Anno Accademico 2011-12
A. CERIOTTI
1
2
Addotto Trifenilfosfina-Borano, H3B PPh3
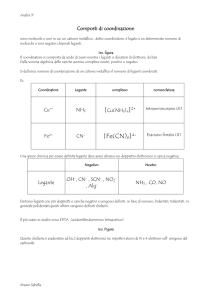
GENERALITA’ SUGLI ADDOTTI CON IL BORANO
Un addotto è un prodotto ottenuto dall’interazione tra una base di Lewis
(specie donatrice di un doppietto elettronico) (trifenilfosfina) e un acido di
Lewis (specie accettrice di un doppietto elettronico) (borano) (Figura 1), in
cui le due specie molecolari mantengono una propria identità.
H3B P(C6H5)3
Il B (Gruppo 13) ha 3 elettroni di valenza e 4 orbitali disponibili quindi i
composti BR3 sono elettronicamente insaturi e tendono a raggiungere la
saturazione in vari modi, comportandosi da acidi di Lewis, mediante a)
un’interazione
intramolecolare
(diborano)
oppure
b)
una
reazione
intermolecolare con basi di Lewis (addotti).
La reazione con basi comporta un processo di cambiamento di
ibridizzazione sull’atomo di boro, da sp2 a sp3 e di geometria intorno all’atomo
di boro (Figura 2).
Le basi (L) che si possono sommare all’acido di Lewis sono diverse: H-,
N(CH3)3, NH3, P(C6H5)3, CO.
Prendendo in esame l’addotto trifenilfosfina-borano occorre ricordare che
il P (Gruppo 15) ha 5 elettroni di valenza e 4 orbitali disponibili, quindi i
composti PR3 sono composti saturi dal punto di vista elettronico e presentano
una coppia di elettroni non condivisa sull’atomo di fosforo in ibridizzazione
sp3. Questi composti si comportano da basi di Lewis e la loro basicità può
essere modulata facendo variare la natura del radicale R (R = alchile, arile)
(Figura 3).
Un esempio di altro addotto è quello tra borano e monossido di carbonio
(detto carbonile-borano), isostrutturale e isoelettronico con CO2 (lineare)
3
(Figura 4). Questo composto presenta la stessa reattività chimica del
biossido di carbonio, ad esempio:
H3B(-)-C(+)O
+
2 OH-
[H3BCO2]2-
+
H2O
L’addotto monossido di carbonio-trialchilborano non è stabile ma evolve a
dialchil-acil borano tramite una reazione di inserzione del CO nel legame B-R
(di solito le inserzioni avvengono sui metalli di transizione).
4
SINTESI E CARATTERIZZAZIONE
La preparazione dell’addotto trifenilfosfina-borano in laboratorio prevede
l’ossidazione di [BH4]- con I2 che genera la specie BH3. Questa specie è
instabile e deve essere “catturata” da un opportuno legante. Questo è il
motivo per cui l’ossidazione è compiuta in presenza di PPh3, (Figura 5).
(reazione redox)
[BH4]- +
1
I2
2
BH3 + I- +
BH3 + PPh3
1
H2
2
H3B PPh3
L’addotto è solubile in solvente tetraidrofurano (THF) ed è di colore
bianco, ossia non assorbe radiazione elettromagnetica nella lunghezza
d’onda dello spettro visibile.
Durante la sintesi devono essere osservate le seguente precauzioni:
1) occorre dare tempo al borano di coordinarsi alla trifenilfosfina,
2) è richiesto, all’inizio della reazione, un ambiente anidro per evitare:
[BH4]- + H2O
[BH3(OH)]- + H2
3) occorre lavare il grezzo di reazione con H2O, per eliminare I-, e con
toluene, per sciogliere l’eccesso di trifenilfosfina.
Per caratterizzare il composto possiamo:
-
determinare il P.F. (relativamente basso, 189°C, essendo un
composto molecolare),
-
registrare uno spettro I.R. (bande a
2240 cm-1,
-
B-P
B-H asymm
= 2370 cm-1,
B-H symm
=
= 608 cm-1),
registrare uno spettro NMR multinucleare; infatti contiene quattro
nuclei NMR attivi: 1H,
osserva il
11
11
B,
10
B e
31
P. Nel boro generalmente si
B che ha una abbondanza isotopica naturale più elevata
e uno spin più favorevole (molteplicità di spin inferiore) (Figura 6).
5
Nucleo
Abbondanza %
1
100
½
2s + 1 = 2
H
Spin
Molteplicità
10
B
18
3
2s + 1 = 7
11
B
32
3/2
2s + 1 = 4
31
P
100
½
2s + 1 = 2
In Figura 7 è riportato lo spettro NMR del 31P (a) disaccoppiato da 1H, b)
accoppiato con 1H) dell’addotto H3P-BCl3, analogo al composto sintetizzato.
In a) ci sono 4 linee intense (80& circa dell’area totale) (accoppiamenti 11B31
P) e 7 linee deboli (20%) (accoppiamenti 10B-31P).
In b) ogni linea dello spettro a è scomposta in un ulteriore quartetto
(accoppiamenti 1H-31P).
UTILIZZO
L’addotto trifenilfosfina-borano viene impiegato nelle reazioni in cui è
necessario disporre facilmente del borano, (Figura 8), ad esempio nella
reazione di idroborazione (anti-Markovnikov), ossia aggiunta del frammento
B-H ad un doppio legame:
Ph3PBH3 + 3 RCH=CH2 + CH3I
[Ph3PCH3]+I- + (RCH2CH2)3B
6
GENERALITA SUL DIBORANO
Il boro forma con l’idrogeno una serie di composti detti borani, il cui studio
ha assunto una notevole importanza teorica e pratica.
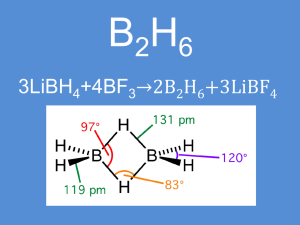
Il diborano, B2H6 , p.e. –92,6°C, che è il più semplice dei borani, si
prepara nel modo migliore per reazione tra l’addotto boro trifluorurodietiletere, Et2O, e l’idruro di litio (Figura 9):
(reazione acido-base)
8 BF3 Et2O
+
6 LiH
B2H6
+
6 LiBF4
+
8 Et2O
Il diborano è stabile in ambiente inerte ma reagisce con ossigeno e con
acqua. La disposizione degli atomi di idrogeno nel diborano è molto diversa
da quella presente nell’etano, C2H6; infatti la struttura elettronica dei due
composti non può essere la stessa in quanto B2H6 non è isoelettronico con
C2H6, ma possiede 2 elettroni in meno. In questo composto non c’è un
legame diretto tra i due atomi di boro e due atomi di idrogeno sono a ponte
tra essi (Figura 10). Non si tratta però di un legame a idrogeno nel senso
classico, in quanto la bassa elettronegatività del boro esclude questa
possibilità. In questo caso, al posto del legame covalente usuale, formato da
2 orbitali su 2 centri e 2 elettroni (2c-2e-), si ha un legame delocalizzato su 3
centri B…H…B, formato da 3 orbitali (2 del tipo sp3, su ciascun atomo di B, e
1 del tipo s, sull’atomo di idrogeno a ponte) e 2 elettroni (3c-2e-) (Figura 11).
In B2H6 gli 8 atomi sono legati da 12e- anziché 16e- pertanto non sono
possibili legami a 2c-2e- tra essi; si formano, oltre a 4 legami a 2c-2e- anche 2
legami a 3c-2e- (Figura 12). La conseguenza è che nella molecola abbiamo
due tipi di legame B-H con diverso ordine di legame:
O.L. B-Hter = 1 ;
O.L. B-Hpon = 0,5
e una diversa distanza di legame B-H (Figura 13).
Il processo di formazione di B2H6 a partire da BH3 è termodinamicamente
favorito (Figura 14):
2 BH3(g)
B2H6(g)
7
H° = -146 kJ / mole
S° = -209 J / mole K
G° = H° - T S° = -146 + 62 = -84 kJ / mole
G° = -2,3RTlogK
-84000 = -2,3x8,31x298logK
K = 5x1014
B2H6, essendo deficiente di elettroni, è elettrofilo e può sommare l’anione
idruro, presente negli idruri dei metalli, ad esempio:
B2H6
+
2 NaH
2 Na[BH4]
Questa reazione porta alla formazione del tetraidroborato di sodio, Na[BH4],
detto commercialmente sodio boro-idruro, [BH4]-.
8
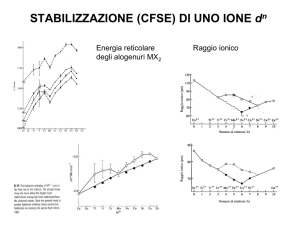
ACIDITA’ DEGLI ALOGENURI DI BORO
Acidi di Lewis molto importanti sono gli alogenuri di boro di formula BX3
(X=F, Cl, Br, I). Una serie di reazioni omologhe tra i trialogenuri di boro e una
stessa base di Lewis ha mostrato che l’acidità di Lewis per questi composti è
nell’ordine BI3 > BBr3 > BCl3 > BF3; tale sequenza è stata individuata da
misure termodinamiche dei calori di formazione degli addotti di formula
generale X3B L.
BX3 + L X3B L
H° < 0
Questo ordine di acidità è inverso a quello prevedibile considerando le
elettronegatività e le dimensioni degli alogeni. Infatti, l’alta elettronegatività
del fluoro dovrebbe rendere la molecola di BF3 più adatta ad acquistare una
coppia di elettroni da parte di un potenziale legante bielettronico e le piccole
dimensioni del fluoro dovrebbero abbassare tutte le barriere legate ad
impedimenti sterici, Figura 15.
I composti BX3 sono dei monomeri con struttura trigonale planare dove
l’atomo di boro è ibridizzato sp2; l’angolo X-B-X = 120°. Gli altri alogenuri
covalenti del Gruppo 13, EX3 (E=Al, Ga, In) sono composti dimerici.
Questo diverso comportamento è spiegabile con il fatto che l’atomo di
boro è troppo piccolo e gli alogeni sono troppo grandi per formare strutture
stabili come quelle richieste da una struttura dimerica, Figura 16.
Gli alogenuri di boro sono monomeri anche per ragioni elettroniche e la
loro diversa acidità è spiegabile con interazioni di legame multiplo. Il boro
raggiunge una configurazione ottezziale mediante interazioni multiple con gli
alogeni usando l’orbitale pz, di adatta simmetria, per sovrapporsi con gli
analoghi orbitali pz pieni degli alogeni, portando alla formazione di un parziale
carattere di doppio legame , Figura 17.
9
Tale interazione sarà tanto più forte quanto più efficacemente possono
sovrapporsi gli orbitali p. Nel caso del legame B-F, che coinvolge gli orbitali
(2p-2p), si ha una sovrapposizione più efficace di quella (2p-3p) del legame
B-Cl che a sua volte è migliore di quella (2p-4p) del legame B-Br che a sua
volte è migliore di quella (2p-5p) del legame B-I.
La conseguenza di questo è che in BF3 c’è un maggior contributo della
struttura con legame multiplo di quanto non ci sia in BCl3, BBr3 e BI3. Il
maggior contributo della forma con un doppio legame nel derivato di fluoro,
nella descrizione della molecola BF3, trova conferma sia da calcoli sia da dati
sperimentali.
Quanto BX3 agisce da acido di Lewis, deve passare da una geometria
trigonale planare a tetraedrica, rompendo la componente
del legame B-X,
un processo che richiede un certo dispendio energetico. Questo dispendio di
energia è maggiore in BF3 che non in BI3: questo è il motivo per cui BF3 è un
acido più debole di BI3.
STRUTTURE DI DERIVATI DI BORO
E’ utile confrontare tra loro le strutture dei derivati di formula BH3, BX3,
B(CH3)3.
La forma stabile di BH3 è il dimero B2H6. I motivi di questo sono stati
ampiamente discussi; riassumendo possiamo affermare che il composto
dimerizza per tentare di raggiungere una configurazione di Lewis
soddisfacente e non ci sono ingombri sterici nella formazione della molecola.
I derivati di formula BX3 e B(CH3)3 sono monomeri. BX3 non dimerizza per
guadagnare elettroni in quanto questi gli sono forniti dai doppi legami B-X;
oltre a questo la reazione di dimerizzazione è impedita per ingombro sterico.
B(CH3)3 non dimerizza per ingombro sterico e recupera elettroni mediante
iperconiugazione tra C-H e B, Figura 18.
10
11
Tris(acetilacetonato)Manganese(III), [Mn(acac)3]
E’
chiamato
anche
tris(2,4-pentandionato)
oppure
tris( -
dichetonato)manganese(III).
E’ un composto di coordinazione, formato da un atomo, circondato da vari
altri atomi, ioni o piccole molecole, chiamati leganti. I composti di
coordinazione, detti anche complessi, hanno un numero di coordinazione
superiore al numero di ossidazione dell’atomo centrale.
Il tris(acetilacetonato)manganese(III) è un composto neutro molecolare,
con tre leganti monoanionici intorno al Mn in stato di ossidazione formale +3;
il metallo ha numero di coordinazione sei, poiché i tre leganti sono bidentati,
ossia hanno due denti per legarsi al metallo. L’anione acetilacetonato è
coordinato al metallo tramite due atomi di ossigeno, ossia è un chelante.
Poichè la parte esterna del complesso consiste di gruppi organici il
composto è idrofobico, ossia insolubile in acqua, ma è solubile in solventi
organici. Il legante anionico si forma per deprotonazione del 2,4-pentadione o
acetilacetone (abbreviato acacH). In presenza di una base acacH perde
facilmente
un
protone
per
formare
l’anione
2,4-pentandionato
o
acetilacetonato (abbreviato acac) Figura 20.
Il legante neutro presenta tautomeria cheto-enolica.
Nella preparazione del complesso l’ambiente basico è fornito dall’idrolisi
dello ione acetato, aggiunto come acetato di sodio.
Ciascun legante dà con il metallo un anello a sei termini.
La formazione di un composto chelato è favorita dal fattore entropico,
perchè l’impiego di un chelante libera un maggior numero di molecole in
soluzione aumentando l’entropia del sistema.
Tre leganti acetilacetonato sono in coordinazione ottaedrica intorno allo
ione metallico centrale. La struttura del complesso è mostrata in Figura 21.
12
Nella sintesi si utilizza una reazione di coproporzionamento tra Mn(II) e
Mn(VII) per formare in situ Mn(III) che viene stabilizzato per complessazione,
Figura 19:
4 Mn2+ + MnO4- + 15 acacH + 7 B
5 [Mn(acac)3] + 7 BH+ + 4 H2O
(B = base)
Il composto è di colore marrone.
Durante la sintesi devono essere osservate le seguente precauzioni
(Figura 22):
1) occorre aggiungere lo ione acetato in due porzioni successive perché
dapprima si forma il bis(acetilacetonato)manganese(II) che
successivamente è ossidato dal MnO4-, e complessato dal rimanente
acac, al tris(acetilacetonato)manganese(III),
2) occorre dare tempo all’acetilacetonato di coordinarsi al Mn, e non
aggiungere il permanganato troppo rapidamente perché durante questa
ossidazione si forma, come sottoprodotto, MnO2,
3) occorre lavare il grezzo di reazione con H2O, per eliminare i sali solubili,
4) occorre ricristallizzare il prodotto mediante solubilizzazione in toluene (a
freddo) e precipitazione con etere di petrolio.
Per caratterizzare il composto possiamo:
- registrare uno spettro I.R. (bande a
C-O =
1588 cm-1,
C-C =
1512 cm-1),
- misurare la suscettività magnetica.
COMPORTAMENTO MAGNETICO DEL MANGANESE(III)
Il manganese, la cui configurazione elettronica allo stato fondamentale è
[Ar]3d54s2, forma lo ione Mn3+ che, sempre allo stato gassoso, ha
configurazione elettronica [Ar]3d4.
13
Secondo la teoria del campo cristallino, sei leganti disposti intorno allo
ione Mn3+ in coordinazione ottaedrica provocano la separazione dei cinque
orbitali 3d del Mn in due gruppi, l’uno, denominato t2g, a energia minore,
comprendente tre orbitali degeneri, e l’altro, denominato
eg, a energia
maggiore, comprendente due orbitali degeneri.
La configurazione d4 può presentare una configurazione elettronica ad
alto spin (t2g)3(eg)1 (campo debole, quattro elettroni spaiati) o a basso spin
(t2g)4 (campo forte, due elettroni spaiati).
La misura della suscettività magnetica permette di ricavare il momento
magnetico del complesso dovuto allo spin degli elettroni spaiati e tale misura
permette di evidenziare che il complesso [Mn(acac)3] ha quattro elettroni
Figura 23.
Una configurazione d4 ad alto spin presenta l’effetto Jahn-Teller, ossia
un’ulteriore separazione energetica (perdita di degenerazione) degli orbitali eg
dovuta ad un riempimento elettronico non omogeneo. La perdita di
degenerazione abbassa l’energia del sistema e si traduce in una distorsione
della geometria molecolare.
14
AROMATICITÀ DELL’ANELLO METALLO-ACETILACETONATO
Gli anelli metallo-acetilacetonato danno facilmente reazioni di sostituzione
elettrofila. La reazione di sostituzione generalmente avviene sull’atomo di
idrogeno legato al carbonio metinico posto tra i due gruppi carbonilici. Queste
reazioni sono analoghe a quelle sugli anelli aromatici (bromurazione,
iodurazione, nitrazione, acetilazione ecc.), Figura 24
[Mn(CH3C(O)CHC(O)CH3)3] + 3 E+
[Mn(CH3C(O)CEC(O)CH3)3] + 3 H+
Il comportamento aromatico dell’anello a sei termini deriva dalla presenza
di un sistema delocalizzato di sei elettroni
cui partecipano cinque orbitali pz
degli atomi di carbonio e di ossigeno, ibridizzati sp2, e un orbitale dxz o dyz del
metallo, Figura 25.
Per il conteggio elettronico il legante può essere considerato anionico,
donatore di quattro elettroni, oppure neutro, donatore di tre elettroni. Pertanto
il numero di elettroni di valenza nel composto di Mn sarà 16e- Figura 26:
Legante anionico, metallo positivo Mn3+ (d4)
4 e- (Mn3+) + 3 x 4 e- (acac) = 16 e-
Legante neutro, metallo neutro Mn0 (d7)
7 e- (Mn0) + 3 x 3 e- (acac) = 16 e-
15
COMPLESSI TRIS-CHELATI CON GEOMETRIA OTTAEDRICA
Con metalli trivalenti l’acetilacetone forma complessi tris-chelati neutri, ad
esempio [Al(acac)3], [Ti(acac)3], [Cr(acac)3] e [Co(acac)3].
Come risultato delle formule di risonanza, le due distanze M-O in ogni
anello sono uguali, e altrettanto lo sono le distanze C-O e C-C dando una
struttura simmetrica, Figura 27.
I complessi tris-chelati, in geometria ottaedrica, esistono in due forme
isomeriche (isomeri ottici o enantiomeri, poiché si comportano diversamente
quando attraversati da un fascio di luce polarizzata). E’ possibile effettuare
misure polarimetriche o di dicroismo circolare sui singoli isomeri ottici.
Senza accorgimenti la sintesi dà luogo ad una miscela equimolare
(racema) dei due isomeri ottici che possono essere separati mediante
particolari tecniche (risoluzione del racemo).
Nel caso del complesso [Mn(acac)3], possono essere separati mediante
l’utilizzo di una colonna cromatografia avente un supporto otticamente attivo.
Una molecola esiste in due isomeri ottici se è dissimmetrica (priva di assi
impropri Sn, S2 centro di inversione, S1 piano di simmetria), Figura 28.
Nel caso di [Mn(acac)3] la molecola ha 1 asse C3 e 3 assi C2 ossia ha
gruppo puntuale D3 .
Con metalli trivalenti l’ossalato forma complessi tris-chelati anionici, ad
esempio [Cr(C2O4)3]3- , [Al(C2O4)3]3-.
La struttura del complesso tris(ossalato)cromato(III), [Cr(C2O4)3]3-, è
mostrata in Figura 29, in cui sono presentati i due isomeri ottici dell’anione.
Nel caso di tali complessi i due isomeri ottici possono essere separati
mediante l’utilizzo di un catione otticamente attivo, ad esempio (+)[Co(en)3]3+,
ossia preparando due diastereoisomeri aventi diversa solubilità.
16
17
Tetra(acetonitrile)rame(I) esafluorofosfato,
[Cu(CH3CN)4][PF6]
Una categoria importante di complessi di rame(I) è quella con leganti
azotati, tra cui i nitrili (es. acetonitrile) e le fenantroline (es. 1,10-fenantrolina,
detta ortofenantrolina).
Il tetra(acetonitrile)rame(I) esafluorofosfato è un sale la cui parte cationica
è rappresentata da un complesso nitrilico di Cu(I), [Cu(CH3CN)4]+, e la cui
parte anionica è rappresentata da un complesso fluorurato di P(V), [PF6]-.
L’acetonitrile è una molecola lineare che, quando funziona da legante,
dona 2e- al metallo tramite il doppietto sull’atomo di azoto:
N C-CH3
La sintesi del complesso è una reazione di coproporzionamento tra un
sale di Cu(II), ossia il carbonato basico rameico, e Cu(0), rame metallico,
(Figura 30):
CuCO3 Cu(OH)2 + 2 Cu + 4 HPF6 + 16 CH3CN
4 [Cu(CH3CN)4][PF6] + 3 H2O + CO2
Lo ione [Cu(CH3CN)4]+ ha struttura tetraedrica con 4 legami Cu-N. Lo ione
Cu+ , in configurazione elettronica d10, e il legante non assorbono nel visibile,
quindi il complesso è di colore bianco. Il trasferimento di carica Cu NCCH3
avviene per assorbimento di radiazione nell’ultravioletto.
La tonalità azzurra che a volte è presente è dovuta alla presenza dello ione
Cu2+, poiché il composto è sensibile all’umidità e all’ossigeno atmosferico.
La struttura del complesso è riportata in Figura 31.
Il complesso [Cu(CH3CN)4]+ ha 18e- di valenza:
0
Leganti neutri, metallo neutro Cu (s1d10)
1 x 11 e- (Cu0) + 4 x 2 e- (acetonitrile) – 1 e- (carica positiva) = 18 ePer questioni di solubilità, viene isolato allo stato solido con un anione molto
grosso [PF6]-.
18
Per caratterizzare il composto possiamo registrarne lo spettro I.R. in nujol
(
C N
= 2303, 2275 cm-1,
P-F
= 840 cm-1).
Lo ione Cu+ (d10) è un centro soft che predilige leganti soft all’azoto (nitrili),
fosforo e zolfo. Il complesso [Cu(CH3CN)4]+ è così stabile che, in acetonitrile
come solvente, lo ione Cu2+ è un buon ossidante e che CuI risulta solubile in
acetonitrile:
Cu2+ + 4 CH3CN + e-
[Cu(CH3CN)4]+
[Cu(CH3CN)4]+ + I-
CuI(s) + 4 CH3CN
Al contrario, lo ione Cu2+ (d9) è un centro hard e preferisce leganti hard
all’ossigeno (acqua, alcoli).
I composti di Cu(I) sono diamagnetici, mentre quelli di Cu(II) sono
paramagnetici e hanno un
1,8 BM.
eff
La stabilità del Cu (I) maggiore in acetonitrile che in acqua e, al contrario, la
stabilità del Cu(II) maggiore in acqua che in acetonitrile, è qualitativamente
spiegata dalla teoria Hard-Soft di Pearson che può in qualche misura essere
correlata alla differenza di elettronegatività del Cu(II) e del Cu(I) nei confronti
dei siti leganti delle molecole di acqua e di acetonitrile.
Dal diagramma di Latimer del rame in acqua:
Cu2+ -------- Cu+ -------- Cu
0,150
0,520
si nota che il Cu+, come ione libero, disproporziona in acqua secondo la
reazione:
2 Cu+
Cu2+ + Cu
E° = 0,520 – 0,150 = 0,370 V,
da cui
K = 10 n
E°/ 0,059
= 106,3 = 2.106
K = [Cu2+] / [Cu+]2
Lo ione Cu+ può essere stabilizzato a contatto con l’acqua, quando è
presente in un sale poco solubile, es. CuI, CuCN
Cu2+ + 2 I-
CuI(s) + ½ I2
19
Cu2+ + 2 CN-
CuCN(s) + ½ (CN)2
Dal diagramma di Latimer del rame in acetonitrile:
Cu2+ -------- Cu+ -------- Cu
E1
E2
dove E1 > E2 , si ha che il Cu2+ e il Cu coproporzionano in acetonitrile,
secondo la reazione:
Cu2+ + Cu
2 Cu+
Quando il legante si coordina al metallo tramite l’atomo di N si hanno i
complessi nitrilici:
M N C-R
Mentre, quando il legante si coordina al metallo tramite l’atomo di C, si hanno
i complessi isonitrilici:
M C=N-R
20
Bis(ortofenantrolina)rame(I) esafluorofosfato,
[Cu(o-phen)2][PF6]
Il bis(ortofenantrolina)rame(I) esafluorofosfato è un sale la cui parte
cationica è rappresentata da un complesso fenantrolinico, [Cu(1,10C12H8N2)2]+ oppure [Cu(o-phen)2]+, dove o-phen = ortofenantrolina.
L’ortofenantrolina, molecola aromatica a 3 anelli condensati, è un legante
all’azoto bidentato chelante, donatore di 4e- (Figura 32).
L’ortofenantrolina è un buon
-donatore e un buon -accettore, quindi è
un legante versatile per diversi stati di ossidazione del metallo.
La sintesi del complesso è una reazione di sostituzione di 4 leganti
monodentati con 2 leganti bidentati (Figura 33):
[Cu(CH3CN)4][PF6] + 2 o-phen
[Cu(o-phen)2][PF6] + 4 CH3CN
(o-phen, 1,10-fenantrolina, C12H8N2)
Lo ione [Cu(o-phen)2]+ ha struttura tetraedrica con 4 legami Cu-N.
Lo ione Cu+, in configurazione elettronica d10, ha orbitali d pieni e quindi non
ha la possibilità di dare bande di assorbimento elettronico d-d ma il
complesso dà una banda intensa a trasferimento di carica M
L, nel visibile,
che impartisce al composto una colorazione bruna con riflessi viola, quando è
in solido, e una colorazione arancio-marrone quando è in soluzione di
acetonitrile.
La struttura del complesso è presentata in Figura 34.
Il complesso [Cu(o-phen)2]+ ha 18e- di valenza:
0
Leganti neutri, metallo neutro Cu (s1d10)
-
1 x 11 e- (Cu0) + 2 x 4 e- (fenantrolina) – 1e- (carica positiva) = 18 e .
Per caratterizzare il composto possiamo registrarne lo spettro elettronico in
alcol metilico (
max
= 438 nm, banda intensa con elevata assorbività molare,
) (vedi legge di Lambert-Beer A= l C).
Confrontiamone le proprietà con un analogo complesso di Cu(II).
21
Un esempio di struttura perfettamente quadrato-planare di un complesso
di Cu(II) è rappresentato dallo ione [Cu(en)2]2+ (en: etilendiammina oppure
1,2-etandiammina) (Figura 35).
Il complesso è paramagnetico per un elettrone spaiato, con 17e- di valenza.
Lo ione Cu2+ in configurazione elettronica d9, con orbitali d parzialmente
occupati ha la possibilità di dare bande deboli di assorbimento elettronico d-d
nel visibile. Il complesso ha colorazione azzurra.
Il bis(ortofenantrolina)rame(II) solfato può essere preparato dal composto
bis(ortofenantrolina)rame(I) esafluorofosfato per reazione con persolfato di
sodio in acqua distillata scaldando la sospensione per favorire l’ossidazione.
La stechiometria della reazione è:
2 [Cu(o-phen)2][PF6] + Na2S2O8
2 [Cu(o-phen)2](SO4) + 2 NaPF6
Il composto di Cu(II) è di color azzurro chiaro.
22
23
Dicloro-bis(trifenilfosfina)nichel(II), [NiCl2(PPh3)2]
Il dicloro-bis(trifenilfosfina)nichel(II), [NiCl2(PPh3)2] è un complesso
covalente, ossia non si dissocia in acqua e quindi non mostra conducibilità.
La sintesi del complesso è una reazione di sostituzione dei leganti a
partire dal sale NiCl2.6H2O, favorita da un aumento di temperatura, ossia
(Figura 36):
[Ni(H2O)6]2+ + 2 Cl- + 2 PPh3
[NiCl2(PPh3)2] + 6 H2O
(PPh3 = P(C6H5)3)
Il complesso ha struttura tetraedrica; assorbe radiazione nello spettro
visibile a
max
= 887 nm, con transizione elettronica d-d. Tale lunghezza
d’onda corrisponde alla frequenza del rosso, quindi il complesso appare
verde (colore complementare).
La struttura del complesso è riportata in Figura 37.
Nel caso dello ione Ni2+ (d8), la misura di suscettività magnetica dà
indicazioni sulla geometria del complesso tetracoordinato.
Infatti,
secondo
la
teoria
del
campo
cristallino
un
complesso
tetracoordinato d8 con due elettroni spaiati (paramagnetico) ha geometria
tetraedrica (Figura 38).
Poiché
t
= 4/9
o,
dove
orbitali d nel caso tetraedrico e
t
è la differenza energetica tra i due set di
o
nel caso ottaedrico e quadrato planare, i
complessi tetraedrici assorbono energie più basse nel visibile, rispetto a quelli
ottaedrici e quadrato planari, pertanto sono colorati più intensamente dei
complessi ottaedrici e quadrato planari.
I
t
e
o
sono dovuti all’effetto repulsivo dei leganti sugli elettroni d del
metallo e, poiché nel tetraedro gli orbitali d non puntano direttamente verso i
leganti come capita nell’ottaedro, si ha che
t
<
o.
24
Inoltre poiché P >
t
, nel tetraedrico risultano favorite le situazioni ad alto
spin.
Il complesso [NiCl2(PPh3)2] ha 16e- di valenza.
Legante anionico, metallo positivo Ni
2+
8
(d )
2+
1 x 8 e- (Ni ) + 2 x 2 e- (fosfina) + 2 x 2 e- (cloruro) = 16 e-
0
Legante neutro, metallo neutro Ni (d10)
0
1 x 10 e- (Ni ) + 2 x 2 e- (fosfina) + 2 x 1 e- (cloro) = 16 e-
25
LEGANTI FOSFINICI
Una categoria importante di leganti è quella delle fosfine monodentate o
bidentate (chelanti), ossia composti organici o inorganici di P(III).
Nel caso di complessi con metalli di transizione, quando il metallo è in
basso stato di ossidazione, può avere luogo un tipo di legame sinergico di
donazione di tipo
P M e di retrodonazione di tipo
L’importanza della donazione
da M P.
e della retrodonazione
dipendono dai
sostituenti sulla fosfina.
Nella serie delle fosfine monodentate PF3 – PCl3 – P(OR)3 – PPh3 - PR3
aumenta la basicità di Lewis, quindi aumenta la donazione
retrodonazione
e diminuisce
La retrodonazione avviene con i bassi stati di ossidazione
del metallo.
Ad esempio, PF3 e PCl3 stabilizzano Ni(0) e Fe(0) nei composti [Ni(PCl3)4]
e [Fe(PF3)5] ; PPh3 stabilizza Ni(II) e Co(I) nei composti [NiCl2(PPh3)2] e
[CoH(PPh3)4] (Figura 39).
Il legante 1,2-bis(difenilfosfina)etano (abbreviato dppe) appartiene alla
categorie delle fosfine bidentate chelanti e forma un anello a 5 termini con il
metallo (Figura 40).
Il complesso [NiCl2(dppe)] ha struttura quadrato planare ed è di colore
arancione, ossia assorbe nello spettro elettronico UV-Vis a max = 460 nm,
una banda dovuta a transizioni elettroniche d-d (Figura 41).
Il complesso assorbe nel blu e appare arancione (colore complementare).
La struttura del complesso è riportata in Figura 42. Gli atomi di cloro sono
forzatamente in cis a causa del chelante che occupa 2 posizioni in cis.
Il complesso ha 16e- di valenza e può essere sintetizzato a partire dal
complesso [NiCl2(PPh3)2] mediante una reazione di scambio di leganti in alcol
etilico all’ebollizione, secondo la stechiometria:
[NiCl2(PPh3)2] + dppe
[NiCl2(dppe)] + 2 PPh3
26
Nel caso dello ione Ni2+ (d8), la misura di suscettività magnetica dà
indicazioni sulla geometria del complesso tetracoordinato.
Infatti,
secondo
la
teoria
del
campo
cristallino
un
complesso
tetracoordinato d8 con zero elettroni spaiati (diamagnetico) ha geometria
quadrato planare (Figura 43).
Allontanando due leganti sui due apici opposti di un ottaedro (esempio
lungo la direzione z) si arriva alla situazione quadrato planare in cui i 3 orbitali
d con componente z si stabilizzano, mentre gli altri 2 orbitali d si
destabilizzano.
Il complesso quadrato planare risulterà tanto più favorito quanto più
l’orbitale ad alta energia d x2
y2
, a carattere di antilegante, risulterà vuoto.
Questo accade spesso nella configurazione d8: Ni2+, Pd2+, Pt2+.
La stabilità dei due complessi di nichel è molto differente.
[NiCl2(PPh3)2] è instabile e decompone rapidamente in soluzione, mentre
[NiCl2(dppe)] è molto stabile.
La struttura tetraedrica è favorita quando i leganti hanno un elevato
ingombro sterico, mentre la struttura quadrato planare è favorita da fattori
elettronici. Il fattore sterico diventa meno importante nella seconda e terza
serie di transizione, pertanto i complessi tetracoordinati di Pd(II) e Pt(II)
hanno quasi esclusivamente la geometria quadrato planare, ad esempio
[PtCl2(PPh3)2].
Inoltre la struttura quadrato planare è favorita con leganti che favoriscono
il campo forte (basso spin) (ossia a destra della serie spettrochimica), ad
esempio [Ni(CN)4]2- è quadrato planare e [NiCl4]2- è tetraedrico.
A volte però, per uno stesso composto, ad esempio [NiBr2(PPh2Et)2] , può
esistere una situazione di equilibrio tra le due forme. L’interconversione tra le
due forme può essere modulata variando la temperatura del composto
(effetto termocromico).
27
A –78°C il complesso è rosso, diamagnetico, con geometria quadrato
planare, mentre a 25°C lo stesso complesso è verde, paramagnetico, con
geometria tetraedrica. Le due forme sono in equilibrio a temperatura
intermedia e l’interconversione tra le due forme può essere monitorata
mediante
varie
tecniche:
misura
del
momento
magnetico
effettivo,
registrazione dello spettro UV-vis e dello spettro 1H-NMR.
28
29
(1,4,8,11-tetraazaciclotetradecano)nichel(II) bis(perclorato)
[Ni(C10H24N4)](ClO4)2
Detto in forma abbreviata: nichel cyclam perclorato
E’ un sale la cui parte cationica è un complesso formato da un
macrociclo coordinato attorno allo ione Ni2+.
N.B. un macrociclico è un composto che, come legante, può circondare
completamente uno ione metallico.
L’ 1,4,8,11-tetraazaciclotetradecano, a formula C10H24N4 , è detto cyclam,
ed è un legante tetradentato all’azoto. In questa sintesi il legante macrociclico
non è un reagente iniziale della reazione ma viene sintetizzato a stadi intorno
al metallo mediante una sintesi templata.
La reazione illustra bene l’effetto templato cinetico, in cui cioè uno ione
metallico viene utilizzato per tenere i gruppi reattivi in una geometria adatta a
far avvenire una reazione a stadi in maniera stereochimicamente selettiva.
Le stechiometrie della reazione complessiva sono le seguenti (Figura 44):
Ni2+ + C8H22N4 [Ni(C8H22N4)]2+
[Ni(C8H22N4)]2+ + C2H2O2 [Ni(C10H20N4)]2+ + 2 H2O
[Ni(C10H20N4)]2+ + 2 BH4- + 2 H+ [Ni(C10H24N4)]2+ + 2 BH3
La reazione è effettuata in tre stadi distinti e il meccanismo è riportato in
Figura 45 a, b.
1° stadio: dapprima si coordina al Ni(II) un’ammina tetradentata a catena
aperta flessibile, ossia la 1,2 bis(3-aminopropil)etandiammina.
30
2° stadio: successivamente si effettua la reazione con una dialdeide,
ossia la gliossale, che in virtù della conformazione dell’ammina intorno
all’atomo metallico, porta alla formazione di una diimmina ciclica coordinata.
3° stadio: infine si effettua una riduzione, con sodio tetraidroborato,
dell’immina coordinata con formazione del macrociclo saturo, ossia 1,4,8,11tetraazaciclotetradecano (cyclam) coordinato al metallo, ottenendo la specie
stabile [Ni(cyclam)]2+ arancione, complesso a 16e- di valenza (8e- da parte del
macrociclo e 8e- da parte del metallo). Il metallo è lo ione Ni2+ (d8).
In alcuni casi il macrociclo può essere chimicamente separato dal metallo
mediante una reazione di sostituzione di leganti, ossia il metallo viene
coordinato con un legante più forte ed il macrociclo viene decoordinato.
La struttura del complesso è riportata in Figura 46 , da cui si nota che la
geometria attorno al Ni2+ è quadrato-planare. La coordinazione del
macrociclo provoca la formazione di 4 anelli chelati, 2 a 6 termini e 2 a 5
termini.
Tale specie cationica viene isolata in stato solido come perclorato,
mediante aggiunta di acido perclorico. Il composto è caratterizzato mediante
uno spettro infrarosso in KBr (
N-H
mediante uno spettro elettronico (
= 3270, 3211 cm-1 ,
max.
= 450 nm,
450
Cl-O
= 1104 cm-1) e
= 64 Lmol-1cm-1).
31
Dicloro(1,4,8,11-tetraazaciclotetradecano)nichel(III) perclorato
[NiCl2(C10H24N4)](ClO4)
Detto in forma abbreviata: nichel dicloro cyclam perclorato
E’ un sale la cui parte cationica è un complesso formato da un macrociclo
(cyclam) coordinato attorno allo ione Ni3+. La coordinazione intorno al metallo
è completata da due leganti cloruro.
La sintesi ha lo scopo di evidenziare la grande stabilità di un complesso
macrociclico (effetto macrociclo termodinamico) con la possibilità di ottenere
un metallo in stato di ossidazione non comune.
Ad esempio vediamo la capacità del cyclam nello stabilizzare Ag(II):
2 Ag+
2 Ag+ + cyclam
Ag(s) + Ag2+
K = 10-20 (in H2O)
Ag(s) + [Ag(cyclam)]2+
K > 1 (in H2O)
Le stechiometrie della reazione complessiva sono le seguenti (Figura 47):
2 [Ni(C10H24N4)]2+ + S2O82- 2 [Ni(C10H24N4)]3+ + 2 SO42[Ni(C10H24N4)]3+ + 2 HCl [NiCl2(C10H24N4)]+ + 2 H+
La reazione è effettuata in due stadi distinti e il meccanismo è riportato in
Figura 48. Il composto di partenza è il complesso [Ni(cyclam)](ClO4)2
ottenuto nella precedente sintesi.
1°stadio: dapprima si effettua un’ossidazione controllata del metallo con lo
ione persolfato, S2O82-, che si riduce a solfato, senza intaccare il legante
macrociclico, ottenendo lo ione [Ni(cyclam)]3+ verde, complesso a 15e- di
valenza; questa specie non viene isolata e caratterizzata.
E° [Ni(cyclam)]3+ / [Ni(cyclam)]2+ = 0.97 V ; E° S2O82- / SO42- = 2.01 V
32
2°stadio: successivamente si aggiunge acido cloridrico per completare la
coordinazione intorno al metallo ed ottenere la specie più stabile
[NiCl2(cyclam)]+ giallo-ocra, complesso a 19e- di valenza (8e- da parte del
macrociclo, 7e- da parte del metallo e 4e- da parte dei leganti alogenuro).
Il metallo è lo ione Ni3+ (d7).
La struttura del complesso è riportata in Figura 49 , da cui si nota che la
geometria attorno al Ni2+ è ottaedrica con i due leganti alogenuro in trans. In
questo caso la distanza Ni-N è minore di circa 5% rispetto alla distanza Ni-N
nel complesso di Ni(II), a causa delle minori dimensioni dello ione Ni3+
rispetto allo ione Ni2+.
Tale specie cationica viene isolata in stato solido come perclorato. Il
composto è caratterizzato mediante uno spettro infrarosso in KBr (
3190, 3143 cm-1 ,
magnetica (
eff
Cl-O
N-H
=
= 1097 cm-1) e mediante una misura di suscettività
= 1,90 BM).
33
34
Pentamminoclorocobalto(III) dicloruro, [CoCl(NH3)5]Cl2
E’ un sale, di colore viola, contenente il catione [CoCl(NH3)5]2+, un
complesso ottaedrico di Co(III) a 18e- di valenza. La struttura del complesso
è riportata in Figura 50.
La sintesi può essere scomposta nei seguenti passaggi (Figura 51):
CoCO3 + 4 HCl + H2O
[CoCl4]2- + CO2 + 2 H3O+
(residuo insolubile di CoO, presente come impurezza nel carbonato di
cobalto, che viene separato per filtrazione)
[CoCl4]2- + 6 NH3
[Co(NH3)6]2+ + 4 Cl-
(si tampona a pH=9 per evitare la precipitazione di Co(OH)2)
2 [Co(NH3)6]2+ + H2O2 + 4 H+
2 [Co(H2O)(NH3)5]3+ + 2 NH4+
(si esegue l’ossidazione con acqua ossigenata)
H2O2 + 2e- + 2H+
2 H2O
E [Co(H2O)(NH3)5]3+ / [Co(NH3)6]2+ = 0,334 V
E H2O2 / H2O = 1,23 V
[Co(H2O)(NH3)5]3+ + 3 HCl
[CoCl(NH3)5]Cl2 + H2O + 3 H+
(si scalda con acido cloridrico concentrato per effettuare la sostituzione
H2O Cl- ; l’ambiente acido catalizza la reazione di sostituzione).
Lo ione Co3+ è in configurazione elettronica (d6), a basso spin (t2g)6(eg)0
(Figura 52).
Il complesso può essere caratterizzato (Figura 53) mediante:
- registrazione dello spettro UV-Vis in acqua:
- determinazione della conducibilità molare
conducibilità specifica
max.
M
= 534, 468(sh), 364 nm
tramite una misura di
della soluzione acquosa 10-3 M di [CoCl(NH3)5]Cl2
(S cm-1) = L(S)K(cm-1)
dove L=conduttanza , K=costante di cella
da cui si calcola la conducibilità molare
M
M
della soluzione acquosa del sale
(S cm2 mole-1) = (S cm-1)1000(cm3/L)/M(moli/L)
35
Si confronta il valore misurato con il valore teorico di
M
(quando l’anione è
un monoanione e quando la soluzione è ca. 10-3 M):
N. di ioni
2
3
4
5
S cm2 mole-1)
90-110
235-250
400-430
ca. 560
Dalla misura di conducibilità molare si osserva se la reazione di
sostituzione H2O Cl- è avvenuta completamente; infatti:
[Co(H2O)(NH3)5]Cl3
4 ioni/mole in soluzione (
[CoCl(NH3)5]Cl2
3 ioni/mole in soluzione (
[Co(H2O)(NH3)5]3+ + 3 ClM
= 400-430 S cm2 mole-1)
[CoCl(NH3)5]2+ + 2 ClM
= 235-250 S cm2 mole-1).
N.B. Si noti inoltre che lasciando in soluzione [CoCl(NH3)5]Cl2 per lungo
tempo, in assenza di HCl, si forma il composto [Co(H2O)(NH3)5]Cl3
e
quindi si misura un valore più elevato di conducibilità molare.
36
Pentamminonitritocobalto(III) dicloruro,[Co(NO2-O)(NH3)5]Cl2
e isomerizzazione a
Pentamminonitrocobalto(III) dicloruro, [Co(NO2-N)(NH3)5]Cl2
In due stadi successivi, si effettua la preparazione di due diversi composti di
coordinazione, tra loro isomeri di struttura (ossia composti aventi la stessa
formula empirica, ma una diversa sequenza di connessioni atomiche).
L’isomeria di struttura in oggetto è più precisamente detta isomeria di
legame, e si verifica quando lo stesso legante si coordina alternativamente al
metallo con due atomi appartenenti ad elementi diversi. Il legante che
possiede queste caratteristiche è detto ambidentato (es. NO2-, SCN-, OCN-,
CN-).
Il legante utilizzato per questa sintesi è lo ione nitrito NO2- che, quando si
coordina tramite l’atomo di ossigeno, forma il complesso nitrito, indicato nella
formula come NO2-O, mentre quando si coordina tramite l’atomo di azoto,
forma il complesso nitro, indicato nella formula come NO2-N.
In entrambi i casi il legante, se considerato come anione, dona 2e- al metallo,
mentre, se considerato come neutro, dona 1e- al metallo.
Dapprima viene sintetizzato il prodotto cinetico, ossia il
pentamminonitritocobalto(III) dicloruro, [Co(NO2-O)(NH3)5]Cl2, che viene poi
trasformato, mediante una reazione di isomerizzazione, nel prodotto
termodinamico, ossia il pentamminonitrocobalto(III) dicloruro,
[Co(NO2-N)(NH3)5]Cl2.
In Figura 54 è esemplificato il concetto di prodotto cinetico e termodinamico
mediante il grafico E vs. coordinata di reazione.
Le strutture dei due isomeri sono riportate rispettivamente in Figura 55 e in
Figura 56.
37
Entrambi i composti, [Co(NO2-O)(NH3)5]Cl2, di colore rosa-arancio, e
[Co(NO2-N)(NH3)5]Cl2, di colore marrone, sono complessi ottaedrici di Co(III)
a 18e- di valenza in configurazione elettronica a basso spin.
La sintesi avviene in due stadi distinti (Figura 57):
1° stadio: sostituzione di Cl- con NO2-:
[CoCl(NH3)5]2+ + NO2-
[Co(NO2-O)(NH3)5]2+ + Cl-
(NO2- viene aggiunto come NaNO2 + HCl; si forma HNO2 che disproporziona
a NO3- e NO e quest’ultimo con O2 dà i vapori nitrosi NO2 + NO),
2°stadio: isomerizzazione, che avviene scaldando il primo composto:
[Co(NO2-O)(NH3)5]2+ [Co(NO2-N)(NH3)5]2+
La seconda reazione è una reazione di equilibrio (K=99 a 20°C) e può essere
seguita tramite spettroscopia infrarossa in KBr:
[Co(NO2-O)(NH3)5]2+ :
N-O
= 1065 cm-1 ;
N=O
= 1453 cm-1 ;
s N-O
= 1316 cm-1
(le ultime due bande sono causate dalla presenza di due isomeri geometrici)
[Co(NO2-N)(NH3)5]2+ :
as N-O
= 1427 cm-1 ;
s N-O
= 1308 cm-1
I due complessi isomeri possono essere caratterizzati mediante registrazione
degli spettri UV-Vis in acqua:
[Co(NO2-O)(NH3)5]2+
max.
= 492 nm
[Co(NO2-N)(NH3)5]2+
max.
= 455 nm
38
Tris(1,2-etandiammina)cobalto(III) triioduro monoidrato,
[Co(en)3]I3 .H2O
La sintesi coinvolge la preparazione di due stereoisomeri (la formula empirica
è identica, come pure la sequenza delle connessioni atomiche, ma è diversa
la distribuzione spaziale degli atomi).
L’esperienza ha per scopo: a) la sintesi di un composto di coordinazione la
cui parte cationica esiste in due isomeri ottici, b) la separazione dei 2 isomeri
(risoluzione del racemo), c) l’isolamento di un isomero puro.
Il complesso che viene preparato come miscela racema (equimolare dei due
isomeri) è lo ione:
[Co(NH2CH2CH2NH2)3]3+
dove il legante NH2CH2CH2NH2 (etilendiammina, 1,2-etandiammina, 1,2
diamminoetano, abbreviato en) è un chelante bidentato all’azoto, donatore di
4e-.
Il complesso è preparato a partire da un sale di Co(II), ione che viene
ossidato con aria in presenza di legante e di carbone attivo. Quest’ ultimo
catalizza la completa sostituzione delle molecole di acqua dello ione Co(III)
con molecole di legante.
Il complesso, con geometria ottaedrica intorno al cobalto, attorno al quale
sono coordinati tre leganti chelanti, ha 18 elettroni totali di valenza. E’ un
complesso a basso spin (d6, 0e- spaiati). La coordinazione avviene tramite
interazioni di tipo
(sp3 dell’azoto con d2sp3 del cobalto). La struttura è
riportata in Figura 58.
La presenza di due enantiomeri è dovuta al fatto che lo ione [Co(en)3]3+ non
possiede assi di simmetria impropri Sn (assenza di centro di inversione S2 e
di piani di simmetria S1). Lo ione appartiene al gruppo puntuale D3 (1 C3, 3
C2).
39
Secondo le convenzioni i due isomeri ottici hanno configurazione assoluta
oppure , che dipende dall’orientazione spaziale dei leganti (vite destrorsa o
sinistrorsa) (Figura 59).
La configurazione assoluta non deve essere confusa con la direzione di
rotazione del piano di luce polarizzata, che viene invece indicata con (+)
oppure (-) se la luce è ruotata rispettivamente a destra oppure a sinistra
rispetto al piano di luce polarizzata.
Nella specifica sintesi e successiva risoluzione del racemo viene isolato
l’isomero (+)-[Co(en)3]3+ , la cui configurazione assoluta, ottenuta mediante
analisi ai raggi X , è risultata essere .
La risoluzione del racemo (±)-[Co(en)3]3+ viene effettuata mediante il metodo
tradizionale della precipitazione frazionata dei sali diastereoisomerici
ottenuti per aggiunta di un enantiomero puro di uno ione avente carica
opposta a quella della miscela racema.
I due sali diastereoisomerici hanno proprietà chimico-fisiche diverse
(solubilità), mentre i due composti presenti nel racemo hanno identiche
proprietà chimico-fisiche.
40
Lo ione di carica opposta a carattere enantiomerico è il tartrato (+)-[C4H4O6]2, che viene aggiunto come tartrato di bario, Ba[(+)-C4H4O6] . Questo sale
viene preparato nella seconda fase della ricetta da BaCO3 e acido tartarico
(+)-[C4H6O6] . ([α]D20 = + 12°), secondo la reazione acido-base (Figura 60):
BaCO3 + (+)-[C4H6O6]
Ba[(+)-C4H4O6] + CO2 + H2O
Lo ione (+)-[C4H4O6]2- ha configurazione 2R, 3R
mentre lo ione (-)-[C4H4O6]2- ha configurazione 2S, 3S ma non viene
utilizzato perché è troppo costoso l’acido tartarico (-)-[C4H6O6] . ([α]D20 = 12°).
Le reazioni coinvolte nella sintesi sono riportate in Figura 61:
2 [Co(H2O)6](SO4) + 6 en
(±)-[Co(en)3](SO4) + 12 H2O
(±)-[Co(en)3](SO4) + ½ O2 + 2 HCl
(±)-[Co(en)3](SO4)Cl + 2 Ba[(+)-C4H4O6]
(±)-[Co(en)3](SO4)Cl + H2O
(+)-[Co(en)3] [(+)-C4H4O6] Cl
+ (-)-[Co(en)3] [(+)-C4H4O6] Cl + 2 BaSO4
(+)-[Co(en)3] [(+)-C4H4O6] Cl + 3 NaI
(+)-[Co(en)3]I3
+ Na2[(+)-C4H4O6] + NaCl
41
Il sale diastereoisomero meno solubile è (+)-[Co(en)3] [(+)-C4H4O6] Cl da cui
viene recuperato l’enantiomero (+)-[Co(en)3]3+ mediante sostituzione del
contro ione con ioduro e successiva cristallizzazione.
La purezza dell’enantiomero (purezza ottica) può essere controllata al
polarimetro.
In teoria l’altro enantiomero (-)-[Co(en)3]3+ potrebbe essere ottenuto dalle
acque madri della prima precipitazione, ma poiché la precipitazione non è
completa, si otterrebbe un prodotto impuro.
Lo spettro UV-Vis.in acqua del complesso (+)-[Co(en)3]3+ mostra due
massimi d’assorbimento λmax. = 467 nm, 331 nm.
Racemizzazione
L’elevata inerzia cinetica dei complessi di Co(III) tris-chelati li rende
abbastanza stabili alla racemizzazione, ossia trasformazione di un
enantiometro nella miscela racema con perdita del potere ottico rotatorio.
Tuttavia, bollendo in acqua un enantiomero in presenza di carbone attivo, si
osserva racemizzazione.
Il ruolo del carbone attivo è quello di promuovere la formazione di Co(II) che
porta a complessi labili, passibili di racemizzazione secondo un meccanismo
di ring opening (rottura di un legame dell’anello chelato) (Figura 62).
42
Attività ottica
Un enantiomero interagisce con un fascio di luce monocromatica (
)
polarizzata in un piano, ruotando tale piano o verso destra (+) o verso sinistra
(-), rispetto alla direzione di propagazione della luce.
N.B. un fascio di luce è polarizzato in un piano, quando il vettore del
campo elettrico ruota di un angolo pari a 2
/ ciclo, intorno alla
direzione di propagazione, con uguale ampiezza e fase, sia verso destra
che verso sinistra (racemato di luce) (Figura 63).
Indice di rifrazione (n):
n = c° / c
c° : velocità della luce nel vuoto
c : velocità della luce nel mezzo attraversato
43
Misura dell’attività ottica (polarimetria)
Per la misura dell’attività ottica si usa la luce emessa da una lampada al
sodio (λ = 589,3 nm). Quando tale luce passa
attraverso un prisma
polarizzatore si ottiene luce polarizzata in un piano. Sostanze otticamente
attive sono in grado di ruotare il piano della luce polarizzata a destra (+)
oppure a sinistra (-) dalla verticale secondo un angolo che dipende dalla
natura della sostanza e da altre variabili. La direzione e l’angolo di rotazione
possono essere misurati sperimentalmente mediante un polarimetro.
L’ampiezza della rotazione dipende anche dalla lunghezza l del campione
attraversato dalla luce e dalla concentrazione c della soluzione del campione.
E’ stato definito come potere ottico rotatorio specifico [ ]D20
di una
sostanza, alla lunghezza D del sodio e alla temperatura di 20°C, il rapporto
(Figura 64):
[ ]D20 =
/lc
(° cm3 / dm g)
: angolo di rotazione (°)
l : lunghezza della cella (dm)
c : concentrazione del soluto (g / cm3) a 20°C
Il potere ottico rotatorio specifico, espresso in unità SI, è il rapporto:
[ ]D20 =
(°m2 / Kg)
/ 100 l c
: angolo di rotazione (°)
l : lunghezza della cella (m)
c : concentrazione del soluto (Kg / m3) a 20°C
Il rapporto tra il potere ottico rotatorio specifico sperimentale e quello
teorico di una sostanza permette di calcolare l’eccesso enantiomerico (ee%):
ee% = ( [ ]
sperimentale
/[ ]
teorico
) x 100
44
Purezza ottica
La purezza ottica di un enantiometro ossia il grado di contaminazione
con l’enantiomero opposto, è espressa come eccesso enantiomerico (ee%),
quantità adimensionale (Figura 65):
ee% = [
dove
(+)
e
(-)
(+) -
(-)]
/[
(+) +
(-)]
x 100
sono le frazioni molari dei 2 enantiomeri, ossia
(+) +
(-)
=1
ee = 1 per un enantiometro puro
ee = 0 per una miscela racema
N.B. normalmente è un numero positivo (0
ee%
100), ossia è riferito
all’enantiomero presente in quantità maggiore.
45
E’ stato anche definito come potere ottico rotatorio specifico molare il
rapporto (Figura 64):
[M]D20 =
/lM
(° cm3 / dm mole)
: angolo di rotazione (°)
l : lunghezza della cella (dm)
M : concentrazione del soluto (moli / cm3) a 20°C
Il potere ottico rotatorio specifico molare, espresso in unità SI, è il
rapporto:
[M]D20 = 10-5
/ 100 l M
(°m2 / mole)
: angolo di rotazione (°)
l : lunghezza della cella (m)
M : concentrazione del soluto (moli / m3)
Da [ ] è possibile calcolare l’angolo di rotazione molare ([M] ):
[M] = PM [ ]
(° cm2 / mole)
PM : peso molecolare (g / mole)
La variazione di [M] con la lunghezza d’onda
è la dispersione ottica
rotatoria (ORD)(Figura 66):
46
47
Tris(N,N’-dietilditiocarbammato)ferro(III) , [Fe(S2CNR2)3]
(R = C2H5)
Il tris(N,N’-dietilditiocarbammato)ferro(III), abbreviato Fe(dttc)3 , è un
composto neutro molecolare, con tre leganti chelanti dietilditiocarbammato
monoanionici intorno al Fe(III), per un totale di 6 interazioni di legame Fe-S. Il
metallo ha numero di coordinazione 6 e stato di ossidazione formale +3.
Il chelante forma con il metallo un anello a quattro termini. La
coordinazione intorno al ferro è ottaedrica distorta. La struttura è mostrata in
Figura 67.
Poichè la parte esterna del complesso consiste di gruppi organici il
composto è idrofobico, insolubile in acqua, ma è solubile in solventi organici.
Nella sintesi il legante monoanionico viene fornito direttamente come sale
sodico, Na(S2CNR2). Si utilizza una reazione di sostituzione di leganti, Figura
68:
[Fe(H2O)6]3+ + 3 (S2CNR2)-
[Fe(S2CNR2)3] + 6 H2O
Per caratterizzare il composto, di colore marrone, possiamo:
- registrare uno spettro I.R. (
C=N =
1500 cm-1,
- misurare la suscettività magnetica (
eff.=
C=S =
990 cm-1),
4,24 BM).
Per il conteggio elettronico il legante può essere considerato anionico,
donatore di 4 e-, oppure neutro, donatore di 3 e-. Il numero di elettroni di
valenza del complesso è 17e-:
Legante anionico, metallo positivo Fe
3+
5
(d )
3+
1 x 5 e- (Fe ) + 3 x 4 e- (dttc) = 17 e0
8
Legante neutro, metallo neutro Fe (d )
0
1 x 8 e- (Fe ) + 3 x 3 e- (dttc) = 17 eComplessi con geometria ottaedrica, aventi tre leganti chelanti del tipo L-L
simmetrici, esistono in due forme isomeriche (isomeri ottici o enantiomeri,
48
poiché si comportano diversamente quando attraversati da un fascio di luce
polarizzata). La sintesi dà luogo ad una miscela equimolare (racema) dei due
isomeri che, nel caso del complesso Fe(dttc)3, potrebbero essere separati
mediante l’utilizzo di una colonna cromatografia avente un supporto
otticamente attivo.
COMPORTAMENTO MAGNETICO DEL FERRO(III)
Il ferro, la cui configurazione elettronica allo stato fondamentale è
[Ar]3d64s2, forma lo ione Fe3+ che, sempre allo stato gassoso, ha la
configurazione elettronica [Ar]3d5.
Secondo la teoria del campo cristallino, sei leganti monodentati o tre
leganti bidentati chelanti, disposti intorno allo ione Fe3+ provocano la
separazione dei cinque orbitali 3d del Fe in due gruppi, l’uno, denominato t2g,
a energia minore, comprendente tre orbitali degeneri, e l’altro, denominato
eg, ad energia maggiore, comprendente due orbitali degeneri.
La configurazione d5, per un composto di coordinazione con geometria
ottaedrica, può presentare una configurazione elettronica ad alto spin
(t2g)3(eg)2 (campo debole, 5e- spaiati) o a basso spin (t2g)5 (campo forte, 1espaiato) (Figura 69).
La misura della suscettività magnetica permette di ricavare il momento
magnetico del complesso dovuto allo spin degli elettroni spaiati; tale misura
evidenzia che Fe(dttc)3, a 20°C, è in una situazione di equilibrio di spin
(Figura 70).
A bassa temperatura (100 K) è prevalentemente in basso spin, mentre ad
alta temperatura (350 K) è in alto spin.
49
EQUILIBRIO DI SPIN
Nei complessi ottaedrici, quando
o
è abbastanza grande o abbastanza
piccolo, a temperatura ambiente, lo stato fondamentale è esclusivamente a
basso spin o ad alto spin rispettivamente.
Nei complessi ottaedrici, quando
o
P, gli stati ad alto spin o a basso spin
hanno una piccola differenza in energia e se tale differenza è paragonabile
all’energia di agitazione termica, la popolazione dei due stati di spin può
essere influenzata variando di poco la temperatura, ossia in soluzione è
presente un equilibrio di spin:
basso spin alto spin
Fe(S2CNR2)3,
tris(N,N’-dialchilditiocarbammato)ferro(III)
R = C2H5 (d5 basso spin
alto spin)
50
Bis(N,N’-dietilditiocarbammato)nitrosilferro (II),
[Fe(S2CNR2)2(NO)]
(R = C2H5)
Il bis(N,N’-dietilditiocarbammato)nitrosilferro, abbreviato Fe(dttc)2(NO) è
un
composto
neutro
molecolare,
con
due
leganti
chelanti
dietilditiocarbammato monoanionici e un legante nitrosilico neutro intorno al
Fe, per un totale di 4 interazioni Fe-S. Il metallo ha numero di coordinazione
5 e stato di ossidazione formale +2.
Due leganti dttc nel piano basale ed un legante nitrosilico nella posizione
apicale completano una coordinazione piramidale-quadrata intorno allo ione
metallico centrale. La struttura dell’analogo derivato [Fe(S2CNR2)2(NO)] (R =
CH3) è mostrata in Figura 71, in cui NO è in coordinazione lineare (FeNO =
174°).
Nella sintesi il legante monoanionico viene fornito direttamente come sale
sodico, Na(S2CNR2), mentre il legante nitrosilico viene generato in situ da
nitrito sodico in ambiente acido.
Nella sintesi del complesso si utilizza una reazione redox con
contemporanea coordinazione di leganti, Figura 72:
4 [Fe(H2O)6]2+ + 6 NO2- + 4 H+
4 [Fe(H2O)5(NO)]2+ + 2 NO3- + 6 H2O
[Fe(H2O)5(NO)]2+ + 2 (S2CNR2)-
[Fe(S2CNR2)2(NO)] + 5 H2O
Per caratterizzare il composto, di colore verde, possiamo:
- registrare uno spettro I.R. (
C=S =
N=O (lineare)
= 1687 cm-1,
C=N
= 1505 cm-1,
1022 cm-1),
- misurare la suscettività magnetica (
eff.=1,73
BM).
Per il conteggio elettronico il legante dttc può essere considerato anionico,
donatore di 4e-, oppure neutro, donatore di 3e-, mentre il legante nitrosilico
può essere considerato cationico, donatore di 2e-, oppure neutro, donatore di
3e-. Il numero di elettroni di valenza del complesso è 17e-:
51
Leganti ionici, metallo positivo Fe
2+
6
(d )
2+
1 x 6 e- (Fe ) + 2 x 4 e- (dttc) + 3 e- (NO) = 17 e0
8
Leganti neutri, metallo neutro Fe (d )
0
1 x 8 e- (Fe ) + 2 x 3 e- (dttc) + 3 e- (NO) = 17 e-
COMPORTAMENTO MAGNETICO DEL FERRO(II)
Il ferro, la cui configurazione elettronica allo stato fondamentale è
[Ar]3d64s2, forma lo ione Fe2+ che, sempre allo stato gassoso, ha la
configurazione elettronica [Ar]3d6.
Secondo la teoria del campo cristallino, nella coordinazione piramidale
quadrata intorno allo ione Fe2+, 3 orbitali d con componente z si stabilizzano,
mentre gli altri 2 orbitali d si destabilizzano.
La configurazione d6 per un composto di coordinazione con geometria
piramidale quadrata presenta una configurazione elettronica a basso spin
(campo forte, 0e- spaiati), Figura 73.
Mediante spettroscopia ESR (electron spin resonance), è stato possibile
osservare che l’elettrone spaiato si trova prevalentemente sul NO e non sul
Fe, e questo porta a considerare il metallo come Fe(II) e non come Fe(I)
(Figura 74).
52
COMPLESSI NITROSILICI
Sono complessi contenenti almeno un legante NO.
Il monossido di azoto libero possiede 11e- di valenza (1e- spaiato in un
orbitale
*
2p)(O.L.=
2,5). La perdita formale di tale elettrone porta allo ione
nitrosonio [NO]+ (O.L.= 3), isoelettronico con CO (Figura 75)
NO
[NO]+ + e-
Il legante NO, coordinato al metallo tramite l’atomo di N, si può disporre in
due modi:
a) lineare
b) piegato.
Quando si coordina in modo lineare, l’interazione di legame, ai fini del
conteggio elettronico del complesso, può essere vista in due modi diversi
(Figura 76):
come legante neutro, NO, donatore di 3e- (1e- da
*
2p
+ 2e- da
2p)
(in tal
caso M non varia il suo stato di ossidazione formale, M0),
come legante ionico, [NO]+, donatore di 2e- (da
preso su di sé 1e- da
*
2p,
2p)
(in tal caso M ha già
diminuendo di un’unità il suo stato di
ossidazione formale, M M- ).
es. Fe S2CN(C2H5)2 2(NO),
bis(N,N’-dietilditiocarbammato)nitrosilferro(II)
Quando si coordina in modo piegato (MNO = 120-140°), l’interazione di
legame può essere vista in due modi diversi:
come legante neutro, NO, donatore di 1e- (da
*
2p)
(in tal caso M non varia
il suo stato di ossidazione formale, M0)
come legante ionico, [NO]-, donatore di 2e- (da
dato 1e- a
*
2p,
*
2p)
(in tal caso M ha già
aumentando di un’unità il suo stato di ossidazione formale,
M M+ ).
es.
[IrCl(CO)(NO)(PPh3)2]+,
carbonilcloronitrosilbis(trifenilfosfina)iridio(II)
53
Per dedurre il modo di coordinazione (lineare o piegato), prima dell’indagine
strutturale, si possono utilizzare sia dati 15N NMR sia dati IR (
N-O lineare
>
N-O
piegato).
Con metalli in basso stato di ossidazione formale, l’interazione M-NO
comporta, oltre alla donazione M NO, anche una retrodonazione M NO,
quest’ultima per rimuovere l’accumulo di elettroni sul metallo,
es.
Co(CO)3(NO)
Fe(CO)2(NO)2
Mn(CO)(NO)3
Cr(NO)4
54