio dell’emissione stimolata, il “MASER all’ammoniaca”.
3
Transizioni fra livelli
Per indurre la transizione fra due livelli energetici, occorre che la perturbazione abbia due
caratteristiche:
-
sia sintonizzata con il sistema,
abbia il corretto accoppiamento fra radiazione e materia, cioè fra il campo elettromagnetico e
gli operatori in grado di indurre la transizione della funzione d’onda dell’elettrone dallo stato
iniziale a quello finale.
Nella derivazione che segue descriveremo quantisticamente gli stati iniziale e finale
dell’elettrone, mentre descriveremo lo stato del fotone in modo classico, cioè attraverso un campo
elettromagnetico non quantizzato. Scopo della derivazione è calcolare la probabilità di transizione
per unità di tempo fra stato iniziale e finale.
Deriveremo l’espressione della probabilità di transizione solo per l’assorbimento e per
l’emissione stimolata, cioè per processi in cui è già presente inizialmente un campo
elettromagnetico che può produrre la “perturbazione” sullo stato iniziale dell’elettrone tale da
indurre la transizione allo stato finale. Inoltre terremo conto solo della perturbazione indotta dal
r
campo elettrico E , perché è il tipo di interazione più forte. Il campo viene scritto come un’onda
caratterizzata da una pulsazione ω=2πf e da un numero d’onda k=2π/λ =2πf/c=ω/c:
rr
rr
r r
r
r r
r
E ( r , t ) = 2 Eo cos(k ⋅ r − ω t ) = E o (ei( k ⋅ r −ω t ) + e− i( k ⋅ r −ω t ) )
(1)
La perturbazione introdotta dalla presenza del campo elettrico dell’onda elettromagnetica
nella
r
zona dello spazio in cui “si trova” l’elettrone, cioè nei punti di coordinate r tali che la funzione
r
d’onda ψ (r ) dell’elettrone sia sensibilmente diversa da zero, viene descritta come una
hamiltoniana di interazione Hint dipendente dal tempo, proporzionale al campo elettrico, piccola
rispetto agli altri termini che compaiono nella hamiltoniana imperturbata Ho, e che soddisfa alle due
condizioni di sintonizzazione e di accoppiamento sopra citate.
Sintonizzazione
Per la sintonizzazione, occorre che la pulsazione ω del campo elettromagnetico sia prossima alla
differenza di frequenza fra il livello alto e quello basso. Se chiamiamo E2= h ω2 l’energia del livello
alto e E1= h ω1 quella del livello basso, occorre che sia ω ≈ ω2 - ω1. Infatti, se, ad esempio, il
sistema è inizialmente sul livello E1, la sua funzione d’onda ha una evoluzione temporale della fase
del tipo e −iω1t ; applicando una hamiltoniana di interazione che contiene un termine che varia nel
tempo come e −iω t , quale è il primo dei due termini della (1), si ottiene una variazione del tipo
e −i (ω1 + ω )t ≈ e −iω 2t . Tale evoluzione è appunto quella aspettata per uno stato sul livello E2 ed è
conseguenza della conservazione dell’energia nel processo di assorbimento di un quanto del campo
elettromagnetico, cioè di un “fotone” di energia Eγ = h ω, che somma la sua energia all’energia E1
dell’elettrone: E2= E γ+ E 1.
Il secondo termine della (1) permette invece la “sintonizzazione” in un processo di emissione
stimolata da parte di un campo elettromagnetico di pulsazione ω che agisce sullo stato di energia
più elevata E2>E1, la cui funzione d’onda ha una evoluzione temporale della fase del tipo e − iω 2t ;
applicando una hamiltoniana di interazione che contiene un termine che varia nel tempo come
e iω t , quale è appunto il secondo dei due termini della (1), si ottiene una variazione del tipo
e − i(ω 2 − ω )t = e − iω1t , che è appunto l’evoluzione aspettata per uno stato sul livello E1. In termini di
conservazione di energia, ciò è proprio quanto accade nel processo di emissione stimolata causata
4
da un quanto del campo elettromagnetico, cioè di un “fotone” di energia Eγ = h ω, che somma la sua
energia all’energia E2 dell’elettrone dando luogo a due fotoni nello stato finale aventi la stessa
energia, E2+E γ = 2Eγ + E1 (che, semplificando, si riduce a E2 = E γ+ E1).
Accoppiamento
Costruiamo l’accoppiamento per analogia con quanto avviene nell’accoppiamento di un circuito
oscillante LC a un campo elettromagnetico. Anche in questo caso è essenziale la sintonizzazione tra
la frequenza del campo elettromagnetico esterno e la frequenza caratteristica della “materia” che qui
è rappresentata dai parametri caratteristici del circuito elettrico, cioè deve essere LC ≈ ω ' .
Tuttavia la sintonizzazione non basta, perché tutti sappiamo che non ogni circuito LC è un buon
“ricevitore”, cioè si accoppia bene con il campo esterno. Per creare un buon accoppiamento, occorre
che ci sia una “antenna”, che si ottiene deformando la geometria del condensatore in modo che si
adatti al campo elettrico esterno attraverso un dipolo elettrico esteso. L’ampiezza d del dipolo
oscilla nel tempo con una pulsazione ω’=√LC e vale d=qrmax cos(ω’t). È questo dipolo oscillante
che si accoppia con il campo elettrico dell’onda, con una energia di accoppiamento
r
r
r r
r
r
Eacc = qr cos(ω ' t ) ⋅ E = qr cos(ω ' t ) ⋅ Eo cos(k ⋅ r − ωt )
(1’)
E
C
k
rmax
L
B
r r
Nella (1’) la sintonizzazione si ottiene con ω prossimo a ω’, l’accoppiamento si ottiene se r ⋅ Eo
è diverso da zero.
Calcolo della probabilità di transizione
Per analogia, possiamo
r scrivere l’hamiltoniana di interazione come termine di accoppiamento di
dipolo elettrico, dove r diventa ora l’operatore che opera sulla funzione d’onda dell’elettrone:
rr
rr
r r
r r
Hint = er ⋅ E = er ⋅ E o (e i( k ⋅r −ω t ) + e − i( k ⋅r −ω t ) )
(2)
Inseriamo l’hamiltoniana di interazione nell’equazione temporale di Schroedinger:
r
r
∂? ( r , t )
( Ho + H int )? ( r , t ) = ih
∂t
(3)
r
p 2 Ze2
dove H o =
−
è l’hamiltoniana imperturbata e ? (r , t ) è l’intera funzione d’onda
2m
r
dell’elettrone, che include anche la dipendenza dal tempo: non si può infatti usare la funzione
d’onda stazionaria perché l’elettrone non è in uno stato di energia definita dato che sta facendo la
transizione fra lo stato 1 e lo stato 2. Mentre infatti l’antenna descritta prima è un sistema classico,
in cui si poteva, almeno in linea di principio, pensare di misurare, istante per istante, la posizione
del baricentro delle cariche positive e delle cariche negative che oscillano nell’antenna, nulla di
questo può essere fatto per l’elettrone atomico, del quale si conosce solo le autofunzioni che
descrivono gli stati stazionari su cui l’elettrone può trovarsi.
5
r
Scriviamo pertanto ? (r , t ) come sovrapposizione delle due funzioni d’onda temporali
r
r
r
r
? 1 ( r , t ) = ψ 1 (r )e − iE1t / h e ? 2 ( r , t ) = ψ 2 ( r )e − iE2t / h associate alle energie E1 = hω1 ed E2 = hω2 :
r
r
r
? (r , t) = c1ψ1 ( r )e− iω1t + c2ψ 2 ( r )e− iω 2t con c1 2 + c2 2 = 1
(4)
dove ψ1 e ψ2 sono autofunzioni di Ho con autovalori rispettivamente E1 ed E2:
r
r
r
H oψ 1 (r ) = E1ψ 1 (r ) = hω1ψ 1 ( r ) ;
r
r
r
H oψ 2 (r ) = E 2ψ 2 ( r ) = hω 2ψ 2 (r )
(5)
Da notare che la funzione d’onda (4) non è uno stato stazionario, con energia definita, ma è una
sovrapposizione di stati stazionari, ognuno dei quali ha una fase che evolve nel tempo con una
propria frequenza: la sovrapposizione dei due ha non solo la fase ma anche il modulo che evolve nel
tempo, come si può calcolare facilmente, e proprio per questo non è uno stato stazionario.
A questo punto si assume che, in presenza di una hamiltoniana di interazione dipendente dal
tempo, i coefficienti di sovrapposizione c1 e c2 dipendano dal tempo, dato che l’interazione causa
una variazione dell’ampiezza di probabilità di ciascuno dei due stati. Sostituendo la (4) nella (3) si
ottiene:
ih
r −iω t
r −iω t
r
r
∂(c1ψ1 (r )e 1 + c2ψ 2 ( r )e 2 )
= ( H o + H int )(c1ψ1 (r )e − iω 1t + c2ψ 2 (r )e − iω 2t )
∂t
r r
dove abbiamo trascurato la dipendenza spaziale del campo elettrico, contenuta nel termine k ⋅ r che
è molto piccolo: infatti è dell’ordine di 2π<r>/λ, cioè del rapporto fra le dimensioni medie
dell’atomo date da <r> (regione in cui le funzioni d’onda ψ1 e ψ2 sono sensibilmente diverse da
zero, dell’ordine di 10-10m) e la lunghezza d’onda del campo elettromagnetico (che è dell’ordine di
10-6m). Sviluppando i calcoli e tenendo conto delle relazioni (5) si ottiene:
r
∂c
r
r
r
∂c
ih 1 ψ1 (r )e − iω 1t + 2 ψ 2 (r )e − iω 2t − iω1c1ψ1 (r )e − iω 1t − iω 2 c2ψ 2 (r )e − iω 2t =
∂t
∂t
r
r
r
r
hω1c1ψ 1 (r )e − iω1t + hω 2 c2ψ 2 ( r )e − iω 2 t + H int (c1ψ 1 ( r )e − iω 1t + c2ψ 2 (r )e − iω 2t )
e, semplificando i termini eguali nei due membri, si ottiene:
(
r
∂c
r
r
r
∂c
ih 1 ψ 1 (r )e −iω1t + 2 ψ 2 ( r )e− iω 2t = H int c1ψ 1 (r )e − iω1t + c2ψ 2 ( r )e− iω 2 t
∂t
∂t
)
(6)
Se ad esempio interessa conoscere l’evoluzione temporale di c2 a partire da un istante iniziale in
cui il sistema è nello stato (1), moltiplichiamo per il “bra” <ψ2(r)| e integriamo su tutto lo spazio,
tenendo conto delle le condizioni di autonormalizzazione:
r
* r
3
∫ψ 2 ( r , t )ψ 2 (r , t ) d r
=1
r
r
; ∫ψ *2 (r , t)ψ 1 ( r , t )d 3 r = 0
Si ottiene così:
∂c
r
r
r r r
r
ih 2 e − iω 2t =< ψ 2 ( r ) | H int | ψ1 (r ) > e − iω1t = e < ψ 2 (r ) | r ⋅ E o (eiωt + e − iωt ) | ψ 1 (r ) > e − iω1t
∂t
6
dove, a secondo membro, si è posto c1≈1 e trascurato il termine proporzionale a c2, supponendo che
per tutto l’intervallo di tempo to in cui si considererà la transizione l’ampiezza dello stato 2 resti
comunque piccola. Esaminiamo prima la dipendenza dal tempo:
∂c2 e
r r r
r
= < ψ 2 (r ) | r ⋅ Eo | ψ 1 ( r ) > e i(ω 2 − ω1 )t (e iωt + e − iωt )
∂t i h
Conviene introdurre l’elemento di matrice di dipolo elettrico M21 fra gli stati 1 e 2:
r
r r
r
M 21 = < ψ 2 (r ) | er |ψ 1 (r ) >
sostituendo si ottiene:
(7)
r
r
∂c2
M 21 ⋅ E o i(ω 2 −ω1 +ω )t
=
e
+ e i(ω 2 −ω1 −ω )t
(8)
∂t
ih
Per calcolare la probabilità di transizione per unità di tempo, occorre:
• calcolare l’ampiezza c2 all’istante to integrando la (8) fra l’istante t=0 in cui viene “accesa” la
perturbazione e l’istante to,
• calcolare il modulo quadro di c2 e dividerlo per to,
• integrare su tutte le pulsazioni ω del campo elettrico fra 0 e infinito, anche se il maggior
contributo verrà dalle pulsazioni intorno al valore ω21 = |ω2 - ω1|.
I dettagli del calcolo sono dati in appendice. Il risultato è:
(
Γ21 =
4p2
3h
2
)
2
M 21 ρ (ω21 ) = B21ρ (ω 21)
(9)
L’equazione è un esempio di regola d’oro di Fermi, secondo la quale la probabilità di
transizione è proporzionale all’elemento di matrice al quadrato del termine di hamiltoniana di
interazione che collega stato iniziale e stato finale.
Il coefficiente B21 è detto “coefficiente di assorbimento di Einstein”.
È chiaro che la stessa equazione descriverebbe l’emissione indotta, cioè la probabilità di
transizione per il processo inverso, il passaggio dallo stato 2 allo stato 1: basta scambiare, in tutte le
equazioni a partire dalla (6) lo stato 2 con lo stato 1. Il risultato che si ottiene è lo stesso, dato che
nella (9) |M21|2=|M12|2 (e anche B21 = B12): infatti lo stesso elemento di matrice di dipolo elettrico
induce la transizione dallo stato 2 allo stato 1 oppure la transizione opposta.
Questo è il cosiddetto principio del bilancio dettagliato.
Emissione spontanea
Si calcola per analogia al calcolo classico di emissione di onde elettromagnetiche da una antenna, in
cui la potenza irradiata è proporzionale al quadrato del momento di dipolo elettrico e alla frequenza
1 ω 4 (qz )2
). Interpretando
4πε o 3c 3
l’emissione in modo quantizzato, cioè calcolando il numero di fotoni emessi nell’unità di tempo alla
frequenza ω21 corrispondente alla differenza di energia fra i due livelli, e tenendo conto che ogni
fotone ha una energia hω 21 , si ottiene la probabilità di emissione spontanea nell’unità di tempo:
alla quarta (vedi, ad esempio, Costa e Predazzi, XV.20, W (ω ) =
3
2
spont 4ω 21
Γ21 =
M 21 = A21
3
(9’)
3hc
dove A21 è chiamato il “coefficiente di Einstein dell’emissione spontanea”.
7
Le regole di selezione
Come si vede dalla (9) la probabilità di transizione, a parità di
intensità dell’onda elettromagnetica, è proporzionale |M21|2. Per
calcolare M21 scriviamo esplicitamente i tre operatori contenuti
nell’espressione data in equazione (7), proporzionali alle componenti
r
rx, ry, rz dell’operatore r , che, scritti in coordinate polari, sono:
rx = r sinθ cosϕ = r sinθ (e iϕ + e -iϕ)/2
ry = r sinθ sinϕ = r sinθ (e iϕ - e -iϕ)/2
rz = r cosθ
z
θ
d3r
r
y
ϕ
x
Esaminiamo per primo l’effetto di rz; supponiamo ad esempio che lo stato 1 sia lo stato 1s
r
dell’idrogeno, per cui ψ 1( r ) = R10Y00 . La funzione d’onda, che rappresenta l’ampiezza di
probabilità di trovare l’elettrone nel volumetto d3r intorno al punto di coordinate r, θ e ϕ, dipende
solo dalla distanza radiale e non dagli angoli θ e ϕ. Applicando allo stato l’operatore rz si ottiene:
r
rzψ1( r ) = rR10 (r )Y00 cos θ
che, chiaramente, non è più uno stato indipendente dai valori di θ e di ϕ, ma ha una dipendenza da θ
e ϕ uguale a quella della funzione sferica Y10 ; si può quindi dire che rz “opera” la trasformazione di
uno stato s in uno stato p avente ml=0. In modo analogo si può calcolare come opererebbe su uno
stato con un l qualunque, ad esempio l=1:
3
1 1
r
rzψ n ,1,0 (r ) = rRn1 (r ) Y10 cosθ = CrRn1 (r ) cos 2 θ = C ' rRn1 (r ) cos 2 θ − +
2 2
2
(
= C ' ' rRn1 (r ) C 2Y20 + C 0Y00
)
Come si vede, rz trasforma lo stato con l=1 in una sovrapposizione di stati con l=2 oppure l=0,
ma sempre con ml=0. In generale, l’operatore rz opera quindi una trasformazione che cambia di una
unità il numero quantico l di momento angolare orbitale mentre lascia invariato il numero quantico
ml, quindi il corrispondente elemento di matrice sarà diverso da zero solo se valgono le regole:
∆l = ± 1 ; ∆ml = 0
(10)
Per calcolare l’elemento di matrice corrispondente, scriviamo esplicitamente la (7) per la
componente z, chiamando n, l, ml i numeri quantici dello stato 2 ed n’, l’, ml’ i numeri quantici dello
stato 1:
∞
1 2π
0
−1 0
r
r
m *
m
M 21, z = < ψ 2 (r ) | erz | ψ1 (r ) >= e ∫ Rnl (r ) Rn 'l ' (r )r 3 dr ∫ ∫ Yl l cos θ Yl ' l ' d cos θ dϕ
L’integrale sugli angoli θ e ϕ è diverso da zero solo se valgono le regole date nella (10), che
sono quindi “regole di selezione” della transizione indotta da rz. Non c’è invece nessuna regola di
selezione particolare che riguardi il numero quantico n, tuttavia il valore dell’integrale sulla
variabile r può essere più o meno grande a seconda delle funzioni Rnl coinvolte e, di conseguenza, la
transizione può essere più o meno probabile e la relativa riga spettrale più o meno intensa.
In una rappresentazione in termini di diagrammi di Grotrian, le transizioni sono quindi sempre
rappresentate da “frecce” che si spostano lateralmente verso destra o verso sinistra di una unità di
momento angolare l:
8
E (eV)
4
-0.85
3
-1.5
2
-3.4
rz
rz
rz
1 -13.6
0
n
-1
0
+1
-2
-1
0
0
1
2
s
p
d
+1
ml
+2
l
In modo analogo si calcola come operano gli operatori rx o ry. Applicando, ad esempio, rx allo
stato 1s, si ottengono gli stati p con ml = ±1:
r
rxψ 1 (r ) = rR10 (r )Y00 sen θ (e iϕ + e − iϕ ) = CrR10 ( r )(Y11 + Y1−1 )
Applicando rx allo stato con l e ml qualunque, si ottiene una sovrapposizione di stati con l e ml
variati di una unità in più o in meno. Conviene definire gli operatori
r+ = (rx+ iry)/2 = r eiϕ
r- = (rx- iry)/2 = r e-iϕ
che variano rispettivamente di +1 e di –1 il valore di ml e di ±1 il valore di l, dando quindi origine a
ben precise transizioni. Come per rz, non ci sono regole che impongano una variazione precisa del
numero quantico radiale n, tuttavia la variazione di n va a influire il valore dell’integrale su r e
quindi il valore di M12.
Alcune transizioni indotte da questi operatori sono mostrate nel seguente diagramma di Grotrian.
9
E (eV)
4
-0.85
3
-1.5
2
-3.4
rz
r-
rz
r-
rz
r-
r+
r+
r+
1 -13.6
0
n
-1
0
-2
+1
-1
0
0
1
2
s
p
d
+1
ml
+2
l
Nessuno dei tre operatori r+, r-, rz può invece indurre variazioni nel valore di ms, perché nessuno
contiene operatori che possano operare sugli spinori, quindi ms si conserva nella transizione.
Riassumendo, le regole di selezione sono:
∆l = ± 1
∆ml = 0, ± 1
∆ms = 0
Regole di selezione nella rappresentazione |n, l, j, mj>
Come opera il dipolo elettrico nella rappresentazione |n, l, j, mj>? Le regole di selezione per i
numeri quantici j, mj sono: ∆j = 0, ± 1; ∆mj = 0, ± 1. Complessivamente, le regole sono quindi:
∆l = ± 1
∆ml = 0, ± 1
∆ms = 0
∆j = 0, ± 1
∆mj = 0, ± 1
(11)
10
Possiamo vedere, su un esempio, come queste nuove regole rientrino nelle precedenti.
Esaminando una transizione fra uno stato d e uno stato p. Secondo le regole ∆l = ± 1 e ∆ml = 0, ±
1 tutte le transizioni sono permesse, mentre secondo la regola ∆j = 0, ± 1 sono proibite tutte le
transizioni fra stati d5/2 e stati p1/2 perché comporterebbero ∆j = 2. Il motivo diventa chiaro se si
esamina la transizione indicata in figura fra lo stato d5/2 avente mj =-5/2 e lo stato p1/2 con mj =-1/2.
Guardando i contenuti in ml e ms dei due stati, si vede infatti che la transizione non può andare nello
stato p1/2 | -1↑> perché occorrerebbe invertire lo spin e quindi violare la regola ∆ms=0, ma non può
neppure andare nello stato | 0 ↓> perché occorrerebbe variare ml di +2. Poiché uno degli stati d5/2
non può transire ad alcun stato p1/2, la transizione è proibita anche per tutti gli altri stati d5/2 aventi
diverso mj, perché l’effetto fisico non può dipendere dalla scelta dell’asse di quantizzazione 1, cioè
dal valore di mj.
Nello stesso modo possiamo discutere perché sono invece permesse le transizioni fra stati con lo
stesso j, come quella indicata in figura fra uno stato d3/2 e uno stato p3/2, perché è immediato
controllare che può avvenire nel rispetto delle regole ∆ml = 0, ± 1 e ∆ms=0.
d5/2
-5/2
-2 ↓
d3/2
-3/2
-1/2
-1 ↓
-1 ↑
0↓
0↑
+1 ↓
mj
-1/2
+1/2
ml ms
ml ms
-1 ↑
0↓
0↑
+1 ↓
mj
ml ms
ml ms
+1/2 +3/2
+1/2 +3/2 +5/2
mj
0↑
+1 ↓
ml ms
ml ms
-3/2
-1/2
-2 ↑
-1 ↓
-1 ↑
0↓
-3/2
-1/2
+1/2 +3/2
mj
-2 ↑
-1 ↓
-1 ↑
0↓
0↑
+1 ↓
ml ms
ml ms
+1 ↑
+2 ↓
+2 ↑
+1 ↑
+2 ↓
p3/2
+1 ↑
p1/2
Appendice: dettagli del calcolo della probabilità di transizione
Partendo dall’equazione (8):
r
r
∂c2
M 21 ⋅ Eo i (ω 2 −ω1 + ω ) t
=
e
+ ei (ω 2 − ω1 −ω )t
∂t
ih
(
)
(A1)
per calcolare l’ampiezza c2 all’istante to occorre integrare fra l’istante t=0 in cui viene “accesa” la
perturbazione e l’istante to,
r
r t
r
r
− +
− −
M 21 ⋅ E o i(ω −ω + ω )t
M 21 ⋅ Eo ei(ω2 ω1 ω )to − 1 ei (ω2 ω1 ω )to − 1
i (ω 2 −ω 1 −ω )t
2
1
c2 (to ) =
(e
+e
)dt =
+
(ω2 − ω1 ) + ω
ih
ih
(ω 2 − ω1 ) − ω
∫
0
r
r i(ω 21 + ω )to / 2 i(ω +ω )t / 2
i (ω 21 − ω ) to / 2
o
− e − i(ω 21 +ω )to / 2 ) e
M 21 ⋅ Eo e
(e 21
(ei (ω 21 −ω )to / 2 − e −i (ω21 −ω )to / 2
=
+
ih
(ω2 − ω1 ) + ω
(ω 2 − ω1 ) − ω
dove si è utilizzata la notazione semplificata ω21=ω2 - ω1. Dei due termini, solo il secondo è
sensibilmente diverso da zero, dato che ω21 è positivo e consente di realizzare la situazione di
1
Per gli altri stati d5/2 la verifica non è così diretta, occorre scrivere per intero gli stati iniziale e finale, tenendo conto
dei coefficienti di Clebsch-Gordan e dell’espressione delle funzioni sferiche Ylm .
11
“sintonizzazione” sopra discussa, che si verifica quando ω ≈ ω21. Teniamo solo il secondo termine e
calcoliamo il modulo:
r
r
iM 21 ⋅ Eo ei (ω2 −ω1 −ω )to / 2 (sen ω2 − ω1 − ω )to / 2
c2 ( t o ) =
(ω2 − ω1 ) − ω
ih
r
r
M 21 ⋅ Eo sen( ω2 − ω1 − ω )to / 2 to
=
h
[(ω2 − ω1 ) − ω ]to / 2 2
da cui ottieniamo la probabilità di transizione per unità di tempo Γ21 fra i due stati:
2 r 2
c2 (to ) 2 M 21 Eo
Γ21 =
=
to
3h 2
2
sen( ω 2 − ω1 − ω )to / 2
to
[(ω2 − ω1 ) − ω ]to / 2
(A2)
dove il fattore 1/3 deriva dalla media fra i valori di cos2(θME), θME essendo l’angolo fra la direzione
r
del momento di dipolo elettrico atomico e il campo Eo .
Quale è il significato del tempo to che compare nella (A2)? È legato alla larghezza di banda del
campo Eo: il campo infatti non è mai rigorosamente monocromatico, ma ha una certa distribuzione
in frequenza e la banda di frequenze di cui tenere conto è, per il principio di indeterminazione,
inversamente proporzionale a to. Per fare il calcolo, si tiene conto che l’energia immagazzinata nel
campo è, per unità di volume, pari a Eo2 / 2 p (nel sistema di unità di Gauss) e quindi, esprimendo
Eo2 come l’integrale su tutte le frequenze della densità di energia elettromagnetica ρ(ω)
nell’intervallo di pulsazione dω, si ottiene:
Eo2
∞
= 2π ∫ ρ (ω ) dω
(A3)
0
Sostituendo nella (A2), si dovrà includere nell’integrazione anche tutta la dipendenza dalla
frequenza contenuta nel termine in parentesi, che anzi è molto più forte della dipendenza di ρ(ω):
2
2
2
∞
2
M 21 2π ∞ sen(ω 2 − ω1 − ω )to / 2
M 21 4π
sen u
Γ21 =
∫ (ω 2 − ω1 − ω )to / 2 to ρ (ω ) dω = 3h 2 ρ (ω 21) ∫ u du
3h 2
0
0
Nell’ultimo passaggio si è introdotta la variabile adimensionale u=(ω2 - ω1 - ω)to/2 e si è estratto il
valore di ρ(ω) per ω=ω2 -ω1, dato che l’integrando tende alla funzione δ di Dirac per u tendente a 0.
Poiché l’integrale vale π, si ottiene la relazione finale:
Γ21 =
4p2
3h
2
2
M 21 ρ (ω21 ) = B21ρ (ω 21)
(A4)
12